

Background: ATG4B mediates the cleavage of pro-LC3 and removes lipid conjugates from LC3 during autophagy.
Results: We determined that defects in phosphorylation of ATG4B reduced its hydrolyase activity and impaired autophagic flux.
Conclusion: Phosphorylation of ATG4B plays an important role in modulating its hydrolyase activity.
Significance: This is the first report showing a role for phosphorylation of an ATG4-family protease in control of autophagy.
Keywords: autophagy, phosphorylation, protease, protein kinase, protein-protein interaction
Abstract
Autophagy is a catabolic cellular mechanism for entrapping cellular macromolecules and organelles in intracellular vesicles and degrading their contents by fusion with lysosomes. Important roles for autophagy have been elucidated for cell survival during nutrient insufficiency, eradication of intracellular pathogens, and counteracting aging through clearance of senescent proteins and mitochondria. Autophagic vesicles become decorated with LC3, a protein that mediates their fusion with lysosomes. LC3 is a substrate of the cysteine protease ATG4B (Autophagin-1), where cleavage generates a C-terminal glycine required for LC3 conjugation to lipids in autophagosomes. ATG4B both cleaves pro-LC3 and also hydrolyzes lipids from cleaved LC3. We show here that phosphorylation of ATG4B at Ser-383 and Ser-392 increases its hydrolyase activity as measured using LC3 as a substrate. Reconstituting atg4b−/− cells with phosphorylation-deficient ATG4B showed a role of ATG4B phosphorylation in LC3 delipidation and autophagic flux, thus demonstrating that the cellular activity of ATG4B is modulated by phosphorylation. Proteolytic conversion of pro-LC3 to LC3-I was not significantly impacted by ATG4B phosphorylation in cells. Phosphorylation-deficient ATG4B also showed reduced interactions with the lipid-conjugated LC3 but not unconjugated LC3. Taken together, these findings demonstrate a role for Ser-383 and Ser-392 phosphorylation of ATG4B in control of autophagy.
Introduction
Autophagy is an evolutionarily conserved catabolic pathway within all eukaryotic cells that plays a critical housekeeping function to remove senescent or damaged proteins and organelles through lysosome-mediated degradation. Autophagy also plays roles in generating substrates for maintaining cellular energy during times of nutrient insufficiency and hypoxia and in host defenses against intracellular pathogens (1). The hallmark of autophagy is the generation of cytoplasmic double-membrane vesicles, termed autophagosomes, which emerge from intracellular membranes (probably the endoplasmic reticulum), engulf cytoplasmic macromolecules, and then fuse with lysosomes to achieve degradation of the contents.
Genetic studies of yeast have identified more than 30 autophagy-related (ATG)3 genes that are required for this process (2). Among the core autophagy machinery of yeast is an ubiquitin-like conjugation network called the Atg8 system that is essential for autophagosome formation (3). The Atg8 protein is synthesized as a cytoplasmic precursor that is cleaved near its C terminus by the cysteine proteinase Atg4 to generate an exposed C-terminal glycine, which becomes conjugated to membrane-bound phosphatidylethanolamine (PE), an event that is important for completion of the autophagosome (3, 4). This reaction is catalyzed by the sequential actions of Atg7 (E1-like activating enzyme), Atg3 (E2-like conjugation enzyme) and the Atg12—Atg5-Atg16 complex (E3-like ligase) (3, 5, 6). Importantly, Atg8-PE is also deconjugated by the protease Atg4, facilitating the release and recycling of Atg8 from membranes. This deconjugation process is also essential for normal autophagy (4). Thus, Atg4 plays dual roles in the progression of autophagy through the irreversible (proteolytic cleavage) and reversible (lipid deconjugation) modification of Atg8.
The Atg8 modification system is well conserved in higher eukaryotes. In mammals, at least 6 orthologs of yeast Atg8 have been identified, belonging to two subfamilies known as the light chain 3 (LC3) and γ-aminobutyric acid (GABA)-receptor-associated protein (GABARAP) proteins, all of which have been shown to participate in autophagy (7, 8). Similar to the Atg8 system in yeast, the C-terminal peptide of LC3 is cleaved by mammalian Atg4 homologues (9, 10). The proteolytically processed form, called LC3-I, has a glycine residue at its C terminus and resides in the cytosol. LC3-I is further modified to LC3-II, which is analogous to the PE-conjugated form in the Atg8 system (7, 11). To date, four human homologues of yeast Atg4 have been identified: ATG4A, ATG4B, ATG4C, and ATG4D (12), also known as autophagins-1, -2, -3, and -4. Among them, ATG4B (autophagin-1) has broad specificity for almost all of the mammalian Atg8 homologues and is the sole enzyme reported to efficiently cleave LC3 precursors and to deconjugate lipid from LC3-PE (7). Of the four autophagins identified, ATG4B (autophagin-1) shows the highest catalytic efficiency for cleaving the C terminus of LC3B (13). Moreover, atg4b−/− mice showed a clear reduction in basal- and starvation-induced autophagy in all tissues, associated with impaired proteolytic cleavage of LC3 orthologs (14).
ATG4B is regulated in a manner that has concomitant effects on LC3 processing. Forced expression of ATG4B has been shown to reduce the amount of cellular lipidated LC3 (LC3-II), and co-expression of ATG4B with LC3 under nutrient depletion conditions reduces the punctate localization pattern of LC3, consistent with removal from autophagic vesicles (7, 15). Similarly, knockdown of ATG4B increases steady-state LC3-II levels. However, increased LC3-II does not translate to increased autophagic flux (15, 16), because ATG4B deficiency also inhibits proteolytic processing of LC3 paralogues and thereby hampers autophagy (14, 17, 18). In embryonic stem cells, ATG4B overexpression resulted in an increase of LC3-I, which was attributed to LC3-II deconjugation (7), whereas up-regulating ATG4B by aplasia Ras homolog member I (ARH1) or stabilizing ATG4B protein levels by inactivation of the E3 ligase RNF5 was associated with increased LC3 processing (19, 20). Collectively, these observations indicate a convoluted and critical role for ATG4B in the mechanisms that control autophagy.
Little is known about the molecular events that govern ATG4B activity in cells. Several studies have highlighted the complex regulation of ATG4B activity at the level of transcription as well as post-translational modifications. For example, CCAAT/enhancer-binding protein β (C/EBPβ) specifically promotes autophagy through transactivation of the expression of ATG4B (21). In addition, the oxidation of cysteine residues of ATG4B reduces its deconjugating activity toward LC3-PE, and this mechanism has been proposed as a signaling switch for ATG4B inactivation (22). Moreover, several large-scale phosphoproteomics studies have suggested that ATG4B is phosphorylated at Ser-34 (23), Ser-383, and Ser-392 (24–26). However, to date, no evidence has clearly shown the functional significance of ATG4B phosphorylation.
Here, we investigated ATG4B phosphorylation and its effect on the relationship between ATG4B activity and LC3 processing. Our studies demonstrate that phosphorylation of ATG4B is required for optimal LC3-directed protease activity and that ATG4B phosphorylation correlates with autophagy induction. We identified two conserved serine residues, Ser-383 and Ser-392, essential for ATG4B phosphoregulation. Moreover, further analyses revealed that phosphorylation of ATG4B regulates LC3 lipidation in cells, and thereby modulates autophagic flux.
Experimental Procedures
Plasmids
The plasmids pcDNA3 expressing Flag-ATG4B WT and Flag-ATG4B C74A mutant were previously reported (27). The cDNAs encoding WT ATG4B and C74A mutant were further subcloned into pNTAP-B (Stratagene, 240103) to fuse the TAP tag. Flag-ATG4B WT and Flag-ATG4B C74A were also subcloned into pCDH (System Biosciences, CD520A-1). To create ATG4B mutants, site-directed mutagenesis was performed following Stratagene QuickChange kit instructions and specificity of mutagenesis was confirmed by direct sequencing. The plasmids encoding substrate reporters of intracellular proteolysis based on non-conventional secretion of Gaussia luciferase actin-dNGLUC and actin-LC3B-dNGLUC in a pLKO vector were a gift from Dr. Brian Seed (Massachusetts General Hospital, Boston, MA). Firefly luciferase (LUC2) was cloned into a pcDNA3 vector. The plasmids pEGFP-LC3 and HA-p62 were described previously (11, 28). The EGFP-LC3 plasmid was also subcloned into pWZL Hygro (Addgene, plasmid 18750). Lentivirus-based shRNAs of ATG4B were purchased from Sigma validated MISSION shRNA.
