

Background: A new method using comprehensive mutagenesis libraries, yeast display, and deep sequencing is proposed to determine fine conformational epitopes for three antibody-antigen interactions.
Results: For three separate antigens, the experimentally determined conformational epitope is consistent with orthogonal experimental datasets.
Conclusion: We conclude that this new methodology is reliable and sound.
Significance: With this new method, four antibody-antigen interactions can be mapped per day.
Keywords: antibody, antibody engineering, Bordetella pertussis, epitope mapping, protein-protein interaction, tumor necrosis factor (TNF), TROP2, conformational epitope mapping
Abstract
Knowledge of the fine location of neutralizing and non-neutralizing epitopes on human pathogens affords a better understanding of the structural basis of antibody efficacy, which will expedite rational design of vaccines, prophylactics, and therapeutics. However, full utilization of the wealth of information from single cell techniques and antibody repertoire sequencing awaits the development of a high throughput, inexpensive method to map the conformational epitopes for antibody-antigen interactions. Here we show such an approach that combines comprehensive mutagenesis, cell surface display, and DNA deep sequencing. We develop analytical equations to identify epitope positions and show the method effectiveness by mapping the fine epitope for different antibodies targeting TNF, pertussis toxin, and the cancer target TROP2. In all three cases, the experimentally determined conformational epitope was consistent with previous experimental datasets, confirming the reliability of the experimental pipeline. Once the comprehensive library is generated, fine conformational epitope maps can be prepared at a rate of four per day.
Introduction
Pinpointing the fine conformational epitope targeted by a given antibody affords a better understanding of the structural basis of its mechanism of protection, which provides an intellectual property basis and can lead to improved prophylactic or therapeutic interventions against human diseases (1–9). Recent technical advances allow an unprecedented look at the adaptive immune response to an immunogen (10–12). For example, single cell isolation methods coupled to deep sequencing have revealed the identification of thousands of patient-specific paired antibody heavy and light chain sequences elicited in response to infection or vaccination, and such information has begun to be used in antibody discovery. Whereas functional or neutralization assays can be used to determine the efficacy of individual members in these repertoires, a full utilization of this wealth of information awaits the development of a high throughput method of determining conformational epitopes targeted by these antibodies (13).
Existing methods either do not identify conformational epitopes (14, 15) or are labor-intensive and costly. Co-crystallization provides unambiguous epitope identification but can require considerable effort and generation of many antigen variants to identify one that is compatible with crystallization (2). Mass spectrometry-based methods utilizing hydrogen/deuterium exchange identify epitopes to a ∼5-amino acid resolution only under rigorous control experiments that limit throughput (16, 17). Competing display-based methods use many sorts (18), identify only partial epitopes (19, 20), or are limited by restricting mutations to alanine (21).
Recently, yeast surface display (22) coupled to deep mutational scanning (23) was used to understand the sequence effects of binding for nearly every single point mutant for two computationally designed proteins targeting a conserved epitope on influenza hemagglutinin (7). This method was used to confirm the paratope for both small proteins, as validated by crystal structures. More recently, other approaches using yeast display and deep sequencing for the purposes of conformational epitope mapping have been demonstrated (18, 20). However, current implementations require several sorting steps that severely hinder throughput. Because additional inefficiencies exist at several stages in the deep sequencing and analysis workflow, we asked whether we could simplify the yeast display-deep sequencing pipeline to increase the method throughput, reduce cost, and improve the ability to resolve complete conformational epitopes for full-length proteins.
Experimental Procedures
Strains
Escherichia coli strains used in this study: XL1-Blue (Agilent, Santa Clara, CA) recA1 endA1 gyrA96 thi-1 hsdR17 supE44 relA1 lac [F′ proAB lacI1qZΔM15 Tn10 (Tetr)]; BL21* (Life Technologies) fhuA2 [lon] ompT gal [dcm] ΔhsdS); BL21(DE3) (New England BioLabs, Ipswich, MA) fhuA2 [lon] ompT gal (λ DE3) [dcm] ΔhsdS λ DE3 = λ sBamHIo ΔEcoRI-B int::(lacI::PlacUV5::T7 gene1) i21 Δnin5. Yeast strain used in this study: EBY100 (American Type Culture Collection, Manassas, VA) MATa AGA1::GAL1-AGA1::URA3 ura3–52 trp1 leu2-delta200 his3-delta200 pep4::HIS3 prb11.6R can1 GAL.
Plasmids
The plasmid pETCON_TNF was created by inserting a codon optimized gene encoding the Gly-57–Leu-233 extracellular portion of tumor necrosis factor (TNF) (GenScript, Piscataway, NJ) into the pETCON plasmid using flanking NdeI/XhoI restriction sites. The plasmid pETCON_PTx-S1-2202 was created by amplifying PTxS1-220 from pAK400_PTx-S1–200K (24) and inserting into pETCON at NdeI/XhoI sites. To create pETCOn_TROP2Ex, DNA was isolated from HeLa cells with the GenElute Mammalian Genomic DNA Miniprep kit (Sigma). This DNA was used as a template for amplification of the ectodomain of Trop2 encompassing residues 27–274, which was then inserted into pETCON at Nde/XhoI sites. Polypeptide sequences of the variable regions for the heavy and light chains of infliximab were obtained (25) and used to generate codon optimized DNA sequences (GenScript). A (Gly4-Ser)3 linker was placed between the C-terminal residues of the heavy and N-terminal residue of the light chains. The plasmid pET29b_inflix_scfv was prepared by inserting the inflix_scFv gene into the pET-29b(+) vector (EMD BioSciences, Billerica, MA) using NdeI/XhoI restriction sites. m7e6 heavy and light chains were subcloned into 293-6E expression vector pTT5 were custom-ordered from GenScript. Plasmids for the yeast display constructs have been deposited in the AddGene plasmid repository.
Preparation of inflix_scFv
pET29_inflix_scFv was transformed into chemically competent BL21*(DE3). Cultures were grown to an A600 of 0.8 and were then induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside and incubated with shaking at 18 °C for ∼18 h. Inflix-scFv was isolated from inclusion bodies and refolded using existing protocols (26, 27). After the refolding procedure the sample was centrifuged at 17,000 × g to remove precipitated protein. The concentration was determined using the Bradford method using BSA as a protein standard and was biotinylated at a molar ratio of 1:20 protein:biotin using the EZ link NHS-biotin kit following the manufacturer's instructions (Life Technologies).
Preparation of Trop2 and Ptx Subunit 1 (PTxS1) Fabs
PTxS1 antibody hu1B7 was prepared according to previous reports.3 Anti-TROP2 monoclonal antibody m7E6 (29) was produced in 293-6E cells and purified by protein A column by GenScript. Fabs were produced using the Pierce Fab Preparation kit (Life Technologies). Concentrations were determined using A280 with the recommended estimated extinction coefficient (1 mg/ml) of 1.4 and was biotinylated at a molar ratio of 1:20 protein:biotin using the EZ link NHS-biotin kit following the manufacturer's instructions (Life Technologies).
Dissociation Constant Determination
Equilibrium dissociation constants (KD) were determined using clonal population yeast display titrations according to Chao et al. (22). Fab concentrations between 50 pm and 1 μm were tested.
Yeast Display Sorts
1 × 107 cells were grown in 2 ml of synthetic dextrose plus casein amino acids (SDCAA) for 6 h at 30 °C and re-inoculated at A600 = 1.0 in 2 ml of synthetic galactose plus casein amino acids (SGCAA) at 20 °C for 18 h. 3 × 107 cells were labeled with biotinylated Fab or scFv for 30 min at room temperature in Dulbecco's phosphate-buffered saline with 1 g/liter BSA at a concentration of half of the experimentally determined dissociation constant on the yeast surface. Cells were then secondarily labeled with anti-cmyc-FITC (Miltenyi Biotec, San Diego, CA) and streptavidin-phycoerythrin (Thermo Fisher, Waltham, MA). Sorting was done on an Influx Cell Sorter (BD Biosciences). FSC/SSC (gate 1), FSC/FITC (gate 2), and phosphatidylethanolamine/FITC (gate 3) gates were set. Three populations were collected: an unselected population satisfying gate 1, a displayed population satisfying gates 1 and 2, and a bound population satisfying all three gates. The number of cells collected for each population was at least 100-fold higher than the theoretical library complexity. Sorting statistics for each population collected are listed in supplemental Table 2. Following the sort, recovered populations were grown for 48 h in 10 ml of SDCAA (22), and 1 × 107 cells from this culture cells were stored in 1 ml of yeast storage buffer at −80 °C.
Deep Sequencing Preparation
Yeast plasmid DNA was prepared for deep sequencing following the protocol in Kowalsky et al. (30) (primers are listed in supplemental Table 3). 5 μl of the PCR products were run on a 2% agarose gel stained with SYBR-GOLD (Thermo Fisher) to ensure one band was obtained at the correct size (∼250–350 bp). Agencourt AMPure XP PCR Purification (Beckman Coulter, Beverly, MA) was used per the manufacturer's protocol to purify the PCR product. Library DNA was sequenced on an Illumina MiSeq using either the 300 × 2 or 250 × 2 Illumina MiSeq kits (Illumina, San Diego, CA) at the Michigan State University Sequencing Core.
