

Abstract
Many proteins can form amyloid-like fibrils in vitro, but only about 30 amyloids are linked to disease, whereas some proteins form physiological amyloid-like assemblies. This raises questions of how the formation of toxic protein species during amyloidogenesis is prevented or contained in vivo. Intrinsic chaperoning or regulatory factors can control the aggregation in different protein systems, thereby preventing unwanted aggregation and enabling the biological use of amyloidogenic proteins. The molecular actions of these chaperones and regulators provide clues to the prevention of amyloid disease, as well as to the harnessing of amyloidogenic proteins in medicine and biotechnology.
Keywords: molecular chaperone, pH regulation, prion, protein domain, protein structure, BRICHOS, lung surfactant, spider silk protein
Introduction
Amyloid fibers are assemblies in which proteins or peptides adopt a β-strand conformation and assemble into elongated fibrils (1). This conversion of soluble proteins is self-propagating, as the presence of a small amount of proteins in amyloid conformation can trigger further refolding of natively folded proteins. The resulting fibrillar structures are hallmarks of severe disorders, such as Alzheimer disease, prion diseases, and diabetes mellitus (2).
Although amyloidogenic proteins natively adopt widely different folds, their aggregated states all share the use of stacked β-sheets as the principal component. Many pathogenic amyloids are composed of short peptides or small proteins and encompass paired β-strands linked by a turn, a structure that is shared by functional assemblies such as the bacterial curli fibers (1, 3). The HET-s prion, implicated in non-genetic inheritance of yeast, forms a β-solenoid with its β-strands arranged in a triangular shape (4). Electron microscopy additionally suggests that yeast prion fibers are capable of forming higher-order structures linked by organizational elements (5). Spider silk, which shares key characteristics with amyloid fibers (6, 7), is composed of β-crystalline blocks connected by unstructured segments, an architecture that gives rise to the exceptional properties of silk (8–10).
The common structural principles of amyloids also convey their double nature as “good” or “bad” assemblies; self-propagation and high stability are essential for functional amyloids, for example those that facilitate the rapid deactivation and storage of transcription factors in yeast or provide scaffolding for reaction intermediates (1). On the other hand, runaway aggregation of highly resistant protein assemblies poses serious threats to cellular function, as evident from protein aggregation diseases. Amyloid and/or intermediates occurring during fiber assembly are cytotoxic, and several underlying mechanisms may apply. Toxic properties appear to be inherent with amyloid formation process independent of the proteins involved (11). Proposed mechanisms include membrane damage (12) and sequestration of intracellular proteins (13). For the amyloid β-peptide (Aβ)4 associated with Alzheimer disease, many studies indicate that pre-fibrillar intermediates present during the aggregation trigger neuronal dysfunction, rather than the fibrils per se (14). However, mature fibrils can also exert more potent toxic effects than pre-fibrillar forms of Aβ in certain experimental systems (15).
Several molecular chaperones, and even non-chaperone proteins (16), have been implicated in the defenses against amyloid (Table 1). This minireview focuses on two chaperones, the BRICHOS domain and CsgC, which appear to have evolved to protect specific amyloidogenic clients, and on regulatory domains that control time and place of formation of functional amyloids.
TABLE 1.
Proteins with anti-amyloid chaperoning activity
| Chaperoning protein | Client protein | Reference |
|---|---|---|
| proSP-C BRICHOS | SP-C, Medin, Aβ40, Aβ42 | 37 and 81 |
| Bri2 BRICHOS | Aβ40, Aβ42 | 37 and 60 |
| Gastrokine-1 BRICHOS | Aβ40 | 36 |
| CsgC | CsgA, CsgB, α-synuclein | 57 |
| Hsp104 | Aβ42, Sup35 | 59 and 82 |
| Hsp70 | Aβ42, α-synuclein | 83 and 84 |
| Hsp90 | Aβ42 | 83 |
| Hsp27 | Aβ40 | 85 |
| DnaJ B6 | Aβ42 | 86 |
| Prefoldin | Aβ42 | 87 |
| αB-crystallin | Aβ40, Aβ42 | 88 |
| Clusterin | Aβ42, calcitonin, lysozyme, α-synuclein, β2-microglobulin, κ-casein | 89 |
| Haptoglobulin α2-macroglobulin | Aβ42, calcitonin, lysozyme | 90 |
| Lysozyme | Aβ40 | 91 |
| Pyruvate kinase | Aβ40 | 16 |
| Catalase | Aβ40 | 16 |
| β-Lactoglobulin | Aβ40 | 16 |
| α-Lactalbumin | Aβ40 | 16 |
| Albumin | Aβ40 | 16 |
Amyloidogenic Sequences Guarded by Dedicated Chaperone Domains
It has been proposed that the ability to form amyloid is a general property of the protein backbone (17). However, some sequences are significantly more aggregation-prone than others (18, 19). As a result, proteins are required to safeguard such segments in their folded state to prevent unwanted aggregation (20). However, some short amyloidogenic peptides lack the ability to conceal their aggregation hot spots due to lack of stable secondary structures and/or a three-dimensional fold. An example is lung surfactant protein C (SP-C), a 35-residue transmembrane peptide that contributes to the control of the surface tension of the alveolar air/liquid interface by affecting the phospholipid bilayer to adopt monolayer transitions (21).
The transmembrane region of mature SP-C has an α-helical conformation in the membrane (22). However, the SP-C transmembrane segment is composed essentially of polyvaline and therefore strongly favors a β-strand conformation. As a consequence, the SP-C helix is unable to refold into the helical structure once it has unfolded, and then instead aggregates (23). The insoluble, aggregated form of SP-C contains a β-sheet structure and shows abundant amyloid-like fibrils (24). In sharp contrast to the native poly-Val structure, a poly-Leu analogue of SP-C has a high helix propensity (19) and results in a dramatic increase in the spontaneous formation of a stable α-helix with no signs of amyloid fibril formation (25). A fully synthetic lung surfactant preparation based on this analogue is efficient in animal models of respiratory disease (26–29). This shows that sequence-dependent amyloidogenic properties can be overcome by protein engineering. It also raises the question of how nature has solved the problem of folding the highly β-prone transmembrane segment of SP-C into an α-helix.