Generation of atg4b−/− MEFs and Stable Reconstitution of Cell Lines
Primary ATG4B knock-out MEFs were generated from matings of ATG4B heterozygous mice generously provided by Dr. Carlos López-Otín (14), then immortalized by transfection with SV40 large T Antigen constructed in pBABE-neo (Addgene, plasmid 1780). The atg4b+/+ and atg4b−/− MEFs were stably infected with either pCDH-GFP control, pCDH-Flag-ATG4B WT, deletion or phosphorylation mutants in the pLKO.1 lentiviral background. Lentiviral particles were produced by cotransfection with psPAX2 and pMD2.G plasmids into HEK293T cells. Viral supernatants were collected from 48 to 60 h and cleared through a 0.45-μm filter and applied every 12 h on target cells for three rounds. Polybrene (4 μg/ml; Sigma) was supplemented into viral supernatants. After final infection, stable populations were obtained by selection with 2 μg/ml puromycin (Invitrogen). The atg4b+/+ and atg4b−/− MEFs stably expressing various Flag-ATG4B constructs or control were then infected with retroviral particles expressing GFP-LC3 generated by transfection of pWZL-EGFP-LC3 into Phoenix packaging cell lines. Following the same infection procedure described above for lentiviral particles, cells were then selected with 2 μg/ml puromycin and 250 μg/ml hygromycin (Invitrogen). Polyclonal populations were screened to identify WT and mutant lines with similar levels of ATG4B and GFP-LC3 expression.
Cell Culture
Immortalized MEFs were cultured in high-glucose DMEM supplemented with 10% FBS (Hyclone), 100 μg/ml streptomycin, 100 IU penicillin, 55 μm β-mercaptoethanol, and 0.1 mm non-essential amino acids. Hanks' Balanced Salt Solutions (HBSS) was purchased from Gibco (Life Technologies). HEK293T, HeLa, and MCF7 cells were grown in similar media omitting the β-mercaptoethanol. Medium was replenished the night before starvation experiments.
Antibodies and Chemicals
Rabbit polyclonal anti-ATG4B, -LC3B, and anti-Flag M2 antibodies were purchased from Sigma-Aldrich. Rabbit polyclonal anti-GFP and anti-p62 were purchased from Santa Cruz Biotechnology. Protein A/G affinity, anti-Flag M2 gels, and 3×Flag peptides were purchased from Sigma-Aldrich. 2-(6-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino) hexanoyl-1-hexadecanoyl-sn-glycero-3-phosphocholine (NBD-C6-HPC) was purchased from Invitrogen.
Protein Expression and Purification in Escherichia coli and Mammalian Cells
Plasmid expressing LC3-PLA2 was transformed into E. coli BL21 (DE3, Invitrogen, C6010-03). The proteins were induced and purified as previously described (27). For TAP-tagged ATG4B protein purification, HEK-293T cells transiently expressing human Atg4B with N-terminal affinity tags, including Streptavidin and Calmodulin binding peptide, were pelleted by centrifugation at 3000 × g and TAP-tagged ATG4B protein was purified using the manufacturer's protocol (InterPlay Mammalian TAP System by Agilent Technologies). For Flag-tagged ATG4B protein purification, HEK293T cells (∼107) transfected with expression plasmids (pcDNA3-FLAG ATG4B WT or catalytic mutant C74A) were harvested and washed with PBS once, then resuspended in 500 μl of lysis buffer containing 25 mm Tris-HCl, pH 8.0, 100 mm NaCl, 1 mm CaCl2, 5 mm MgCl, 5% Glycerol, 0.1% Nonidet P-40, 1 mm PMSF, and 1 mm DTT on ice followed by sonication 2 × 2 s to fracture cells. Lysates were centrifuged at 16,000 × g for 15 min and 250 μg of supernatant proteins were used for immunoprecipitation using anti-Flag M2 gel following the manufacturer's protocol. After washing with PBS 3∼4 times, Flag-tagged Atg4B was eluted using 25 μg of 3×Flag peptides. Purified TAP-tagged or Flag-tagged ATG4B proteins were treated with alkaline phosphatase (CIP, 0.5 units, New England Biolabs) with or without phosphatase inhibitor mixture (PhosSTOP, Roche Applied Bioscience) at 37 °C for 1 h, followed by in vitro ATG4B protease assay or immunoblotting.
In Vitro ATG4B Activity Assay
Atg4B activity was measured with 890 nm LC3B-PLA2 fusion protein in 20 μl of PLA2 reaction buffer containing 20 mm Tris-HCl, pH 8.0, 2 mm CaCl2, 1 mm DTT, and 20 μm NBD-C6-HPC (Invitrogen). Fluorescence intensity was measured within 80 min using an AnalystTM HT (Molecular Devices Corp) at room temperature with excitation and emission wavelength of 485 and 530, respectively (27).
Luciferase Assay
Cellular ATG4B activity was assayed by measuring the cellular release of an N-terminally deleted form of Gaussia luciferase (dNGLUC) as described by Ketteler et al. (29). Briefly, MEFs were seeded in 24-well plates and co-transfected with 200 ng of plasmid expressing the appropriate actin-LC3B-dNGLUC (or actin-dNGLUC control) and 50 ng of plasmid expressing Firefly luciferase (LUC2, as normalization control). Transfections were performed using Lipofectamine 2000 (Invitrogen) according to the standard protocol. After 48 h, the Renilla luciferase and Firefly luciferase assays were carried out according to the manufacturer's instructions for the dual luciferase reporter assay system and the Firefly luciferase assay system, respectively (Promega).
Co-immunoprecipitation and Immunoblotting
For co-immunoprecipitation studies, cell lysates were prepared in lysis buffer (50 mm Tris-HCl, pH 8.0, 100 mm NaCl, 1 mm CaCl2, 5 mm MgCl, 5% glycerol, 0.1% Nonidet P-40, 1 mm PMSF, and 1 mm DTT) containing protease inhibitor mixture (Roche Applied Bioscience) and phosphatase inhibitor mixture (Sigma). Cellular debris were removed and the supernatants were incubated with antibody for 2 h and then incubated with protein A/G affinity gels for either 2 h or overnight at 4 °C. Beads were washed three times with lysis buffer and the captured proteins were resolved by SDS-PAGE. For evaluation of cellular proteins, cell lysates were prepared using RIPA buffer (1% Nonidet P-40, 50 mm Tris-HCl pH 7.5, 150 mm NaCl, 0.25% sodium deoxycholate 0.1% SDS, 1 mm EDTA) containing protease inhibitor mixture (Roche Applied Bioscience) and phosphatase inhibitor mixture (Sigma). Equal amounts of lysates were subjected to SDS-PAGE. Blots were probed using the indicated primary antibodies. The band intensities were quantified by Image J (National Institutes of Health).
Calf intestinal alkaline phosphatase (CIP, New England Biolabs) with or without phosphatase stop (PS) were added to beads and incubated at 37 °C for 1 h, followed by heat inactivation for 30 min and analyzed by immunoblotting. For Phos-tag experiments, we followed the manufacturer's instructions. Briefly, 10% SDS-PAGE gels were prepared using Phos-tag acrylamide (50 μm) and an equal volume of 10 mm MnCl2. After electrophoresis, the samples were transferred to PVDF membranes and washed with 10 mm EDTA, followed by immunoblotting analysis.
Results
ATG4B Is Phosphorylated in Cells
To determine whether the cysteine protease ATG4B is phosphorylated in cells, we expressed functional ATG4B tagged with Flag peptide in HEK293T cells. The Flag-ATG4B protein was recovered from cells by anti-Flag antibody immunoprecipitation and its mobility was assessed using both conventional and Phos-tag SDS-PAGE by immunoblotting with anti-ATG4B antibody. By Phos-tag analysis, we observed two distinct bands corresponding to Flag-ATG4B (Fig. 1, A and B). Phosphatase treatment of transfected HEK293T cell lysates abolished the upper band, leaving a single band that was cross-reactive with both anti-ATG4B and anti-Flag epitope tag antibodies (Fig. 1, A and B). Moreover, co-treatment with phosphatase and chemical phosphatase inhibitor prevented the loss of the upper band (Fig. 1B). These results indicate that ATG4B protein is present in both phosphorylated and non-phosphorylated forms in mammalian cells under normal culture conditions.
FIGURE 1.