Data Analysis
A modified version of Enrich-0.2 as described in Kowalsky et al. (30) was used to compute enrichment ratios of individual mutants from the raw Illumina sequencing files. To normalize the data across the multiple tiles we define the fitness metric for variant i (ζi) as the binary logarithm of the fluorescence of variant i to the fluorescence of the wild-type sequence (Fwt) (30),
 |
This results in the following equation.
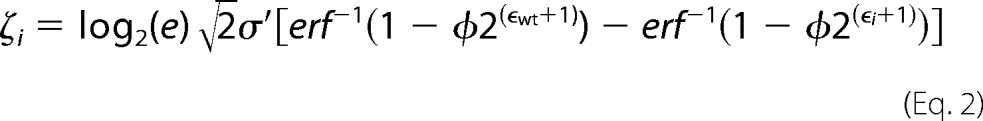 |
where φ is the percentage of cells collected, ϵi is the enrichment ratio for variant i, σ′ is the log normal standard deviation of a clonal population, and the subscript wt denotes the wild type. Custom python scripts used to calculate the fitness metric and statistics are at Github. The full deep sequencing datasets are provided at figshare. Sorting parameters needed for each tile normalization are listed in Supplemental Table 1.
Shannon (sequence) entropy values at a given position j in the protein sequence (Ej) were calculated by
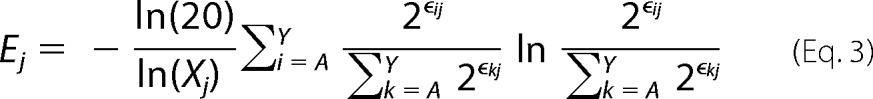 |
where ϵij is the enrichment ratio of substitution i at position j, and Xj is the number of mutants with adequate sequencing counts in the unselected population at position j. The derivation for Equation 3 is shown under “Theory.” We excluded residues with an Xj < 12 from analysis. Enrichment ratios for stop codons were not included in the Shannon entropy analysis.
Soluble Expression of PTxS1
BL21(DE3) cells containing pAK400_ PTx-S1-220K (24) with an added C-terminal lysine residue were grown in terrific broth at 25 °C to an A600 of 1.5, then induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside for 5 h. Cell pellets were collected, and the outer membrane was lysed by osmotic shock. Lysate was purified by immobilized metal affinity chromatography followed by size exclusion chromatography in PBS (S75, AKTA FPLC) as previously reported (24).Yields were between 0.2 and 0.6 mg/liter. 3 μg of pertussis toxin (PTx; 26.1 KDa) or PTx-S1-220K (25.9 KDa) were run on a 12% polyacrylamide gel at 130 V and stained with GelCode blue.
PTxS1 ELISA Binding Assay
A high binding 96-well plate (Costar, Corning, NY) was coated with either 4 nm PTx or 4 nm PTx-S1-220K in PBS and incubated overnight at 4 °C. The plate was blocked with a solution of 5% nonfat dry milk in PBS with 0.05% Tween (PBSTM) for 1 h at room temperature. hu1B7 was serially diluted across the plate in duplicate with a starting concentration of 5 μg/ml in PBSTM and incubated for 1 h at room temp. Secondary antibody was GαhFc-HRP prepared at 1:2500 in PBSTM and incubated for 1 h at room temp. The plate was developed with tetramethylbenzidine and quenched with HCl, and absorbance was read on a plate reader at 450 nm.
PTxS1 Western Blot
0.3 μg of PTx or PTx-S1-220K were run on a 12% poly acrylamide gel at 130 V and transferred to a PVDF membrane. The membrane was blocked for 1 h at room temperature with PBSTM, then incubated for 1 h at room temperature with 1 μg/ml hu1B7A in PBSTM. The secondary antibody was GαhFc-HRP prepared at 1:10,000 in PBSTM and incubated for 1 h at room temperature followed by washing and development using SuperSignal West Pico Chemiluminescent Substrate (Pierce) and a 30-s exposure time to x-ray film.
Cell Culture and Transfection
MCF10A human breast epithelial cell lines were obtained from Dr. Kathleen Gallo at Michigan State University. MCF10A cells were cultured in Dulbecco's modified Eagle's media (DMEM)/F-12(1:1) (Life Technologies) with 5% horse serum, 10 μg/ml insulin, 20 ng/ml EGF, 100 ng/ml cholera toxin, 0.5 μg/ml hydrocortisone, and penicillin/streptomycin (Invitrogen). MDA-MB-231 human breast cancer cell lines were obtained from the ATCC and cultured in DMEM with 10% fetal bovine serum (FBS), 2 mm glutamine, and penicillin/streptomycin (Invitrogen) as described (31, 32). Cells were maintained at 37 °C and 5% CO2 environment.
SiGENOMESMARTpool ON-TARGETplusTROP2siRNA and scramblesiRNA were purchased from Thermo Scientific (Asheville, NC) and Ambion (Grand Island, NY). The SMARTpoolsiRNAs and the transfection reagent Lipofectamine were diluted with Opti-MEM (Invitrogen) as described (33, 34). The diluted SMARTpoolsiRNAs were mixed with RNAiMAX to form siRNA-RNAiMAX complexes. The cell culture medium was replaced with antibiotic-free medium containing the siRNA-RNAiMAX complexes at a final concentration of 10 nm siRNA. Media were changed after 12 h, and the cells were incubated in fresh media.
TROP2 Western Blot Analysis
The protein concentrations of the cell extracts were measured by the Bradford method. Protein samples of 15–30 μg were subjected to Western blot analysis as previously described (34, 35) using TROP2 antibody (Abcam, Cambridge, MA), β-actin (Sigma), anti-mouse, and anti-rabbit HRP-conjugated secondary antibodies (Thermo Scientific) and donkey anti-goat IgG-HRP (Santa Cruz Biotechnology, Dallas, TX). The blots were visualized by SuperSignal West Femto maximum sensitivity substrate (Thermo Scientific).
Total mRNA Extraction and Quantitative Real Time PCR
Total mRNA from cells was extracted using the RNeasy Plus kit (QiaGen, Valencia, CA) according to the manufacturer's instructions. The total mRNA was reverse-transcribed into cDNA using the cDNA synthesis kit (Bio-Rad) as previously described (36, 37). The primer sets (Operon, Huntsville, AL) used for PCR:human actin were 5′-tggacttcgagcaagagatg-3′ and 5′-aggaaggaaggctggaagag-3′ and for human TROP2were 5′-gagattcccccgaagttctc-3′ and 5′-aactcccccagttccttgat-3′. Quantitative real-time PCR was performed using iQSYBR Green Supermix and the real-time PCR detection system (Bio-Rad). The cycle threshold values were determined by the MyIQ software (Bio-Rad).
Transwell Migration and Invasion Assays
Chemotactic migration or invasion was quantified using a Boyden chamber transwell assay (8-μm pore size; Corning Costar, Cambridge, MA) with either uncoated or Matrigel-coated filters, respectively. Cells were deprived of serum overnight, trypsinized, and introduced into the upper chamber. Mitomycin C (Sigma) was added to the cultured media. The chemoattractant in the lower chamber was medium-supplemented with 5% FBS. After 8 h of incubation at 37 °C, the cells were fixed and stained. Migrated cells in five randomly chosen fields were counted. The experiments were performed in triplicate wells, and each experiment was performed three times as indicated.
Wounding-healing Assay
MDA-MB-231 cells were grown to confluence. The growth medium was replaced with fresh medium containing 5% FBS and supplemented with mitomycin C (1 mg/ml) (Sigma), and the monolayer of cells was subsequently scratched using a 200-μl pipette tip. Wound width was monitored over time by microscopy.
Cytotoxicity and Proliferation Assays
Cytotoxicity and proliferation were assessed using lactate dehydrogenase and Alamar Blue microplate assays, respectively. Cells were seeded in 96-well plates and treated with either PBS (control) or various concentrations of m7EG IgG or Fab for 48 h. Cytotoxicity was detected using the lactate dehydrogenase cytotoxicity detection kit (Roche Applied Science) after the manufacturer's protocol. Proliferation was determined by AN Alamar Blue assay (Pierce) following the manufacturer's protocol.
Confocal Microscopy
MDA-MB-231 cells were seeded in glass-bottom 24-well plates (In Vitro Scientific, Sunnyvale, CA) and treated with either PBS (control), m7E6 IgG, or Fab. After treatment, cells were washed 2× with ice-cold PBS, fixed with 3.7% formaldehyde for 15 min at 37 °C, and washed 3× with PBS. Next, the samples were incubated in blocking buffer (1% BSA, 0.5% Triton X-100 in PBS) for 1 h at 37 °C. The cells were incubated with TROP2 intracellular domain-specific primary antibodies (EMD Millipore #ABC425, 1:500 dilution) in incubation buffer (0.5% BSA, 0.5% Triton X-100) overnight at 4 °C. The samples were then washed 3× with PBS and incubated in the respective secondary antibodies (AlexaFluor 488) diluted in PBS for 1 h at 37 °C in the dark. Cells were washed 2× with PBS and incubated in nuclear counter stain Hoechst 3342 (Invitrogen) for 10 min at room temperature. After the final incubation cells were washed twice with PBS and covered with anti-fade solution for imaging. Images were recorded with an Olympus FluoView 1000 Inverted IX81 microscope using a 10× or 60× oil objective using identical exposure and photomultiplier tube settings for each primary antibody-fluorophore pair across the different treatment conditions.