BRICHOS: A Multi-purpose Anti-amyloid Chaperone
In vivo, SP-C is synthesized as a proprotein (proSP-C) that includes a BRICHOS domain. BRICHOS domains were first identified in multiple sequence alignments of the BRI proteins that are related to British and Danish dementia, as well as chondromodulin and proSP-C (30). Subsequently, BRICHOS-containing proteins have been identified in 10 protein families with relations to a range of human diseases such as lung fibrosis and cancer (31, 32). The proSP-C BRICHOS domain displays anti-amyloidogenic chaperone activity, and mutations in this domain lead to the accumulation of SP-C amyloid (33). The proSP-C BRICHOS domain hence may be a chaperone tailored to interact with a particularly amyloidogenic sequence (34, 35). Other BRICHOS homologues possess similar chaperone activity (36, 37). Most importantly, the BRICHOS-containing Bri2 protein (also known as ITM2B), which is expressed in the central nervous system, is closely linked to protein aggregation diseases. Mutations in the C-terminal part of Bri2 give rise to familial Danish and British dementias (38, 39), whereas its BRICHOS domain is associated with amyloid plaques in patients with Alzheimer disease (40). Moreover, Bri2 suppresses Aβ deposition, suggesting that the levels and/or activity of Bri2 can play a role in Alzheimer disease (41). Bri2 is believed to be a physiological inhibitor of Aβ precursor protein processing, probably by masking the cleavage sites for the processing enzymes. Consequently, it has been suggested that the loss of wild-type Bri2 affects the levels of Aβ precursor protein metabolites, causing similar pathobiology in familial Danish dementia and Alzheimer disease (43).
ProSP-C BRICHOS lacks strict sequence specificity (44) and is also able to inhibit amyloid formation of other peptides and proteins, including Aβ (37). Expression of Aβ42 in the brain of transgenic Drosophila flies decreases longevity and impairs locomotor activity, correlated with Aβ aggregation and deposition in the brain (45). Crossing Aβ42 transgenic flies with transgenic flies overexpressing the BRICHOS domain from proSP-C alleviates the lifespan deficit and improves the locomotor activity (46). Aβ42 and BRICHOS were found to co-localize in the brain, which can explain the slower Aβ42 aggregation. Together with in vitro data showing that the BRICHOS domain binds to the surface of Aβ fibrils and blocks secondary nucleation (47) (see below), this strongly suggests that the BRICHOS domain constitutes a direct example of a proprotein domain with the specific ability in vitro and in vivo to prevent toxicity from amyloid formation.
Chaperones in the Regulation of Functional Amyloids
Although constant safekeeping can be sufficient to control amyloidogenic proteins in a cellular environment, the requirements for functional amyloids are different. Here, aggregation has to be initiated in response to external cues, meaning that any chaperoning or regulatory factors present must be tunable. At the same time, the potentially toxic effects of these assemblies have to be kept at a minimum. This can be achieved via dedicated chaperone proteins that can prevent or promote the aggregation of their targets, as exemplified by the chaperone network that controls the generation of prion protomers, fibers, and seeds in yeast. Because soluble and aggregated yeast prions represent transcription factors in their active and inactive states, respectively, their distribution during cell division results in non-genetic inheritance (48). Prion assembly is regulated by parts of the proteostasis network responsible for protein folding and quality control in general (49). Its chief component is heat shock protein (Hsp)104, an ATP-driven disaggregase with the unique ability to dissociate amyloid fibrils (50). Hsp104 is able to break down prion fibrils to produce aggregate “seeds” that accelerate the conversion of soluble prions into fibrils. Interestingly, it has recently been reported that Hsp104 and the co-chaperone Sis1 exhibit differential interactions with their amyloid system, depending on which parts of the amyloidogenic sequences are accessible in different fibril morphologies (51, 52). This illustrates how chaperones can be adapted to fulfill specific amyloid-related functions. However, yeast disaggregases are not specific for amyloids alone and still retain a role in the protein folding and quality control machinery of the organism.
A different strategy involving a dedicated anti-amyloid chaperone can be found in enteric bacteria that utilize protein components under the control of the curli operons to produce functional amyloid fibers (53). The resulting curli fibers are assembled extracellularly and contribute to biofilm production for surface colonization. The main component of the fibers, CsgA, is secreted into the extracellular space in an unstructured state, where the membrane-associated CsgB acts as aggregation template (54). CsgA and CsgB each have five stacked β-strand-turn-β-strand motifs formed by imperfect repeats. In the moreamyloidogenic CsgA, aggregation propensities of the repeats are fine-tuned by gatekeeper residues that reduce self-assembly (55). Recently, the CsgC protein was identified as a chaperone that prevents the intracellular aggregation of unsecreted CsgA. Its production is correlated with the other components of the Csg system (56), and it likely functions as a specific anti-amyloid chaperone in the production of curli fibers. Interestingly, CsgC was found to inhibit amyloid formation not only of CsgA, but also the Parkinson-associated α-synuclein, while not affecting the aggregation of the highly amyloidogenic Aβ peptide (57). In contrast, for the BRICHOS domain, it appears to be the other way around; it efficiently reduces Aβ aggregation and toxicity (see above), whereas it only marginally inhibits α-synuclein aggregation.5
Mechanisms of Anti-amyloid Chaperones
Because a significant part of the proteome possesses the ability to form amyloid fibers (20), the general protein folding and quality control systems, supplemented with client-specific chaperones, are likely to constitute the first line of defense against amyloid formation (58). Hsp104 and its co-chaperones, in addition to their specific role in prion regulation, also work as an all-purpose system to remove protein aggregates, and to act on intermediates at all stages of aggregation (59).
However, there are instances where a highly targeted chaperoning approach is essential, as illustrated by the non-native conformational preference of SP-C (33). Additionally, functional amyloids require regulated partitioning between soluble and assembled states to avoid precocious aggregation. A comparison of the amyloid-specific chaperones BRICHOS and CsgC provides clues to how they exert anti-amyloid activities and prevent toxic effects (Fig. 1). Both are closely associated with their targets; proSP-C BRICHOS is synthesized together with SP-C as part of the same proprotein (30), and CsgC is co-expressed together with CsgA and CsgB under the control of the csgBAC operon (56).
FIGURE 1.

