

Background: Ac2PIM signals via TLR2 to direct both pro- and anti-inflammatory responses.
Results: Ac2PIM induces miR-150/143 via SRC-FAK-PYK2-CREB-P300 signaling to target RIP2 and TAK1 and subdues MDP-stimulated PI3K-PKC-MAPK-β-catenin axis.
Conclusion: Ac2PIM-mediated TLR2 signaling suppresses the NOD2-induced immunomodulators viz. COX-2, SOCS-3, and MMP-9.
Significance: TLR2-NOD2 crosstalk accentuated the utilities of Ac2PIM and MDP as vaccine adjuvant.
Keywords: innate immunity, macrophage, microRNA (miRNA), Nod-like receptor (NLR), signal transduction, toll-like receptor (TLR)
Abstract
Specific and coordinated regulation of innate immune receptor-driven signaling networks often determines the net outcome of the immune responses. Here, we investigated the cross-regulation of toll-like receptor (TLR)2 and nucleotide-binding oligomerization domain (NOD)2 pathways mediated by Ac2PIM, a tetra-acylated form of mycobacterial cell wall component and muramyl dipeptide (MDP), a peptidoglycan derivative respectively. While Ac2PIM treatment of macrophages compromised their ability to induce NOD2-dependent immunomodulators like cyclooxygenase (COX)-2, suppressor of cytokine signaling (SOCS)-3, and matrix metalloproteinase (MMP)-9, no change in the NOD2-responsive NO, TNF-α, VEGF-A, and IL-12 levels was observed. Further, genome-wide microRNA expression profiling identified Ac2PIM-responsive miR-150 and miR-143 to target NOD2 signaling adaptors, RIP2 and TAK1, respectively. Interestingly, Ac2PIM was found to activate the SRC-FAK-PYK2-CREB cascade via TLR2 to recruit CBP/P300 at the promoters of miR-150 and miR-143 and epigenetically induce their expression. Loss-of-function studies utilizing specific miRNA inhibitors establish that Ac2PIM, via the miRNAs, abrogate NOD2-induced PI3K-PKCδ-MAPK pathway to suppress β-catenin-mediated expression of COX-2, SOCS-3, and MMP-9. Our investigation has thus underscored the negative regulatory role of Ac2PIM-TLR2 signaling on NOD2 pathway which could broaden our understanding on vaccine potential or adjuvant utilities of Ac2PIM and/or MDP.
Introduction
Pattern recognition receptor (PRR)3 signaling is known to orchestrate the innate immune responses in an antigen-presenting cell like macrophage (1). Since the microbial pathogens usually harbor several pathogen-associated molecular patterns (PAMPs), it is likely that multiple PRRs are activated during infection of macrophages. Hence, signaling cascades downstream of the PRRs and their possible crosstalks essentially and effectively regulate the cell-fate and immune responses (2, 3). Among the PRRs, reports suggest extensive crosstalks of toll-like receptors (TLRs) with other PRRs (2, 4, 5). Despite several known nodes of interaction of TLRs with other PRRs, presence of multiple ligands/PAMPs for some of the TLRs like TLR2 presents an interesting ligand-specific response that could differentially fine-tune immune responses (6–8).
Phosphatidylmyo-inositol mannosides (PIMs) are the mycobacterial cell wall glycolipids recognized by TLR2. PIM2 and PIM6 constitute the abundant classes of biosynthetic intermediates of lipomannan in mycobacteria and tetra-acylated PIMs like Ac2PIM (previously called as Ac4PIM2) are abundant forms of PIMs (9, 10). During infection, PIMs traffic out of the macrophages and mediate signaling in the bystander uninfected cells, thus act as an important modulator of immune responses during mycobacterial infection (10, 11). Functional consequences of PIM-induced TLR2 signaling span both pro- and anti-inflammatory capacities (10, 12–15). Importantly, stimulation of PIM-TLR2 pathway down-regulates TLR4-induced pro-inflammatory signals (13). However, mechanistic insights of such PIM-mediated crosstalk of TLR2 with other PRRs need extensive investigations. Interactions of TLR2 and nucleotide-binding oligomerization domain (NOD)2 signaling are reported in several contexts. Surprisingly, both synergistic and antagonistic crosstalks of the two PRRs are known (7, 8, 16).
NOD2 senses the bacterial peptidoglycan, muramyl dipeptide (MDP) (17, 18). On recognition of the cognate ligand, NOD2 oligomerizes and activates the specific adaptor, receptor-interacting protein 2 (RIP2). RIP2 then recruits and activates the transforming growth factor beta-activated kinase 1 (TAK1) complex to mediate the downstream signaling including MAPK and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) which in turn orchestrate NOD2-induced immune responses (18, 19). NOD2 activation translates to the expression of several immunomodulators like cyclooxygenase (COX)-2 (20), suppressor of cytokine signaling (SOCS)-3 (21), matrix metalloproteinase (MMP)-9 (22), inducible nitric-oxide synthase catalyzed NO (23), cytokines like TNF-α (10, 24), VEGF-A (25), and IL-12 (24, 26). While NO and cytokines like TNF-α, IL-12 mediate the NOD2-responsive pro-inflammatory responses, COX-2, SOCS-3, MMP-9, and VEGF-A constitutes the anti-inflammatory arm of the NOD2 responses. COX-2 catalyzes the rate-limiting step of conversion of arachidonic acid to prostaglandin E2. Prostaglandin E2 directs several anti-inflammatory responses in macrophages (27). SOCS-3 functions as a negative regulator of several cytokines or a negative feedback for various signaling pathways including the TLRs (28). Zn2+- and Ca2+-dependent MMP-9 is a class of endopeptidase that mediate both pro- and anti-inflammatory functions (29).
In the current study, we attempted to unravel the crosstalk, if any, between Ac2PIM-mediated TLR2 signaling and MDP-induced NOD2 pathway. Interestingly, we found that macrophages that were stimulated with Ac2PIM displayed marked reduction in its ability to express NOD2-responsive immunomodulators like COX-2, SOCS-3, and MMP-9 but not NO, TNF-α, VEGF-A, or IL-12. This underscores the differential regulatory abilities of Ac2PIM-induced TLR2 pathway on NOD2 signaling. Ac2PIM indeed suppressed NOD2 responses by downregulating the expression of RIP2 and TAK1, adaptors of NOD2 signaling. Importantly, we identified Ac2PIM-induced post-transcriptional mechanism presented by microRNAs, miR-150 and miR-143, that targeted RIP2 and TAK1, respectively. Deciphering the molecular mechanism, we found that Ac2PIM signals via the TLR2-SRC-focal adhesion kinase (FAK)-protein tyrosine kinase 2 (PYK2)-cAMP response element-binding protein (CREB) pathway to mediate the recruitment of a coactivator complex with intrinsic histone acetyltransferase (HAT) functions, CREB-binding protein (CBP)/P300 to the promoters of miR-150 and miR-143. Further, NOD2-induced PI3K-PKC-MAPK-β-catenin signaling axis, that was found to be crucial for the expression of the immunomodulators, was significantly inhibited in the presence of Ac2PIM. Together, this study has generated avenues to evaluate the vaccine potential and adjuvant utilities of Ac2PIM and/or MDP.
Experimental Procedures
Cells and Mice
Brewer thioglycollate (8%)-elicited primary macrophages were obtained from peritoneal exudates of wild-type (C3H/HeJ or C57BL/6J) or tlr2-KO mice. Murine RAW 264.7 macrophages cell line was obtained from National Center for Cell Sciences, Pune, India. Macrophages were cultured in DMEM (Gibco, Life Technologies) supplemented with 10% heat-inactivated FBS (Gibco, Life Technologies) and maintained at 37 °C in 5% CO2 incubator. All strains of mice were purchased from The Jackson Laboratory and maintained in the Central Animal Facility (CAF), Indian Institute of Science (IISc). All studies involving mice were performed after the approval from the Institutional Ethics Committee for animal experimentation as well as from Institutional Biosafety Committee.