Statistical Analysis
All experiments were performed at least three times. Representative results are shown as the mean ± S.D. Statistical analysis were performed using unpaired, two-tail Student's t tests. * indicates p < 0.05, ** indicates p < 0.01, and *** indicates p < 0.001.
Theory
Derivation of Sequence Entropy Metric and Calculation of Estimated Errors
At any given position j in a protein sequence, Shannon entropy for that position (Ej) is defined as,
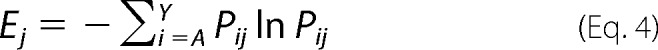 |
Here, Pij is the probability of finding amino acid i at position j after sorting given an equal representation of all 20 amino acids in the starting population. To determine pij, we first define pij for a single substitution at position j using the frequency of mutant i in the initial (fo,ij) and final (ff,ij) populations,
 |
This can be written in terms of an enrichment ratio of a given substitution i at position j (ϵij) such that
pij gives the probability of a variant with a mutation i at position j passing through the sort. Because the summation of these probabilities will not necessarily sum to unity, we can normalize the probabilities over a single residue such that
 |
Combining with the definition of Shannon Entropy in Equation 1,
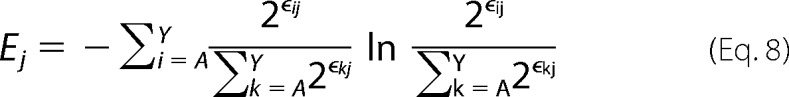 |
Because some positions do not have adequate sequencing counts for all 20 amino acids, the sequence entropy metric must be normalized by the maximum possible Shannon entropy,
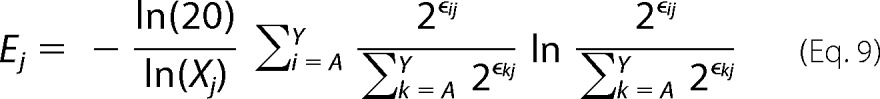 |
where Xj is the number of mutants with adequate sequencing counts in the unselected population at position j. Equation 9 is the final form of the sequence entropy metric used in the manuscript. In practice, we excluded residues with an Xj < 12. This removed 13/647 (2.0%) positions tested in the present work.
To estimate the expected error in Ej, we can define the variance in Ej as,
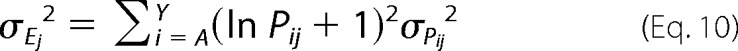 |
Similarly, the variance on Pij can be defined as,
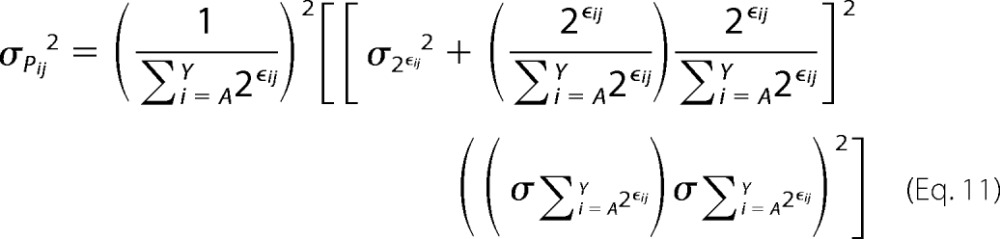 |
Because the minimal error associated with counting sequences approximates Poisson noise (7, 30), we can write the variance for the two unknowns as,
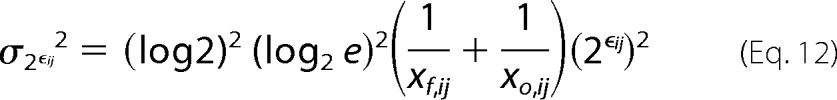 |
Here, xf,ij and xo,ij are the raw sequencing counts of mutation i at position j in the final and initial population, respectively. We can write a similar derivation for the variance of the denominator in the probability term,
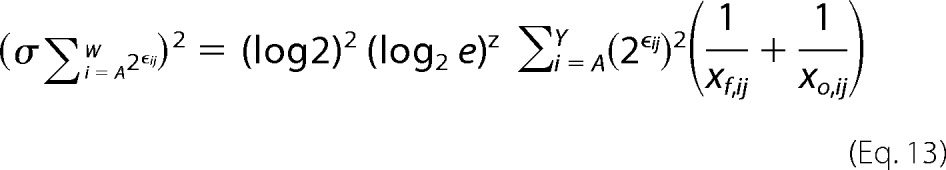 |
Accurate calculation of the variance on the sequence entropy for a given position requires the raw sequencing counts for each position in the unselected and selected populations. Using these numbers, Equations 12 and 13 can be solved, which can then be plugged into Equations 10 and 11 to solve for the unknown. The variance on sequence entropy should be maximized when the raw sequencing counts are just above the inclusion threshold in the unselected populations. Even in this case, the S.D. of sequence entropy metric is 0.02. Accordingly, we are justified in not including sequence entropy errors in the determination of the conformational epitope.
Calculation of Relative Dissociation Constants from Sequencing Counts
For a clonal population of yeast cells displaying variant i and labeled with antibody at a labeling concentration of [Lo], we can write the mean fluorescence (Fi) as,
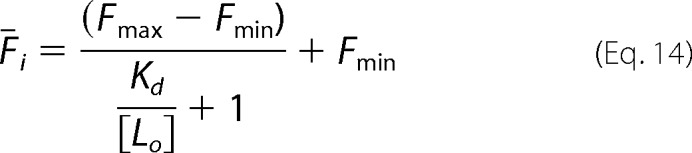 |
Here Fmax and Fmin are, respectively, the maximum and minimum average fluorescence for clone i in the fluorescence channel used for antibody binding. Using the labeling conditions [Lo] = ½Kd, wt, Equation 14 becomes
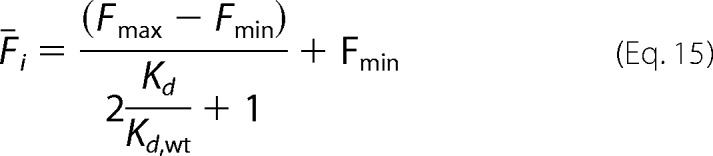 |
Similarly, the mean fluorescence for the wild-type variant can be expressed as,
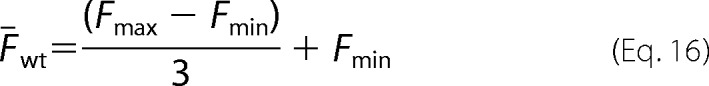 |
The ratio of mean fluorescence for a variant to wild-type can be written in terms of the fitness metric (ζi) derived from the sequencing data (30),
 |
Substitution of Equations 15 and 16 into Equation 17 leads to,
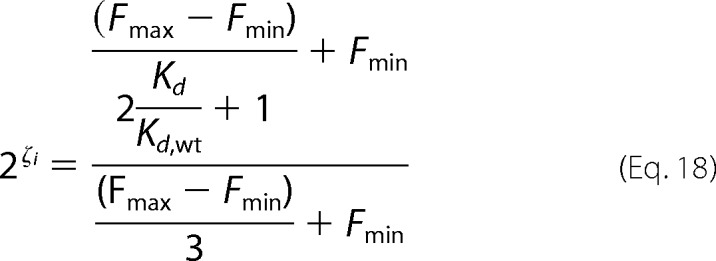 |
If we assume that Fmax ≫ Fmin, Equation 18 simplifies to,
 |
This assumption is valid for the ratio of Fmax/Fmin typically seen in yeast display. The ratio depends on multiple factors, including the sensitivity of the fluorescent detection on the cell sorter, the quantum yield of the fluorescent dye used, the biotin labeling per antibody, and the surface expression of a given antigen. In our hands we observe a range from 50 to 2000 for this ratio. Here the range of Kd values that we can see is relative to the interval of fitness metrics observed from the sequencing counts. Practically speaking, the range on the lower end of fitness metrics should be the average fitness metric for the stop codons (−1.1 to −0.75 depending on the percent collected), which gives a relative dissociation constant of ∼2.7. This highlights the sensitivity of the method for differentiating small energetic changes in binding activity (ΔΔGbinding ∼ 0.1–0.4 kcal/mol). The drawback, however, is we are unable to discriminate smaller perturbations with larger energetic changes typically associated with interface “hot spots” (ΔΔGbinding ∼ 1–2 kcal/mol).
Another important consideration is the error associated with digital counting of variants from deep sequencing data, as this will also introduce error on both the fitness metric and corresponding relative dissociation constants for variants with low numbers of counts in the unselected population. We have previously shown that digital counting of variants from deep sequencing error using our methods result in minimal error associated with Poisson noise (30).