ATG4B protease is phosphorylated in cells and phosphorylation modulates protease activity in vitro. A and B, Flag-tagged vector or Flag-tagged ATG4B WT or C74A catalytic mutant proteins were expressed in HEK293T cells. Cell lysates were prepared and normalized for total protein content. Flag-ATG4B was then immunoprecipitated, followed by treatment with or without CIP, in the presence or absence of PS. Immunoprecipitates of Flag-ATG4B (WT) were fractionated by phos-tag gel versus regular (conventional) SDS-PAGE gel electrophoresis and then analyzed by immunoblotting using anti-ATG4B antibody. C, plasmids encoding TAP-tagged Atg4B or catalytic mutant Atg4B C74A were transfected into HEK293T cells for 48 h. The TAP-tagged proteins were purified using streptavidin and calmodulin binding beads, followed by elution with calmodulin elution buffer. Purified TAP-tagged ATG4B proteins were treated with or without CIP at 37 °C for 1 h, followed by incubation with the synthetic substrate LC3B-PLA2 to monitor ATG4B activity. Fluorescence intensity was monitored for 80 min to measure ATG4B activity. ATG4B activity at 30 min was within the linear phase of the reaction and was normalized relative to ATG4B WT without CIP treatment (set as 100%; mean ± S.D.; n = 3; ***, p < 0.001). Simultaneously, aliquots of these protein samples were subjected to immunoblotting using anti-ATG4B or anti-phosphoserine antibodies. D, immunoprecipitated Flag-ATG4B WT was treated with or without CIP, in the presence or absence of PS. ATG4B protease activity was measured using the LC3-PLA2 assay (relative fluorescence units, RFU) over time (0–80 min) (mean ± S.D.; n = 3; *, p < 0.05; **, p < 0.01; ***, p < 0.001).
Phosphorylation of ATG4B Promotes Protease Activity
We sought to determine whether phosphorylation of ATG4B modulates its protease activity. To assess ATG4B catalytic activity we turned to a novel and quantitative assay developed previously in our laboratory, involving a synthetic substrate comprised of LC3B fused to the N terminus of phospholipase A2 (LC3B-PLA2). Upon cleavage by ATG4B, active PLA2 is released for fluorogenic assay (27). We overexpressed TAP-tagged ATG4B (either wild-type or a catalytic site cysteine→alanine mutant) under the control of the constitutive CMV promoter in HEK293T cells. The purified fraction from calmodulin-Sepharose affinity chromatography containing both phosphorylated and non-phosphorylated forms of TAP-tagged ATG4B was assessed using the LC3B-PLA2 assay. Wild-type ATG4B protein showed a robust ATG4B protease activity (Fig. 1C). In contrast, vector alone or the catalytic ATG4B mutant of protein, C74A, showed essentially no activity using the LC3B-PLA2 substrate for assessing protease activity. Upon phosphatase treatment, wild-type ATG4B showed a dramatic reduction in the protease activity toward LC3B-PLA2 (Fig. 1C). The concurrent immunoblot probed with an anti-phosphoserine antibody showed reduced phosphorylation of the ATG4B protein after phosphatase treatment (Fig. 1C). These data suggest that ATG4B activity correlates with the level of ATG4B phosphorylation.
To exclude the possibility that phosphorylation of ATG4B was caused by the TAP tag of the protein, we overexpressed functional Flag-ATG4B under the control of constitutive CMV promoter in HEK293T cells. Immunoprecipitated Flag-ATG4B proteins were eluted with 3×Flag peptides, followed by treatment with phosphatase either with or without phosphatase inhibitor. Again, we observed a significant reduction in ATG4B protease activity upon phosphatase treatment (Fig. 1D). Furthermore, co-treatment with phosphatase and phosphatase inhibitor prevented this reduction and showed even more robust ATG4B protease activity than non-treated control, suggesting the possibility that the phosphatase inhibitor may protect phospho-ATG4B from endogenous phosphatases that co-purify with Flag-ATG4B (Fig. 1D). Taken together, these experiments suggest that the protease activity of ATG4B toward the recombinant substrate LC3B-PLA2 is enhanced by phosphorylation.
Phosphorylation of ATG4B Correlates with the Induction of Autophagy
Next, we sought to determine the physiological significance of ATG4B phosphorylation. Cells expressing Flag-ATG4B were exposed to various environmental conditions known to induce autophagy (30), and the phosphorylation of Flag-ATG4B was assessed. When cells were exposed to nutrient starvation (HBSS) or treated by the mTOR inhibitor rapamycin for 4 h, a clear increase was observed in the phosphorylated form of ATG4B relative to the non-phosphorylated form of ATG4B protein (Fig. 2, A and B). To rule out the possibility that the observed increase in phosphorylation of ATG4B might be due to cell death caused by nutrient stress, we shifted the cells back to nutrient-rich medium for another 4 h (recovery period). Following recovery from nutrient stress we observed a decrease in the ratio of phosphorylated ATG4B to the non-phosphorylated form (Fig. 2C). These experiments suggest that ATG4B phosphorylation increases upon autophagy induction.
FIGURE 2.
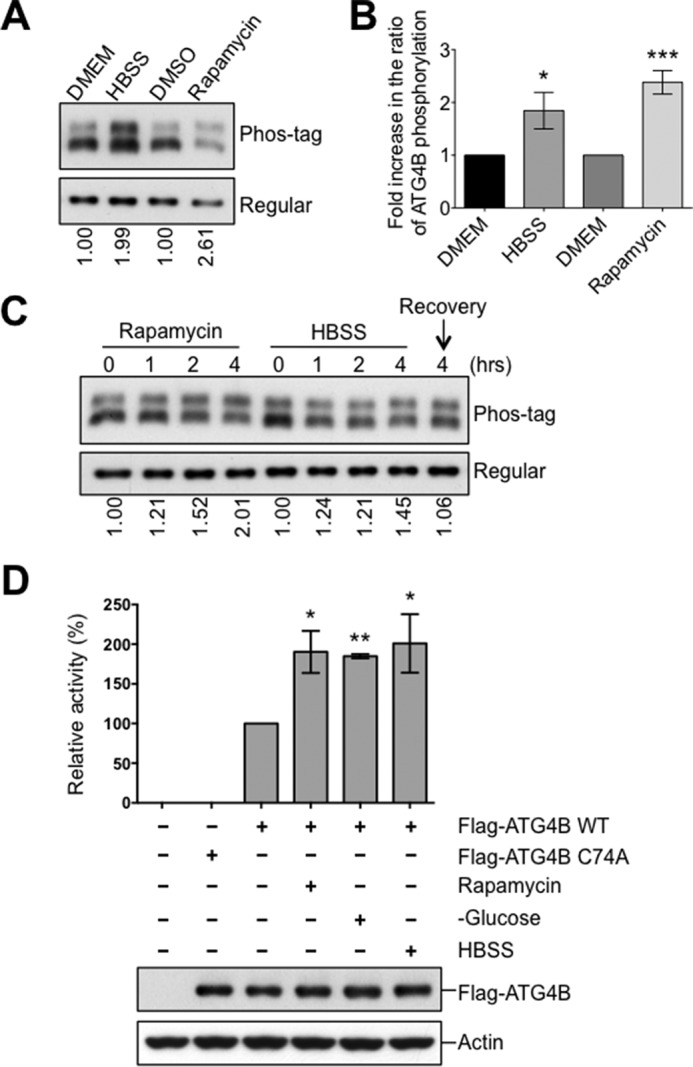
Increase of ATG4B phosphorylation and protease activity upon the induction of autophagy. A–C, HEK293T cells expressing Flag-ATG4B WT were cultured with or without rapamycin (25 μg/ml) or HBSS starvation for 4 h. In C, after 4 h HBSS starvation, cells were recovered by introducing nutrient medium (DMEM) for 4 h. Cell lysates were subjected directly to phos-tag versus regular SDS-PAGE and then analyzed by immunoblotting using anti-ATG4B antibody. The fold increase in the ratio of phosphorylated/non-phosphorylated ATG4B relative to the DMEM condition (or time 0) was quantified using scanning densitometry (values shown below lanes and plotted in B, *, p < 0.05; ***, p < 0.001). D, HeLa cells expressing Flag-tagged empty vector or Flag-ATG4B WT or C74A mutant proteins were cultured under various nutrient conditions for 5 h. Immunoprecipitated Flag-ATG4B was analyzed by immunoblotting with anti-ATG4B and anti-Actin antibodies. Protease activity of immunoprecipitated ATG4B was measured by LC3-PLA2 assay over time (0–80 min). ATG4B activity at 30 min was within the linear phase of the reaction and was further normalized relative to ATG4B WT from nutrient-rich conditions (set as 100%; mean ± S.D.; n = 3; *, p < 0.05; **, p < 0.01).
Our findings indicate that ATG4B protease activity is regulated by phosphorylation and upon autophagy induction ATG4B phosphorylation is increased. Thus, we hypothesized that ATG4B activity would also be increased upon initiation of autophagy. Consistent with this idea, when autophagy was induced by nutrient deprivation (HBSS or no glucose) or rapamycin treatment, a significant increase was observed in the protease activity of ATG4B as determined by the LC3-PLA2 assay (Fig. 2D). Altogether, our results show a clear correlation between ATG4B phosphorylation levels and its protease activity under conditions that induce autophagy.
