

Background: MAPK cascade proteins bind to each other selectively via docking interactions.
Results: The high selectivity of JNK family MAPKs for cognate binding partners is controlled by two key hydrophobic residues in the docking site.
Conclusion: This contrasts with other proposed models of docking specificity.
Significance: This has implications for drug design and for the evolution of signaling specificity.
Keywords: c-Jun N-terminal kinase (JNK), cell signaling, dual-specificity kinase, mitogen-activated protein kinase (MAPK), phosphorylation, protein complex, protein kinase, protein phosphorylation, substrate specificity, docking site
Abstract
MAPKs bind to many of their upstream regulators and downstream substrates via a short docking motif (the D-site) on their binding partner. MAPKs that are in different families (e.g. ERK, JNK, and p38) can bind selectively to D-sites in their authentic substrates and regulators while discriminating against D-sites in other pathways. Here we demonstrate that the short hydrophobic region at the distal end of the D-site plays a critical role in determining the high selectivity of JNK MAPKs for docking sites in their cognate MAPK kinases. Changing just 1 or 2 key hydrophobic residues in this submotif is sufficient to turn a weak JNK-binding D-site into a strong one, or vice versa. These specificity-determining differences are also found in the D-sites of the ETS family transcription factors Elk-1 and Net. Moreover, swapping two hydrophobic residues between these D-sites switches the relative efficiency of Elk-1 and Net as substrates for ERK versus JNK, as predicted. These results provide new insights into docking specificity and suggest that this specificity can evolve rapidly by changes to just 1 or 2 amino acids.
Introduction
Changes in protein kinase activity are associated with many human diseases. As such, there is considerable interest in targeting protein kinases with drugs and in understanding kinase signaling networks (1–3). To advance both of these goals, it is important to better understand how protein kinases recognize and select their substrates and regulators.
A key aspect of kinase-substrate recognition is the interaction of the kinase active site with the target phosphosite (4, 5). Many kinases, however, augment the limited selectivity of this interaction in various ways. For example, in cyclin-dependent kinases (CDKs),2 the non-catalytic cyclin subunit is critical for substrate recognition (6, 7). A similar function has also been proposed for scaffold proteins, which bridge transient interactions between kinases and their substrates (3, 8). Finally, other kinases, as exemplified by mitogen-activated protein kinases (MAPKs), bind directly to short docking motifs on substrates that are located at various distances from the target phosphosite (4, 5, 9–11). These docking interactions are thought to dynamically tether the catalytic domain within range of appropriate target sites (12). There is increasing interest in targeting docking interactions as a possible drug development strategy (13–17).
MAPK cascades are crucial to the regulation of cell division, differentiation, and death and are also important regulators of metabolic, inflammatory, and stress responses (18–20). The core of a MAPK cascade consists of a MAPK kinase (MKK or MEK) that phosphorylates and activates a cognate MAPK. The activated MAPK then phosphorylates multiple downstream substrates, including transcription factors, such as Elk-1, c-Jun, and ATF2 (21). Docking interactions are critical for both of these steps; MAPK kinases contain a MAPK-docking site that promotes binding/phosphorylation of their cognate MAPKs, and MAPK-regulated transcription factors and other substrates typically contain docking sites that enhance their binding/phosphorylation by MAPKs. Moreover, docking interactions mediate the binding of MAPKs with scaffolds and phosphatases (3, 9, 22). These networks of MAPK docking interactions are broadly conserved in eukaryotes (23–25).
Although there are several different classes of MAPK docking site, the most abundant class is that designated the “D-site” or “D-domain” (9). The D-site consensus consists of a cluster of about 2 or 3 basic residues, a short spacer, and a hydrophobic-X-hydrophobic submotif ((K/R)2–3X1–6ΦXΦ, where X is any residue and Φ is a hydrophobic residue) (26, 27). D-sites are found in MAPK kinases, in MAPK phosphatases, in MAPK substrates, and in MAPK scaffold proteins. The D-sites in these proteins bind to a small surface region of the MAPK that consists of closely spaced acidic patches and shallow hydrophobic pockets, which collectively form a “docking groove” (11, 14). The docking groove is located on the opposite side of the kinase structure from the kinase active site.
Major MAPK pathways in mammalian cells include the MEK1/2 → ERK1/2 pathway, which primarily regulates growth and developmental signaling and is dysregulated in many types of cancer (28–31); the MKK3/6 → p38 pathway, which mainly regulates stress responses and inflammation (32, 33); and the MKK4/7 → JNK pathway, which regulates cell life-death decisions and many other disease-relevant processes and plays a central role in the pathogenesis of diabetes and related metabolic disorders (34, 35).
As stated above, the ERK1/2, JNK, and p38 MAPK families are activated by distinct MKKs. This interaction is highly specific, and D-sites located near the N termini of MKKs help determine this specificity. For example, JNK proteins bind strongly to D-sites found in their activators, MKK4 and MKK7, yet bind weakly or not at all to D-sites found in MKKs that do not activate JNK (36). A similar situation is seen in MAPK-substrate interactions: ERK1/2, JNK, and p38 phosphorylate distinct but overlapping sets of substrates, and differences in docking affinities are thought to influence these substrate preferences (37, 38). Our understanding of the rules that determine such family preferences (e.g. why some D-sites bind preferentially to JNK, whereas other bind better to p38) is incomplete (11, 39–42). What makes this problem particularly challenging is that D-sites found in ERK, JNK, and p38 binding partners share a core consensus, and there are no obvious differences in the non-consensus residues that suggest how they might influence family preferences.
Here we investigated the docking preferences for the JNK family of MAPKs, using the striking preference of JNK for cognate MKK-derived D-sites as a starting point. We found that the selectivity of D-sites for JNK versus ERK/p38 was largely determined by the composition of the hydrophobic submotif and that selectivity could be flipped (with weakly binding D-sites becoming strong ones and vice versa) by changing just 1 or 2 residues in this element. This work provides significant new insights into MAPK docking specificity.
Experimental Procedures
Proteins
Fusions of glutathione S-transferase (GST) to human c-Jun(1–89) and ATF2(19–96) were purchased from Cell Signaling Technology. Activated human JNK1α1 and JNK2α2 were purchased from Upstate Cell Signaling Solutions/Millipore. Activated mouse ERK2 was purchased from New England Biolabs. GST fusion proteins used in Figs. 4 and 8 were expressed in bacteria and purified by affinity chromatography using glutathione-Sepharose (GE Healthcare) and quantified as described elsewhere (26).
FIGURE 4.