The variance on the fitness metric can be defined as,
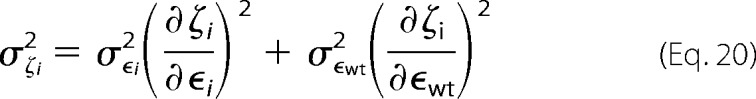 |
where ϵi is the enrichment ratio for variant i. The variance for ϵi (σϵi2) can be estimated from Poisson noise as
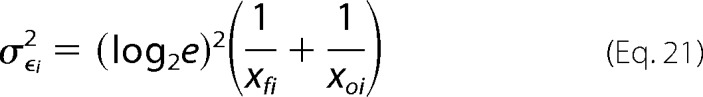 |
where xoi and xfi are the number of counts in the unselected and selected populations respectively. For variants with many counts the error approaches zero, highlighting the importance of sequencing depth of coverage in these experiments. The fitness metric is defined as (30),
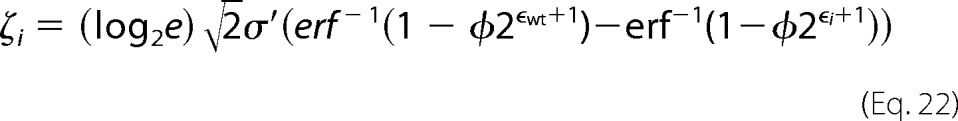 |
where φ is the percentage of cells collected from the respective gate(s), σ′ is the log normal standard deviations of a clonal population, and the subscript wt denotes the wild type.
Combining these we can estimate the variance of the fitness metric as,
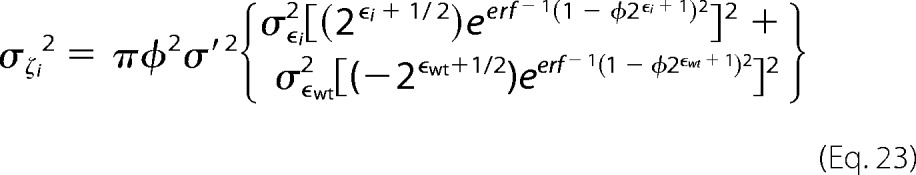 |
The error here is largest with variants with low fitness metrics and few counts in the unselected population.
The error for the relative dissociation constant is defined as,
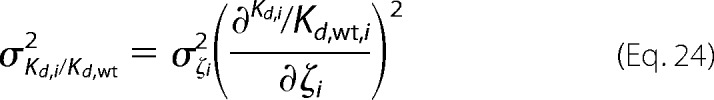 |
which can be written as
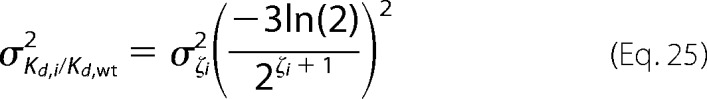 |
Results
The streamlined method is shown schematically in Fig. 1. In the first step, single-site saturation mutagenesis (SSM) libraries for 250–300-nucleotide contiguous sections of the gene of interest are prepared by PFunkel mutagenesis (39) and transformed into yeast. These yeast libraries are labeled with a biotinylated Fab or scFv and sorted once by FACS. The labeling concentration and FACS gates are set such that the capture probability of any given variant is a monotonically increasing function of its binding affinity. Three distinct populations are collected: an unselected population of cells that passed through a cell-size gate (unselected population), a displayed population of cells passing through the previous gate as well as a gate confirming display of the C-terminal c-myc epitope tag (displayed population), and a binding population of cells satisfying these two previous gates as well as a gate on the fluorescence channel associated with antibody binding (bound population). The DNA from each population is prepared and sequenced on an Illumina platform. Then the frequencies of each variant in the population are compared and merged into a single fitness metric (30) that allows direct, quantitative comparisons across different mutational libraries. Together, this approach allows for the rapid and comprehensive reconstruction of the sequence binding determinants of full-length proteins for a given antibody.
FIGURE 1.

Schematic of streamlined conformational epitope mapping process. 1, SSM libraries were made for 250–300-nucleotide contiguous sections along the gene of interest. Each library contains mutations in a different section of the gene. 2, sorting conditions were determined such that there was a higher probability of capturing stronger binding cells. 3, yeast libraries were labeled with biotinylated Fab and sorted by FACS. Three different gates are drawn: a gate on light scattering parameters SSC/FSC (top; unselected population), a gate set on FSC and fluorescence channel corresponding to display of antigen (middle; displayed population), and a binding gate that collects the top 5–10% of cells by fluorescence corresponding to channel for bound antibody (bottom; bound population). 4, DNA from each population was extracted and prepared for deep sequencing on an Illumina platform. The frequency of each variant in the bound and displayed populations is compared against the unselected population and used to calculate a fitness metric. For each residue, sequence entropy (bottom) for bound (black) and displayed (green) sorts is used to determine the degree of conservation. 5, sequence entropy is used to identify conserved and non-conserved residues that are used to determine the conformational epitope (orange).
As a first test of our approach we chose to evaluate the binding of yeast-displayed TNF (TNFα) on the monoclonal antibody infliximab (marketed by Johnson & Johnson as RemicadeTM). A structure for this complex is known (40), allowing assessment of the ability of the sequence-function method to demarcate discontinuous conformational binding epitopes. TNF is a homo-trimeric, multi-disulfide linked, marginally stable protein and thus represents a stringent test of the ability of yeast to surface-display complicated proteins. We ordered a codon-optimized gene encompassing the Gly-57–Leu-233 extracellular portion of TNF and subcloned it into the yeast display plasmid pETCON (6). Next, we created three SSM libraries of TNF and induced yeast surface expression of library variants (22). For each library we performed a single FACS sort using cells labeled with the biotinylated scFv of infliximab (inflix_scFv) at 32 nm, which is half of the observed dissociation constant for the interaction. Approximately 200,000 cells from each library were collected for the unselected, displayed, and bound populations (Supplemental Table 1). Deep sequencing was used to determine the enrichment ratios of the bound and displayed populations compared with the unselected population. These enrichment ratios were then transformed to a fitness metric that allows direct comparisons across the different mutational libraries, allowing the sequence determinants of binding to be evaluated for nearly every possible single point mutant in the protein sequence (Fig. 2a, supplemental Fig. 1). Overall, we observed 95.1% coverage of all possible single non-synonymous mutants in the extracellular TNF sequence (n = 2985/3140) (supplemental Table 2).
FIGURE 2.

TNF-infliximab conformational epitope determination. a, a subset (41/177 residues) of the fitness-metric heat map for bound population of the TNF-inflix_scFv interaction. Sequence entropy for the display (green) and bound population (black) is plotted below with their respective cut-offs (dashed lines). b, subtractive sequence entropy analysis for TNF-infliximab interaction. Conserved residues (orange) are found mainly within the binding footprint of the TNF-infliximab interaction (cyan). Non-conserved residues (purple) can also be mapped onto structure and fall outside of the footprint (middle). These non-conserved residues can be used to find regions where false positive conserved residues appear. For clarity, only one TNF monomer is shown. c, close-up view of the structural interface between TNF (ribbon) and infliximab (cyan surface). TNF residues are colored according to sequence conservation as in panel b.
To identify the conformational epitope, we reasoned that residues essential to the protein-protein interaction would be conserved in the bound population. Conversely, residues that do not participate at the protein-protein interface would be mostly non-conserved. To discriminate among these positions we introduced a positional Shannon (sequence) entropy metric that is calculated using the enrichment ratios of every variant at a given position (see “Theory”). Because sequence conservation will depend strongly on the stringency of the sorting conditions, we next asked for cutoffs to discriminate between a conserved and non-conserved position. We considered a position to be conserved if the sequence entropy in the bound population compared with the unselected population was less than or equal to the midpoint of the sequence entropy range. Even using this stringent cutoff, 56/177 TNF residues were identified as conserved. Many of these residues are buried in the core of TNF and presumably disrupt the fold of the protein. Positions located at the epitope can be partially discriminated from those that disrupt protein stability by calculating the sequence entropy of the displayed sort and using a cutoff of the midpoint of the sequence entropy range. This analysis removed 22 of the 56 residues from consideration as epitope positions. The removed residues were almost all buried, with a mean fraction solvent-accessible surface area of 0.03 (range 0.00–0.22). The 34 remaining residues were a combination of surface positions (using a fraction accessible surface area cutoff of 0.10, n = 18) and buried positions (Fig. 2b).
The conserved surface positions clustered in three regions on TNF (all subsequent residues use PDB numbering): the AB loop (Asn-19, Pro-20, Gly-24), the EF loop (Gln-102, Glu-104, Thr-105, Glu-107, Ala-111), and the GH loop (Asn-137–Tyr-141). There are also a handful of noncontiguous, partially surface-exposed positions scattered throughout (Fig. 2b). To further discriminate epitope from non-epitope positions, we reasoned that the epitope would be depleted in non-conserved positions. Identifying non-conserved positions as the upper quintile of the sequence entropy range removed 48/177 positions from consideration. Of these, only Glu-110 was within 4 Å of infliximab in the bound complex. However, the Cα-Cβ vector of Glu-110 was pointed away from the interface, and its side chain did not make significant interactions to infliximab (Fig. 2c), suggesting that its mutation to other amino acids would not disrupt the affinity of the TNF-infliximab complex.