Architectures of proSP-C and the bacterial curli system. Left side, ProSP-C is shown with its transmembrane SP-C part in red. The proSP-C BRICHOS domain, located in the endoplasmic reticulum lumen, prevents the aggregation of the amyloidogenic SP-C segment. Hydrophobic residues in the putative client-binding face A of the central β-sheet and its opposing helix 1 are highlighted in red. Right side, for the curli system, the amyloidogenic segments in CsgA and the Gln residues in CsgC are highlighted in green. CsgC keeps CsgA in a soluble state for export through the secretion channel CsgG. In the extracellular space, CsgB nucleates the assembly of CsgA into curli fibers.
Mode of Action of the BRICHOS Domain
Peptide binding studies have revealed that proSP-CBRICHOS lacks strict sequence dependence, but preferentially recognizes hydrophobic amino acid residues with high β-strand propensity. The related BRICHOS domain from Bri2, in contrast, preferentially binds Tyr and charged residues (44, 60, 61). Hydrophobic, β-prone residues are commonly found in amyloid-forming segments (19), and consequently, the ability of proSP-C BRICHOS to bind SP-C conveys an anti-amyloid activity that goes beyond SP-C (44). Analysis of the high-resolution structure of proSP-C BRICHOS shows a unique fold composed of a five-stranded β-sheet flanked by α-helical segments on each side (33). This fold is likely shared by other BRICHOS domains, although low sequence identities make it difficult to model the unknown structures (32). In proSP-C BRICHOS, the interface between one of the β-sheet faces and α-helix 1 is lined with hydrophobic residues (Fig. 1, left side). The SP-C segment in an unfolded or β-hairpin conformation could be embedded in the resulting hydrophobic groove (33). BRICHOS can thereby prevent SP-C aggregation by reducing the entropic cost of acquiring the polyvaline helical fold and by preventing protein-protein contacts that lead to amyloid formation (62). Structural modeling and sequence comparisons suggest that the features of the β-sheet/helix 1 interface differ between BRICHOS homologues and that this design may be the basis of their client preference (32).
Sub-stoichiometric amounts of recombinant BRICHOS domains of human proSP-C and Bri2 prevent fibril formation of both Aβ40 and Aβ42 in vitro (37). Recently, it was shown that proSP-C BRICHOS specifically inhibits the secondary nucleation of Aβ42 aggregation (47). BRICHOS binds to the fibril surface and blocks the sites, where otherwise the secondary nucleation takes place, thus leading to significantly less oligomer formation. By preventing the major source of oligomers, BRICHOS slows the exponential growth of the fibril and reduces the toxicity associated with Aβ aggregation. Indeed, using hippocampal slice models, proSP-C BRICHOS was shown to interfere with Aβ fibril formation through suppression of the generation of toxic aggregates (47).
Insights into the Molecular Mechanism of the CsgC Chaperone
The molecular action of the curli chaperone CsgC is not yet clearly established, but its features provide some clues. The CsgC structure is homologous to the N-terminal region of the redox protein DsbD that forms a β-sandwich with a hydrophobic core (63). It is possible that CsgC utilizes the same mechanism as most chaperones by displaying hydrophobic segments to interact with the unfolded substrate. However, it was not found to interact with Aβ, which harbors hydrophobic and aromatic residues with high β-strand propensity in its amyloid core region (64). Instead, the preferred target sequence for CsgC was identified as a DQWXGKNSE motif located at the end of repeat 3 of CsgA. However, the glutamine- and asparagine-rich segments in other repeats as well as in CsgB may also be recognized (57). CsgC contains a high number of glutamine residues evenly distributed on its surface (Fig. 1, right side). Because the glutamine-rich repeats in CsgA act as zippers that bind the β-strands together during aggregation, it is tempting to speculate that the glutamine-containing segments exposed by CsgC may bind partially unfolded CsgA oligomers, in this manner blocking self-assembly (3, 57).
Spider Silk Utilizes Alternative Strategies to Control Amyloid Assembly
The presence of specialized chaperones to control assembly is not common for functional amyloids. Instead, the role of a regulatory system can be fulfilled by conformational switches in the protein itself that prevent or promote self-assembly (65). For example, spidroins, the principal components of spider silk, undergo a rapid change from a soluble to an insoluble state. In their soluble state, they are stored in the sac of the silk gland as an extremely concentrated fluid dope, with spidroin concentrations equal to the total protein concentration of the cytosol (30–50 weight %) (66). During its travel through the duct of the gland, the dope experiences changes in pH, CO2 pressure, and salt concentration that mediate the assembly into extendable fibers with β-crystalline blocks (9, 67, 68). This process is controlled via conserved N- and C-terminal domains (NT and CT), which are unique to spidroins. NT and CT are stably folded and confer solubility under storage conditions, but through external stimuli, they can be switched to act as triggers for assembly (Fig. 2) (68–70). Protons and CO2 affect the stabilities and properties of NT and CT in different but specific ways. For the CT, decreasing the pH and interacting with CO2 contribute to further destabilization (68). The pH-dependent destabilization is likely initiated by breakage of a conserved salt bridge, liberating an amyloidogenic segment that forms amyloid-like fibrils, which in turn seed the aggregation of the repetitive segments. This is a hitherto unknown functional application of amyloid seeding (71).
FIGURE 2.
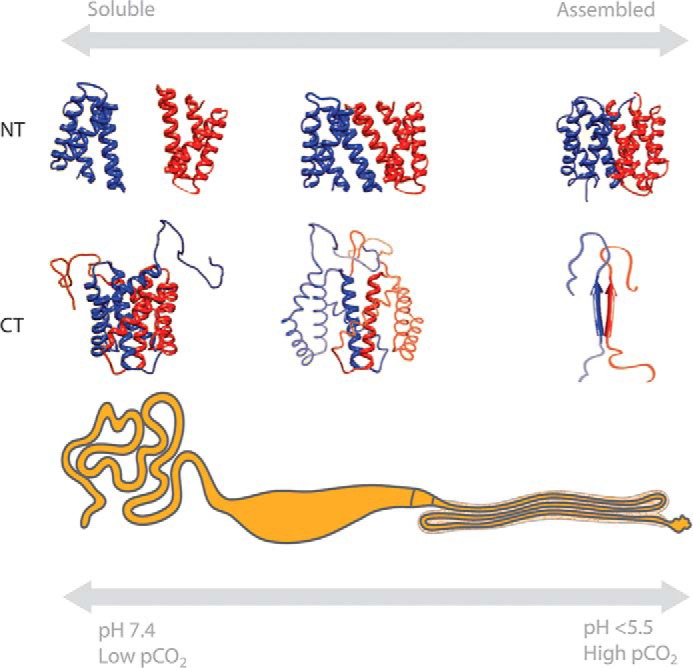
Concerted structural changes of NT and CT regulate spidroin assembly. At the bottom, a spider silk gland with its tail, central sac, and three-limbed duct is shown, below which the pH and CO2 pressures in the tail and distal duct regions are indicated. Structural changes of NT and CT between soluble and assembled states are depicted above the gland; NT acts as a lock and forms dimers that initially are flexible but become increasingly stable as pH continues to drop, and CT gets destabilized, unfolds, and forms amyloid-like fibrils that may trigger fiber formation. See text for details.