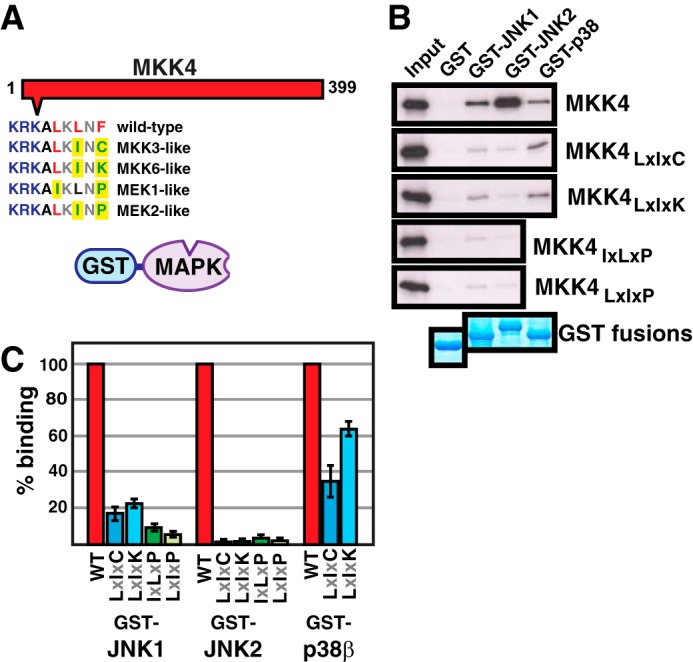
Non-cognate hydrophobic residue substitutions into the D-site of full-length MKK4 reduce JNK binding. A, 35S-radiolabeled full-length human MKK4 protein and mutant derivatives thereof were produced by in vitro translation and tested for binding to full-length, purified human GST-MAPK proteins in a pull-down assay. Results were quantified by SDS-PAGE followed by PhosphorImager analysis. The sequences of the D-sites of the wild-type and mutant proteins are shown below a schematic of full-length MKK4. B, MKK4 (∼1 pmol) was tested for binding to 20 μg of GST (lane 2), GST-JNK1α1 (lane 3), GST-JNK2α2 (lane 4), or GST-p38β (lane 5). Lane 1 shows 10% of the total MKK4 input. The bottom panel shows the Coomassie Blue staining of the sedimented GST fusion proteins. C, quantification of MAPK-MKK4 binding based on several experiments, such as that shown in B. Error bars, S.E. (n ≥ 3).
FIGURE 8.

Switching the MAPK preference of Elk-1 and Net. A, comparison of Elk-1 and Net proteins, showing the D-site (triangle), a region subjected to multiple MAPK-mediated phosphorylations (circled P), and the FXFP site (pink stripe). Also shown are the GST fusion protein derivatives of Elk-1 and Net used in this study, which contain the D-site and phosphorylation site but lack the FXFP site. Sequences shown are human. See “Results” for further details. B, ERK efficiently phosphorylates both Elk-1 and Net, whereas JNK prefers Elk-1 (left schematic). We asked if switching just 2 hydrophobic residues in the D-site would switch specificity as predicted in the right schematic. C, phosphate incorporation into Elk-1 derivatives was determined in a standard protein kinase assay, using ERK2, JNK1, or JNK2, as indicated. Results for each kinase were normalized relative to phosphorylation of wild-type Elk-1. Error bars, S.E. (n ≥ 3). Representative data are shown below the graph. The band images shown were compiled from several different experiments; images were adjusted to make the exposures equivalent. D, phosphate incorporation into Net derivatives was determined and analyzed as in C.
Peptides
The soluble peptides used in this study were synthesized by Mimotopes. The MEK peptides are composed of the following residues of the full-length protein; MEK1, residues 1–17; MEK2, residues 1–20; MKK3, residues 17–33; MKK4, residues 37–52; MKK6, residues 2–21; MKK7-D2, residues 37–51.
Plasmids for the Production of GST Fusion Proteins
The vector used for generating GST fusion proteins was pGEX-LB, a derivative of pGEX-4T-1 (Amersham Biosciences) (26). In pGEX-LB, an encoded Pro residue is replaced with a Gly-Gly-Gly-Gly-Gly-Ser-Gly coding sequence to promote the independent functioning of the GST and fusion moieties. Plasmids encoding GST-JNK1α1, GST-JNK2α2, and GST-p38β have been described elsewhere (43). To generate GST-Elk1(310–393) and GST-Net(288–367), PCR was used to amplify the specific fragments and introduce an EcoRI site at the N terminus and a SalI site at the C terminus; human ELK1 and NET cDNA clones were used as templates. The PCR products were digested with EcoRI and SalI and subsequently inserted into the appropriate sites on pGEX-LB.
Protein Kinase Assays
Protein kinase reactions (20 μl) contained kinase assay buffer (50 mm Tris-HCl (pH 7.5), 10 mm MgCl2, 1 mm EGTA, and 2 mm dithiothreitol), 0.1 mg/ml BSA, 1 μm substrate, active MAPK (2 milliunits (∼17 ng) of JNK1 or JNK2 or 10 units (∼1 ng) of ERK2), 50 μm ATP, 1 μCi of [γ-32P]ATP, and (for peptide competition assays, see below) varying concentrations of particular D-site peptides. Reactions were for 20 min at 30 °C. Substrate phosphorylation was quantified by SDS-PAGE (12% gels), followed by analysis of relative incorporation using a PhosphorImager. All data points shown are averages from experiments repeated 3–7 times, with duplicate points in each experiment. The S.E. between experiments was typically <10% of the mean.
Binding Assay
The binding assay shown in Fig. 4 was performed as described previously (26). The human genes used in this experiment were MKK4 (MAP2K4; NCBI accession number NM_003010), JNK1α1 (MAPK8; NM_002750), JNK2α2 (MAPK9; NM_002752), and p38β (MAPK11; NM_002751).
Competition Assay for Measuring D-site Selectivity
D-sites in MKKs and D-sites in substrates compete for MAPK binding. This competition can be demonstrated by using MKK-derived D-site peptides to inhibit MAPK-mediated phosphorylation of substrates containing D-sites (44). For example, the D-site from MKK4 inhibits JNK-mediated phosphorylation of c-Jun and ATF2 (45, 46). We have found that this competitive inhibition assay provides a highly sensitive method for quantifying the binding of a given D-site peptide to a target MAPK (36, 44–46) and have used it extensively in the present study. To perform this assay, protein kinase reactions are set up as described above but with peptide competitor included in the reaction. Under the conditions of our assay, reaction velocity responds approximately linearly to changes in [substrate]; thus, [substrate] ≪ Km. (Furthermore, under the conditions of our assay, velocity is approximately linear in [E]). Assuming a competitive inhibition scheme (47–49), and given that [substrate] ≪ Km, the IC50 (the concentration of D-site peptide that inhibits substrate phosphorylation by 50%) is a measure of the dissociation constant (Kd) of the peptide-kinase binding interaction, with a lower IC50 indicating tighter binding (50, 51). We have found that IC50 values measured by this protocol are reproducible within about 2–3-fold, even when remeasured several years later using different lots of all components. Furthermore, IC50 values measured using this procedure have generally correlated well with Kd values measured in direct binding assays (45, 46). IC50 estimates were obtained by nonlinear fitting of the quantified data to a competitive inhibition isotherm.