Considering both the conserved and non-conserved positions highlights the EF loop and GH loop as essential to the interaction. The single most conserved section documented by sequence entropy analysis is on the EF loop between Asn-137–Tyr-141, and these residues mapped neatly to the center of the experimentally determined binding region (Fig. 2c). Additionally, conservation of several residues on the EF loop were consistent with the infliximab-TNF structure, including Glu-107, which made a salt bridge interaction across the interface. Furthermore, the importance of these two loops to the energetics of the interaction has been confirmed by mutagenesis (40).
Examination of non-conserved residues located at the interface identifies limitations in using a single metric for epitope determination. For example, Pro-70, Ser-71, and His-73 on the CD loop and Thr-77 in strand D are interface residues that are potentially energetically important but are above the sequence entropy cutoff (Fig. 2c). For the CD loop residues the positions were conserved, but the sequence entropy were just slightly above the cutoff. Proline at position 70 and serine 71 were the most favored amino acids, whereas a substitution of H73K was slightly favored. Thr-77 was removed from consideration of the epitope as it was relatively conserved in the displayed population. Additional epitope residues that were not identified include the above-mentioned Glu-110 and Gln-67. However, neither was expected to be energetically significant as determined by alanine scanning mutagenesis and its position in the bound complex. Indeed, mutation of Gln-67 to aromatic residue increased binding affinity for the infliximab interaction (Fig. 2a). Based on this comprehensive mutagenesis dataset enabled by deep sequencing, we conclude that this improved yeast display-deep sequencing pipeline is effective in identifying fine conformational epitopes for antibody-antigen interactions.
We next asked whether the automated identification of the epitope using sequence entropy could be used to map binding footprints of other antibody-antigen interactions. To accomplish this, we evaluated the binding of yeast-displayed PTxS1 against a single humanized neutralizing antibody. Whooping cough, a respiratory disease caused by the bacteria Bordetella pertussis, remains a major cause of infant mortality in both the developing and industrialized countries despite widespread vaccination (41). Recently, Nguyen et al.4 demonstrated the ability of a binary antibody mixture to halt whooping cough disease progression in a baboon model. Although one of the mixture antibodies, hu1B7, is able to bind to the S1 subunit on a Western blot, indicating a linear component of the epitope, previous studies using 15-mer peptides covering the entire S1 sequence were unable to identify a peptide showing binding activity against murine 1B7 (42). Further information about the epitope on S1 targeted by hu1B7, one of the mixture antibodies, will help elucidate its neutralizing mechanism.
After a similar procedure to that of TNF, we used PFunkel mutagenesis to create an SSM library of nearly all possible single point mutants in the Asp-1–Gly-220 fragment of PTxS1 (PTx-S1-220) and performed a single FACS sort collecting 400,000 cells in each of the three populations. Soluble PTx-S1-220 can be expressed in E. coli and retains affinity for hu1B7 (Fig. 3). Positional Shannon entropy was used to determine the most conserved residues at the interface using the same cutoffs identified in the TNF test case (Fig. 4a, supplemental Fig. 2). As before, epitope residues were discriminated from residues that result in disruption of the protein fold by analysis of sequence conservation in the displaying population. Altogether, this procedure identified 16 residues at the proposed antibody-antigen conformational epitope: Glu-75, Gly-78–His-83, Ile-85–Tyr-87, Ala-93, Tyr-148, Asn-150–Ile-152, and Asn-163. Mapping these residues onto the structure of the PTx (43) shows that 14 of 16 residues are located in a spatially contiguous location (Fig. 4, b and c). This proposed epitope is consistent with a previous alanine scanning dataset developed by Sutherland and Maynard (24). The two conserved residues outside of this region, Ala-93 and Asn-163, are most likely of structural importance for the conformational epitope as they are buried in the protein core. The epitope surface is typical of antibody-antigen interactions with charged and aromatic residues along with hydrophobic patches (Fig. 4c). Partial conservation of buried residues Phe-84, Gly-86, and His-149 near the identified epitope indicate that the hu1B7 binding affinity depends somewhat on the conformation in the PTxS1 folded state.
FIGURE 3.
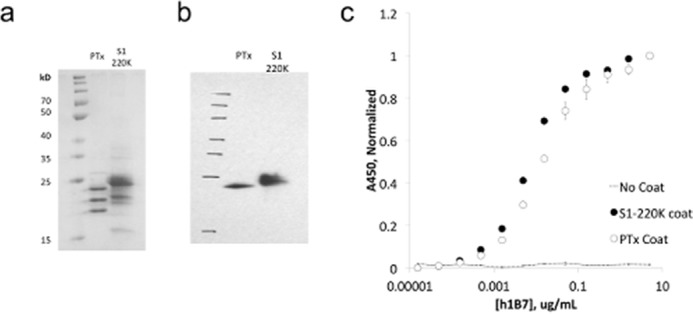
A soluble version of the pertussis toxin S1 subunit can be expressed in E. coli and retains affinity for hu1B7. Truncated S1 in a pAK400 expression vector was produced in BL21(DE3), harvested by osmotic shock, and purified by immobilized metal affinity chromatography and size exclusion. a, SDS-PAGE of truncated S1 (S1-220K, 25.9 kDa) and full-length PTx (26.1 kDa). b, Western blot of S1-220K and PTx, probed by hu1B7 and GαhFc-HRP. c, ELISA of hu1B7 on a 4 nm coat of PTx or S1-220K, detected by GαhFc-HRP.
FIGURE 4.

PTxS1 conformational epitope determination. a, a subset (29/220 residues) of the fitness-metric heat map for the PTxS1-hu1B7 interaction. Sequence entropy for the unselected/display population (green) and unselected/bound population (black) is plotted below with their respective cut-offs (dashed lines). b, subtractive sequence entropy analysis for PTxS1-hu1B7interaction. The light gray surface represents the S1 subunit, and the dark gray represents other subunits of PTx. Conserved residues (orange) are found on the S1 subunit proximal to the S5 and S6 subunits. Non-conserved residues (purple) are found over most of the solvent-accessible surface area. c, close-up view of the conserved residues at the epitope interface. PTxS1 is represented with a schematic and sticks format, whereas the other subunits are represented as the dark gray surface.
In principle, relative dissociation constants (Kd,i/Kd,WT) for antibody-antigen interactions can be calculated directly from the digital counting (21) (see “Theory”). However, our method relies on counting individual sequences after a single cell sort, resulting in a limited dynamic range. To determine whether dissociation constants calculated from deep sequencing results are quantitative, we compared our results with an alanine scanning dataset for in vitro binding of PTx-S1-220 and murine 1B7 (24). Our results are consistent with scanning data for 16 of the 17 mutations (Table 1). The one discrepancy, R39A, is not in spatial proximity of the highlighted epitope, potentially indicating different long range interactions between PTx-S1-220 and the murine and humanized 1B7 antibodies used in the separate experiments. Consistent with the limited dynamic range of the deep sequencing method, the relative dissociation constants for hot spot residues Arg-79, His-83, Tyr-148, and Asn-150 are significantly underestimated in the deep sequencing datasets compared with in vitro measurements (Table 1). This limitation caused by digital counting a handful of sequences restricts the experimentally determined range of relative dissociation constants to 0.4–2.5 (Fig. 5).
TABLE 1.
Comparison of fitness metric-based dissociation constant calculations with published experimentally determined dissociation constants
| Variant |
In vitro binding |
Deep sequencing data |
|
|---|---|---|---|
| Kd,i/Kd,wt | Kd,i/Kd,wt | Fitness metric | |
| WT | 1.00 ± 0.10 | 1.00 | 0 |
| R146A | 1.07 ± 0.10 | 1.08 ± 0.02 | −0.074 |
| E155A | 0.79 ± 0.09 | 0.97 ± 0.02 | 0.034 |
| T156A | 0.79 ± 0.09 | 1.04 ± 0.04 | −0.037 |
| T159A | 1.00 ± 0.16 | 1.06 ± 0.03 | −0.053 |
| Y161A | 0.93 ± 0.43 | 0.88 ± 0.04 | 0.116 |
| N176A | 0.79 ± 0.09 | 0.81 ± 0.03 | 0.199 |
| E210A | 1.14 ± 0.16 | 1.05 ± 0.02 | −0.052 |
| E16A | 1.14 ± 0.11 | 1.30 ± 0.08 | −0.266 |
| T81A | 1.21 ± 0.65 | 1.19 ± 0.03 | −0.168 |
| T158A | 0.93 ± 0.16 | 0.98 ± 0.03 | 0.020 |
| Y166A | 1.00 ± 0.16 | 1.10 ± 0.02 | −0.094 |
| R39A | 2.14 ± 0.32 | 0.84 ± 0.06 | 0.165 |
| T153A | 1.43 ± 0.30 | 1.48 ± 0.03 | −0.403 |
| R79A | 17.9 ± 3.1 | 2.62 ± 0.25 | −1.058 |
| H83A | 7.1 ± 1.5 | 2.14 ± 0.14 | −0.826 |
| Y148A | 20.7 ± 4.5 | 1.69 ± 0.16 | −0.546 |
| N150A | 5.7 ± 0.8 | 1.89 ± 0.08 | −0.670 |
| T81K | No bindinga | 4.25 ± 0.88 | −1.664 |
| T81H | 1.51 ± 0.43a | 2.55 ± 0.24 | −1.025 |
| I152M | 0.68 ± 0.23a | 0.67 ± 0.11 | 0.359 |
| I152P | 1.36 ± 0.46a | 2.12 ± 0.06 | −0.806 |
a Reported numbers are experimental relative EC50 values.