Reagents and Antibodies
General laboratory chemicals were obtained from Sigma-Aldrich, Merck Millipore, HiMedia, or Promega. Tissue culture plasticware was purchased from Corning Inc. or Tarsons Products Pvt Ltd. MDP was purchased from Sigma-Aldrich. Ac2PIM was procured from Dr. Martine Gilleron and Dr. Germain Puzo (IPBS, France). Anti-MMP-9 and HRP-conjugated anti-β-ACTIN antibodies were purchased from Sigma-Aldrich and HRP-conjugated anti-rabbit IgG antibody was purchased from Jackson ImmunoResearch. Anti-COX-2 was from Calbiochem. Anti-P300 was from Merck Millipore. Anti-Ser176 phospho-RIP2, anti-RIP2, anti-Ser412 phospho-TAK1, anti-TAK1, anti-SOCS-3, anti-Tyr458/199 phospho-p85, anti-p85, anti-Thr70 phospho-4EBP1, anti-4EBP1, anti-Thr505 phospho-PKCδ, anti-Thr202/Tyr204 phospho-ERK1/2, anti-ERK1/2, anti-Thr180/Tyr182 phospho-p38, anti-p38, anti-Tyr701 phospho-STAT1, anti-Tyr705 phospho-STAT3, anti-Ser33/37/Thr41 phospho-β-catenin, anti-Ser9 phospho-GSK-3β, anti-Tyr397 phospho-FAK, anti-Tyr402 phospho-PYK2, anti-Tyr416 phospho-SRC, anti-Ser133 phospho-CREB, and anti-H3K18ac antibodies were obtained from Cell Signaling Technology. TNF-α, VEGF-A and IL-12 ELISA kits were purchased from PeproTech.
Treatment with Pharmacological Reagents
Cells were treated with the given pharmacological inhibitors (all from Calbiochem) 1 h prior to the experimental treatments at following concentrations: PP2 (10 μm), LY294002 (50 μm), Rapamycin (100 nm), Rottlerin (10 μm), U0126 (10 μm), SB203580 (10 μm), SP600125 (50 μm), β-catenin inhibitor (15 μm), LiCl (5 mm), AG490 (10 μm), HAT inhibitor (5 μm), and FAK inhibitor (10 μm). DMSO at 0.1% concentration was used as the vehicle control. In all experiments involving pharmacological reagents, a tested concentration was used after careful titration experiments assessing the viability of the macrophages using the MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay.
Ripk2 and Map3k7 3′-UTR WT and Mutation Generation
The 3′-UTRs of Ripk2 and Map3k7 were PCR amplified and cloned into pmirGLO vector using the restriction enzymes SacI and XbaI. Primer pairs used: WT Ripk2 3′UTR forward 5′-cgagctcgcaccgccttcaaatttccc-3′, reverse 5′-gctctagagcaacgtcatgggaagagact-3′; WT Map3k7 3′-UTR forward 5′-cgagctcagcagatgatggcacctgt-3′, reverse 5′-gctctagaagtcagtaaacttgctctcct-3′. The miR-150 and miR-143 binding sites were mutated in Ripk2 and Map3k7 3′-UTR, respectively by nucleotide replacements through site-directed mutagenesis using the megaprimer inverse PCR method. The forward primer comprised the desired mutation and respective reverse primer was used to generate megaprimers. Primer pairs used: Ripk2 megaprimer forward 5′-ccttctggtttaggaagtca-3′, reverse 5′-caacgtcatgggaagagact-3′; Map3k7 megaprimer forward 5′-aaagtgctcgcttcaaaaatct-3′, reverse 5′-ctgccaccactcaccttta-3′. The megaprimer was further used to amplify the WT Ripk2 and Map3k7 3′UTR plasmid and generate the miR-150Δ Ripk2 and miR-143Δ Map3k7 3′UTR plasmids, respectively.
Transfection Studies
Murine RAW 264.7 macrophages were transfected with WT Ripk2 3′-UTR, WT Map3k7 3′-UTR, miR-150Δ Ripk2 3′UTR, miR-143Δ Map3k7 3′-UTR, β-galactosidase, 100 nm miRNA inhibitors (miR-150, miR-143, or NC inhibitor from Ambion, Life Technologies) or miRNA mimics (miR-26a, miR-150, miR-143, or NC mimics from Ambion, Life Technologies) using low m. w. polyehtylenimine (Sigma-Aldrich) as indicated. In all cases, 36 h post-transfection, the cells were treated as indicated and processed for analysis.
Luciferase Assay
Cells were lysed in Reporter lysis buffer (Promega) and assayed for luciferase activity using Luciferase Assay Reagent (Promega) as per the manufacturer's instructions. The results were normalized for transfection efficiencies measured by β-galactosidase activity. O-nitrophenol β-d-galactopyranoside (HiMedia) was utilized for the β-galactosidase assay.
RNA Isolation and Real-Time qRT-PCR
Total RNA from macrophages was isolated using TRI reagent (Sigma-Aldrich). First strand cDNA synthesis was done with 1 μg of total RNA using First Strand cDNA synthesis kit (Applied Biological Materials Inc.). Expression of target gene was assessed by Real-Time quantitative Reverse Transcription-PCR (qRT-PCR) using SYBR Green PCR mix (KAPA Biosystems). All the experiments were repeated at least three times independently to ensure the reproducibility of the results. Gapdh was used as internal control. The primers used for Real-Time qRT-PCR amplification were as follows: Gapdh forward 5′-gagccaaacgggtcatcatct-3′, reverse 5′-gaggggccatccacagtctt-3′; Ripk2 forward 5′-gccattgagattccgcatcct-3′, reverse 5′-aacttcgtgattgagagagtgac-3′ and Map3k7 forward 5′-cggatgagccgttacagtatc, reverse 5′-actccaagcgtttaatagtgtcg-3′. All the primers were purchased from Eurofins Genomics.
miRNA Expression Profiling
Total RNA was isolated from untreated, MDP-treated, and Ac2PIM and MDP co-treated macrophages (n = 2). Sample and reference RNAs were labeled with Hy3 and Hy5, respectively using miRCURY LNATM array power labeling kit (Exiqon). Sample and reference RNA hybridization was carried out in Tecan HS4800 hybridization station (Tecan). The miRCURY LNATM array microarray slides were scanned using a G2565BA microarray scanner system (Agilent) and ImaGene (version 7.0) software (BioDiscovery) was used for image analysis. The log median ratio of Hy3/Hy5 intensity for replicative spots of each miRNA and the fold change in the log median ratio for each sample was calculated. miRNAs that exhibited increased fold expression in the Ac2PIM-MDP co-treated samples when compared with MDP alone were clustered and represented in the heat map. Data obtained were analyzed by significance analysis of microarrays (SAM) to identify differentially regulated miRNAs.
Quantification of miRNA Expression
Total RNA was isolated from macrophages using TRI reagent. Real-Time qRT-PCR for miR-26a, miR-150, and miR-143 was performed using specific TaqMan miRNA assays (Ambion, Life Technologies) as per manufacturer's instructions. U6 snRNA was used as internal control.
Immunoblotting Analysis
Cells were washed with 1× PBS and lysed in RIPA buffer (50 mm Tris-HCl (pH 7.4), 1% Nonidet P-40, 0.25% sodium deoxycholate, 150 mm NaCl, 1 mm EDTA, 1 mm PMSF, 1 μg/ml each of aprotinin, leupeptin, pepstatin, 1 mm Na3VO4, 1 mm NaF) on ice for 30 min. Whole cell lysates were collected. After estimation of total protein by Bradford reagent, equal amount of protein from each cell lysate was resolved on 12% SDS-PAGE and transferred onto PVDF membranes (Millipore) by semi-dry immunoblotting method (Bio-Rad). 5% nonfat dry milk powder in TBST (20 mm Tris-HCl (pH 7.4), 137 mm NaCl, and 0.1% Tween 20) was used for blocking nonspecific binding for 60 min. After washing with TBST, the blots were incubated overnight at 4 °C with primary antibody diluted in TBST with 5% BSA. After washing with TBST, blots were incubated with anti-rabbit secondary antibody conjugated to HRP for 2 h. The immunoblots were developed with enhanced chemiluminescence detection system (PerkinElmer) as per manufacturer's instructions. β-Actin was used as loading control.
Enzyme Immunoassay (ELISA)
Cell-free culture supernatants were used for performing ELISA for TNF-α, VEGF-A, and IL-12 (kits from PeproTech) as per the manufacturer's instructions. Briefly, 96-well flat bottom plates (Nunc MaxiSorp, Thermo Scientific) were coated with specific capture antibodies overnight at 4 °C followed by three washes with 1× PBST (1× PBS with 0.05% Tween 20). After blocking with 1% BSA for 1 h at room temperature, wells were washed, and incubated with cell-free culture supernatants for 2 h. After three washes with 1× PBST, wells were incubated with respective detection antibodies for 2 h at room temperature. Further, the wells were washed and incubation with streptavidin-HRP antibody for 30 min at room temperature. The reactions were developed with 3,3′,5,5′-tetramethylbenzidine (Sigma-Aldrich) and the absorbance was measured at 450 nm using an ELISA reader (Molecular Devices).
Estimation of NO
Cell-free culture supernatants were used for estimating NO produced by macrophage. Greiss reagent (Promega) was used to assay NO production according to the manufacturer's instructions. Briefly, nitrite standards and cell-free supernatants were added to 96-well flat bottom plates (Nunc MaxiSorp, Thermo Scientific). The samples were incubated with equilibrated sulfanilamide solution for 10 min in dark at room temperature. Further, N-1-napthylethylenediamine dihydrochloride solution was added and incubated in dark for 10 min before measuring the absorbance at 520 nm.