ATG4B Phosphorylation at the C Terminus Affects Its Protease Activity
Previous large-scale phosphoproteomics studies have suggested that ATG4B is phosphorylated at Ser-34 (23), Ser-383, and Ser-392 (24–26). We performed mass spectrometry analysis of purified Flag-ATG4B and identified additional putative phosphorylated sites, including Ser-138, Tyr-143, and Ser-368 (data not shown). We then compared the sequences around these putative phosphorylation sites among the ATG4B orthologs of different organisms by ClustalW2 analysis. The phosphorylated region at the C terminus of ATG4B, which includes Ser-383 and Ser-392, is well conserved across species (Fig. 3A), whereas Ser-138, Tyr-143, and Ser-368 are not conserved (not shown).
FIGURE 3.

Phosphorylation of the C-terminal region of ATG4B. A, protein sequence alignment of the C-terminal region of ATG4B in zebrafish (Danio rerio), frog (Xenopus tropicalis), chicken (Gallus gallus), human (Homo sapiens), mouse (Mus musculus), and rat (Rattus norvegicus) using the ClustalW2 program. Gray shading indicates the identities of residues among proteins of different species, generated by the Percentage Identity option in Jalview, with darker gray signifying identity in more species. Amino acid residues known to become phosphorylated in mammalian cells are shown by arrows. B, Flag-ATG4B WT and various mutants (as labeled) were expressed in HEK293T cells. Cell lysates were prepared and normalized for total protein content. Flag-tagged ATG4B was then immunoprecipitated and treated in vitro with or without CIP. Flag-ATG4B proteins were fractionated by phos-tag versus regular SDS-PAGE then analyzed by immunoblotting using anti-ATG4B antibody. C, HEK293T cells expressing Flag-ATG4B WT were cultured with or without rapamycin for 5 h. Cell lysates were subjected directly to phos-tag versus regular SDS-PAGE and immunoblotting was performed using anti-ATG4B antibody. The fold increase in the ratio of phosphorylated/non-phosphorylated ATG4B before and after rapamycin treatment was quantified using scanning densitometry (values shown below lanes). D, full-length Flag-ATG4B and various C-terminal-truncated mutants were expressed in HEK293T cells. Cell lysates were fractionated by phos-tag versus conventional SDS-PAGE and analyzed by immunoblotting using anti-ATG4B antibody. E, protease activity of immunoprecipitated Flag-ATG4B WT or various mutants was measured using the LC3-PLA2 assay over time (0–80 min). ATG4B activity at 30 min was within the linear phase of the reaction and was normalized relative to ATG4B WT (set as 100%; mean ± S.D.; n = 3; **, p < 0.01; ***, p < 0.001.). F, ATG4B Ser-383 and Ser-392 residues are important for LC3 cleavage in vivo. Actin-LC3B-dNGLUC (the reporter plasmid for LC3 cleavage) or Actin-dNGLUC (control plasmid) were co-transfected with CMV-Luc2 into atg4b−/− MEF cells stably expressing GFP control, or ATG4B WT or various mutants (as labeled). After 48 h, the activity of Gaussia luciferase in the culture medium was measured, and normalized to Firefly luciferase activity in the cells, setting the activity for ATG4B WT as 100%. Error bars indicate the S.D. of three independent experiments. *, p < 0.05; **, p < 0.01. Simultaneously, aliquots of these protein samples were analyzed by immunoblotting using anti-ATG4B and anti-Actin antibodies.
Next, to test these putative ATG4B phosphorylation sites in vivo, we mutated each of the potential phosphorylated serine residues to alanine, and examined each variant by Phos-tag analysis after expression in HEK293T cells (Fig. 3B). All of the ATG4B mutants were expressed at similar levels in transfected HEK293T cells, suggesting that these phosphorylatable residues do not overtly control ATG4B protein stability. We found that S383A mutation caused a loss of the upper ATG4B band seen by Phos-tag analysis, as well as increasing the mobility of the ATG4B band resolved by conventional SDS-PAGE (Fig. 3, B and C). The S392A mutant also displayed a reduction in the ATG4B upper band seen by Phos-tag SDS-PAGE/immunoblot analysis (Fig. 3, B and C), suggesting that this site may also be phosphorylated to some extent. Mutation of the additional putative ATG4B phosphorylation sites Ser-34, Ser-138, and Ser-368, had no effect on migration of the ATG4B bands observed by Phos-tag analysis (Fig. 3B). (Note that, as a control for comparisons, we used WT ATG4B recovered by immunoprecipitation from cells that was treated in vitro with phosphatase.) The Phos-tag results were even more striking when autophagy was induced by treating the transfected HEK293T cells with Rapamycin prior to recovering ATG4B mutants by immunoprecipitation, especially for the S383A mutant (Fig. 3C). Thus, clearly Ser-383 and to some extent Ser-392 appear to be sites of phosphorylation of ATG4B.
Because the putative phosphorylation sites Ser-383 and Ser-392 are located near the C terminus of ATG4B, we created several C-terminal truncation mutants and examined their phosphorylation status and effect on ATG4B protease activity. To date, three groups have reported crystal structures of ATG4B, all of which have shown that the C-terminal 39 residues (residues 355–393) are highly flexible and thus not structurally resolved (31–33). One group used the region comprised of residues 1–354 of ATG4B for the analysis of the crystal structure of the ATG4B-LC3 complex (33). The flexible C-terminal region (residues 354–393) was thus missing in their structural analysis. Considering that Ser-383 and Ser-392 are located in this region, we generated ATG4B1−389, ATG4B1−379, and ATG4B1−354 mutants, and found that deletion of the C-terminal four residues (1–389) was sufficient to disrupt the appearance of the phosphorylated form of ATG4B as detected by Phos-tag SDS-PAGE analysis (Fig. 3D).
To correlate this apparent change in phosphorylation with protease activity, the C-terminal truncation mutants were recovered from mammalian cells by immunoprecipitation and analyzed using the LC3B-PLA2 substrate (Fig. 3E). ATG4B protease activity was almost completely abolished in these C-terminal truncation mutants. ATG4BS392A and ATG4BS383A/S392A also showed significantly decreased protease activity, whereas ATG4BS383A displayed a non-significant decrease in protease activity as measured using the LC3-PLA2 assay. Note that mutation of the other putative ATG4B phosphorylation sites Ser-138, Tyr-143, and Ser-368, displayed no effect on ATG4B protease activity using the LC3-PLA2 assay (data not shown).
To investigate what effect the loss of ATG4B phosphorylation has on cellular protease activity of ATG4B, we utilized immortalized atg4b−/− mouse embryonic fibroblasts (MEFs) for reconstitution experiments, introducing control, wild-type (WT) or various deletion or serine/alanine mutants of ATG4B. We then employed a cellular ATG4B protease assay for quantifying the intracellular proteolysis of LC3 based on non-conventional secretion of Gaussia luciferase (dNGLUC) (29, 34). In this system, the LC3B-dNGLUC substrate is anchored inside the cell by fusion to β-actin and the cleaved product is secreted after release from the cytoskeleton. Thus, ATG4B activity can be monitored by detecting extracellular luciferase activity resulting from the proteolytic cleavage of the LC3 site situated between Gaussia luciferase and β-actin. Using this cellular assay method, we observed a strong increase in the cleavage of LC3 as measured by the Gaussia luciferase activity in culture supernatants of the immortalized atg4b−/− MEFs stably expressing ATG4B WT (Fig. 3F). In contrast, atg4b−/− MEFs stably expressing control (empty) vector or the C74A catalytic mutant of ATG4B showed no increase, indicating a deficiency of ATG4B protease activity. In atg4b−/− MEFs stably expressing variants of ATG4B mutants, dramatic decreases were observed in the intracellular proteolysis of the engineered LC3 substrate, compared with ATG4B WT, with the most significant decrease in ATG4BS383A/S392A, ATG4B1–389, ATG4B1–379, and ATG4B1–354 mutants (Fig. 3F). Cells expressing single site mutants S383A or S392A showed intermediate decreases in ATG4B activity using the luciferase assay method. All of the ATG4B mutants were expressed at comparable levels in reconstituted atg4b−/− cells, suggesting that these phosphorylatable sites do not have an overt impact on ATG4B protein stability. Taken together, these results suggest that C-terminal phosphorylation of ATG4B contributes to the regulation of its ability to cleave LC3 in intact cells.