Spiders apparently have adopted the amyloid seeding phenomenon (72, 73) to control fiber formation. The structural transitions of CT into β-sheet fibrils in the duct of the silk gland result in nuclei that trigger the conversion of the repetitive regions into β-sheet polymers (68). The N-terminal domain, on the other hand, is stabilized by charge interactions at low pH and undergoes a multistep monomer-to-dimer transition that crosslinks the spidroins in the nascent fiber (68, 74). Fast polymerization kinetics are required for the ability of spiders to spin silk at >1 m/s. However, it is also vital for the spider to ensure that the polymerization process is confined to the duct and not spread up to the sac where it could prematurely aggregate the contents of the gland. Loosely associated NT dimers at the beginning of the duct provide a solution to both these problems; they ensure pre-alignment of the NTs so that the interlocking of the silk proteins in the distal parts of the duct is independent of diffusion (i.e. they associate ultra-fast) (75). At the same time, they act as a safety mechanism that keeps the pulling forces from propagating up to the gland (68, 71, 74). Through the concerted actions of NT and CT, mature silk can be generated at high speeds. However, the terminal domains of spidroins are not the only factors that confer temporal and spatial control over silk fiber formation. The spiders pull the fiber out of the silk gland, and this may promote refolding of helical/random repetitive segments into extended, β-sheet conformations. The passage of the spidroins through the narrowing duct will also cause shearing, which has been shown to contribute to the transition of CT into β-sheet nuclei (70).
Chaperones and Regulatory Domains for the Prevention of Amyloid Disease and Design of New Biomaterials
Dedicated anti-amyloid chaperones provide insights into the strategies that can be employed to prevent amyloid disease or allow the functional use of the unique amyloid fold. Common to all systems is their ability to overcome amyloid toxicity. They can confine aggregation to a specific compartment, as exemplified by the pH dependence of spider silk assembly through the well regulated pH gradient in the silk gland (68). Alternatively, control mechanisms can take the shape of chaperone components like the BRICHOS domain or CsgC that are co-produced with their amyloidogenic targets and prevent self-association and the generation of toxic, misfolded species.
It has been proposed that the same strategies may be harnessed to prevent the detrimental effects of disease-related amyloids in vivo (76). To achieve this, a chaperone should sequester aggregation from the sensitive cellular environment or modulate aggregation pathways to suppress the generation of toxic species altogether. In fact, chaperone systems have been successfully used to modulate neurodegenerative diseases by overexpression of chaperones in the affected tissue, which illustrates the power of this approach (77). The recent discovery that BRICHOS specifically blocks the secondary nucleation step in Aβ42 fibril formation (47) (see above) suggests novel ways to combat Alzheimer disease, as the addition of BRICHOS may efficiently block generation of toxic oligomers. Chaperone-based strategies are hampered by the need to deliver high amounts of functional proteins to the affected tissues or cells. To circumvent this problem, it might instead be possible to use endogenous chaperones that are already present at the site where the toxic protein species originate. The production of the BRICHOS domain from the neuronal BRI2 together with Aβ42 in transgenic mice results in no detectable cognitive decline although plaques were formed (78), suggesting that BRICHOS can block Aβ42 secondary nucleation and consequently also the formation of toxic oligomers in vivo (46).
An alternative strategy is the targeted inhibition of chaperones of the cellular quality control system. Although counterintuitive at first sight, this strategy effectively increases the proteolytic turnover of the chaperone's target proteins. It has been shown that inhibition of ATP binding to Hsp90 and Hsp70 by small molecules is able to reduce the load of aggregation-prone polyglutamine proteins. Due to the broad specificity of Heat shock proteins, however, this interference potentially affects the entire proteostasis network (79). It will be interesting to see whether similar approaches can be adapted to amyloid-specific chaperones.
Another application would be the production of amyloid-like biomaterials or aggregation-prone proteins for biomedical applications (80). In this case, inclusion of chaperones or regulatory components from functional amyloids or anti-amyloid chaperones in the recombinant production of aggregation-prone proteins may provide a cost-effective and less invasive alternative to protein engineering strategies.
This work was supported by the Swedish Research Council and by the Mobilitas Project (MTT16). This is the fifth article in the Thematic Minireview series “Protein Interactions, Structures, and Networks.” The authors declare that they have no conflicts of interest with the contents of this article.
J. Presto and J. Johansson, unpublished data.
- Aβ
- amyloid β-peptide
- CT
- spidroin C-terminal domain
- NT
- spidroin N-terminal domain
- Hsp
- heat shock protein
- SP-C
- surfactant protein C.