Chromatin Immunoprecipitation (ChIP) Assay
ChIP assays were carried out using a protocol provided by Upstate Biotechnology and Sigma-Adrich with certain modifications. Briefly, macrophages were fixed with 1.42% formaldehyde for 15 min at room temperature followed by quenching of formaldehyde with addition of 125 mm glycine. Nuclei were lysed in 0.1% SDS lysis buffer (50 mm Tris-HCl (pH 8.0), 200 mm NaCl, 10 mm HEPES (pH 6.5), 0.1% SDS, 10 mm EDTA, 0.5 mm EGTA, 1 mm PMSF, 1 μg/ml of each aprotinin, leupeptin, pepstatin, 1 mm Na3VO4, and 1 mm NaF). Chromatin was sheared using Bioruptor Plus (Diagenode) at high power for 70 rounds of 30 s pulse ON/45 s OFF cycle at 4 °C. Chromatin extracts containing DNA fragments with an average size of 200–500 bp were immunoprecipitated using P300- or pCREB- or H3K18ac-specific antibodies or rabbit preimmune sera complexed with protein A-agarose beads (Merck Millipore). Immunoprecipitated complexes were sequentially washed (Wash Buffer A: 50 mm Tris-HCl (pH 8.0), 500 mm NaCl, 1 mm EDTA, 1% Triton X-100, 0.1% Sodium deoxycholate, 0.1% SDS, and protease/phosphatase inhibitors; Wash Buffer B: 50 mm Tris-HCl (pH 8.0), 1 mm EDTA, 250 mm LiCl, 0.5% Nonidet P-40, 0.5% sodium deoxycholate, and protease/phosphatase inhibitors; TE: 10 mm Tris-HCl (pH 8.0), 1 mm EDTA with protease inhibitors) at 4 °C and eluted in elution buffer (1% SDS, 0.1 m NaHCO3) at 65 °C. The eluted sample was treated with RNase A and Proteinase K, DNA was precipitated using phenol-chloroform-ethanol method. Purified DNA was analyzed for CREB-binding sites on the promoters of miR-150 and miR-143 by Real-Time qRT-PCR. Primers used: at miR-150 promoter forward 5′-gaactgaatcctttgacctctac-3′, reverse 5′-gaactgaatcctttgacctctac-3′; at miR-143 promoter forward 5′-gacaaagaggcaggggacg-3′, reverse 5′-cagtaagtagctaggagtggtg-3′. All values in the test samples were normalized to amplification of the specific gene in Input and IgG pull down and represented as fold change in enrichment or modification.
Statistical Analysis
Levels of significance for comparison between samples were determined by the Student's t test distribution and one-way ANOVA. The data in the graphs are expressed as the mean ± S.E. for values from three independent experiments and p values < 0.05 were defined as significant. GraphPad Prism 5.0 software (GraphPad Software) was used for all the statistical analysis.
Results
Ac2PIM Inflects NOD2-induced Immunomodulators
Confounding to several studies where PIM2 was found to be pro-inflammatory in its function (10, 12), several members of PIM family that are TLR2 agonists are majorly anti-inflammatory in nature and can suppress pro-inflammatory responses mediated by other PRRs (13–15). We sought to analyze the cross-regulation, if any, between PIM-induced TLR2 signaling and another PRR signaling like NOD2 pathway. While Ac2PIM, one of the abundant PIMs of mycobacteria (9), was utilized as TLR2 agonist, MDP was utilized as the NOD2 agonist. Canonical NOD2 activation leads to the recruitment and active phosphorylation of the cytosolic adaptor proteins, RIP2 and TAK1. Interestingly, prior activation of TLR2 signaling by Ac2PIM significantly suppressed MDP-induced active phosphorylation of RIP2 and TAK1 (Fig. 1A). To establish if such inhibition had effects on the downstream responses, we chose to analyze the known NOD2-responsive immunomodulatory genes such as COX-2, SOCS-3, MMP-9. In accordance with the existing literature, MDP-NOD2 failed to induce the expression of COX-2, SOCS-3, and MMP-9 in presence of the RIP2 inhibitor, PP2 (Fig. 1B). Surprisingly, though MDP or Ac2PIM treatment alone induced the expression of COX-2, SOCS-3, and MMP-9 in macrophages, prior engagement of TLR2 with Ac2PIM significantly down-regulated the ability of NOD2 to induce these genes (Fig. 1C). Ability of Ac2PIM to signal through TLR2 to mediate such functions was confirmed in macrophages derived from tlr2-KO mice wherein pretreatment of Ac2PIM failed to suppress the expression of the candidate genes (Fig. 1D). However, other NOD2-responsive immunomodulators like NO, TNF-α, VEGF-A, and IL-12 remained unchanged during co-treatment with Ac2PIM (Fig. 1, E–H). These results collectively indicate a differential regulation of NOD2 responses by Ac2PIM-induced TLR2 signaling.
FIGURE 1.

Ac2PIM differentially regulates NOD2-induced immune response. A, peritoneal macrophages were treated with Ac2PIM followed by MDP for the indicated time points. Lysates were analyzed for pRIP2 and pTAK1 by immunoblotting. B-D, immunoblot analysis of COX-2, SOCS-3, and MMP-9 was performed on lysates obtained from macrophages after the following treatments: treatment with pharmacological inhibitor PP2 prior to 12 h MDP addition (B), treatment with indicated concentrations of Ac2PIM followed by 12 h treatment of MDP (C), and pretreatment of macrophages from C57BL/6J WT and tlr2-KO mice with Ac2PIM followed by MDP for 12 h (D). E–H, macrophages were pretreated with indicated concentration of Ac2PIM followed by MDP for 12 h. Cell-free supernatants were analyzed for NO by Greiss reagent (E), TNF-α (F), VEGF-A (G), and IL-12 (H) by ELISA. All data represent the mean ± S.E. from three independent experiments, ***, p < 0.001 (one-way ANOVA) and all blots are representative of three independent experiments. The cells were treated with 2 μg/ml Ac2PIM unless mentioned otherwise for 2 h followed by 200 ng/ml MDP. Med, medium; WT, wild-type; KO, knock-out; DMSO, dimethyl sulfoxide.
Ac2PIM-responsive miRNAs Target RIP2 and TAK1 to Down-regulate NOD2 Responses
Further, we investigated the molecular mechanism that govern the Ac2PIM-induced suppression of NOD2 signaling. As Ac2PIM abrogated the active phosphorylation of NOD2-induced RIP2 and TAK1 (Fig. 1A), transcript and total protein of RIP2 and TAK1 were assessed. Interestingly, while no significant difference in Ripk2 and Map3k7 transcripts were observed with individual or co-treatment of Ac2PIM and MDP (Fig. 2A), marked reduction in the total RIP2 and TAK1 protein was observed when the cells were treated with Ac2PIM prior to MDP (Fig. 2B). This indicated a possible involvement of post-transcriptional regulation, like those mediated by miRNAs, of RIP2 and TAK1 by Ac2PIM-induced TLR2 signaling. In this regard, we carried out a genome-wide expression profiling of miRNAs in macrophages treated with MDP alone or co-treated with Ac2PIM. Among the various differentially regulated miRNAs, we identified and clustered the miRNAs that exhibited increased expression in the Ac2PIM-MDP co-treated samples when compared with MDP alone (Fig. 2C). Extensive bioinformatic analysis (TargetScan, miRWalk, miRanda and RNAhybrid) identified Ripk2 as a potential target of miR-26a and miR-150 and Map3k7 as a potential target of miR-143 (Fig. 2D). The target sites located at the residues spanning from 867 to 873 of the coding sequence of Ripk2 (for miR-26a), 12 to 18 of the 3′-UTR of Ripk2 (for miR-150) and 380 to 387 of the 3′-UTR of Map3k7 (for miR-143) were identified as critical for miRNA-CDS/3′-UTR interactions.
FIGURE 2.