Results
Selectivity of Docking Sites in MAPK Kinases
MKKs efficiently phosphorylate their cognate, within-pathway MAPKs and do not appreciably phosphorylate MAPKs in other pathways (Fig. 1A). In particular, MEK1 and MEK2 phosphorylate ERK (meaning ERK1 and ERK2) but not JNK or p38. Likewise, MKK3 and MKK6 phosphorylate p38 but not ERK or JNK (52, 53), and MKK7 phosphorylates JNK but not ERK or p38 (54, 55). MKK4 is an exception in that it phosphorylates both JNK and p38 (52, 56).
FIGURE 1.
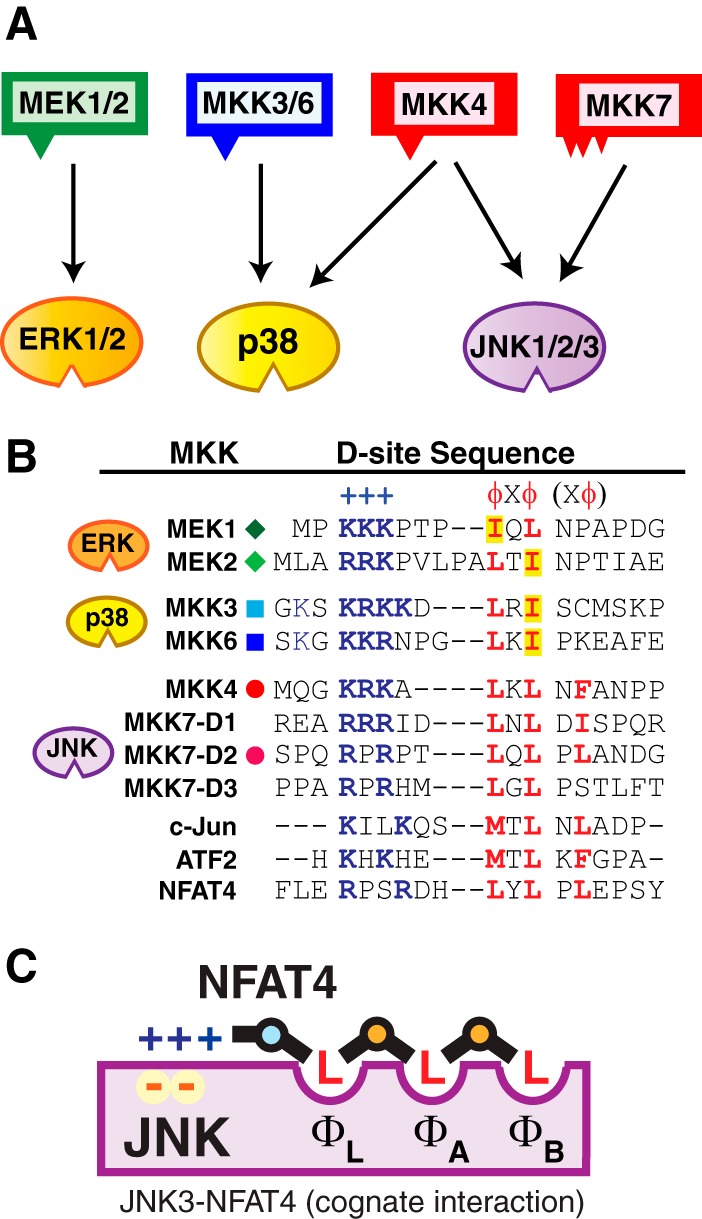
MAPK pathways and docking sites in MKKs. A, the schematic shows six of the seven human MAPK kinases (MKKs or MEKs) with their cognate, within-pathway MAPKs indicated by the arrows. The triangles on the MKKs represent their D-sites. Whereas most MKKs contain a single, higher-affinity D-site near their N termini, MKK7 contains three lower-affinity D-sites in its N-terminal domain (46). B, comparison of D-sites found in the human MKKs shown in A. Residues comprising the basic submotif (+++) are shown in boldface blue type; residues comprising the hydrophobic submotif (φXφ(Xφ)) are shown in boldface red type. The last “(Xφ)” indicates the extended hydrophobic submotif found in some D-sites. Isoleucine residues in the core “φXφ” are also highlighted in yellow. Gaps have been introduced to maximize alignment of functionally similar residues; spaces are for visual clarity. C, schematic interpretation of the co-crystal structure of the D-site of the NFAT4 transcription factor binding to the JNK1 MAPK (42), an example of a cognate docking interaction. The interaction of the basic submotif of the D-site with negatively charged acidic residues in the MAPK is depicted on the left, whereas the interaction of the hydrophobic submotif of the D-site with the three hydrophobic pockets (ΦL, ΦA, and ΦB) in the docking groove of the MAPK is shown on the right. The illustration is inspired by a similar representation by Reményi et al. (58); the pockets are labeled according to Peti and Page (11).
The selectivity of MEK-MAPK docking interactions largely parallels the specificity of MEK-MAPK enzymatic transactions. In other words, D-sites from MKKs bind to their cognate MAPKs with about a 10-fold higher affinity than they bind to non-cognate MAPKs (36). In particular, JNK1 and JNK2 are highly selective, binding to their cognate D-sites (from MKK4 and MKK7) between 5- and 50-fold tighter than to non-cognate D-sites (36, 45, 46). Similarly, p38 also exhibits selective binding to its cognate D-sites in MKK3, MKK4, and MKK6 (36). In contrast, ERK2 exhibits a promiscuous ability to bind to D-sites of non-cognate MKKs (36).
Comparison of the Basic Submotif in Strong Versus Weak JNK-binding D-sites
MKK-derived D-sites that bind efficiently to JNK include the single high-affinity D-site in the N terminus of MKK4 (45) and the three interacting, lower affinity D-sites in the N terminus of MKK7 (46) (Fig. 1B); as stated above, MKK4 and MKK7 are the cognate, within-pathway upstream kinases for JNK family MAPKs. MKK-derived D-sites that bind to JNK weakly (or not at all) consist of the D-sites in MEK1, MEK2, MKK3, and MKK6 (Fig. 1B); these D-sites/kinases are in different MAPK cascades; in other words, they are non-cognate with respect to the JNK pathway. To restate, D-sites in proteins that are in the JNK pathway bind tightly to JNK, whereas D-sites in proteins in other pathways bind weakly to JNK.