References
- 1.Greenwald J., and Riek R. (2010) Biology of amyloid: structure, function, and regulation. Structure 18, 1244–1260 [DOI] [PubMed] [Google Scholar]
- 2.Eisenberg D., and Jucker M. (2012) The amyloid state of proteins in human diseases. Cell 148, 1188–1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tian P., Boomsma W., Wang Y., Otzen D. E., Jensen M. H., and Lindorff-Larsen K. (2015) Structure of a functional amyloid protein subunit computed using sequence variation. J. Am. Chem. Soc. 137, 22–25 [DOI] [PubMed] [Google Scholar]
- 4.Wasmer C., Lange A., Van Melckebeke H., Siemer A. B., Riek R., and Meier B. H. (2008) Amyloid fibrils of the HET-s(218–289) prion form a β solenoid with a triangular hydrophobic core. Science 319, 1523–1526 [DOI] [PubMed] [Google Scholar]
- 5.Saibil H. R., Seybert A., Habermann A., Winkler J., Eltsov M., Perkovic M., Castaño-Diez D., Scheffer M. P., Haselmann U., Chlanda P., Lindquist S., Tyedmers J., and Frangakis A. S. (2012) Heritable yeast prions have a highly organized three-dimensional architecture with interfiber structures. Proc. Natl. Acad. Sci. U.S.A. 109, 14906–14911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Slotta U., Hess S., Spiess K., Stromer T., Serpell L., and Scheibel T. (2007) Spider silk and amyloid fibrils: a structural comparison. Macromol. Biosci. 7, 183–188 [DOI] [PubMed] [Google Scholar]
- 7.Kenney J. M., Knight D., Wise M. J., and Vollrath F. (2002) Amyloidogenic nature of spider silk. Eur. J. Biochem. 269, 4159–4163 [DOI] [PubMed] [Google Scholar]
- 8.Xiao S., Xiao S., and Gräter F. (2013) Dissecting the structural determinants for the difference in mechanical stability of silk and amyloid β-sheet stacks. Phys. Chem. Chem. Phys. 15, 8765–8771 [DOI] [PubMed] [Google Scholar]
- 9.Vollrath F., and Knight D. P. (2001) Liquid crystalline spinning of spider silk. Nature 410, 541–548 [DOI] [PubMed] [Google Scholar]
- 10.Knowles T. P., Fitzpatrick A. W., Meehan S., Mott H. R., Vendruscolo M., Dobson C. M., and Welland M. E. (2007) Role of intermolecular forces in defining material properties of protein nanofibrils. Science 318, 1900–1903 [DOI] [PubMed] [Google Scholar]
- 11.Bucciantini M., Giannoni E., Chiti F., Baroni F., Formigli L., Zurdo J., Taddei N., Ramponi G., Dobson C. M., and Stefani M. (2002) Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 416, 507–511 [DOI] [PubMed] [Google Scholar]
- 12.Quist A., Doudevski I., Lin H., Azimova R., Ng D., Frangione B., Kagan B., Ghiso J., and Lal R. (2005) Amyloid ion channels: a common structural link for protein-misfolding disease. Proc. Natl. Acad. Sci. U.S.A. 102, 10427–10432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Olzscha H., Schermann S. M., Woerner A. C., Pinkert S., Hecht M. H., Tartaglia G. G., Vendruscolo M., Hayer-Hartl M., Hartl F. U., and Vabulas R. M. (2011) Amyloid-like aggregates sequester numerous metastable proteins with essential cellular functions. Cell 144, 67–78 [DOI] [PubMed] [Google Scholar]
- 14.Hardy J., and Selkoe D. J. (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297, 353–356 [DOI] [PubMed] [Google Scholar]
- 15.Kurudenkandy F. R., Zilberter M., Biverstål H., Presto J., Honcharenko D., Strömberg R., Johansson J., Winblad B., and Fisahn A. (2014) Amyloid-β-induced action potential desynchronization and degradation of hippocampal γ oscillations is prevented by interference with peptide conformation change and aggregation. J. Neurosci. 34, 11416–11425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luo J., Wärmländer S. K., Gräslund A., and Abrahams J. P. (2014) Non-chaperone proteins can inhibit aggregation and cytotoxicity of Alzheimer amyloid β peptide. J. Biol. Chem. 289, 27766–27775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fändrich M., Fletcher M. A., and Dobson C. M. (2001) Amyloid fibrils from muscle myoglobin. Nature 410, 165–166 [DOI] [PubMed] [Google Scholar]
- 18.Sawaya M. R., Sambashivan S., Nelson R., Ivanova M. I., Sievers S. A., Apostol M. I., Thompson M. J., Balbirnie M., Wiltzius J. J., McFarlane H. T., Madsen A. Ø., Riekel C., and Eisenberg D. (2007) Atomic structures of amyloid cross-β spines reveal varied steric zippers. Nature 447, 453–457 [DOI] [PubMed] [Google Scholar]
- 19.Johansson J., Nerelius C., Willander H., and Presto J. (2010) Conformational preferences of non-polar amino acid residues: an additional factor in amyloid formation. Biochem. Biophys. Res. Commun. 402, 515–518 [DOI] [PubMed] [Google Scholar]
- 20.Goldschmidt L., Teng P. K., Riek R., and Eisenberg D. (2010) Identifying the amylome, proteins capable of forming amyloid-like fibrils. Proc. Natl. Acad. Sci. U.S.A. 107, 3487–3492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Parra E., and Pérez-Gil J. (2015) Composition, structure and mechanical properties define performance of pulmonary surfactant membranes and films. Chem. Phys. Lipids 185, 153–175 [DOI] [PubMed] [Google Scholar]
- 22.Johansson J., Szyperski T., Curstedt T., and Wüthrich K. (1994) The NMR structure of the pulmonary surfactant-associated polypeptide SP-C in an apolar solvent contains a valyl-rich α-helix. Biochemistry 33, 6015–6023 [DOI] [PubMed] [Google Scholar]
- 23.Szyperski T., Vandenbussche G., Curstedt T., Ruysschaert J. M., Wüthrich K., and Johansson J. (1998) Pulmonary surfactant-associated polypeptide C in a mixed organic solvent transforms from a monomeric α-helical state into insoluble β-sheet aggregates. Protein Sci. 7, 2533–2540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gustafsson M., Thyberg J., Näslund J., Eliasson E., and Johansson J. (1999) Amyloid fibril formation by pulmonary surfactant protein C. FEBS Lett. 464, 138–142 [DOI] [PubMed] [Google Scholar]