miR-150 and miR-143 target RIP2 and TAK1 kinases. A and B, peritoneal macrophages were pretreated with Ac2PIM followed by MDP treatment for 2 h. Transcript (A) and protein (B) levels of RIP2 and TAK1 were determined by Real-Time qRT-PCR and immunoblotting respectively. C, genome-wide miRNA microarray profiling was done in macrophages treated as indicated. A heat map comparison of miRNAs that exhibited increased fold expression in the Ac2PIM-MDP co-treated samples when compared with MDP alone (n = 2). D, putative miR-26a, miR-150 and miR-143 binding sites in the CDS of Ripk2, 3′-UTR of Ripk2 and 3′-UTR of Map3k7, respectively. E and F, peritoneal macrophages were treated with Ac2PIM alone for 4 h (E) or with Ac2PIM for 2 h prior to 2 h MDP treatment (F). Real-Time qRT-PCR was performed on total RNA isolated using miRNA-specific primers. G, RAW 264.7 macrophages were transfected with specific miRNA mimics as indicated to assess the total expression levels of RIP2 and TAK1 by immunoblotting. H and I, RAW 264.7 macrophages were transfected with WT Ripk2 3′-UTR or miR-150Δ Ripk2 3′-UTR (H) or WT Map3k7 3′-UTR or miR-143Δ Map3k7 3′UTR (I) with miR-150 mimics (H) or miR-143 mimics (I) as indicated. Transfected macrophages were further treated with MDP or Ac2PIM or both and luciferase assay was performed. All data represent the mean ± S.E. from three independent experiments, *, p < 0.05; **, p < 0.005; ***, p < 0.001; ns, non-significant (t test in E, one-way ANOVA in F, H, and I). All blots are representative of three independent experiments. The cells were treated with 2 μg/ml Ac2PIM for 2 h unless mentioned otherwise followed by 200 ng/ml MDP. Med, medium; NC, negative control.
Validating the microarray results, macrophages treated with Ac2PIM alone (Fig. 2E) or co-treated with MDP (Fig. 2F) displayed increased expression of miR-26a, miR-150, and miR-143. Importantly, MDP treatment alone did not alter the expression of these miRNAs (Fig. 2F). To establish the effect of these miRNAs on the identified targets, miRNA-specific mimics were utilized. While miR-26a mimic failed to down-regulate RIP2 expression, miR-150 and miR-143 were identified as the Ac2PIM-responsive miRNAs that targeted RIP2 and TAK1 respectively (Fig. 2G). To further establish that Ripk2 and Map3k7 are the bonafide targets of miR-150 and miR-143, we utilized the classical 3′-UTR luciferase assays. In line with the previous results, Ac2PIM, or co-treatment of Ac2PIM with MDP or transfection with miR-150 (in case of Ripk2) or miR-143 (in case of Map3k7) mimics markedly reduced WT Ripk2 and Map3k7 3′-UTR luciferase activity. However, the reduction was not significant when mutant constructs for miR-150 binding on Ripk2 3′UTR and miR-143 binding site on Map3k7 3′-UTR were utilized (Fig. 2, H and I). These results thus validate that Ripk2 and Map3k7 are direct targets of miR-150 and miR-143 respectively. Further, macrophages transfected with miR-150- or miR-143-specific inhibitors failed to down-regulate RIP2 and TAK1 expression in presence of Ac2PIM (Fig. 3, A and B). Corroborating these results, macrophages transfected with miR-150- or miR-143-specific inhibitors also failed to exhibit Ac2PIM-mediated suppression of MDP-NOD2 signaling-induced expression of COX-2, SOCS-3, and MMP-9 (Fig. 3, C and D).
FIGURE 3.

Ac2PIM-induced miR-150 and miR-143 target RIP2 and TAK1 to down-regulate MDP responses. A and B, RAW 264.7 cells were transfected with miR-150- (A) or miR-143- (B) specific miRNA inhibitors followed by 4 h treated with Ac2PIM. RIP2 (A) and TAK1 (B) expression were analyzed by immunoblotting. C and D, miR-150- (C) or miR-143- (D) specific miRNA inhibitor-transfected RAW 264.7 macrophages were treated with Ac2PIM prior to MDP treatment for 12 h. Lysates were assessed for COX-2, SOCS-3, and MMP-9 by immunoblotting. All blots are representative of three independent experiments. The cells were treated with 2 μg/ml Ac2PIM for 2 h unless mentioned otherwise followed by 200 ng/ml MDP. Med, medium; NC, negative control; Inhi., inhibitor.
SRC-FAK-PYK2-CREB Signaling Mediates Ac2PIM-induced Expression of miR-150 and miR-143 via CBP/p300 Recruitment
Further, we explored the possible molecular mechanism of Ac2PIM-induced miR-150 and miR-143 expression. Role for TLR2 in mediating the expression of these miRNAs was validated in primary macrophages obtained from tlr2-KO mice. Ac2PIM stimulation failed to induce both miR-150 and miR-143 in tlr2-KO macrophages (Fig. 4A). Of note, TLR2 activates multiple signaling cascades to regulate immune responses in macrophages including several tyrosine kinase receptor family receptors like SRC, FAK, and PYK2 (30). Interesting reports also indicate that FAK signaling could mediate the activation of a cellular transcription factor, CREB and its binding to the DNA (31, 32). Hence, we explored the role of TLR2-dependent activation of the SRC-FAK-PYK2 complex and a possible downstream CREB-dependent CBP/P300 functions in the current scenario. While macrophages obtained from WT mice exhibited activation of SRC-FAK-PYK2 complex and CREB on Ac2PIM stimulation as assessed by the respective activatory phosphorylations, tlr2-KO macrophages failed to do so (Fig. 4B). Importantly, stimulation of macrophages with MDP alone did not induce the pathway (Fig. 4B). We also confirmed the Ac2PIM-induced FAK-dependent CREB activation by utilizing FAK-specific pharmacological inhibitor (Fig. 4C). Activation of CREB leads to its binding to the CBP/P300 coactivator complex that is recruited to the DNA to bring about transcriptional activation via its HAT functions (33). To establish the role for the above mentioned pathway during the Ac2PIM-induced expression of miR-150 and miR-143, macrophages were treated with FAK- or HAT-specific pharmacological inhibitors prior to Ac2PIM treatment. Macrophages failed to induce the expression of both miR-150 and miR-143 on Ac2PIM stimulation in presence of these inhibitors (Fig. 4D). The role for CREB-CBP/P300 in expression of Ac2PIM-induced miR-150 and miR-143 was further validated by ChIP experiments. Corroborating the previous results, Ac2PIM-stimulation of macrophages resulted in significant recruitment of pCREB, P300, and corresponding increased H3K18 acetylation at both miR-150 and miR-143 promoters (Fig. 4E). This suggests that Ac2PIM activates TLR2-SRC-FAK-PYK2 complex, which in turn effectuates CREB activation, recruitment of CREB-CBP/P300 at the promoters of miR-150 and miR-143 and their expression. Analyzing the functional significance of the identified pathway, we found that primary macrophages pretreated with FAK or HAT inhibitors failed to exhibit Ac2PIM-mediated suppression of MDP-NOD2 signaling-induced expression of COX-2, SOCS-3, and MMP-9 (Fig. 4F).
FIGURE 4.

Ac2PIM induces the expression of miR-150 and miR-143 via SRC-FAK-PYK2-CREB-CBP/P300 axis. A, macrophages from C57BL/6J WT and tlr2-KO mice were treated with Ac2PIM for 4 h and real-time qRT-PCR was performed on total RNA isolated using miRNA-specific primers. B, peritoneal macrophages from C57BL/6J WT and tlr2-KO mice were treated with Ac2PIM or MDP for 1 h as indicated. Total cell lysates were assessed for pFAK, pPYK2, pSRC, and pCREB by immunoblotting. C, macrophages were treated with FAK-specific inhibitor for 1 h prior to 1 h Ac2PIM treatment and lysates were analyzed for pCREB by immunoblotting. D, primary macrophages pretreated with either FAK inhibitor or HAT inhibitor for 1 h were treated with Ac2PIM for 4 h. Real-Time qRT-PCR was performed using miRNA-specific primers. E, pCREB and P300 recruitment and H3K18ac modification at the promoters of miR-150 and miR-143 was evaluated by ChIP in macrophages treated with Ac2PIM for 4 h. F, peritoneal macrophages were treated with indicated inhibitors for 1 h prior to 2 h Ac2PIM treatment followed by a 12 h MDP treatment. Lysates were assessed for COX-2, SOCS-3, and MMP-9 by immunoblotting. All data represent the mean ± S.E. from three independent experiments, *, p < 0.05; **, p < 0.005; ***, p < 0.001 (one-way ANOVA in A and D, t test in E). All blots are representative of three independent experiments. All blots are representative of three independent experiments. The cells were treated with 2 μg/ml Ac2PIM for 2 h unless mentioned otherwise followed by 200 ng/ml MDP. Med, medium; KO, knock-out; Inhi., inhibitor.
Ac2PIM Down-regulates PI3K-PKC-MAPK Signaling to Modulate NOD2 Responses
Having established that miR-150 and miR-143 target RIP2 and TAK1 to suppress NOD2 responses, we sought to identify the molecular mechanism downstream to RIP2-TAK1 that mediates COX-2, SOCS-3, and MMP-9 expression. Pharmacological inhibition of PI3K-PKC-MAPK signaling significantly down-regulated NOD2-induced COX-2, SOCS-3, and MMP-9 expression in macrophages (Fig. 5A). In line with this result, we found that macrophages treated with Ac2PIM failed to activate NOD2-induced PI3K-PKC-MAPK signaling (Fig. 5, B and C). Importantly, peritoneal macrophages pretreated with FAK or HAT inhibitors or in cells transfected with miR-150- or miR-143-specific inhibitors, Ac2PIM was not effective to subdue NOD2-activated PI3K-PKC-MAPK signaling(Fig. 5D).