What are the key differences between the cognate D-sites in MKK4 and MKK7 that bind tightly to JNK and the non-cognate sites in MEK1/2 and MKK3/6 that bind weakly to JNK? D-sites are composed of two main elements: an N-terminal basic submotif and a C-terminal hydrophobic submotif, which are separated by a spacer of variable length (Fig. 1B). Mutagenesis experiments have established that the integrity of both of these submotifs is crucial for MAPK binding (26, 45). There is no obvious difference between the basic submotifs of MKK-derived D-sites that bind to JNK and those that do not (Fig. 1B). Consistent with this, engineering conservative substitution mutations (i.e. lysine for arginine or vice versa) into the D-site of MKK4 did not diminish binding to JNK1, whereas alanine substitutions had a more dramatic effect (Table 1) (45). These results suggest that the precise chemical identity of a given basic residue does not dramatically influence binding efficiency and thus likewise does not determine binding specificity. This is consistent with the idea that the basic residues may be able to bind to corresponding acidic patches in the docking groove in a flexible or “fuzzy” manner, accounting for the lack of resolution of this portion of the D-site in many co-crystal structures (11, 42).
TABLE 1.
Lysine to arginine substitutions in basic subdomain do not affect MKK4-JNK binding
Shown is a summary of MKK4-JNK binding assay data. Experiments were performed with 35S-radiolabeled full-length MKK4 protein and mutants thereof, which were tested for binding to purified GST-JNK1 as in Fig. 4, or with 5S- radiolabeled full-length JNK3 protein, which was tested for binding to GST- MKK4(37–94) and mutants thereof as described in Ho et al.(45). Each plus sign represents approximately 20% of wild-type binding after background subtraction.

Comparison of the Hydrophobic Submotif in Strong Versus Weak JNK-binding D-sites
The hydrophobic submotif of the D-site has the consensus sequence ΦXΦ, where Φ is a hydrophobic residue and X is a non-hydrophobic residue. Furthermore, many D-sites contain an “extended” ΦXΦXΦ hydrophobic submotif. The “extra” hydrophobic residue in such D-sites can be accommodated in an additional hydrophobic pocket in the docking groove, as was first seen in the co-crystal structure of the MEF2A D-site peptide with p38α (57) and has since been observed in several other D-site·MAPK co-crystal structures, including Msg5·Fus3 (58), Ste7·Fus3 (58), JNK1·NFAT4 (42), and JNK3·ATF2 (59). A schematic depiction of the JNK1·NFAT4 structure is shown in Fig. 1C.
When we compared the hydrophobic submotifs of the D-sites in the six human MKKs (Fig. 1B), we found two intriguing differences between the JNK-binding versus non-binding groups.
The first difference we noticed was in the first 3 residues of the hydrophobic submotif. For the JNK-binding group of MKKs, the D-site of MKK4 contains LXL, as do all three of the D-sites in MKK7. In contrast, for the MKKs in other pathways, the D-sites of the ERK pathway activators MEK1 and MEK2 contain IXL and LXI, respectively, and the D-sites of the p38 activators MKK3 and MKK6 both contain LXI. In other words, the cognate MKK D-sites for JNK are LXL, whereas the non-cognate D-sites are IXL or LXI.
This first observation suggested a straightforward hypothesis. Perhaps JNK dislikes isoleucine in the hydrophobic submotif, and this contributes to its poor binding to D-sites from non-cognate MKKs.
MKK D-sites That Bind JNK Weakly Lack an Extended Hydrophobic Motif
The second difference we noticed between JNK binders and non-binders was whether or not the hydrophobic submotif was extended to include a third hydrophobic residue. First, examination of MKK D-site sequences revealed the presence of a putative extended hydrophobic submotif in the single high-affinity JNK-docking D-site in MKK4, which has the sequence LKLNF (Fig. 1B). This interpretation was predicated on the assumption that the phenylalanine at position 48 in MKK4 is a suitable hydrophobic residue; in support of this assumption, leucine is most often substituted with isoleucine/methionine, phenylalanine, and valine, in that order, according to the PAM250 (percent accepted substitution) matrix, which records the likelihood of change from one amino acid to another in homologous protein sequences during evolution (60). Furthermore, in the crystal structure of JNK3 co-crystalized with a D-site peptide derived from ATF2, the phenylalanine in the extended hydrophobic submotif of ATF2 (MTLKF) occupies the ΦB hydrophobic pocket of the docking groove (59).
Examination of the three D-sites present in the N terminus of MKK7 revealed that MKK7-D1 and MKK7-D2 also contain putative extended hydrophobic motifs (Fig. 1B). MKK7-D2, the site with the highest JNK binding affinity of the three (46), contains the sequence LQLPL. MKK7-D1, the site with the next highest binding affinity for JNK, contains the sequence LNLDI. Only MKK7-D3, which has the lowest affinity for JNK of the three MKK7 D-sites, appears to lack an extended hydrophobic submotif, having instead the sequence LGLPS.
In contrast, examination of the p38 pathway kinases MKK3 and MKK6 revealed that neither of them feature an extended hydrophobic submotif. Instead, MKK3 contains the residue cysteine, which would be expected to be polar in the reducing intracellular environment and is seldom substituted for leucine according to the PAM250 matrix. Strikingly, MKK6 contains the highly polar, positively charged residue lysine at the position where the third hydrophobic residue would sit. Based on these observations, we hypothesized that the presence or absence of a third hydrophobic residue might contribute to the ability of JNK to discriminate between its cognate D-sites in MKK4 and MKK7 and the non-cognate D-sites in the p38 pathway kinases MKK3 and MKK6.
The ERK pathway kinases MEK1 and MEK2 contain a proline at the position where the third hydrophobic residue should be (Fig. 1B). Although proline is non-polar, it is infrequently substituted for leucine, isoleucine, or phenylalanine according to the PAM250 matrix. Thus, we considered the possibility that this position might contribute to the ability of JNK to discriminate against MEK1 and MEK2.
For the purpose of increased clarity, we will adopt the following terminology in the remainder of this paper. We will refer to the first and second hydrophobic residues in the D-site using the terms “first position” and “second position.” Also, we will use the term “third position” to refer to the residue that is generally hydrophobic in MKK4/7 but not hydrophobic in MEK1/2 and MKK3/6. In addition, we will often abbreviate the sequence of a particular hydrophobic submotif as LXLXF (for MKK4) or LXIXK (for MKK6), etc., to emphasize the identity of the residues in the first, second, and third positions.