FIGURE 5.

Fitness metric and relative dissociation constant error. a, relative dissociation constant as a function of fitness metrics. The vertical dashed line represents the average fitness metric for stop codon positions. b, standard error as a function of fitness metric for different numbers of unselected counts (red, 10; blue, 20; green, 30; orange, 50; purple, 100; black, 500). Fitness metrics associated with lower number of counts have higher error. c, relative dissociation constant error as a function of relative dissociation constant for different numbers of unselected counts. As the relative dissociation constant increases the amount of error increases.
For further validation we tested four additional mutations identified from our deep mutational scanning datasets. PTxS1-220 variants T81K, T81H, I152M, and I152P were produced in E. coli and purified. A polyclonal anti-PTx antibody preparation was titrated against ELISA wells coated with 5 nm PTx S1 variants. Similar binding to all variants suggests that no variant has severe folding defects. Relative binding dissociation constants were calculated from observed EC50 values by titration of the hu1B7 antibody on an ELISA plate coated with 5 nm concentrations of the truncated PTx S1 or each variant (Table 1). In vitro binding for variants T81H, I152M, and I152P were quantitatively predicted by deep sequencing data. In contrast and consistent with the limited dynamic range highlighted above, the relative binding for binding knock-out variant T81K is significantly underestimated in the deep sequencing datasets. We conclude that although relative dissociation constants calculated directly from the deep sequencing data are consistent with in vitro measurements, care must be exercised when calculating a quantitative energetic contribution.
Next, we asked whether the method could be used to map the conformational epitope of an antibody targeting tumor-associated calcium signal transducer 2 (TACST2, also known as TROP2), a 323-amino acid, 36-kDa transmembrane glycoprotein. TROP2 is overexpressed in numerous human epithelial cancers (44) and identified as an oncogene in colon cancer, with metastatic and invasive abilities (36, 45). Studies have linked Trop2 to increased tumor growth, as ectopic expression of Trop2 in cancer cell lines causes them to become highly tumorigenic when implanted in mice, whereas silencing Trop2 inhibits cell proliferation in vitro (46). Furthermore, silencing Trop2 in breast cancer cell line MDA-MB-231 decreased migration as observed by transwell migration and wound-healing assays (Fig. 6, a–e).
FIGURE 6.

TROP2 expression in MDA-MB-231 cells enhances migration and invasion. a and b, MDA-MB-231 cells were transfected with scramble siRNA or siRNA targeting TROP2 for 24 h. The mRNA (a) and protein levels (b) of TROP2 were measured by real-time PCR and Western blotting. n = 3. **, p < 0.01; ***, p < 0.001 versus control. c, suppression of MDA-MB-231 migration by silencing TROP2. MDA-MB-231 cells were transfected with scramble siRNA or siRNA targeting TROP2 and then subjected to a transwell migration assay. The cells were allowed to migrate toward serum for 8 h. Triplicate wells were used for each condition in three independent experiments. ***, p < 0.001 versus control. d, silencing TROP2 reduced the migration of highly invasive breast cancer cells. MDA-MB-231 cells were transfected with scramble siRNA or TROP2 siRNA and subjected to a wound-healing assay. Representative photographs at the indicated time points from three independent experiments, with each performed in triplicate wells. Magnification, 10×. e, suppression of MDA-MB-231 invasion by silencing TROP2. MDA-MB-231 cells were transfected with scramble siRNA or siRNA targeting TROP2 and then seeded in a Matrigel-coated Boyden chamber and subjected to a transwell invasion assay. The cells were allowed to migrate toward serum for 20 h. Triplicate wells were used for each condition in three independent experiments. ***, p < 0.001 versus control.
The extracellular portion of TROP2 (TROP2Ex) contains three domains: an N-terminal domain (ND), a middle TY domain, and a C-terminal domain (CD) (Fig. 7a). Like its close paralogue EpCAM (epithelial cell adhesion molecule), TROP2 is a nuclear signal transducer activated by regulated intramembrane proteolysis (47, 48). TROP2Ex is first shed by proteolysis followed by intramembrane cleavage to release intracellular TROP2 (TROP2Ic). Because recombinant TROP2Ex forms a homodimer in solution (49), it has been speculated that destabilization of the extracellular region by local environmental changes or an agonist leads to regulated intramembrane proteolysis. However, neither the proteolytic cleavage sites nor potential agonist(s) has been unambiguously identified.
FIGURE 7.

TROP2 conformational epitope determination. a, the domains of membrane protein TROP2. The extracellular portion of TROP2 (TROP2Ex) contains an N-terminal domain (ND, green), TY domain (brick red), and C-terminal domain (CD, cyan). TM indicates the membrane spanning portion, and Trop2Ic is the intracellular domain. b, homology model of TROP2Ex homodimer shown in surface view. One subunit is colored by sequence entropy based on m7e6 binding (orange, conserved; purple, non-conserved; gray, intermediate). The other subunit is colored by domain using the same coloring scheme as in panel a. The fine epitope is located on the membrane distal face of the C-terminal domain. The center of the epitope on one subunit is separated by 4.1 nm from the epitope on the adjacent subunit. c, bar graphs showing the number of migrating cells in MDA-MB-231 cells treated with PBS (control) or m7EG IgG. d, representative bright field images of Boyden's chamber inserts showing cells that migrated across the 8-μm membrane after 24 h of treatment. e, bar graphs showing number migrating cells with control or m7E6 Fab treatment. f, representative bright field images showing migrating cells across the membrane. p values indicate significance of difference in mean (n = 3) determined using Student's two-tailed t tests.
A recently developed mAb m7E6 (mouse7E6) targeting TROP2Ex inhibited tumor growth in the A431 xenograft model, and m7E6 drug conjugates induced long term regression in the BxPC3 xenograft model (29). To investigate the structural basis of m7E6 efficacy, we prepared SSM libraries covering 95.8% of possible single non-synonymous mutations for the yeast-displayed TROP2Ex (residues Thr-28–Thr-274) (supplemental Table 2) and performed a single FACS sort against the biotinylated Fab of m7E6 at a labeling concentration of 22 nm. After the experimental workflow, the same sequence conservation analysis as above was used to determine residues contributing to the epitope. The subtractive sequence entropy measure completely removed most portions of TROP2Ex from consideration of the epitope, resulting in unambiguous determination of the binding footprint. In contrast to most previously described mAbs that bind at or near the N-terminal cysteine-rich domain, residues Asp-171, Arg-178, and the ridge on CD (RCD) loop Gly-241–Pro-250 were identified as contributing to the m7e6-TROP2 interaction (Fig. 7, a and b). This epitope were in agreement with m7e6 binding affinity results from domain swapping experiments (29). Using a homology model of TROP2 guided by the structure of the paralogue epithelial cell adhesion molecule EpCAM (38), these residues mapped to a membrane-distal region opposite to the face on the CD domain that putatively makes specific interdimer contacts with the TY loop (Fig. 7b).
There are several reasons why targeting this epitope could be effective. For example, m7e6 could partially block the agonist(s) binding site on TROP2, preventing activation. Alternatively, m7e6 could prevent destabilization of the extracellular region by sterically blocking proteolytic cleavage or by preventing dimer dissociation after proteolytic cleavage. Because 4.1-nm separates the centers of the proposed epitope between the dimer subunits (Fig. 7b), an IgG could occupy both subunits of the dimer. In this scenario we speculate that the antibody could prevent destabilization of the extracellular region or prevent an agonist site by the steric bulk of the IgG. As a test of this hypothesis, we reasoned that, because of its monovalency, m7e6 Fab would not be as effective as the corresponding IgG in preventing dimer destabilization. We performed Boyden's chamber assays to investigate the influence of m7E6 IgG and Fab on migration rates in MDA-MB-231 cells. Whereas m7e6 IgG treatment inhibited migration (p = 0.004) (Fig. 7, c and d), m7e6 Fab was unable to inhibit migration (p = 0.187) at the highest tested concentration (40 μg/ml) (Fig. 7, e and f). We confirmed that both m7e6 Fab and IgG were able to label breast cancer cell line MDA-MB-231 (Fig. 8a). Additionally, we tested the influence of m7E6 IgG on inducing proliferation (metabolic activity) and cytotoxicity in MDA-MB-231 cells. We found that the treatment did not result in a statistically significant decrease in proliferation rates (one way analysis of variance p = 0.384) or increase in cytotoxicity levels (one way analysis of variance p = 0.141), indicating that the mechanism by which m7EG IgG resulted in reduced migration rates was independent of reduced proliferation or cell death (Fig. 8, b–c). We further investigated the localization of the TROP2 intracellular domain in response to the IgG treatment using confocal microscopy. We found that nuclear expression levels of TROP2Ic were retained in both the IgG- and Fab-treated cells (Fig. 9). This scenario suggests that the effect of m7E6 binding to TROP2 on reducing migration rates may be mediated by blocking the agonist binding site or by influencing the downstream signaling cascade. Whatever the exact mechanism behind m7e6 efficacy, the conformational epitope uncovered by the present method was used to predict that m7e6 Fab could not inhibit migration.