FIGURE 5.

Ac2PIM regulates MDP-induced activation of PI3K-PKCδ-MAPK pathway. A, peritoneal macrophages were treated with specific pharmacological inhibitors of PI3K-PKCδ-MAPK pathway such as LY294002 (PI3K inhibitor), Rapamycin (mTOR inhibitor), Rottlerin (PKCδ inhibitor), U0126 (ERK1/2 inhibitor), SB203580 (p38 inhibitor), and SP600125 (JNK1/2 inhibitor) prior to 12 h MDP treatment. Total cell lysates were analyzed for COX-2, SOCS-3, and MMP-9 by immunoblotting. B and C, activation of PI3K pathway (B) and PKCδ-MAPK pathway (C) was analyzed in macrophages pretreated with Ac2PIM followed by MDP for the indicated time points. D, activation of PI3K-PKCδ-MAPK pathway was assessed under the following conditions: Peritoneal macrophages were pretreated with the indicated inhibitors for 1 h followed by Ac2PIM and MDP treatment for 2 h (D, left panel), RAW264.7 cells transfected with miR-150- (D, middle panel), or miR-143- (D, right panel) specific miRNA inhibitors were treated with Ac2PIM followed by 2 h of MDP treatment. All blots are representative of three independent experiments. The cells were treated with 2 μg/ml Ac2PIM for 2 h followed by 200 ng/ml MDP. Med, medium; NC, negative control; Inhi., inhibitor; DMSO, dimethyl sulfoxide.
β-Catenin Activation by MDP Facilitates COX-2, SOCS-3, and MMP-9 Expression
To further elucidate the downstream regulators of NOD2 responses, we performed MatInspector analysis of the promoters of mouse COX-2, SOCS-3, and MMP-9. STATs and β-catenin were among the common transcription factors that could regulate COX-2, SOCS-3, and MMP-9. Phosphorylation of STAT1 at Tyr-701 and STAT3 at Tyr-705 renders them active whereas phosphorylation of β-catenin at Ser-33/37/Thr-41 and glycogen synthase kinase-3β (GSK-3β) at Ser-9 renders them inactive. Active GSK-3β phosphorylates β-catenin at Ser-33/37/Thr-41 to turn it inactive. Here, while MDP-triggered NOD2 did not signal STAT activation (Fig. 6A), NOD2-induced suppression of GSK-3β and hence activation of β-catenin (Fig. 5B, left panel) was subverted in the presence of Ac2PIM (Fig. 6B, right panel). In support of our previous data, MDP-NOD2-induced β-catenin activation was found to be PI3K-MAPK pathway dependent (Fig. 6C). Substantiating these observations, expression of NOD2-responsive immunomodulators COX-2, SOCS-3, and MMP-9 were not altered by JAK-STAT pathway inhibition but significantly down-regulated in the presence of β-catenin inhibitor and significantly up-regulated in the presence of GSK-3β inhibitor (Fig. 6D). Importantly, macrophages pretreated with FAK or HAT inhibitors (Fig. 6E) or macrophages transfected with miR-150- or miR-143-specific inhibitors failed to exhibit Ac2PIM-mediated suppression of β-catenin activation on NOD2 stimulation (Fig. 6F). Together, these results suggest that Ac2PIM-responsive miR-150 and miR-143 suppressed the NOD2-induced β-catenin activation and β-catenin-dependent gene expression.
FIGURE 6.

Ac2PIM regulates NOD2-β-catenin-mediated COX-2, SOCS-3, and MMP-9 expression. A, peritoneal macrophages were pretreated with a pharmacological inhibitor of JAK kinase (AG490) followed by treatment with MDP or IFN-γ (200 units/ml) to analyze phosphorylation status of STAT1 and STAT3. B and C, inhibitory phosphorylation status of β-catenin and GSK-3β during the following conditions: treatment of macrophages with MDP for the indicated time points (B), pretreatment of macrophages with PI3K-MAPK pathway-specific pharmacological inhibitors followed by MDP treatment for 6 h (C). D, peritoneal macrophages were treated with AG490, β-catenin inhibitor or LiCl (GSK-3β inhibitor) prior to 12 h MDP treatment. Lysates were assessed for COX-2, SOCS-3, and MMP-9 by immunoblotting. E and F, Phosphorylation status of β-catenin and GSK-3β was assessed by immunoblotting under following conditions: Peritoneal macrophages were pretreated with the indicated inhibitors for 1 h followed by Ac2PIM and MDP treatment for 6 h (E), miR-150- (left panel) or miR-143- (right panel) specific miRNA inhibitor-transfected RAW 264.7 macrophages were treated with Ac2PIM prior to MDP treatment for 6 h (F). All blots are representative of three independent experiments. The cells were treated with 2 μg/ml Ac2PIM for 2 h followed by 200 ng/ml MDP. Med, medium; DMSO, dimethyl sulfoxide; NC, negative control; Inhi., inhibitor; LiCl, lithium chloride.
Discussion
Commonly, a concerted inter-regulatory network of PRR signaling determines the immune responses mounted against the pathogen/PAMPs (1, 34). In the current investigation, we analyzed the crosstalk of two important families of PRR, TLRs, and NLRs. Specifically, a mycobacterial cell wall glycolipid Ac2PIM and component of bacterial peptidoglycan MDP were utilized as cognate ligands for TLR2 and NOD2 pathway. Interestingly, Ac2PIM-stimulated TLR2 signaling was found to abrogate NOD2-responsive immune modulators. This was in accordance with the previous observations of antagonistic regulation between TLR2-NOD2 signaling wherein NOD2 was found to negatively regulate TLR2-induced Th1 responses (6, 7). The agonist used in the study was peptidoglycan, a bacterial PAMP common for both pathways. However, NOD2-TLR2 can also synergistically induce inflammatory responses (16, 35). Further, there exists a cooperative regulation of NOD2- and TLR2-specific ligands for mediating immune cytokines (8, 36, 37). NOD2-TLR2 synergy was also found in different cellular contexts (38, 39). Thus, the present study underscores the fact that though multiple PAMPs recognize a single PRR, there exist regulatory mechanisms to orchestrate the ligand-specific immune responses.
Ac2PIM was potent to inhibit COX-2, SOCS-3, and MMP-9 expression but did not alter the levels of NO, TNF-α, VEGF-A, and IL-12 during co-treatment with MDP. This was surprising as Ac2PIM alone, like MDP, could upregulate these immunomodulators. Supporting this observation, monoacyl form of PIM2 was previously reported to have induced the expression of these immunomodulators (40, 41).
Of note, though classical NOD2 responses via RIP2 and TAK1 are well established, various previous investigations have suggested that MDP-induced NOD2 responses could be RIP2/TAK1-independent (42, 43). However, in the current study, we found role for the classical NOD2 responses. Deciphering the mechanism of Ac2PIM-arbitrated inhibition of MDP-NOD2 responses, we found miRNA-mediated regulation of RIP2 and TAK1 expression. Extensive studies on TLR-responsive miRNAs suggest a role for miRNAs in not only orchestrating innate immune responses or a negative feed-back loop (44–48) but also negatively regulate responses mediated by other PRRs including NOD2 (49). Here, miR-150 and miR-143 were found to be induced by Ac2PIM-stimulation of macrophages that targeted the NOD2 adapters, RIP2 and TAK1, respectively. Though several investigations have implicated miR-150 in regulating innate immune responses (45, 50–52), no reports on miR-150 and regulation of RIP2 or NOD2 signaling exists. Interestingly, supporting our data, miR-143 has been previously reported to regulate immune responses in various cellular contexts (53–55) and target TAK1 in mesenchymal stem cells (56) and adipocytes (57).
Though few reports have indicated the crosstalk between TLR2 signaling and SRC-FAK-PYK2 complex (30, 58, 59), no reports exist on the molecular mechanism induced by Ac2PIM via TLR2. We found Ac2PIM activated TLR2-SRC-FAK-PYK2 cascade to induce expression of miR-150 and miR-143. FAK and PYK2 kinases were previously reported for their possible abilities to activate CREB responses (31, 32, 60). Other investigations have implicated CREB activation to mediate TLR2-mediated immune functions (47, 61). In line with these, we found SRC-FAK-PYK2 signals to activate CREB and induce CREB-CBP/P300 recruitment to miR-150 and miR-143 promoters and epigenetically regulate their expression via the intrinsic HAT activity.