ATG4B Phosphorylation Modulates Autophagy
To determine the biological function of ATG4B phosphorylation, GFP-LC3 was introduced into WT MEFs or atg4b−/− MEFs stably expressing ATG4B WT or various ATG4B mutants to monitor the autophagy using a GFP-LC3 processing assay. During autophagy, autophagosomes containing GFP-LC3 fuse with the lysosomes where autophagosome cargo molecules such as GFP-LC3 are degraded. The GFP moiety is removed proteolytically from LC3 in the lysosome. The released GFP remains relatively stable from lysosomal degradation. Thus, the appearance of free GFP is often used as a marker to monitor the delivery of autophagosomes into lysosomes and the degradation of autophagic cargo (30).
A gradual increase of free GFP was observed in normal MEFs over 18 h of rapamycin treatment. Conversely, atg4b−/− MEFs showed dramatically reduced levels of free GFP, indicative of defective GFP-LC3 processing (Fig. 4A). Reconstitution of WT ATG4B rescued the defect in GFP-LC3 processing in the atg4b−/− MEFs, whereas expression of the ATG4B catalytic C74A mutant had an inhibitory effect on autophagy flux, consistent with a previous report (17)(Fig. 4A). Interestingly, the free GFP accumulation induced by rapamycin was significantly reduced in the atg4b−/− MEFs expressing ATG4BS383A/S392A, ATG4B1–379, or ATG4B1–354 mutants compared with the atg4b−/− MEFs expressing ATG4B WT (Fig. 4, B and C).
FIGURE 4.

ATG4B phosphorylation modulates autophagic flux. A–C, WT MEF (atg4b+/+) or atg4b−/− MEF cells stably expressing GFP-LC3 and ATG4B WT or C74A, S383A, S392A, S383A-S392A (S-S) mutants, or various C-terminal truncation mutants of ATG4B (as indicated) were cultured with rapamycin (25 μg/ml) for the indicated times. Cell lysates were analyzed by immunoblotting using anti-GFP, anti-ATG4B and anti-actin antibodies. The relative amount of free GFP was quantified and normalized relative to cells expressing ATG4B WT after rapamycin treatment for 18 h. D–F, atg4b−/− MEF cells stably expressing ATG4B WT or C47A, S383A, S392A, S383A-S392A (S-S) mutants, or various truncation mutants of ATG4B (as indicated) were transfected with HA-p62 for 48 h. After treatment with rapamycin (50 μg/ml) for the indicated times, immunoblotting was performed with anti-p62, anti-ATG4B, and anti-actin antibodies.
Next, we turned to the p62 degradation assay as an additional method to monitor autophagy. In this assay, p62 is a known autophagy substrate and inhibition of autophagy correlates with increased levels of p62 (30). To this end, HA-tagged p62 was introduced into WT MEFs or the atg4b−/− MEFs stably expressing WT or various ATG4B mutants. Rapamycin treatment induced degradation of HA-p62 in the atg4b−/− MEFs expressing WT ATG4B (Fig. 4D). In contrast, the introduction of ATG4B catalytic C74A mutant strongly inhibited the degradation of HA-p62. Consistent with results from the GFP-LC3 processing assay, a significant reduction of HA-p62 degradation upon rapamycin treatment was observed in the cells expressing ATG4BS392A, ATG4BS383A/S392A, ATG4B1–389, ATG4B1–379, or ATG4B1–354 mutants, suggesting that autophagy was defective in these cells (Fig. 4, E and F). Taken together, we conclude that both Ser-383 and Ser-392 dependent phosphorylation play important roles in promoting autophagy.
Phosphoregulation of ATG4B Affects LC3 Status in Cells
To further explore the cellular effects of ATG4B phosphorylation, we analyzed the main ATG4B cellular substrate LC3. As explained above, ATG4B is responsible for the initial cleavage of precursor LC3 (pro-LC3) to LC3-I, which is required for the subsequent conjugation of LC3 with membrane-bound phosphatidylethanolamine (PE) (9–11). However, ATG4B also deconjugates LC3-PE (LC3-II), facilitating the release of LC3 from autophagic/lysosomal membranes (7), and thus converts LC3-II into LC3-I. Normally, LC3-II levels can be used to assess autophagic flux, by comparing LC3-II ratios in cells cultured with or without lysosomal inhibitors, with the assumption that if flux is occurring, then the amount of LC3-II will be higher in the presence of lysosome inhibitors (30). However, this assay is not valid as a measure of autophagic flux in cells lacking ATG4B, because of the roles of this protease in cleaving pro-LC3 and more importantly because of the role of ATG4B for hydrolyzing lipids from LC3-II.
Nevertheless, we sought to gain insights about how phosphorylation of ATG4B impacts its regulation of LC3 in cells. To set the stage for these experiments, we examined the consequences of reducing ATG4B expression by shRNA on LC3. First, we tested the effects of ATG4B-targeting shRNAs on cleavage of the engineered substrate actin-LC3B-dNGLUC, finding reduced cleavage (Fig. 5A). Second, we examined endogenous LC3 in cells with partial loss of ATG4B caused by shRNA knockdown. We observed increased steady-state levels of LC3-II levels (>2-fold) when lysosomal inhibitors were applied, without appreciable differences in LC3-I levels (Fig. 5B). Since autophagy is reduced in these cells, we interpret these data to indicate that shRNA-mediated reductions in ATG4B cause an accumulation of the lipidated form of ATG4B, consistent with reduced lipid deconjugation activity.
FIGURE 5.

Phosphorylation-defective ATG4B mutants show alterations in regulation of LC3. A, knocking down ATG4B reduces autophagy as measured by the actin-LC3B-dNGLUC substrate. MCF7 cells expressing a scrambled non-targeting shRNA or two different ATG4B targeting shRNA (1 and 2) were co-transfected with plasmids encoding Actin-LC3B-dNGLUC or Actin-dNGLUC together with CMV-Luc2. After 48 h, the activity of Gaussia luciferase in the culture medium was measured, and normalized to Firefly luciferase activity in the cells, setting the activity for ATG4B WT as 100%. Error bars indicate the S.D. of three independent experiments. ***, p < 0.001. B, knocking down ATG4B increases levels of endogenous LC3-II. Cells in A were treated with lysosomal inhibitors for the indicated times. Cell lysates were analyzed by immunoblotting using anti-LC3, anti-ATG4B, and anti-actin antibodies. The relative amount of LC3-II was quantified by scanning densitometry, normalized to actin and further normalized to cells expressing non-targeting shRNA with lysosomal inhibitor at 4 h (values shown below lanes). C, atg4b−/− MEF cells stably expressing GFP alone, ATG4B WT or ATG4B C74A mutant were cultured in nutrient-rich medium (DMEM), followed by treatment with HBSS starvation for the indicated times. Cell lysates were analyzed by immunoblotting using anti-LC3, anti-ATG4B, and anti-actin antibodies. D, C-terminal truncation or phosphorylation mutants of ATG4B significantly increase the level of endogenous LC3-PE (LC3-II). atg4b−/− MEF cells stably expressing ATG4B WT or various indicated ATG4B mutants were cultured in nutrient-rich medium (DMEM), followed by treatment with or without rapamycin (25 μg/ml) or HBSS starvation, in the presence or absence of lysosomal inhibitors (±L.I., 20 mm NH4Cl and 100 μm leupeptin) for 4 h. Cell lysates normalized for total protein were analyzed by immunoblotting using anti-LC3, anti-ATG4B and anti-actin antibodies. The relative amount of LC3-II in the presence of lysosomal inhibitor was quantified by scanning densitometry, normalized to actin and further normalized to cells expressing ATG4B WT in nutrient-rich, rapamycin treatment or HBSS starvation conditions, respectively, setting the level of LC3-II for ATG4B WT as 1. Error bars indicate the S.D. of three independent experiments. *, p < 0.05; **, p < 0.01.