FIGURE 8.

a, 105 MDA-MB-231 cells were labeled with 50 nm biotinylated m7e6 IgG (orange), 50 nm biotinylated m7e6 Fab (cyan), or nude (blue) in buffer PBS plus 1 g/L bovine serum albumin for 30 min at room temperature. After washing cells were secondarily labeled with streptavidin-phycoerythrin and processed by flow cytometry. b, bar graphs indicating the average (n = 3) Alamar Blue assay absorbance (570 nm) in cells treated with PBS (control) or increasing concentration of m7E6 IgG (10–40 μg/ml). c, bar graphs indicating the average (n = 3) lactate dehydrogenase absorbance (590 nm) in control and m7EG IgG-treated cells. p values indicate the significance levels of the influence of increasing concentrations of the IgG treatment on proliferation or cytotoxicity levels determined using one-way analysis of variance.
FIGURE 9.

Confocal images showing expression and localization of TROP2 intracellular domain represented by green fluorescence in MDA-MB-231 cells treated with PBS (control), m7E6 IgG, or Fab with nuclear counter-staining indicated by blue fluorescence. The individual panels were recorded at 60× magnification (scale bar = 50 μm) with identical image acquisition parameters between different conditions.
Discussion
The sequence-function mapping pipeline using a yeast-displayed antigen can be used to elucidate conformational, discontinuous epitopes of complex proteins. As demonstrated with TROP2, a solved structure of the antigen is not essential for identification of the conformational epitope. The methodological improvements developed in this paper allowed us to complete the pipeline using a single cell sorter in 14 days for 24 different antibody antigen complexes at an approximate material and supply cost of $330 per antibody-antigen epitope. The cost and speed of this method offer significant advantages compared with competing display-based protocols. Notably, our method requires only a few micrograms of the starting antibody and so can be used directly downstream of immortalized B cell or hybridoma screening. Additionally, the ability to comprehensively map sequence determinants to binding may help elucidate potential escape mutants and be used to predict whether the antibody will maintain affinity for antigen homologs from model organisms. The sequence-function maps may also be integrated into computational prediction software to improve the predictions of specific antibody-antigen structural contacts at the atomic level or improve computational predictions of individual mutations on protein-protein interactions (28).
There are minor limitations in the current protocol. For example, antigens requiring multiple subunits to fold may be difficult to express on the yeast surface. Additionally, conformational epitopes requiring heterogeneous peptide-glycosyl surfaces will not be able to be mapped. Nevertheless, our results show that antibody binding surfaces for complicated homodimer and homotrimer human proteins TROP2 and TNF can be assessed, and we speculate that similar proteins can be mapped. Although our approach can be used as is to interrogate antibody panels of 10–50 members, further improvements in speed and cost must be addressed for integration of the method with other high throughput or single cell technologies. Methodological advances should focus on replacing the bottleneck FACS step with a more high throughput sorting technique and removing the need to prepare multiple libraries for each antigen.
Author Contributions
C. A. K. and T. A. W. conceived the method and developed the analytical equations; C. A. K., M. S. F., A. N., H. E. D., V. W. K., L. L., P. S., and E. K. W. designed and performed experiments; J. A. M., C. C., and T. A. W. designed experiments, and C. A. K. and T. A. W. wrote the manuscript with input from all co-authors.
Supplementary Material
Acknowledgments
We thank Justin Klesmith for writing normalization scripts used in this manuscript and Lani Hack for preparation of the pETCON-TROP2 plasmid.
This work was supported, in whole or in part, by National Institutes of Health Grant 5R21CA176854-02 (to T. A. W. and C. C.). This work was also supported by National Science Foundation CAREER Award CBET-1254238 (to T. A. W.) and a grant from Synthetic Biologics (to J. A. M.). The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Figs. 1–3 and Tables 1–3.
A. W. Nguyen, E. K. Wagner, J. R. Laber, L. Goodfield, W. E. Smallridge, E. T. Harvill, J. F. Papin, R. F. Wolk, E. A. Padlan, A. Bristol, M. Kaleko, and J. A. Maynard, submitted for publication.
J. R. Klesmith, J. P. Bacik, R. Michalczyk, and T. A. Whitehead, submitted for publication.
- PTx
- pertussis toxin
- SSM
- single site saturation mutagenesis
- PTxS1
- Ptx subunit 1
- TROP2Ic
- intracellular TROP2
- FSC
- forward scatter
- SSC
- side scatter.
References
- 1.McLellan J. S., Chen M., Joyce M. G., Sastry M., Stewart-Jones G. B., Yang Y., Zhang B., Chen L., Srivatsan S., and Zheng A. (2013) Structure-based design of a fusion glycoprotein vaccine for respiratory syncytial virus. Science 342, 592–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McLellan J. S., Chen M., Leung S., Graepel K. W., Du X., Yang Y., Zhou T., Baxa U., Yasuda E., and Beaumont T. (2013) Structure of RSV fusion glycoprotein trimer bound to a prefusion-specific neutralizing antibody. Science 340, 1113–1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Correia B. E., Bates J. T., Loomis R. J., Baneyx G., Carrico C., Jardine J. G., Rupert P., Correnti C., Kalyuzhniy O., and Vittal V. (2014) Proof of principle for epitope-focused vaccine design. Nature 507, 201–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Throsby M., van den Brink E., Jongeneelen M., Poon L. L., Alard P., Cornelissen L., Bakker A., Cox F., van Deventer E., and Guan Y. (2008) Heterosubtypic neutralizing monoclonal antibodies cross-protective against H5N1 and H1N1 recovered from human IgM+ memory B cells. PloS ONE 3, e3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sui J., Hwang W. C., Perez S., Wei G., Aird D., Chen L.-M., Santelli E., Stec B., Cadwell G., Ali M., Wan H., Murakami A., Yammanuru A., Han T., Cox N. J., Bankston L. A., Donis R. O., Liddington R. C., and Marasco W. A. (2009) Structural and functional bases for broad-spectrum neutralization of avian and human influenza A viruses. Nat. Struct. Mol. Biol. 16, 265–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fleishman S. J., Whitehead T. A., Ekiert D. C., Dreyfus C., Corn J. E., Strauch E.-M., Wilson I. A., and Baker D. (2011) Computational design of proteins targeting the conserved stem region of influenza hemagglutinin. Science 332, 816–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Whitehead T. A., Chevalier A., Song Y., Dreyfus C., Fleishman S. J., De Mattos C., Myers C. A., Kamisetty H., Blair P., Wilson I. A., and Baker D. (2012) Optimization of affinity, specificity and function of designed influenza inhibitors using deep sequencing. Nat. Biotechnol. 30, 543–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krammer F., Pica N., Hai R., Margine I., and Palese P. (2013) Chimeric hemagglutinin influenza virus vaccine constructs elicit broadly protective stalk-specific antibodies. J. Virol. 87, 6542–6550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wei C.-J., Boyington J. C., McTamney P. M., Kong W.-P., Pearce M. B., Xu L., Andersen H., Rao S., Tumpey T. M., and Yang Z.-Y., and Nabel G. J. (2010) Induction of broadly neutralizing H1N1 influenza antibodies by vaccination. Science 329, 1060–1064 [DOI] [PubMed] [Google Scholar]
- 10.Georgiou G., Ippolito G. C., Beausang J., Busse C. E., Wardemann H., and Quake S. R. (2014) The promise and challenge of high-throughput sequencing of the antibody repertoire. Nat. Biotechnol. 32, 158–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Laserson U., Vigneault F., Gadala-Maria D., Yaari G., Uduman M., Vander Heiden J. A., Kelton W., Taek Jung S., Liu Y., and Laserson J., Chari R., Lee J. H., Bachelet I., Hickey B., Lieberman-Aiden E., Hanczaruk B., Simen B. B., Egholm M., Koller D., Georgiou G., Kleinstein S. H., and Church G. M. (2014) High-resolution antibody dynamics of vaccine-induced immune responses. Proc. Natl. Acad. Sci. U.S.A. 111, 4928–4933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Benichou J., Ben-Hamo R., Louzoun Y., and Efroni S. (2012) Rep-Seq: uncovering the immunological repertoire through next-generation sequencing. Immunology 135, 183–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sela-Culang I., Ofran Y., and Peters B. (2015) Antibody specific epitope prediction: emergence of a new paradigm. Curr. Opin. Virol. 11, 98–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frank R. (2002) The SPOT-synthesis technique: synthetic peptide arrays on membrane supports: principles and applications. J. Immunol. Methods 267, 13–26 [DOI] [PubMed] [Google Scholar]