Further, we also established that NOD2-induced immunomodulators were mediated by the classical PI3K-PKCδ-MAPK cascade in tandem with β-catenin signaling. While MDP-triggered NOD2 signals to activate PI3K-PKC-MAPK pathways in macrophages and dendritic cells (8, 18, 20), PI3K-PKC-MAPK pathway was also previously found to regulate the immunomodulators like COX-2, SOCS-3, and MMP-9 in macrophages on TLR2 stimulation by mycobacteria (62–64). Of note, NOD2-responsive β-catenin signaling exacerbates several pathologies including inflammatory disorders and arthritis (42, 65). β-Catenin also regulates COX-2 and SOCS-3 expression during mycobacterial infection (66). Ac2PIM-induced miR-150 and miR-143 via the TLR2-SRC-FAK-PYK2-CREB-CBP/P300 axis targeted RIP2 and TAK1 to abrogate NOD2-reponsive PI3K-PKCδ-MAPK-β-catenin signaling-mediated expression of COX-2, SOCS-3, and MMP-9 (Fig. 7). The PRR agonists like PIM2 (15, 67–69) and MDP (70, 71) are potent vaccine adjuvant candidates. Thus, our study has accentuated the cross-regulation of the respective PRR in a coactivation scenario, which can contribute to better understanding of the adjuvant utilities of Ac2PIM and MDP.
FIGURE 7.
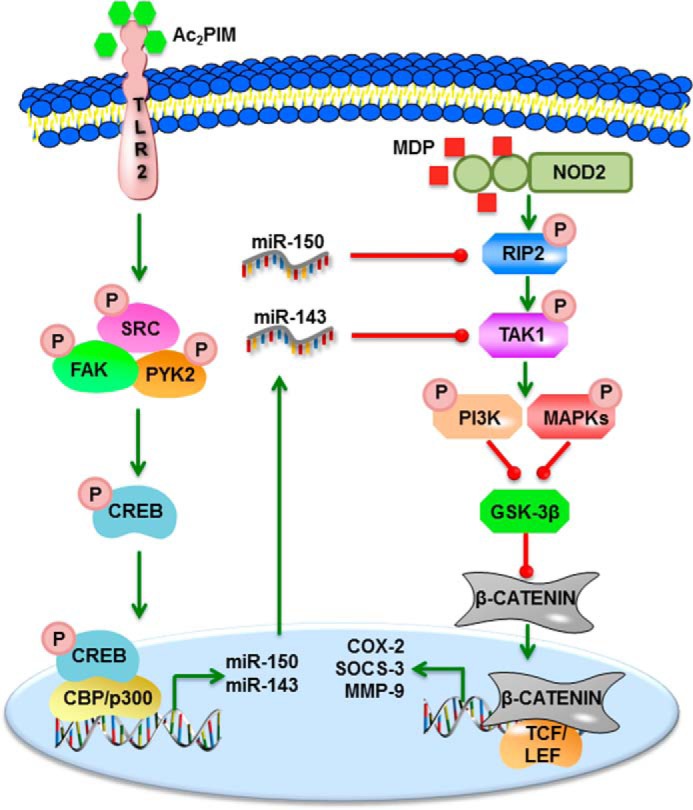
Model. Ac2PIM-responsive, TLR2-SRC-FAK-PYK2-CREB-CBP/P300-dependent miRNAs, miR-150 and miR-143, target RIP2 and TAK1, respectively to suppress NOD2-induced PI3K-PKCδ-MAPK-β-catenin-mediated expression of immunomodulators like COX-2, SOCS-3, and MMP-9.
Author Contributions
P. P., S. H., and D. S. G designed and performed experiments and analyzed data. M. G., and G. P. provided Ac2PIM for the study. V. U. performed experiments. P. P., S. H., and K. N. B. wrote the manuscript. K. N. B. designed experiments, analyzed data, and supervised the study.
Acknowledgments
We thank the Central Animal Facility (CAF), IISc for providing mice for experimentation. We acknowledge Dr. Sandhya S. Visweswariah for providing anti-Tyr416 phospho-SRC antibody and Dr. N. Ravi Sundaresan for providing anti-P300 antibody.
This study is supported by funds from the Dept. of Biotechnology (DBT), Dept. of Science and Technology (DST), Council for Scientific and Industrial Research (CSIR), Indian Council of Medical Research (ICMR), and Indo-French Center for Promotion of Advanced Research (IFCPAR/CEFIPRA). Infrastructure support from ICMR (Center for Advanced Study in Molecular Medicine), DST (FIST) and UGC (special assistance) (to K. N. B.) and fellowship from IISc (to P. P. and S. H.) and CSIR (to D. S. G.) are acknowledged. The authors declare that they have no conflicts of interest with the contents of this article.
- PRR
- pattern recognition receptor
- PAMP
- pathogen-associated molecular pattern
- TLR
- toll-like receptor
- PIM
- phosphatidylmyo-inositol mannoside
- Ac2PIM
- tetraacyl phosphatidylmyo-inositol dimannoside
- NOD2
- nucleotide oligomerization domain 2
- MDP
- muramyl dipeptide
- RIP2
- receptor-interacting protein kinase 2
- TAK1
- transforming growth factor beta-activated kinase 1
- NF-κB
- nuclear factor kappa-light-chain-enhancer of activated B cells
- COX-2
- cyclooxygenase-2
- SOCS-3
- suppressor of cytokine signaling-3
- MMP-9
- matrix metalloproteinase-9
- GSK-3β
- glycogen synthase kinase-3beta
- FAK
- focal adhesion kinase
- PYK2
- protein tyrosine kinase 2
- CREB
- cAMP responsive element-binding protein
- CBP
- CREB-binding protein.
References
- 1.Lee M. S., and Kim Y. J. (2007) Signaling pathways downstream of pattern-recognition receptors and their cross talk. Annu. Rev. Biochem. 76, 447–480 [DOI] [PubMed] [Google Scholar]
- 2.Kawai T., and Akira S. (2011) Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34, 637–650 [DOI] [PubMed] [Google Scholar]
- 3.Mogensen T. H. (2009) Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev 22, 240–273, Table of Contents [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tan R. S., Ho B., Leung B. P., and Ding J. L. (2014) TLR cross-talk confers specificity to innate immunity. Int Rev Immunol 33, 443–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Trinath J., Holla S., Mahadik K., Prakhar P., Singh V., and Balaji K. N. (2014) The WNT signaling pathway contributes to dectin-1-dependent inhibition of Toll-like receptor-induced inflammatory signature. Mol. Cell. Biol. 34, 4301–4314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Watanabe T., Kitani A., and Strober W. (2005) NOD2 regulation of Toll-like receptor responses and the pathogenesis of Crohn's disease. Gut 54, 1515–1518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Watanabe T., Kitani A., Murray P. J., and Strober W. (2004) NOD2 is a negative regulator of Toll-like receptor 2-mediated T helper type 1 responses. Nat Immunol 5, 800–808 [DOI] [PubMed] [Google Scholar]
- 8.Ghorpade D. S., Kaveri S. V., Bayry J., and Balaji K. N. (2011) Cooperative regulation of NOTCH1 protein-phosphatidylinositol 3-kinase (PI3K) signaling by NOD1, NOD2, and TLR2 receptors renders enhanced refractoriness to transforming growth factor-beta (TGF-beta)- or cytotoxic T-lymphocyte antigen 4 (CTLA-4)-mediated impairment of human dendritic cell maturation. J. Biol. Chem. 286, 31347–31360 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 9.Gilleron M., Ronet C., Mempel M., Monsarrat B., Gachelin G., and Puzo G. (2001) Acylation state of the phosphatidylinositol mannosides from Mycobacterium bovis bacillus Calmette Guerin and ability to induce granuloma and recruit natural killer T cells. J. Biol. Chem. 276, 34896–34904 [DOI] [PubMed] [Google Scholar]
- 10.Gilleron M., Quesniaux V. F., and Puzo G. (2003) Acylation state of the phosphatidylinositol hexamannosides from Mycobacterium bovis bacillus Calmette Guerin and mycobacterium tuberculosis H37Rv and its implication in Toll-like receptor response. J. Biol. Chem. 278, 29880–29889 [DOI] [PubMed] [Google Scholar]
- 11.Beatty W. L., Rhoades E. R., Ullrich H. J., Chatterjee D., Heuser J. E., and Russell D. G. (2000) Trafficking and release of mycobacterial lipids from infected macrophages. Traffic 1, 235–247 [DOI] [PubMed] [Google Scholar]
- 12.Gilleron M., Nigou J., Nicolle D., Quesniaux V., and Puzo G. (2006) The acylation state of mycobacterial lipomannans modulates innate immunity response through toll-like receptor 2. Chem. Biol. 13, 39–47 [DOI] [PubMed] [Google Scholar]
- 13.Doz E., Rose S., Court N., Front S., Vasseur V., Charron S., Gilleron M., Puzo G., Fremaux I., Delneste Y., Erard F., Ryffel B., Martin O. R., and Quesniaux V. F. (2009) Mycobacterial phosphatidylinositol mannosides negatively regulate host Toll-like receptor 4, MyD88-dependent proinflammatory cytokines, and TRIF-dependent co-stimulatory molecule expression. J. Biol. Chem. 284, 23187–23196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vergne I., Gilleron M., and Nigou J. (2014) Manipulation of the endocytic pathway and phagocyte functions by Mycobacterium tuberculosis lipoarabinomannan. Front. Cell Infect. Microbiol. 4, 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Court N., Rose S., Bourigault M. L., Front S., Martin O. R., Dowling J. K., Kenny E. F., O'Neill L., Erard F., and Quesniaux V. F. (2011) Mycobacterial PIMs inhibit host inflammatory responses through CD14-dependent and CD14-independent mechanisms. PLoS ONE 6, e24631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gandotra S., Jang S., Murray P. J., Salgame P., and Ehrt S. (2007) Nucleotide-binding oligomerization domain protein 2-deficient mice control infection with Mycobacterium tuberculosis. Infect. Immun. 75, 5127–5134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Strober W., Murray P. J., Kitani A., and Watanabe T. (2006) Signalling pathways and molecular interactions of NOD1 and NOD2. Nat Rev Immunol 6, 9–20 [DOI] [PubMed] [Google Scholar]