Swapping MKK6 Residues into the MKK4 D-site Reduces JNK Binding
To begin to explore the hypothesis that differences in the hydrophobic submotif underlie the ability of JNK to discriminate cognate from non-cognate D-sites, we measured the ability of MKK4-derived D-site peptides to bind to JNK and thereby inhibit JNK-mediated phosphorylation of the D-site-containing substrates c-Jun and ATF2. We have shown that this “peptide competition” assay provides a highly sensitive method for quantifying the binding of a given D-site peptide to a target MAPK (36, 44–46) (Fig. 2A; see “Experimental Procedures” for details). The wild-type MKK4 peptide corresponds to residues 37–52 of full-length MKK4 protein and hence contains leucines at positions corresponding to MKK4 residues 44 and 46 and a phenylalanine at the position corresponding to MKK4 residue 48 (Fig. 2B). Thus, its hydrophobic submotif can be abbreviated as LXLXF. As shown in Fig. 2, C–E, this peptide was an effective inhibitor of JNK-mediated phosphorylation, as we have reported previously (36, 45, 46). The peptide concentration required for 50% inhibition of JNK1 phosphorylation of ATF2 (i.e. the IC50) was 2 μm. The IC50 was 6 μm when the same peptide was used to inhibit JNK2 phosphorylation of c-Jun. In contrast, the MKK6 D-site peptide (hydrophobic submotif: LXIXK) exhibited no detectable binding to JNK; the IC50 was much greater than 100 μm for both the JNK1·ATF2 and JNK2·c-Jun reactions.
FIGURE 2.

Effect of “MKK6-like” hydrophobic residue substitutions on MKK4-JNK binding. A, peptide competition assay. D-site peptides (triangles) were used to inhibit JNK phosphorylation of the D-site-containing substrates c-Jun or ATF2. B, the MKK4 D-site peptide (red) binds with high affinity to JNK and inhibits JNK-mediated phosphorylation of D-site-containing substrates. The MKK6 D-site peptide (blue) does not bind to JNK and therefore does not inhibit JNK-mediated phosphorylation. The experiment was designed to measure the binding of MKK4 peptides that were substituted with residues taken from MKK6. Will these hybrid D-site peptides bind to JNK or not? C, sequence of D-site peptides used in this experiment; all sequences used in this paper are human. Substituted residues are indicated in green and underlined. To the left of the sequences, the IC50 (the peptide concentration that inhibits phosphorylation by 50%; see “Experimental Procedures” for more details) for each kinase/substrate combination tested is shown. For the mutant MKK4 peptides, the -fold increase in IC50, normalized to wild-type MKK4, is also shown. Lower IC50 values indicate stronger D-site·JNK binding; thus, “-fold increase in IC50” quantifies the factor by which JNK binding has been reduced by the mutations indicated. D, purified GST-ATF (1 μm) was incubated with purified active JNK1 (∼50 nm) and [γ-32P]ATP for 20 min in the absence or presence of the specific concentrations of the indicated peptides. In the graph, results are plotted as percent phosphorylation relative to that observed in the absence of any added peptide. Phosphate incorporation into ATF2 was analyzed by SDS-PAGE and quantified on a PhosphorImager. Data are the average of 3–8 experiments, with duplicate data points in each experiment. The S.E. between experiments was typically <10% of the mean; error bars are omitted for visual clarity. E, representative autoradiogram of experiments averaged and graphed in D. F and G, similar to D and E, except the kinase was JNK2, and the substrate was GST-c-Jun.
We next designed mutant MKK4-derived peptides in which position 2 or position 3 residues in MKK4 were exchanged with the equivalent residues in MKK6. In other words, we made the MKK4 peptide more “MKK6-like” by swapping key residues in the hydrophobic submotif. Both MKK4 and MKK6 (as well as MEK2 and MKK3) contain a leucine at the first position; however, MKK6 has an isoleucine at the second position and a lysine at the third position. The L46I substitution makes the MKK4 D-site resemble MEK2, MKK3, and MKK6 at the second position. The F48K mutation makes the D-site of MKK4 resemble MKK6 at the third position. As shown in Fig. 2, both “MKK6-like” single substitutions in MKK4 (L46I and F48K) reduced the ability of the MKK4 peptide to inhibit JNK, with the F48K change showing the more substantial effect. Strikingly, the peptide containing both substitutions (designated MKK4 LXIXK) was as ineffective at binding to JNK as the native MKK6 D-site peptide. Moreover, these conclusions were consistent regardless of whether JNK1 or JNK2 was used as the kinase and whether c-Jun or ATF2 was used as the substrate (Fig. 2).
Swapping MEK1 Residues into the MKK4 D-site Reduces JNK Binding
We next made the MKK4 D-site peptide more “MEK1-like,” again by swapping key residues in the hydrophobic submotif. The D-site of MEK1 is the only MKK-derived D-site that contains an isoleucine at the first position. In addition, MEK1 contains a proline at the third position (as does MEK2). The L44I substitution makes the MKK4 D-site resemble MEK1 at the first position, whereas the F48P substitution makes the MKK4 D-site resemble MEK1 (and MEK2) at the third position. As shown in Fig. 3, both the L44I and the F48P substitutions reduced the ability of the MKK4 peptide to inhibit JNK. Similar to the results presented in Fig. 2, the change in the third position (F48P) exhibited the more substantial effect. Furthermore, the peptide containing both substitutions (designated MKK4 IXLXP) was even less effective at binding to JNK than the wild-type MEK1 D-site peptide. Once again, these conclusions were consistent regardless of whether JNK1 or JNK2 was used as the kinase and whether c-Jun or ATF2 was used as the substrate (Fig. 3).
FIGURE 3.

Effect of “MEK1-like” hydrophobic residue substitutions on MKK4-JNK binding. A, the MKK4 D-site peptide (red) binds with high affinity to JNK and inhibits JNK-mediated phosphorylation of D-site-containing substrates. The MEK1 D-site peptide (green) does not bind to JNK and therefore does not inhibit JNK-mediated phosphorylation. The experiment was designed to measure the binding of MKK4 peptides that were substituted with residues taken from MEK1. Will these hybrid D-site peptides bind to JNK or not? B, peptide sequences used in A. C and D, inhibition of JNK1 phosphorylation of ATF2 by the D-site peptides shown in B. E and F, inhibition of JNK2 phosphorylation of c-Jun by the D-site peptides shown in B. Other details are as in Fig. 2.
Binding Assay
In order to determine the effect of D-site substitutions on the binding of full-length MKK4 to JNK, we produced full-length human MKK4 protein by in vitro transcription/translation and measured its binding to purified, full-length GST-JNK1, JNK2, or p38 proteins. As shown in Fig. 4, wild-type MKK4 protein bound to all three of these cognate MAPKs, as expected, exhibiting the highest affinity binding for JNK2, followed by JNK1 and then p38. (It is interesting to note that MKK7 binds to JNK1 preferentially over JNK2 and does not bind to p38 (46).)
We also produced four mutant versions of full-length human MKK4 protein, by altering just 2 amino acids, and measured the binding of these derivatives to JNK1/2 and p38. In the first of these mutant MKK4 “alleles,” the first and third positions of the hydrophobic submotif of the D-site were exchanged with the corresponding residues of MEK1. In the next three, the second and third positions were exchanged with the corresponding residues in MEK2, MKK3, or MKK6. In other words, we created MEK1-like, MEK2-like, MKK3-like, and MKK6-like alleles of MKK4 by changing 2 residues in the MKK4 D-site (Fig. 4A).