Next, we explore the impact of ATG4B phosphorylation mutants on endogenous LC3. In agreement with a previous study, atg4b−/− MEFs presented very low levels of LC3-II and an accumulation of pro-LC3 (which has approximately the same electrophoretic mobility as LC3-I), indicating a very significant but not complete defect in LC3 processing (14) (Fig. 5C). Accumulation of pro-LC3 in atg4b−/− MEFs was also observed in these cells expressing GFP-LC3 (Fig. 4A). Note that cleavage of pro-LC3 is required for exposure of the C-terminal residue where lipid conjugation occurs. Introduction into atg4b−/− MEFs of WT ATG4B, but not the C74A catalytic mutant, restored LC3 processing (Figs. 4A and 5C). In cells expressing various ATG4B mutants, LC3-II was present, indicating that these phosphorylation-site mutants retain sufficient protease activity to process pro-LC3 and allow for LC3-I's subsequent lipid conjugation to generate LC3-II (Figs. 4, B and C and 5D). We infer therefore that the cellular phosphorylation of ATG4B does not appear to have a substantially rate-limiting effect on the conversion of pro-LC3 to LC3-I. Surprisingly, when we examined LC3-II content in the atg4b−/− MEFs stably expressing WT ATG4B versus ATG4B phosphorylation site mutants, we found that the steady-state levels of lipidated LC3 were significantly higher in cells expressing the phosphorylation-deficient mutants ATG4BS383A/S392A, ATG4B1–389, ATG4B1–379, and ATG4B1–354 compared with cells expressing ATG4B WT (Fig. 5D). Note that lysosomal protease inhibitors were used to arrest autophagic flux. The S392A mutant showed similar behavior, while Ser-383 had little difference from WT ATG4B. These differences in LC3-II content were more evident when cells were cultured in the rapamycin or HBSS (starvation, Fig. 5D). Since ATG4B is the sole enzyme reported to deconjugate LC3-PE (7), the most likely explanation for these findings with ATG4B phosphorylation site and C-terminal truncation mutants was that the delipidation step that converts LC3-II to LC3-I was defective. Taken together, the main role of cellular phosphorylation of ATG4B may be to control the rate of delipidation of LC3-II, rather than the proteolytic conversion of pro-LC3 to LC3-I.
ATG4B Phosphorylation Affects Interaction with Lipidated LC3
Our studies have demonstrated that loss of ATG4B phosphorylation leads to accumulation of PE-conjugated LC3, which we hypothesized may be caused by defective delipidation of LC3-II. To probe this hypothesis, we investigated whether loss of ATG4B phosphorylation affected its interaction with LC3-II, the lipidated form of LC3. Immunoprecipitation of exogenously expressed WT ATG4B confirmed its association with both non-lipidated and lipidated forms of exogenous LC3 (Fig. 6A). The ATG4BS383A/S392A double phosphorylation mutant also associated with the non-lipidated form of LC3 (LC3-I). However, its interaction with lipidated LC3 (LC3-II) was significantly reduced (Fig. 6A). Furthermore, this difference in interaction between lipidated LC3 with WT versus ATG4BS383A/S392A mutant was also evident upon inducing autophagy by rapamycin treatment or starvation (Fig. 6B) as well as when cells were cultured in the presence of lysosomal inhibitors (Fig. 6C). These results suggest that ATG4B phosphorylation aids its interaction with lipidated LC3. We were unsuccessful with attempts to differentiate interactions of wild-type versus phosphorylation-defective ATG4B with non-lipidated and lipidated GABARAP (not shown).
FIGURE 6.

Phosphorylation-deficient ATG4B mutants show impaired interaction with lipidated LC3. A–C, S383A and S392A mutations attenuate the interaction between ATG4B and PE-conjugated LC3 (LC3-II). Flag-ATG4B or S383A, S392A, S383A-S392A (S-S) mutants were co-transfected with GFP-LC3 for 48 h in HEK293T cells. After transfection, cells were treated with rapamycin or HBSS for 4 h in B, or with or without lysosomal inhibitors for 4 h in C. Cell lysates were subjected to immunoprecipitation with anti-Flag M2 affinity gel. The IP complexes and cell lysates were analyzed by immunoblotting as indicated using anti-LC3, anti-ATG4B, and anti-GFP antibodies. In B and C, the relative amount of pulled-down LC3-II was quantified by scanning densitometry, normalized to the amount in the total lysate, and further normalized to ATG4B WT (values shown below lanes). D, C-terminal region of Atg4B is essential for cellular interaction with LC3. Plasmids encoding Flag-ATG4B full-length or various truncation mutants of ATG4B were co-transfected with GFP-LC3 into HEK293T cells. After 48 h, cell lysates were prepared, subjected to anti-Flag immunoprecipitation, and the immune-complexes were analyzed by immunoblotting as described above.
We similarly sought to determine whether the C-terminal tail of ATG4B is crucial for its interaction with the lipidated forms of LC3. Immunoprecipitation of exogenously expressed ATG4B C-terminal truncated mutants, ATG4B1–389, ATG4B1–379, or ATG4B1–354, showed dramatically reduced association with both LC3-I and LC3-II (Fig. 6D). These results suggest that the C-terminal 39 residues that include Ser-383 and Ser-392 make important contributions to cellular association of ATG4B with both cytosolic and membrane-bound LC3.
Discussion
ATG4B (autophagin-1) is an enzyme that is essential for autophagy in yeast and mammals (3, 4, 15). Though the genomes of mice and humans contain at least four genes encoding ATG4-family proteases, ATG4B (autophagin-1) is the sole enzyme reported to efficiently cleave LC3 precursors and to deconjugate lipid from LC3 (7). However, little is known about the molecular events that control ATG4B enzymatic activity and its cellular functions. In this study, we documented phosphorylation of ATG4B in mammalian cells and characterized the molecular function of the phosphorylated residues with respect to the enzymatic and cellular activities of ATG4B. Our data suggest that phosphorylation of residues located in the C terminus of ATG4B plays a role in promoting autophagy, particularly enhancing the ability of ATG4B to delipidate its endogenous substrate LC3.
One of the significant findings of our study is that phosphorylation regulates ATG4B protease activity against its natural substrate LC3. The evidence for an important role of phosphorylation in controlling ATG4B's LC3-directed protease activity included experiments in which ATG4B recovered from cells was dephosphoryated in vitro using phosphatases and studies of phosphorylation site mutants (particularly the S383A, S392A double mutant) recovered from cells. For these experiments, the protease activity of ATG4B was measured in vitro using an engineered substrate LC3-PLA2 and in intact cells using the substrate actin-LC3B-dNGLUC assay (27, 29, 34). Using these assays, we found that phosphorylation-site mutants of ATG4B showed reduced cellular protease activity. It remains to be defined at the enzymological level how phosphorylation of residues near the C terminus of ATG4B contributes to its protease activity against the substrate LC3. The reported 3D-structure of a fragment of ATG4B (residues 1–354) in complex with LC3 does not provide immediate clues because the C terminus where these phosphorylation sites reside was excluded due to its flexibility that interferes with protein crystallization (33).
Our data suggest that ATG4B phosphoregulation is a starvation response, based on the observation that starvation increased ATG4B phosphorylation. Thus, the endogenous kinases/phosphatases that regulate ATG4B phosphorylation are predicted to be sensors of cellular nutrient status. We also found a significant increase in the overall protease activity of ATG4B under autophagy-inducing conditions, such as starvation, by utilizing the in vitro LC3-PLA2 assay to measure ATG4B activity in cell lysates. Importantly, previous studies have reported a similar result using different assays to measure ATG4B activity, including in vivo actin-LC3B-dNGLUC assay and peptide-conjugated polymeric nanoparticles (29, 35). Our result, however, seems contradictory to another study, which showed that starvation regulated ATG4B activity through a redox mechanism by which starvation induces ROS, thereby attenuating ATG4B activity and blocking LC3 delipidation (22). It is conceivable that both types of post-translational mechanisms, phosphorylation versus oxidation, are relevant under different circumstances, operating to maintain a proper level of LC3 lipidation and thus ensure autophagosome maturation (Fig. 7).
FIGURE 7.
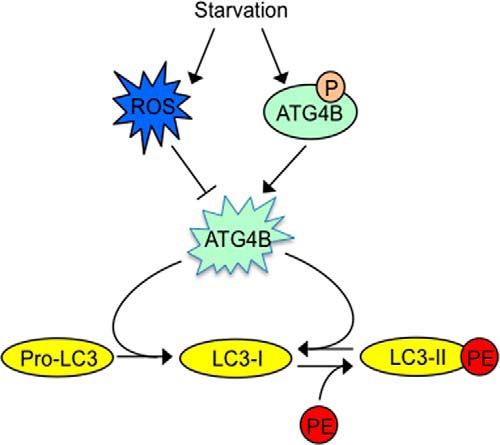
Proposed schematic models for ATG4B regulation of autophagy. We propose a model whereby the rate of autophagic flux is impacted by the activity of ATG4B, and where ATG4B activity is impacted by post-translational modifications that include phosphorylation (positive) and oxidation (negative). Previous studies proposed a model whereby ROS is induced under starvation, thereby attenuating ATG4B activity and causing reduced LC3 delipidation (22). The data reported here reveal that starvation increases phosphorylation of ATG4B, thereby augmenting ATG4B activity, promoting LC3 delipidation. Thus, a well-balanced activation of ATG4B, that maintains the proper level of LC3 lipidation becomes a critical factor for autophagosome maturation.