- 18.Philpott D. J., Sorbara M. T., Robertson S. J., Croitoru K., and Girardin S. E. (2014) NOD proteins: regulators of inflammation in health and disease. Nat Rev Immunol 14, 9–23 [DOI] [PubMed] [Google Scholar]
- 19.Caruso R., Warner N., Inohara N., and Nunez G. (2014) NOD1 and NOD2: signaling, host defense, and inflammatory disease. Immunity 41, 898–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bansal K., and Balaji K. N. (2011) Intracellular pathogen sensor NOD2 programs macrophages to trigger Notch1 activation. J. Biol. Chem. 286, 5823–5835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zheng S., Hedl M., and Abraham C. (2015) TAM receptor-dependent regulation of SOCS3 and MAPKs contributes to proinflammatory cytokine downregulation following chronic NOD2 stimulation of human macrophages. J. Immunol. 194, 1928–1937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vissers M., Hartman Y., Groh L., de Jong D. J., de Jonge M. I., and Ferwerda G. (2014) Recognition of Streptococcus pneumoniae and muramyl dipeptide by NOD2 results in potent induction of MMP-9, which can be controlled by lipopolysaccharide stimulation. Infect Immun 82, 4952–4958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Landes M. B., Rajaram M. V., Nguyen H., and Schlesinger L. S. (2015) Role for NOD2 in Mycobacterium tuberculosis-induced iNOS expression and NO production in human macrophages. J Leukoc Biol [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ghorpade D. S., Sinha A. Y., Holla S., Singh V., and Balaji K. N. (2013) NOD2-nitric oxide-responsive microRNA-146a activates Sonic hedgehog signaling to orchestrate inflammatory responses in murine model of inflammatory bowel disease. J. Biol. Chem. 288, 33037–33048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Juarez-Verdayes M. A., Rodriguez-Martinez S., Cancino-Diaz M. E., and Cancino-Diaz J. C. (2013) Peptidoglycan and muramyl dipeptide from Staphylococcus aureus induce the expression of VEGF-A in human limbal fibroblasts with the participation of TLR2-NFkappaB and NOD2-EGFR. Graefes Arch. Clin. Exp. Ophthalmol. 251, 53–62 [DOI] [PubMed] [Google Scholar]
- 26.Beynon V., Cotofana S., Brand S., Lohse P., Mair A., Wagner S., Mussack T., Ochsenkuhn T., Folwaczny M., Folwaczny C., Glas J., and Torok H. P. (2008) NOD2/CARD15 genotype influences MDP-induced cytokine release and basal IL-12p40 levels in primary isolated peripheral blood monocytes. Inflamm. Bowel Dis. 14, 1033–1040 [DOI] [PubMed] [Google Scholar]
- 27.Ricciotti E., and FitzGerald G. A. (2011) Prostaglandins and inflammation. Arterioscler. Thromb. Vasc. Biol. 31, 986–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carow B., and Rottenberg M. E. (2014) SOCS3, a Major Regulator of Infection and Inflammation. Front. Immunol. 5, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Malemud C. J. (2006) Matrix metalloproteinases (MMPs) in health and disease: an overview. Front. Biosci. 11, 1696–1701 [DOI] [PubMed] [Google Scholar]
- 30.Nag K., and Chaudhary A. (2009) Mediators of Tyrosine Phosphorylation in Innate Immunity: From Host Defense to Inflammation onto Oncogenesis. Curr. Signal Transduct. Ther. 4, 76–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang Y. C., Ma Y. L., Chen S. K., Wang C. W., and Lee E. H. (2003) Focal adhesion kinase is required, but not sufficient, for the induction of long-term potentiation in dentate gyrus neurons in vivo. J. Neurosci. 23, 4072–4080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cho M. K., Cho Y. H., Lee G. H., and Kim S. G. (2004) Induction of cyclooxygenase-2 by bovine type I collagen in macrophages via C/EBP and CREB activation by multiple cell signaling pathways. Biochem. Pharmacol. 67, 2239–2250 [DOI] [PubMed] [Google Scholar]
- 33.Goodman R. H., and Smolik S. (2000) CBP/p300 in cell growth, transformation, and development. Genes Dev. 14, 1553–1577 [PubMed] [Google Scholar]
- 34.Abdullah Z., and Knolle P. A. (2014) Scaling of immune responses against intracellular bacterial infection. EMBO J. 33, 2283–2294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moreira L. O., El Kasmi K. C., Smith A. M., Finkelstein D., Fillon S., Kim Y. G., Nuñez G., Tuomanen E., and Murray P. J. (2008) The TLR2-MyD88-NOD2-RIPK2 signalling axis regulates a balanced pro-inflammatory and IL-10-mediated anti-inflammatory cytokine response to Gram-positive cell walls. Cell Microbiol. 10, 2067–2077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Uehara A., Yang S., Fujimoto Y., Fukase K., Kusumoto S., Shibata K., Sugawara S., and Takada H. (2005) Muramyldipeptide and diaminopimelic acid-containing desmuramylpeptides in combination with chemically synthesized Toll-like receptor agonists synergistically induced production of interleukin-8 in a NOD2- and NOD1-dependent manner, respectively, in human monocytic cells in culture. Cell Microbiol. 7, 53–61 [DOI] [PubMed] [Google Scholar]
- 37.Wolfert M. A., Murray T. F., Boons G. J., and Moore J. N. (2002) The origin of the synergistic effect of muramyl dipeptide with endotoxin and peptidoglycan. J. Biol. Chem. 277, 39179–39186 [DOI] [PubMed] [Google Scholar]
- 38.Wu J., Zhang Y., Xin Z., and Wu X. (2015) The crosstalk between TLR2 and NOD2 in Aspergillus fumigatus keratitis. Mol. Immunol. 64, 235–243 [DOI] [PubMed] [Google Scholar]
- 39.Chen K., Zhang L., Huang J., Gong W., Dunlop N. M., and Wang J. M. (2008) Cooperation between NOD2 and Toll-like receptor 2 ligands in the up-regulation of mouse mFPR2, a G-protein-coupled Aβ42 peptide receptor, in microglial cells. J. Leukoc. Biol. 83, 1467–1475 [DOI] [PubMed] [Google Scholar]
- 40.Bansal K., Kapoor N., Narayana Y., Puzo G., Gilleron M., and Balaji K. N. (2009) PIM2 Induced COX-2 and MMP-9 expression in macrophages requires PI3K and Notch1 signaling. PLoS ONE 4, e4911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Narayana Y., Bansal K., Sinha A. Y., Kapoor N., Puzo G., Gilleron M., and Balaji K. N. (2009) SOCS3 expression induced by PIM2 requires PKC and PI3K signaling. Mol. Immunol. 46, 2947–2954 [DOI] [PubMed] [Google Scholar]
- 42.Singh V., Holla S., Ramachandra S. G., and Balaji K. N. (2015) WNT-Inflammasome Signaling Mediates NOD2-Induced Development of Acute Arthritis in Mice. J. Immunol. 194, 3351–3360 [DOI] [PubMed] [Google Scholar]
- 43.Travassos L. H., Carneiro L. A., Ramjeet M., Hussey S., Kim Y. G., Magalhães J. G., Yuan L., Soares F., Chea E., Le Bourhis L., Boneca I. G., Allaoui A., Jones N. L., Nuñez G., Girardin S. E., and Philpott D. J. (2010) Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat. Immunol. 11, 55–62 [DOI] [PubMed] [Google Scholar]
- 44.O'Neill L. A., Sheedy F. J., and McCoy C. E. (2011) MicroRNAs: the fine-tuners of Toll-like receptor signalling. Nat. Rev. Immunol. 11, 163–175 [DOI] [PubMed] [Google Scholar]