As shown in Fig. 4, all four of the mutants displayed a dramatic decrease in MKK4-JNK binding. For JNK1, binding of the MKK3- and MKK6-like mutants was reduced about 5-fold, and binding of the MEK1- and MEK2-like mutants was reduced by over 10-fold. Even more dramatically, the binding of all four non-cognate mutants to JNK2 was reduced over 20-fold (Fig. 4).
The cognate MKKs for p38 are MKK3, MKK4, and MKK6 (52, 56). Thus, it was not clear what would happen with regard to p38 binding when the MKK4 D-sites were changed to be MKK3- or MKK6-like, because this could be viewed as changing cognate residues into different cognate residues. Indeed, as shown in Fig. 4, these changes only resulted in a modest decrease in MKK4-p38 binding.
In summary (Figs. 2–4), the MKK4 D-site, which normally binds JNK with high affinity, can be converted into a D-site that binds to JNK as poorly as the non-cognate D-sites in MEK1, MEK2, MKK3, and MKK6. Furthermore, this conversion can be accomplished by exchanging just 1 or 2 residues with the equivalent residues in the hydrophobic submotif of the non-cognate D-site.
Hydrophobic Substitutions into MKK3 and MKK6 Increase JNK Binding
Having shown that binding to JNK was reduced by changing the MKK4 D-site into the non-cognate, MKK6-like sequence LXIXK (Fig. 2), we next asked whether the converse were true; would changing 1 or 2 residues of the D-site of MKK6 into those of MKK4 (LXLXF) or MKK7-D2 (LXLXL) be sufficient to enable these non-cognate D-sites to bind efficiently to JNK?
The MKK6 D-site contains the hydrophobic submotif LKIPK. We made a double substitution in MKK6, I15L/K17L, which converted the MKK6 hydrophobic submotif to LKLPL (compare with LQLPL in MKK7-D2). As shown in Fig. 5, whereas the wild-type MKK6 D-site peptide exhibits no detectable binding to JNK, the “MKK7-like” MKK6 D-site bound to JNK with an affinity comparable with the wild-type MKK7-D2 and MKK4 D-sites. In contrast, the single I15L (MKK4- and MKK7-like) or K17L (MKK7-like) changes in the MKK6 D-site exhibited a much less dramatic increase in JNK binding than the double substitution.
FIGURE 5.

Hydrophobic residue substitutions into the MKK6 D-site increase JNK binding. A, the cognate MKK4 D-site peptide (red) binds with high affinity to JNK, whereas the non-cognate MKK6 D-site peptide (blue) does not. The experiment was designed to measure the binding of MKK6 peptides that were substituted with residues taken from MKK4. Will these hybrid D-site peptides bind to JNK or not? B, peptides used in this experiment; MKK7-D2 is shown for comparison. C and D, inhibition of JNK1 phosphorylation of ATF2 by the D-site peptides shown in B. E and F, inhibition of JNK2 phosphorylation of c-Jun by the D-site peptides shown in B. G, IC50 values of the various D-site peptides, normalized to the IC50 of MKK4, plotted in bar graph format, for the JNK1·ATF2 peptide competition assay shown in C. Lower IC50 values indicate stronger D-site·JNK binding. H, as in G, except the data are from the JNK2/c-Jun peptide competition assay shown in E. Other details are as in Fig. 2.
The MKK3 D-site contains the hydrophobic submotif LRISC. As shown in Fig. 6, changing the isoleucine in this D-site to leucine (I27L), the residue found at the equivalent position in MKK4 and MKK7-D2, modestly improved JNK binding. In addition, changing the cysteine in the MKK3 D-site to leucine (C29L), the residue found at the equivalent position in MKK7-D2, substantially improved the binding of the MKK3 D-site to JNK. Finally, when the I27L and C29L substitutions were combined, converting the hydrophobic submotif of MKK3 to LRLSL (again, compare with LQLPL in MKK7-D2), this further increased the binding of the substituted MKK3 peptide to JNK, to a level that was comparable with the cognate MKK4 peptide (Fig. 6).
FIGURE 6.

Hydrophobic residue substitutions into the MKK3 D-sites increase JNK binding. A, the cognate MKK4 D-site peptide (red) binds with high affinity to JNK, whereas the non-cognate MKK3 D-site peptide (blue) binds more weakly. The experiment was designed to measure the binding of MKK3 peptides that were substituted with residues taken from MKK4. B, peptides used in this experiment. C and D, inhibition of JNK2 phosphorylation of c-Jun by the D-site peptides shown in B. E, normalized IC50 values, taken from data in C. Other details are as in Figs. 2 and 5.
Making MEK2 More MKK4/7-like Greatly Increases JNK Binding
The MEK2 D-site contains the hydrophobic submotif LTINP. As shown in Fig. 7, changing the proline in this D-site to leucine (P16L), the residue found at the equivalent position in MKK7-D2, dramatically improved the binding of the MEK2 D-site to JNK. Moreover, the addition of a further I14L substitution, which converted the MEK2 hydrophobic submotif to LTLNL, similar to the LQLPL in MKK7-D2, further increased the binding of the substituted MEK2 peptide to JNK. Indeed, the MEK2 LXLXL D-site bound to JNK essentially just as well as the wild-type MKK7-D2 and MKK4 D-sites. Although these changes in the MEK2 D-site dramatically enhanced non-cognate binding to JNK, they had little or no effect on cognate binding to ERK2 (Table 2).
FIGURE 7.

Hydrophobic residue substitutions into the MEK2 D-sites increase JNK binding. A, the cognate MKK4 D-site peptide (red) binds with high affinity to JNK, whereas the non-cognate MEK2 D-site peptide (green) binds more weakly. The experiment was designed to measure the binding of MEK2 peptides that were substituted with residues taken from MKK4. B, peptides used in this experiment; MKK7-D2 is shown for comparison. C and D, inhibition of JNK1 phosphorylation of ATF2 by the D-site peptides shown in B. E, normalized IC50 values, taken from data in C. Data on the MKK7-D2 peptide is shown for comparison. Other details are as in Figs. 2 and 5.
TABLE 2.
IC50 values for inhibition of ERK2 by the indicated D-site peptides
The concentration of D-site peptide required to inhibit ERK2 phosphorylation of Elk-1 by 50% (the IC50 value) is shown for the indicated peptides.
| D-site peptide | IC50 for inhibition of ERK2 phosphorylation of Elk-1 | Increase in IC50 (relative to WT) |
|---|---|---|
| μm | -fold | |
| MEK2 WT (LXIXP) | 9 | |
| MEK2 I14L | 11 | 1.2 |
| MEK2 P16L | 13 | 1.4 |
| MEK2 LXLXL | 14 | 1.5 |
In summary, the results from the experiments described in Figs. 2–7 all support a crucial role for particular residues in the hydrophobic submotif of the D-site in determining the specificity of MKK-JNK interactions; JNK prefers leucine over isoleucine and greatly prefers D-sites with an extended ΦXΦXΦ hydrophobic submotif.