- 25.Kallberg Y., Gustafsson M., Persson B., Thyberg J., and Johansson J. (2001) Prediction of amyloid fibril-forming proteins. J. Biol. Chem. 276, 12945–12950 [DOI] [PubMed] [Google Scholar]
- 26.Johansson J., Some M., Linderholm B. M., Almlén A., Curstedt T., and Robertson B. (2003) A synthetic surfactant based on a poly-Leu SP-C analog and phospholipids: effects on tidal volumes and lung gas volumes in ventilated immature newborn rabbits. J. Appl. Physiol. 95, 2055–2063 [DOI] [PubMed] [Google Scholar]
- 27.Seehase M., Collins J. J., Kuypers E., Jellema R. K., Ophelders D. R., Ospina O. L., Perez-Gil J., Bianco F., Garzia R., Razzetti R., and Kramer B. W. (2012) New surfactant with SP-B and C analogs gives survival benefit after inactivation in preterm lambs. PLoS ONE 7, e47631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sato A., and Ikegami M. (2012) SP-B and SP-C containing new synthetic surfactant for treatment of extremely immature lamb lung. PLoS ONE 7, e39392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Salvesen B., Curstedt T., Mollnes T. E., and Saugstad O. D. (2014) Effects of natural versus synthetic surfactant with SP-B and SP-c analogs in a porcine model of meconium aspiration syndrome. Neonatology 105, 128–135 [DOI] [PubMed] [Google Scholar]
- 30.Sánchez-Pulido L., Devos D., and Valencia A. (2002) BRICHOS: a conserved domain in proteins associated with dementia, respiratory distress and cancer. Trends Biochem. Sci. 27, 329–332 [DOI] [PubMed] [Google Scholar]
- 31.Hedlund J., Johansson J., and Persson B. (2009) BRICHOS: a superfamily of multidomain proteins with diverse functions. BMC Res. Notes 2, 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Knight S. D., Presto J., Linse S., and Johansson J. (2013) The BRICHOS domain, amyloid fibril formation, and their relationship. Biochemistry 52, 7523–7531 [DOI] [PubMed] [Google Scholar]
- 33.Willander H., Askarieh G., Landreh M., Westermark P., Nordling K., Keränen H., Hermansson E., Hamvas A., Nogee L. M., Bergman T., Saenz A., Casals C., Åqvistg J., Jörnvall H., Berglund H., Presto J., Knight S. D., and Johansson J. (2012) High-resolution structure of a BRICHOS domain and its implications for anti-amyloid chaperone activity on lung surfactant protein C. Proc. Natl. Acad. Sci. U.S.A. 109, 2325–2329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Willander H., Hermansson E., Johansson J., and Presto J. (2011) BRICHOS domain associated with lung fibrosis, dementia and cancer: a chaperone that prevents amyloid fibril formation? FEBS J. 278, 3893–3904 [DOI] [PubMed] [Google Scholar]
- 35.Nerelius C., Fitzen M., and Johansson J. (2010) Amino acid sequence determinants and molecular chaperones in amyloid fibril formation. Biochem. Biophys. Res. Commun. 396, 2–6 [DOI] [PubMed] [Google Scholar]
- 36.Altieri F., Di Stadio C. S., Severino V., Sandomenico A., Minopoli G., Miselli G., Di Maro A., Ruvo M., Chambery A., Quagliariello V., Masullo M., Rippa E., and Arcari P. (2014) Anti-amyloidogenic property of human gastrokine 1. Biochimie 106, 91–100 [DOI] [PubMed] [Google Scholar]
- 37.Willander H., Presto J., Askarieh G., Biverstål H., Frohm B., Knight S. D., Johansson J., and Linse S. (2012) BRICHOS domains efficiently delay fibrillation of amyloid β-peptide. J. Biol. Chem. 287, 31608–31617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vidal R., Frangione B., Rostagno A., Mead S., Révész T., Plant G., and Ghiso J. (1999) A stop-codon mutation in the BRI gene associated with familial British dementia. Nature 399, 776–781 [DOI] [PubMed] [Google Scholar]
- 39.Vidal R., Revesz T., Rostagno A., Kim E., Holton J. L., Bek T., Bojsen-Møller M., Braendgaard H., Plant G., Ghiso J., and Frangione B. (2000) A decamer duplication in the 3′ region of the BRI gene originates an amyloid peptide that is associated with dementia in a Danish kindred. Proc. Natl. Acad. Sci. U.S.A. 97, 4920–4925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Del Campo M., Hoozemans J. J., Dekkers L. L., Rozemuller A. J., Korth C., Müller-Schiffmann A., Scheltens P., Blankenstein M. A., Jimenez C. R., Veerhuis R., and Teunissen C. E. (2014) BRI2-BRICHOS is increased in human amyloid plaques in early stages of Alzheimer's disease. Neurobiol. Aging 35, 1596–1604 [DOI] [PubMed] [Google Scholar]
- 41.Kim J., Miller V. M., Levites Y., West K. J., Zwizinski C. W., Moore B. D., Troendle F. J., Bann M., Verbeeck C., Price R. W., Smithson L., Sonoda L., Wagg K., Rangachari V., Zou F., Younkin S. G., Graff-Radford N., Dickson D., Rosenberry T., and Golde T. E. (2008) BRI2 (ITM2b) inhibits Aβ deposition in vivo. J. Neurosci. 28, 6030–6036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Deleted in proof
- 43.Matsuda S., Giliberto L., Matsuda Y., McGowan E. M., and D'Adamio L. (2008) BRI2 inhibits amyloid β-peptide precursor protein processing by interfering with the docking of secretases to the substrate. J. Neurosci. 28, 8668–8676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Johansson H., Nerelius C., Nordling K., and Johansson J. (2009) Preventing amyloid formation by catching unfolded transmembrane segments. J. Mol. Biol. 389, 227–229 [DOI] [PubMed] [Google Scholar]