The Ser-383 and Ser-392 residues located at the C terminus of ATG4B have previously been identified as phosphorylation sites in three separate large-scale phosphoproteomics studies. However, the functional significance of these phosphorylation events was unknown (24–26). By using mutagenesis coupled with Phos-tag SDS-PAGE analysis we confirmed these phosphorylation data. Mutation of Ser-392 to alanine significantly reduces, but not completely abolished, ATG4B phosphorylation on Phos-tag SDS-PAGE, suggesting the presence of additional ATG4B phosphorylation site, presumably Ser-383. However, ATG4BS383A and the C-terminal truncation mutant ATG4B1–389 had a complete disappearance of upper phosphorylation band on Phos-tag, suggesting that Ser-383 and Ser-392 are critical residues for ATG4B phosphorylation. We can surmise that the additional putative phosphorylation sites found by mass spectrometry analysis were perhaps phosphorylated on only a minority of the total pool of ATG4B molecules and thus were not observed in our Phos-tag analysis or they simply were not resolved by the Phos-tag method. Nonetheless, mutagenesis studies suggested those other sites are not critical regulators of ATG4B activity under the conditions we tested. Recognizing that the Phos-tag method does not consistently detect all phosphorylation sites and is non-quantitative, these data nevertheless provide additional support for phosphorylation of ATG4B at Ser-383 and Ser-392, thus supplementing previous proteome-wide phosphorylation analysis.
Using atg4b−/− MEFs as a background to study the function of individual ATG4B phosphorylation mutants, we observed that atg4b−/− MEFs reconstituted with the ATG4BS383A and ATG4BS392A single and ATG4BS383A/S392A double mutant had reduced cellular ATG4B activity and attenuated autophagic flux. Although S383A mutation had a greater effect in ATG4B phosphorylation on the Phos-tag SDS-PAGE, compared with that of S392A mutation, the S392A mutation showed more pronounced reductions in ATG4B activity and atg4b−/− MEFs carrying the S392A mutation had a greater accumulation of LC3-II. Double mutation of S383A and S392A also reduced autophagic flux more than either single mutation, as assessed by using the GFP-LC3 cleavage assay and p62 measurements as biomarkers. The amino-acids sequences encompassing Ser-383 and Ser-392 are evolutionarily conserved across many species (Fig. 3A). The kinase or kinases responsible for phosphorylation of ATG4B at Ser-383 and Ser-392 await identification.
By reconstituting atg4b−/− cells with wild-type versus phosphorylation-site mutants of ATG4B, we also obtained evidence that phosphorylation may be particularly important for enhancing the ability of ATG4B to delipidate LC3. In this regard, while atg4b−/− cells reconstituted with either wild-type or phosphorylation-deficient (S383A/S392A double mutant) ATG4B both produced LC3-I and LC3-II, the relative amount of LC3-II was substantially higher in cells expressing phosphorylation-deficient ATG4B, thus providing evidence of accumulation of the lipidated form of LC3. The accumulation of LC3-II was particularly pronounced when cells expressing phosphorylation-deficient ATG4B were cultured under autophagy-inducing conditions. These conditions normally would accelerate the cycles of LC3 conjugation/deconjugation. In contrast, our data suggest that the residual protease activity of phosphorylation-deficient ATG4B is sufficient for proteolytic conversion of pro-LC3 into LC3-I in cells. We thus speculate that the deconjugation of LC3-PE may require a quantitative or qualitative difference in ATG4B's lipid hydrolyase activity compared with its proteolytic activity against pro-LC3. Our results obtained with the in vitro LC3-PLA2 protease assay suggest that these ATG4B mutants have reduced protease activity, which would support the notion of quantitatively reduced enzyme activity - which presumably includes ATG4B's hydrolyase activity as pertains both to its proteolysis of pro-LC3 and to its delipidation of LC3-II. We propose that when a minimal level of ATG4B remains in the cell, it may be sufficient for the initial proteolytic cleavage step (pro-LC3 → LC3-I) but rate-limiting for the deconjugation step (LC3II → LC3-I). In support of this idea, we and several other groups consistently showed that silencing of ATG4B by siRNA or shRNA significantly increased the steady-state levels of LC3-II (10, 16).
As ATG8-PE is anchored to the membranes, ATG4 has to access the membranes prior to ATG8-PE deconjugation. Therefore, ATG4 seems to possess some structural features suitable for membrane targeting. However, the molecular mechanism of LC3 recognition by ATG4B, especially recognition of the membrane-anchored LC3-PE, remains elusive. Serine to alanine mutation on Ser-383 and Ser-392 significantly inhibited the association of ATG4B with LC3-II but not LC3-I (Fig. 6), and the ATG4BS383A/S392A double mutant had increased levels of LC3-II (Fig. 5), suggesting a role for ATG4B phosphorylation in promoting the binding of ATG4B with LC3-PE and hence aiding in LC3 delipidation. Using the Actin-LC3B-dNGLUC assay, which serves as a cell-based assay to detect the cleavage of both pro-LC3 and LC3-PE (31, 32, 34), we showed that activity of the ATG4BS383A/S392A mutant toward LC3 cleavage was greatly decreased. It is possible that phosphorylation of the flexible C-terminal region of ATG4B induces a conformation where accessibility of ATG4B to its membrane-associated substrate, LC3-PE is more favorable.
The conjugation of LC3 to PE results in the association of LC3 with membrane structures (11). Conversely, the deconjugation of LC3-PE releases LC3 back to the cytosol and ensures a proper supply of LC3 (4, 36). It has previously been shown that overexpression of ATG4B negatively affects LC3 lipidation, indicating that excess ATG4B efficiently deconjugates the LC3 from PE and decreases membrane association of LC3, hampering autophagy (7, 15). On the other hand, studies in yeast showed that a failure in this deconjugation process leads to accumulation of PE-conjugated ATG8, and also results in a reduction in LC3 recycling and hence a reduction in autophagy (37, 38). In this study, we found that loss of ATG4B phosphorylation attenuated ATG4B activity, leading to a defect in the deconjugation of LC3-PE and hence accumulation of excess LC3-PE, resulting in a decrease in autophagy flux. Therefore, optimal autophagosome formation requires a proper balance between LC3 lipidation and delipidation reactions and ATG4B phosphoregulation may be among the mechanisms by which this critical balance is controlled.
In summary, our work has provided novel insights into the mechanism of autophagy regulation, identifying phosphorylation of the evolutionarily conserved C-terminal region of ATG4B as a means for controlling the balance between lipidated and non-lipidated LC3 and thus impacting autophagic flux.
Author Contributions
Z. Y., R. W. G., S. M., and J. C. R. designed the study and wrote the paper. Z. Y., R. W. G., T. Y., and C. W. S. performed and analyzed the experiments. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Dr. Carlos López-Otín for providing ATG4B heterozygous mice. We thank Dr. Ze'ev Ronai for providing atg4b+/+ and atg4b−/− MEFs and helpful discussions. We thank Yasuko Matsuzawa for technical assistance and Dr. Khatereh Motamedchaboki for mass spectrometry-based proteomic analysis.
This work was supported by National Institutes of Health Grant AI-091967. The authors declare that they have no conflicts of interest with the contents of this article.
- ATG
- autophagin
- CIP
- calf intestinal alkaline phosphatase
- PS
- phosphatase inhibitor
- PE
- phosphatidylethanolamine
- ARH
- aplasia Ras homolog.