Comparison of the D-sites in Elk-1 and Net
Given the importance of the hydrophobic submotif in determining the specificity of MKK-MAPK interactions, we looked for other examples where differences in the hydrophobic submotifs of D-sites in paralogous proteins might underlie the specificity of MAPK targeting. We found examples in the ternary complex factor (TCF) subfamily of ETS domain transcription factors.
The ETS domain family of transcription factors, originally identified on the basis of homology to the ets-1 proto-oncogene, is found in metazoan organisms from sponges to humans (61). The TCFs are a subfamily of ETS domain factors that are distinguished by sequence homology and by the biochemical ability to bind to serum response factor. There are three mammalian TCFs: Elk-1, Elk-3/Net/Erp/Sap2, and Elk-4/Sap1. The TCFs share several regions of primary sequence similarity, including an N-terminal ETS DNA-binding domain and a C-terminal transcriptional regulation domain (designated the C-domain), which is a target for MAPK-mediated phosphorylation (62, 63). The C-domain includes both a D-site and multiple downstream target phosphosites (Fig. 8A). MAPK-mediated phosphorylation of a TCF C-domain enhances its DNA binding and also alters its interactions with other transcriptional regulators, promoting transcriptional activation.
The different members of the TCF subfamily respond differentially to the three MAPK pathways. For example, whereas Elk-1 is an efficient substrate for ERK and JNK, the C-domain of Net has been shown to be a good ERK substrate but a relatively poor JNK substrate, with important physiological consequences (39, 64, 65). Because the structural basis of this discrimination is not known, we examined the sequences of the D-sites of human Elk-1 and Net for differences in their hydrophobic submotifs that might contribute to the differential targeting by ERK versus JNK. As shown in Fig. 8B, the Elk-1 D-site has the hydrophobic submotif LELPL, with leucines at the first, second, and third hydrophobic positions. Based on the results obtained in this study with MKK D-sites, this D-site would be predicted to bind strongly to both ERK and JNK, consistent with published binding and kinase assays that demonstrate that Elk-1 is an efficient substrate for both of these MAPKs (39). In contrast, the Net D-site has a hydrophobic submotif of sequence LEISA. Notably, the Net D-site contains an isoleucine as one of its hydrophobic residues and also lacks a third hydrophobic residue. Based on the results obtained in this study with MKK D-sites, both of these characteristics should negatively impact JNK binding and phosphorylation relative to ERK binding and phosphorylation. Thus, we hypothesized that these two single-residue differences might determine the specificity of Elk and Net interactions with ERK and JNK.
Switching the MAPK Selectivity of Elk-1 and Net
To test the hypothesis that two residues in the hydrophobic submotifs of the D-sites of Elk-1 and Net were important in determining the specificity of MAPK phosphorylation, we fused the C-domains of these two transcription factors to Schistosoma japonicum GST; the resulting fusion proteins were then expressed in bacteria and purified by adsorption to glutathione-Sepharose beads. We chose to express the C-domains of the two transcription factors, rather than the full-length proteins, in order to focus solely on the role of the D-sites in directing C-domain phosphorylation. (Net is known to be phosphorylated by JNK in another part of the protein (64).) For the same reason, an FQFP sequence that abuts the C-terminal end of the C-domains of both proteins was omitted from the expressed fragments (Fig. 8A); FXFP is the consensus sequence for DEF-type MAPK-docking sites, which contact a different part of the MAPK surface than D-sites bind to (66, 67).
First, to verify previous observations that the Net C-domain was a less efficient JNK substrate than the Elk-1 C-domain, these two substrates were incubated with purified active JNK1, JNK2, or ERK2 and radiolabeled ATP in a standard in vitro kinase assay (Fig. 8, C and D). We first titrated the amount of JNK1, JNK2, or ERK2 added so that the amount of Elk-1 phosphorylation by the three kinases was roughly equivalent. Next, the same amounts of kinase were incubated with Net. When Net was used as the substrate rather than Elk-1, the level of Net phosphorylation catalyzed by ERK was about 60% of that seen with Elk-1, indicating that Net is a somewhat less efficient ERK substrate than Elk-1. In contrast, Net was a much less efficient JNK substrate than Elk-1, being phosphorylated to less than 20% of the level of Elk-1. Mutant versions of Elk-1 and Net lacking the D-site (Elk-1Dmut and NetDmut) were uniformly poor substrates for all three kinases, verifying the requirement of the D-site for efficient phosphorylation of the C-domain (64, 66, 68). In summary, the C-domain of Elk-1 was phosphorylated effectively by both ERK and JNK, whereas the C-domain of Net was phosphorylated effectively by ERK but not JNK.
To test the role of critical hydrophobic residues in the D-sites of Elk and Net in determining this targeting specificity, double-point mutant versions of the two C-domains were constructed, expressed, and purified. The mutant Elk-1LXIXA has the “Net-like” hydrophobic submotif LEIPA instead of the wild-type Elk-1 sequence LELPL. As shown in Fig. 8, this 2-residue change was sufficient to decrease JNK-mediated phosphorylation by >2-fold. In contrast, ERK-mediated phosphorylation was barely different between wild-type Elk-1 and Elk-1LXIXA. Very similar results were obtained with another Elk-1 mutant, Elk-1LXIXK, which has the sequence LEIPK. This mutant resembles MKK6, in that a positively charged lysine residue occupies the position where the third hydrophobic residue is found in Elk-1 and MKK4/7. Thus, we hypothesized that this mutant would show decreased phosphorylation by JNK. Consistent with this expectation, the Elk-1LXIXK mutation dramatically reduced Elk phosphorylation by JNK1 and JNK2 (Fig. 8C). In addition, this mutation had virtually no effect on Elk phosphorylation by ERK2, consistent with our previous finding that ERK is not particularly selective for cognate D-sites and can bind to both the MEK1 D-site (cognate) and MKK6 D-site (non-cognate) with comparable affinities (36).
The mutant NetLXLXL has the “Elk1-like” hydrophobic submotif LELSL instead of the wild-type Net sequence LEISA. As shown in Fig. 8D, this 2-residue change was sufficient to increase JNK-mediated phosphorylation by about 3-fold. ERK-mediated phosphorylation was also increased by this change, to about 1.5-fold higher.
To summarize, exchanging two positions in the hydrophobic submotifs of Elk-1 and Net was in large part sufficient to switch the specificity for JNK targeting, making Elk a poor JNK substrate and Net a good JNK substrate. In contrast, ERK targeting was largely unaffected by these changes.