- 45.Luheshi L. M., Tartaglia G. G., Brorsson A. C., Pawar A. P., Watson I. E., Chiti F., Vendruscolo M., Lomas D. A., Dobson C. M., and Crowther D. C. (2007) Systematic in vivo analysis of the intrinsic determinants of amyloid β pathogenicity. PLoS. Biol. 5, e290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hermansson E., Schultz S., Crowther D., Linse S., Winblad B., Westermark G., Johansson J., and Presto J. (2014) The chaperone domain BRICHOS prevents CNS toxicity of amyloid-β peptide in Drosophila melanogaster. Dis. Model Mech. 7, 659–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cohen S. I., Arosio P., Presto J., Kurudenkandy F. R., Biverstål H., Dolfe L., Dunning C., Yang X., Frohm B., Vendruscolo M., Johansson J., Dobson C. M., Fisahn A., Knowles T. P., and Linse S. (2015) A molecular chaperone breaks the catalytic cycle that generates toxic Aβ oligomers. Nat. Struct. Mol. Biol. 22, 207–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Uptain S. M., and Lindquist S. (2002) Prions as protein-based genetic elements. Annu. Rev. Microbiol. 56, 703–741 [DOI] [PubMed] [Google Scholar]
- 49.Doyle S. M., Genest O., and Wickner S. (2013) Protein rescue from aggregates by powerful molecular chaperone machines. Nat. Rev. Mol. Cell Biol. 14, 617–629 [DOI] [PubMed] [Google Scholar]
- 50.True H. L. (2006) The battle of the fold: chaperones take on prions. Trends Genet. 22, 110–117 [DOI] [PubMed] [Google Scholar]
- 51.Frederick K. K., Debelouchina G. T., Kayatekin C., Dorminy T., Jacavone A. C., Griffin R. G., and Lindquist S. (2014) Distinct prion strains are defined by amyloid core structure and chaperone binding site dynamics. Chem. Biol. 21, 295–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stein K. C., and True H. L. (2014) Extensive diversity of prion strains is defined by differential chaperone interactions and distinct amyloidogenic regions. PLoS Genet. 10, e1004337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chapman M. R., Robinson L. S., Pinkner J. S., Roth R., Heuser J., Hammar M., Normark S., and Hultgren S. J. (2002) Role of Escherichia coli curli operons in directing amyloid fiber formation. Science 295, 851–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Evans M. L., and Chapman M. R. (2014) Curli biogenesis: order out of disorder. Biochim. Biophys. Acta 1843, 1551–1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang X., Zhou Y., Ren J. J., Hammer N. D., and Chapman M. R. (2010) Gatekeeper residues in the major curlin subunit modulate bacterial amyloid fiber biogenesis. Proc. Natl. Acad. Sci. U.S.A. 107, 163–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hammar M., Arnqvist A., Bian Z., Olsén A., and Normark S. (1995) Expression of two csg operons is required for production of fibronectin- and congo red-binding curli polymers in Escherichia coli K-12. Mol. Microbiol. 18, 661–670 [DOI] [PubMed] [Google Scholar]
- 57.Evans M. L., Chorell E., Taylor J. D., Åden J., Götheson A., Li F., Koch M., Sefer L., Matthews S. J., Wittung-Stafshede P., Almqvist F., and Chapman M. R. (2015) The bacterial curli system possesses a potent and selective inhibitor of amyloid formation. Mol. Cell 57, 445–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hartl F. U., Bracher A., and Hayer-Hartl M. (2011) Molecular chaperones in protein folding and proteostasis. Nature 475, 324–332 [DOI] [PubMed] [Google Scholar]
- 59.Arimon M., Grimminger V., Sanz F., and Lashuel H. A. (2008) Hsp104 targets multiple intermediates on the amyloid pathway and suppresses the seeding capacity of Aβ fibrils and protofibrils. J. Mol. Biol. 384, 1157–1173 [DOI] [PubMed] [Google Scholar]
- 60.Peng S., Fitzen M., Jörnvall H., and Johansson J. (2010) The extracellular domain of Bri2 (ITM2B) binds the ABri peptide (1–23) and amyloid β-peptide (Aβ1–40): implications for Bri2 effects on processing of amyloid precursor protein and Aβ aggregation. Biochem. Biophys. Res. Commun. 393, 356–361 [DOI] [PubMed] [Google Scholar]
- 61.Fitzen M., Alvelius G., Nordling K., Jörnvall H., Bergman T., and Johansson J. (2009) Peptide-binding specificity of the prosurfactant protein C Brichos domain analyzed by electrospray ionization mass spectrometry. Rapid Commun. Mass Spectrom. 23, 3591–3598 [DOI] [PubMed] [Google Scholar]
- 62.Johansson H., Eriksson M., Nordling K., Presto J., and Johansson J. (2009) The Brichos domain of prosurfactant protein C can hold and fold a transmembrane segment. Protein Sci. 18, 1175–1182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Taylor J. D., Zhou Y., Salgado P. S., Patwardhan A., McGuffie M., Pape T., Grabe G., Ashman E., Constable S. C., Simpson P. J., Lee W. C., Cota E., Chapman M. R., and Matthews S. J. (2011) Atomic resolution insights into curli fiber biogenesis. Structure 19, 1307–1316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lührs T., Ritter C., Adrian M., Riek-Loher D., Bohrmann B., Döbeli H., Schubert D., and Riek R. (2005) 3D structure of Alzheimer's amyloid-β(1–42) fibrils. Proc. Natl. Acad. Sci. U.S.A. 102, 17342–17347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Landreh M., Johansson J., Rising A., Presto J., and Jörnvall H. (2012) Control of amyloid assembly by autoregulation. Biochem. J. 447, 185–192 [DOI] [PubMed] [Google Scholar]
- 66.Hijirida D. H., Do K. G., Michal C., Wong S., Zax D., and Jelinski L. W. (1996) 13C NMR of Nephila clavipes major ampullate silk gland. Biophys. J. 71, 3442–3447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dicko C., Vollrath F., and Kenney J. M. (2004) Spider silk protein refolding is controlled by changing pH. Biomacromolecules 5, 704–710 [DOI] [PubMed] [Google Scholar]
- 68.Andersson M., Chen G., Otikovs M., Landreh M., Nordling K., Kronqvist N., Westermark P., Jörnvall H., Knight S., Ridderstråle Y., Holm L., Meng Q., Jaudzems K., Chesler M., Johansson J., and Rising A. (2014) Carbonic anhydrase generates CO2 and H+ that drive spider silk formation via opposite effects on the terminal domains. PLoS. Biol. 12, e1001921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Askarieh G., Hedhammar M., Nordling K., Saenz A., Casals C., Rising A., Johansson J., and Knight S. D. (2010) Self-assembly of spider silk proteins is controlled by a pH-sensitive relay. Nature 465, 236–238 [DOI] [PubMed] [Google Scholar]