References
- 1.Levine B., and Klionsky D. J. (2004) Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev. Cell 6, 463–477 [DOI] [PubMed] [Google Scholar]
- 2.Yang Z., and Klionsky D. J. (2010) Eaten alive: a history of macroautophagy. Nature Cell Biol. 12, 814–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ichimura Y., Kirisako T., Takao T., Satomi Y., Shimonishi Y., Ishihara N., Mizushima N., Tanida I., Kominami E., Ohsumi M., Noda T., and Ohsumi Y. (2000) A ubiquitin-like system mediates protein lipidation. Nature 408, 488–492 [DOI] [PubMed] [Google Scholar]
- 4.Kirisako T., Ichimura Y., Okada H., Kabeya Y., Mizushima N., Yoshimori T., Ohsumi M., Takao T., Noda T., and Ohsumi Y. (2000) The reversible modification regulates the membrane-binding state of Apg8/Aut7 essential for autophagy and the cytoplasm to vacuole targeting pathway. J. Cell Biol. 151, 263–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hanada T., Noda N. N., Satomi Y., Ichimura Y., Fujioka Y., Takao T., Inagaki F., and Ohsumi Y. (2007) The Atg12–Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J. Biol. Chem. 282, 37298–37302 [DOI] [PubMed] [Google Scholar]
- 6.Fujita N., Itoh T., Omori H., Fukuda M., Noda T., and Yoshimori T. (2008) The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol. Biol. Cell 19, 2092–2100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kabeya Y., Mizushima N., Yamamoto A., Oshitani-Okamoto S., Ohsumi Y., and Yoshimori T. (2004) LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J. Cell Sci. 117, 2805–2812 [DOI] [PubMed] [Google Scholar]
- 8.Weidberg H., Shvets E., Shpilka T., Shimron F., Shinder V., and Elazar Z. (2010) LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J. 29, 1792–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hemelaar J., Lelyveld V. S., Kessler B. M., and Ploegh H. L. (2003) A single protease, Apg4B, is specific for the autophagy-related ubiquitin-like proteins GATE-16, MAP1-LC3, GABARAP, and Apg8L. J. Biol. Chem. 278, 51841–51850 [DOI] [PubMed] [Google Scholar]
- 10.Tanida I., Ueno T., and Kominami E. (2004) Human light chain 3/MAP1LC3B is cleaved at its carboxyl-terminal Met121 to expose Gly120 for lipidation and targeting to autophagosomal membranes. J. Biol. Chem. 279, 47704–47710 [DOI] [PubMed] [Google Scholar]
- 11.Kabeya Y., Mizushima N., Ueno T., Yamamoto A., Kirisako T., Noda T., Kominami E., Ohsumi Y., and Yoshimori T. (2000) LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19, 5720–5728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mariño G., Uría J. A., Puente X. S., Quesada V., Bordallo J., and López-Otin C. (2003) Human autophagins, a family of cysteine proteinases potentially implicated in cell degradation by autophagy. J. Biol. Chem. 278, 3671–3678 [DOI] [PubMed] [Google Scholar]
- 13.Li M., Hou Y., Wang J., Chen X., Shao Z. M., and Yin X. M. (2011) Kinetics comparisons of mammalian Atg4 homologues indicate selective preferences toward diverse Atg8 substrates. J. Biol. Chem. 286, 7327–7338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mariño G., Fernández A. F., Cabrera S., Lundberg Y. W., Cabanillas R., Rodríguez F., Salvador-Montoliu N., Vega J. A., Germanà A., Fueyo A., Freije J. M., and López-Otin C. (2010) Autophagy is essential for mouse sense of balance. J. Clin. Invest. 120, 2331–2344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tanida I., Sou Y. S., Ezaki J., Minematsu-Ikeguchi N., Ueno T., and Kominami E. (2004) HsAtg4B/HsApg4B/autophagin-1 cleaves the carboxyl termini of three human Atg8 homologues and delipidates microtubule-associated protein light chain 3- and GABAA receptor-associated protein-phospholipid conjugates. J. Biol. Chem. 279, 36268–36276 [DOI] [PubMed] [Google Scholar]
- 16.Martinez-Lopez N., Athonvarangkul D., Mishall P., Sahu S., and Singh R. (2013) Autophagy proteins regulate ERK phosphorylation. Nature Commun. 4, 2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fujita N., Hayashi-Nishino M., Fukumoto H., Omori H., Yamamoto A., Noda T., and Yoshimori T. (2008) An Atg4B mutant hampers the lipidation of LC3 paralogues and causes defects in autophagosome closure. Mol. Biol. Cell 19, 4651–4659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fujita N., Noda T., and Yoshimori T. (2009) Atg4B(C74A) hampers autophagosome closure: a useful protein for inhibiting autophagy. Autophagy 5, 88–89 [DOI] [PubMed] [Google Scholar]
- 19.Lu Z., Luo R. Z., Lu Y., Zhang X., Yu Q., Khare S., Kondo S., Kondo Y., Yu Y., Mills G. B., Liao W. S., and Bast R. C. Jr. (2008) The tumor suppressor gene ARHI regulates autophagy and tumor dormancy in human ovarian cancer cells. J. Clin. Invest. 118, 3917–3929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuang E., Okumura C. Y., Sheffy-Levin S., Varsano T., Shu V. C., Qi J., Niesman I. R., Yang H. J., López-Otin C., Yang W. Y., Reed J. C., Broday L., Nizet V., and Ronai Z. A. (2012) Regulation of ATG4B stability by RNF5 limits basal levels of autophagy and influences susceptibility to bacterial infection. PLoS genetics 8, e1003007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guo L., Huang J. X., Liu Y., Li X., Zhou S. R., Qian S. W., Liu Y., Zhu H., Huang H. Y., Dang Y. J., and Tang Q. Q. (2013) Transactivation of Atg4b by C/EBPbeta promotes autophagy to facilitate adipogenesis. Mol. Cell. Biol. 33, 3180–3190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scherz-Shouval R., Shvets E., Fass E., Shorer H., Gil L., and Elazar Z. (2007) Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 26, 1749–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dai J., Jin W. H., Sheng Q. H., Shieh C. H., Wu J. R., and Zeng R. (2007) Protein phosphorylation and expression profiling by Yin-yang multidimensional liquid chromatography (Yin-yang MDLC) mass spectrometry. J. Proteome Res. 6, 250–262 [DOI] [PubMed] [Google Scholar]
- 24.Olsen J. V., Blagoev B., Gnad F., Macek B., Kumar C., Mortensen P., and Mann M. (2006) Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127, 635–648 [DOI] [PubMed] [Google Scholar]
- 25.Villén J., Beausoleil S. A., Gerber S. A., and Gygi S. P. (2007) Large-scale phosphorylation analysis of mouse liver. Proc. Natl. Acad. Sci. U.S.A. 104, 1488–1493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gnad F., Ren S., Cox J., Olsen J. V., Macek B., Oroshi M., and Mann M. (2007) PHOSIDA (phosphorylation site database): management, structural and evolutionary investigation, and prediction of phosphosites. Genome Biol. 8, R250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shu C. W., Drag M., Bekes M., Zhai D., Salvesen G. S., and Reed J. C. (2010) Synthetic substrates for measuring activity of autophagy proteases: autophagins (Atg4). Autophagy 6, 936–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fan W., Tang Z., Chen D., Moughon D., Ding X., Chen S., Zhu M., and Zhong Q. (2010) Keap1 facilitates p62-mediated ubiquitin aggregate clearance via autophagy. Autophagy 6, 614–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ketteler R., Sun Z., Kovacs K. F., He W. W., and Seed B. (2008) A pathway sensor for genome-wide screens of intracellular proteolytic cleavage. Genome Biol. 9, R64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Klionsky D. J., Abdalla F. C., Abeliovich H., Abraham R. T., Acevedo-Arozena A., Adeli K., Agholme L., Agnello M., Agostinis P., Aguirre-Ghiso J. A., et al (2012) Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 8, 445–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kumanomidou T., Mizushima T., Komatsu M., Suzuki A., Tanida I., Sou Y. S., Ueno T., Kominami E., Tanaka K., and Yamane T. (2006) The crystal structure of human Atg4b, a processing and de-conjugating enzyme for autophagosome-forming modifiers. J. Mol. Biol. 355, 612–618 [DOI] [PubMed] [Google Scholar]
- 32.Sugawara K., Suzuki N. N., Fujioka Y., Mizushima N., Ohsumi Y., and Inagaki F. (2005) Structural basis for the specificity and catalysis of human Atg4B responsible for mammalian autophagy. The J. Biol. Chem. 280, 40058–40065 [DOI] [PubMed] [Google Scholar]
- 33.Satoo K., Noda N. N., Kumeta H., Fujioka Y., Mizushima N., Ohsumi Y., and Inagaki F. (2009) The structure of Atg4B-LC3 complex reveals the mechanism of LC3 processing and delipidation during autophagy. EMBO J. 28, 1341–1350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ketteler R., and Seed B. (2008) Quantitation of autophagy by luciferase release assay. Autophagy 4, 801–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Choi K. M., Nam H. Y., Na J. H., Kim S. W., Kim S. Y., Kim K., Kwon I. C., and Ahn H. J. (2011) A monitoring method for Atg4 activation in living cells using peptide-conjugated polymeric nanoparticles. Autophagy 7, 1052–1062 [DOI] [PubMed] [Google Scholar]
- 36.Yu Z. Q., Ni T., Hong B., Wang H. Y., Jiang F. J., Zou S., Chen Y., Zheng X. L., Klionsky D. J., Liang Y., and Xie Z. (2012) Dual roles of Atg8-PE deconjugation by Atg4 in autophagy. Autophagy 8, 883–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nair U., Yen W. L., Mari M., Cao Y., Xie Z., Baba M., Reggiori F., and Klionsky D. J. (2012) A role for Atg8-PE deconjugation in autophagosome biogenesis. Autophagy 8, 780–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakatogawa H., Ishii J., Asai E., and Ohsumi Y. (2012) Atg4 recycles inappropriately lipidated Atg8 to promote autophagosome biogenesis. Autophagy 8, 177–186 [DOI] [PubMed] [Google Scholar]