- 15.Spatola B. N., Murray J. A., Kagnoff M., Kaukinen K., and Daugherty P. S. (2013) Antibody repertoire profiling using bacterial display identifies reactivity signatures of celiac disease. Anal. Chem. 85, 1215–1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Casina V. C., Hu W., Mao J., Lu R., Hanby H. A., Pickens B., Kan Z., Lim W. K., Mayne L., Ostertag E., Kacir S., Siegel D. L., Englander S. W., and Zheng X. L. (2015) High-resolution epitope mapping by HX MS reveals the pathogenic mechanism and a possible therapy for autoimmune TTP syndrome. Proc. Natl. Acad. Sci. U. S. A. 112, 9620–9625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pandit D., Tuske S. J., Coales S. J., E. S. Y., Liu A., Lee J. E., Morrow J. A., Nemeth J. F., and Hamuro Y. (2012) Mapping of discontinuous conformational epitopes by amide hydrogen/deuterium exchange mass spectrometry and computational docking. J. Mol. Recognit. 25, 114–124 [DOI] [PubMed] [Google Scholar]
- 18.Van Blarcom T., Rossi A., Foletti D., Sundar P., Pitts S., Bee C., Melton Witt J., Melton Z., Hasa-Moreno A., and Shaughnessy L., Telman D., Zhao L., Cheung W. L., Berka J., Zhai W., Strop P., Chaparro-Riggers J., Shelton D. L., Pons J., and Rajpal A. (2015) Precise and efficient antibody epitope determination through library design, yeast display and next-generation sequencing. J. Mol. Biol. 427, 1513–1534 [DOI] [PubMed] [Google Scholar]
- 19.Mata-Fink J., Kriegsman B., Yu H. X., Zhu H., Hanson M. C., Irvine D. J., and Wittrup K. D. (2013) Rapid conformational epitope mapping of anti-gp120 antibodies with a designed mutant panel displayed on yeast. J. Mol. Biol. 425, 444–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Doolan K. M., and Colby D. W. (2015) Conformation-dependent epitopes recognized by prion protein antibodies probed using mutational scanning and deep sequencing. J. Mol. Biol. 427, 328–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weiss G. A., Watanabe C. K., Zhong A., Goddard A., and Sidhu S. S. (2000) Rapid mapping of protein functional epitopes by combinatorial alanine scanning. Proc. Natl. Acad. Sci. U.S.A. 97, 8950–8954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chao G., Lau W. L., Hackel B. J., Sazinsky S. L., Lippow S. M., and Wittrup K. D. (2006) Isolating and engineering human antibodies using yeast surface display. Nat. Protoc. 1, 755–768 [DOI] [PubMed] [Google Scholar]
- 23.Fowler D. M., and Fields S. (2014) Deep mutational scanning: a new style of protein science. Nat. Methods 11, 801–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sutherland J. N., and Maynard J. A. (2009) Characterization of a key neutralizing epitope on pertussis toxin recognized by monoclonal antibody 1B7. Biochemistry 48, 11982–11993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Le J., Vilcek J., Dadonna P., Ghrayeb J., Knight D., and Siegel S. A. (August 12, 1997) Methods of treating TNF-α-mediated Crohn's disease using chimeric anti-TNF antibodies. U. S. Patent 5,656,272
- 26.Chen L.-H., Huang Q., Wan L., Zeng L.-Y., Li S.-F., Li Y.-P., Lu X.-F., and Cheng J.-Q. (2006) Expression, purification, and in vitro refolding of a humanized single-chain Fv antibody against human CTLA4 (CD152). Protein Expr. Purif. 46, 495–502 [DOI] [PubMed] [Google Scholar]
- 27.Ong Y. T., Kirby K. A., Hachiya A., Chiang L. A., Marchand B., Yoshimura K., Murakami T., Singh K., Matsushita S., and Sarafianos S. G. (2012) Preparation of biologically active single-chain variable antibody fragments that target the HIV-1 GP120 v3 loop. Cell. Mol. Biol. 58, 71–79 [PMC free article] [PubMed] [Google Scholar]
- 28.Aizner Y., Sharabi O., Shirian J., Dakwar G. R., Risman M., Avraham O., and Shifman J. (2014) Mapping of the binding landscape for a picomolar protein-protein complex through computation and experiment. Structure 22, 636–645 [DOI] [PubMed] [Google Scholar]
- 29.Liu S.-h., Strop P., Dorywalska M. G., Rajpal A., Shelton D. L., and Tran T.-t. (August 15, 2014) Antibodies specific for trop-2 and their uses. U. S. Patent 20,140,357,844
- 30.Kowalsky C. A., Klesmith J. R., Stapleton J. A., Kelly V., Reichkitzer N., and Whitehead T. A. (2015) High-resolution sequence-function mapping of full-length proteins. PloS ONE 10, e0118193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bilgin B., Liu L., Chan C., and Walton S. P. (2013) Quantitative, solution-phase profiling of multiple transcription factors in parallel. Anal. Bioanal. Chem. 405, 2461–2468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu M., Liu L., Hijazi H., and Chan C. (2013) A multi-layer inference approach to reconstruct condition-specific genes and their regulation. Bioinformatics 29, 1541–1552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu L., and Chan C. (2014) IPAF inflammasome is involved in interleukin-1β production from astrocytes, induced by palmitate; implications for Alzheimer's Disease. Neurobiol. Aging 35, 309–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu L., Martin R., and Chan C. (2013) Palmitate-activated astrocytes via serine palmitoyltransferase increase BACE1 in primary neurons by sphingomyelinases. Neurobiol. Aging 34, 540–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Patil S. P., Tran N., Geekiyanage H., Liu L., and Chan C. (2013) Curcumin-induced upregulation of the anti-tau cochaperone BAG2 in primary rat cortical neurons. Neurosci. Lett. 554, 121–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu M., Liu L., and Chan C. (2011) Identification of novel targets for breast cancer by exploring gene switches on a genome scale. BMC Genomics 12, 547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang L., Seitz L. C., Abramczyk A. M., Liu L., and Chan C. (2011) cAMP initiates early phase neuron-like morphology changes and late phase neural differentiation in mesenchymal stem cells. Cell. Mol. Life Sci. 68, 863–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pavšič M., Gunčar G., Djinović-Carugo K., and Lenarčič B. (2014) Crystal structure and its bearing towards an understanding of key biological functions of EpCAM. Nat. Commun. 5, 4764. [DOI] [PubMed] [Google Scholar]
- 39.Firnberg E., and Ostermeier M. (2012) PFunkel: efficient, expansive, user-defined mutagenesis. PloS ONE 7, e52031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liang S., Dai J., Hou S., Su L., Zhang D., Guo H., Hu S., Wang H., Rao Z., and Guo Y. (2013) Structural basis for treating tumor necrosis factor α (TNFα)-associated diseases with the therapeutic antibody infliximab. J. Biol. Chem. 288, 13799–13807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Black R. E., Cousens S., Johnson H. L., Lawn J. E., Rudan I., Bassani D. G., Jha P., Campbell H., Walker C. F., and Cibulskis R. (2010) Global, regional, and national causes of child mortality in 2008: a systematic analysis. Lancet 375, 1969–1987 [DOI] [PubMed] [Google Scholar]
- 42.Kim K. J., Burnette W. N., Sublett R.D., Manclark C. R., and Kenimer J. G. (1989) Epitopes on the S1 subunit of pertussis toxin recognized by monoclonal antibodies. Infect. Immun. 57, 944–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stein P. E., Boodhoo A., Armstrong G. D., Cockle S. A., Klein M. H., and Read R. J. (1994) The crystal structure of pertussis toxin. Structure 2, 45–57 [DOI] [PubMed] [Google Scholar]
- 44.Fong D., Moser P., Krammel C., Gostner J. M., Margreiter R., Mitterer M., Gastl G., and Spizzo G. (2008) High expression of TROP2 correlates with poor prognosis in pancreatic cancer. Br. J. Cancer 99, 1290–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ohmachi T., Tanaka F., Mimori K., Inoue H., Yanaga K., and Mori M. (2006) Clinical significance of TROP2 expression in colorectal cancer. Clin. Cancer Res. 12, 3057–3063 [DOI] [PubMed] [Google Scholar]
- 46.McDougall A. R., Tolcos M., Hooper S. B., Cole T. J., and Wallace M. J. (2015) Trop2: from development to disease. Dev. Dyn. 244, 99–109 [DOI] [PubMed] [Google Scholar]
- 47.Maetzel D., Denzel S., Mack B., Canis M., Went P., Benk M., Kieu C., Papior P., Baeuerle P. A., and Munz M. (2009) Nuclear signalling by tumour-associated antigen EpCAM. Nat. Cell Biol. 11, 162–171 [DOI] [PubMed] [Google Scholar]
- 48.Stoyanova T., Goldstein A. S., Cai H., Drake J. M., Huang J., and Witte O. N. (2012) Regulated proteolysis of Trop2 drives epithelial hyperplasia and stem cell self-renewal via β-catenin signaling. Genes Dev. 26, 2271–2285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vidmar T., Pavšič M., and Lenarčič B. (2013) Biochemical and preliminary X-ray characterization of the tumor-associated calcium signal transducer 2 (Trop2) ectodomain. Protein Expr. Purif. 91, 69–76 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.