- 70.Hagn F., Eisoldt L., Hardy J. G., Vendrely C., Coles M., Scheibel T., and Kessler H. (2010) A conserved spider silk domain acts as a molecular switch that controls fibre assembly. Nature 465, 239–242 [DOI] [PubMed] [Google Scholar]
- 71.Rising A., and Johansson J. (2015) Toward spinning artificial silk. Nat. Chem. Biol. 11, 309–315 [DOI] [PubMed] [Google Scholar]
- 72.Lundmark K., Westermark G. T., Nyström S., Murphy C. L., Solomon A., and Westermark P. (2002) Transmissibility of systemic amyloidosis by a prion-like mechanism. Proc. Natl. Acad. Sci. U.S.A. 99, 6979–6984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jarrett J. T., and Lansbury P. T. Jr. (1992) Amyloid fibril formation requires a chemically discriminating nucleation event: studies of an amyloidogenic sequence from the bacterial protein OsmB. Biochemistry 31, 12345–12352 [DOI] [PubMed] [Google Scholar]
- 74.Kronqvist N., Otikovs M., Chmyrov V., Chen G., Andersson M., Nordling K., Landreh M., Sarr M., Jörnvall H., Wennmalm S., Widengren J., Meng Q., Rising A., Otzen D., Knight S. D., Jaudzems K., and Johansson J. (2014) Sequential pH-driven dimerization and stabilization of the N-terminal domain enables rapid spider silk formation. Nat. Commun. 5, 3254. [DOI] [PubMed] [Google Scholar]
- 75.Schwarze S., Zwettler F. U., Johnson C. M., and Neuweiler H. (2013) The N-terminal domains of spider silk proteins assemble ultrafast and protected from charge screening. Nat. Commun. 4, 2815. [DOI] [PubMed] [Google Scholar]
- 76.Muchowski P. J., and Wacker J. L. (2005) Modulation of neurodegeneration by molecular chaperones. Nat. Rev. Neurosci. 6, 11–22 [DOI] [PubMed] [Google Scholar]
- 77.Auluck P. K., Chan H. Y., Trojanowski J. Q., Lee V. M., and Bonini N. M. (2002) Chaperone suppression of α-synuclein toxicity in a Drosophila model for Parkinson's disease. Science 295, 865–868 [DOI] [PubMed] [Google Scholar]
- 78.Kim J., Chakrabarty P., Hanna A., March A., Dickson D. W., Borchelt D. R., Golde T., and Janus C. (2013) Normal cognition in transgenic BRI2-Aβ mice. Mol. Neurodegener. 8, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.van der Putten H., and Lotz G. P. (2013) Opportunities and challenges for molecular chaperone modulation to treat protein-conformational brain diseases. Neurotherapeutics 10, 416–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rising A. (2014) Controlled assembly: a prerequisite for the use of recombinant spider silk in regenerative medicine? Acta Biomater. 10, 1627–1631 [DOI] [PubMed] [Google Scholar]
- 81.Nerelius C., Gustafsson M., Nordling K., Larsson A., and Johansson J. (2009) Anti-amyloid activity of the C-terminal domain of proSP-C against amyloid β-peptide and medin. Biochemistry 48, 3778–3786 [DOI] [PubMed] [Google Scholar]
- 82.Shorter J., and Lindquist S. (2004) Hsp104 catalyzes formation and elimination of self-replicating Sup35 prion conformers. Science 304, 1793–1797 [DOI] [PubMed] [Google Scholar]
- 83.Evans C. G., Wisén S., and Gestwicki J. E. (2006) Heat shock proteins 70 and 90 inhibit early stages of amyloid β-(1–42) aggregation in vitro. J. Biol. Chem. 281, 33182–33191 [DOI] [PubMed] [Google Scholar]
- 84.Dedmon M. M., Christodoulou J., Wilson M. R., and Dobson C. M. (2005) Heat shock protein 70 inhibits α-synuclein fibril formation via preferential binding to prefibrillar species. J. Biol. Chem. 280, 14733–14740 [DOI] [PubMed] [Google Scholar]
- 85.Wilhelmus M. M., Boelens W. C., Otte-Höller I., Kamps B., de Waal R. M., and Verbeek M. M. (2006) Small heat shock proteins inhibit amyloid-β protein aggregation and cerebrovascular amyloid-β protein toxicity. Brain Res. 1089, 67–78 [DOI] [PubMed] [Google Scholar]
- 86.Månsson C., Arosio P., Hussein R., Kampinga H. H., Hashem R. M., Boelens W. C., Dobson C. M., Knowles T. P., Linse S., and Emanuelsson C. (2014) Interaction of the molecular chaperone DNAJB6 with growing amyloid-β 42 (Aβ42) aggregates leads to sub-stoichiometric inhibition of amyloid formation. J. Biol. Chem. 289, 31066–31076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sörgjerd K. M., Zako T., Sakono M., Stirling P. C., Leroux M. R., Saito T., Nilsson P., Sekimoto M., Saido T. C., and Maeda M. (2013) Human prefoldin inhibits amyloid-β (Aβ) fibrillation and contributes to formation of nontoxic Aβ aggregates. Biochemistry 52, 3532–3542 [DOI] [PubMed] [Google Scholar]
- 88.Raman B., Ban T., Sakai M., Pasta S. Y., Ramakrishna T., Naiki H., Goto Y., and Rao Ch. M. (2005) αB-crystallin, a small heat-shock protein, prevents the amyloid fibril growth of an amyloid β-peptide and β2-microglobulin. Biochem. J. 392, 573–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yerbury J. J., Poon S., Meehan S., Thompson B., Kumita J. R., Dobson C. M., and Wilson M. R. (2007) The extracellular chaperone clusterin influences amyloid formation and toxicity by interacting with prefibrillar structures. FASEB J. 21, 2312–2322 [DOI] [PubMed] [Google Scholar]
- 90.Yerbury J. J., Kumita J. R., Meehan S., Dobson C. M., and Wilson M. R. (2009) α2-Macroglobulin and haptoglobin suppress amyloid formation by interacting with prefibrillar protein species. J. Biol. Chem. 284, 4246–4254 [DOI] [PubMed] [Google Scholar]
- 91.Luo J., Wärmländer S. K., Gräslund A., and Abrahams J. P. (2013) Human lysozyme inhibits the in vitro aggregation of Aβ peptides, which in vivo are associated with Alzheimer's disease. Chem. Commun. (Camb.) 49, 6507–6509, 10.1039/c3cc42325e [DOI] [PubMed] [Google Scholar]