- 45.Ghorpade D. S., Holla S., Kaveri S. V., Bayry J., Patil S. A., and Balaji K. N. (2013) Sonic hedgehog-dependent induction of microRNA 31 and microRNA 150 regulates Mycobacterium bovis BCG-driven toll-like receptor 2 signaling. Mol. Cell. Biol. 33, 543–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sheedy F. J., Palsson-McDermott E., Hennessy E. J., Martin C., O'Leary J. J., Ruan Q., Johnson D. S., Chen Y., and O'Neill L. A. (2010) Negative regulation of TLR4 via targeting of the proinflammatory tumor suppressor PDCD4 by the microRNA miR-21. Nat. Immunol. 11, 141–147 [DOI] [PubMed] [Google Scholar]
- 47.Ghorpade D. S., Leyland R., Kurowska-Stolarska M., Patil S. A., and Balaji K. N. (2012) MicroRNA-155 is required for Mycobacterium bovis BCG-mediated apoptosis of macrophages. Mol. Cell. Biol. 32, 2239–2253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Holla S., Kurowska-Stolarska M., Bayry J., and Balaji K. N. (2014) Selective inhibition of IFNG-induced autophagy by Mir155- and Mir31-responsive WNT5A and SHH signaling. Autophagy 10, 311–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brain O., Owens B. M., Pichulik T., Allan P., Khatamzas E., Leslie A., Steevels T., Sharma S., Mayer A., Catuneanu A. M., Morton V., Sun M. Y., Jewell D., Coccia M., Harrison O., Maloy K., Schönefeldt S., Bornschein S., Liston A., and Simmons A. (2013) The intracellular sensor NOD2 induces microRNA-29 expression in human dendritic cells to limit IL-23 release. Immunity 39, 521–536 [DOI] [PubMed] [Google Scholar]
- 50.Tsitsiou E., and Lindsay M. A. (2009) microRNAs and the immune response. Curr. Opin. Pharmacol. 9, 514–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhou R., O'Hara S. P., and Chen X. M. (2011) MicroRNA regulation of innate immune responses in epithelial cells. Cell Mol. Immunol. 8, 371–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ghorpade D. S., Holla S., Sinha A. Y., Alagesan S. K., and Balaji K. N. (2013) Nitric oxide and KLF4 protein epigenetically modify class II transactivator to repress major histocompatibility complex II expression during Mycobacterium bovis bacillus Calmette-Guerin infection. J. Biol. Chem. 288, 20592–20606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang X., Liu S., Hu T., He Y., and Sun S. (2009) Up-regulated microRNA-143 transcribed by nuclear factor kappa B enhances hepatocarcinoma metastasis by repressing fibronectin expression. Hepatology 50, 490–499 [DOI] [PubMed] [Google Scholar]
- 54.Raisch J., Darfeuille-Michaud A., and Nguyen H. T. (2013) Role of microRNAs in the immune system, inflammation and cancer. World J. Gastroenterol. 19, 2985–2996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ma X., Becker Buscaglia L. E., Barker J. R., and Li Y. (2011) MicroRNAs in NF-kappaB signaling. J. Mol. Cell. Biol. 3, 159–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhao X., Liu D., Gong W., Zhao G., Liu L., Yang L., and Hou Y. (2014) The toll-like receptor 3 ligand, poly(I:C), improves immunosuppressive function and therapeutic effect of mesenchymal stem cells on sepsis via inhibiting MiR-143. Stem Cells 32, 521–533 [DOI] [PubMed] [Google Scholar]
- 57.He Z., Yu J., Zhou C., Ren G., Cong P., Mo D., Chen Y., and Liu X. (2013) MiR-143 is not essential for adipose development as revealed by in vivo antisense targeting. Biotechnol. Lett. 35, 499–507 [DOI] [PubMed] [Google Scholar]
- 58.Toubiana J., Rossi A. L., Belaidouni N., Grimaldi D., Pene F., Chafey P., Comba B., Camoin L., Bismuth G., Claessens Y. E., Mira J. P., and Chiche J. D. (2015) Src-family-tyrosine kinase Lyn is critical for TLR2-mediated NF-κB activation through the PI 3-kinase signaling pathway. Innate Immun. 21, 685–697 [DOI] [PubMed] [Google Scholar]
- 59.Mehta P. K., and Griendling K. K. (2007) Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am. J. Physiol. Cell Physiol 292, C82–97 [DOI] [PubMed] [Google Scholar]
- 60.Katsume A., Okigaki M., Matsui A., Che J., Adachi Y., Kishita E., Yamaguchi S., Ikeda K., Ueyama T., Matoba S., Yamada H., and Matsubara H. (2011) Early inflammatory reactions in atherosclerosis are induced by proline-rich tyrosine kinase/reactive oxygen species-mediated release of tumor necrosis factor-alpha and subsequent activation of the p21Cip1/Ets-1/p300 system. Arterioscler. Thromb. Vasc. Biol. 31, 1084–1092 [DOI] [PubMed] [Google Scholar]
- 61.Wen A. Y., Sakamoto K. M., and Miller L. S. (2010) The role of the transcription factor CREB in immune function. J. Immunol. 185, 6413–6419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kapoor N., Narayana Y., Patil S. A., and Balaji K. N. (2010) Nitric oxide is involved in Mycobacterium bovis bacillus Calmette-Guerin-activated Jagged1 and Notch1 signaling. J. Immunol. 184, 3117–3126 [DOI] [PubMed] [Google Scholar]
- 63.Bansal K., Narayana Y., Patil S. A., and Balaji K. N. (2009) M. bovis BCG induced expression of COX-2 involves nitric oxide-dependent and -independent signaling pathways. J. Leukoc. Biol. 85, 804–816 [DOI] [PubMed] [Google Scholar]
- 64.Narayana Y., and Balaji K. N. (2008) NOTCH1 up-regulation and signaling involved in Mycobacterium bovis BCG-induced SOCS3 expression in macrophages. J. Biol. Chem. 283, 12501–12511 [DOI] [PubMed] [Google Scholar]
- 65.Koslowski M. J., Teltschik Z., Beisner J., Schaeffeler E., Wang G., Kübler I., Gersemann M., Cooney R., Jewell D., Reinisch W., Vermeire S., Rutgeerts P., Schwab M., Stange E. F., and Wehkamp J. (2012) Association of a functional variant in the Wnt co-receptor LRP6 with early onset ileal Crohn's disease. PLoS Genet. 8, e1002523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bansal K., Trinath J., Chakravortty D., Patil S. A., and Balaji K. N. (2011) Pathogen-specific TLR2 protein activation programs macrophages to induce Wnt-beta-catenin signaling. J. Biol. Chem. 286, 37032–37044 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 67.Sprott G. D., Dicaire C. J., Gurnani K., Sad S., and Krishnan L. (2004) Activation of dendritic cells by liposomes prepared from phosphatidylinositol mannosides from Mycobacterium bovis bacillus Calmette-Guerin and adjuvant activity in vivo. Infect. Immun. 72, 5235–5246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wedlock D. N., Denis M., Painter G. F., Ainge G. D., Vordermeier H. M., Hewinson R. G., and Buddle B. M. (2008) Enhanced protection against bovine tuberculosis after coadministration of Mycobacterium bovis BCG with a Mycobacterial protein vaccine-adjuvant combination but not after coadministration of adjuvant alone. Clin. Vaccine Immunol. 15, 765–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Faisal S. M., Chen J. W., McDonough S. P., Chang C. F., Teng C. H., and Chang Y. F. (2011) Immunostimulatory and antigen delivery properties of liposomes made up of total polar lipids from non-pathogenic bacteria leads to efficient induction of both innate and adaptive immune responses. Vaccine 29, 2381–2391 [DOI] [PubMed] [Google Scholar]
- 70.Kornbluth R. S., and Stone G. W. (2006) Immunostimulatory combinations: designing the next generation of vaccine adjuvants. J. Leukoc. Biol. 80, 1084–1102 [DOI] [PubMed] [Google Scholar]
- 71.Pavot V., Rochereau N., Rességuier J., Gutjahr A., Genin C., Tiraby G., Perouzel E., Lioux T., Vernejoul F., Verrier B., and Paul S. (2014) Cutting edge: New chimeric NOD2/TLR2 adjuvant drastically increases vaccine immunogenicity. J. Immunol. 193, 5781–5785 [DOI] [PubMed] [Google Scholar]