Discussion
This study examined the specificity of MAPK docking interactions. We chose to focus on the interaction of MAPKs with their upstream activators, the MAPK kinases (MKKs), because it is clear that these interactions have evolved to be highly specific. In particular, we focused on the very high selectivity of JNK family MAPKs for their cognate docking sites (D-sites) in MKK4 and MKK7. We found that the identity of 2 residues in the hydrophobic submotif of the D-site plays a crucial role in selective binding to JNK. We then extended these findings to the Elk-1 and Net transcription factors and confirmed their importance. Our findings thus highlight a key role for core conserved hydrophobic residues in driving the specificity of MAPK docking interactions.
Two-residue Specificity
We found the hydrophobic submotif of the D-site plays a crucial role in MAPK-binding specificity, as follows. First, JNK prefers leucine to isoleucine in the hydrophobic submotif, preferring LXL (MKK4, MKK7-D1, MKK7-D2, Elk-1) to IXL (MEK1) or LXI (MEK2, MKK3, MKK6, Net). Second, and more importantly, JNK prefers D-sites with extended hydrophobic submotifs (e.g. LXLXL in MKK7-D2 and Elk-1 and LXLXF in MKK4) to those without (e.g. IXLXP in MEK1, LXIXP in MEK2, LXIXC in MKK3, LXIXK in MKK6, and LXIXA in Net). We showed that JNK binding specificity could be switched (a weak JNK-binding D-site could be turned into a strong one, or vice versa) by changing 1 or 2 hydrophobic residues. For example, the high-affinity JNK binding of the MKK4 D-site could be abrogated by changing its hydrophobic submotif from LXLXF to LXLXK; this change replaced the third hydrophobic residue in MKK4 with a positively charged lysine residue found in the equivalent position of MKK6, a poor JNK binder (Fig. 1). Conversely, the MKK6 D-site (which exhibits no detectable binding to JNK) could be converted to a strong JNK binder by switching just 2 residues in its hydrophobic submotif, from LXIXK to LXLXL (Fig. 5). Similar results were obtained for the MEK2 and MKK3 D-sites; they could be switched from weak to strong JNK binders by changing just 2 residues in order to convert their hydrophobic submotifs to LXLXL (Figs. 6 and 7).
In general, the change in the third position appeared to be more important in driving the changes to specific binding. The L versus I difference in the first or second positions clearly contributed to the observed effect, however, and in some cases there was clear synergism (e.g. Figs. 2C and 5). The synergism can be understood as follows. Each of the single mutations causes roughly equal increase in the free energy of binding; these ΔG values synergize in the double mutant because of the exponential relationship between free energy and the equilibrium constant.
When we examined the ternary complex factor subfamily of ETS domain transcription factors, we found that the D-sites in Elk-1 and Net differed in precisely the 2 residues that directed specificity in JNK-MKK transactions. Moreover, these D-site differences are consistent with the known kinase preferences of Net and Elk; LXIXA is present in Net, which is a better substrate for ERK than JNK, whereas LXLXL is present in Elk-1, an efficient substrate of both kinases. Importantly, we showed that switching the two key residues between Elk and Net switched their kinase preferences as predicted, making Elk-1 a poorer substrate for JNK (but not for ERK) while making Net a better substrate for JNK (Fig. 8).
A Model of Docking Specificity
We propose that the weak binding of MEK1/2, MKK3/6, and Net to JNK is attributable to the fact that these D-sites do not have extended hydrophobic submotifs. Moreover, this shortcoming is compounded by the fact that they also contain isoleucine in one of the two hydrophobic positions that they do have. In contrast, ERK and p38 can apparently bind effectively to D-sites that lack an extended hydrophobic submotif and can tolerate isoleucines more readily than JNK. There are no published crystal structures of JNK complexed to any of the D-sites studied in this paper. Nevertheless, a structure-inspired interpretation of our results, which is based on some of the relevant co-crystal structures that do exist, is presented in Fig. 9.
FIGURE 9.
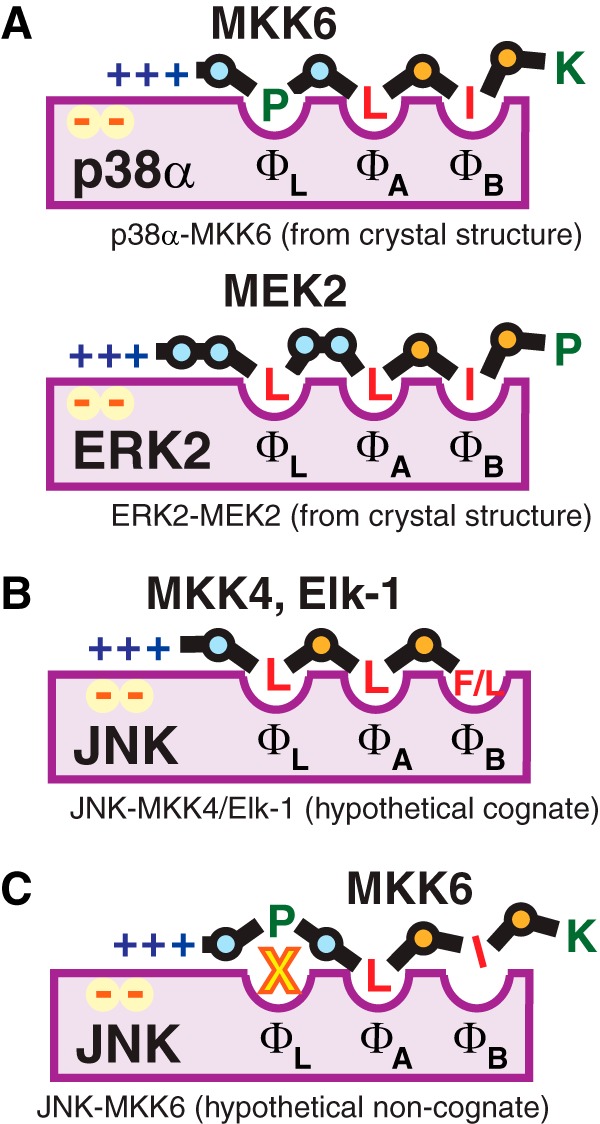
Role of the hydrophobic submotif in selective MAPK binding. A, schematic representation of published cognate D-site·MAPK co-crystal complexes of MKK6·p38α and MEK2·ERK2 (42, 69), displaying the interaction of the D-site hydrophobic residues, and of a proline or leucine in the spacer region, with hydrophobic pockets in the docking groove. The structures suggest how MEK2 and MKK6 can bind effectively to their cognate D-sites in the absence of an extended hydrophobic submotif, because a residue in the spacer fills the ΦL hydrophobic pocket. Other details are as in Fig. 1C. B, hypothesized cognate structures of the MKK4 and Elk-1 D-sites bound to JNK. The MKK4 and Elk-1 D-sites cannot fill the ΦL pocket with a spacer residue, because their spacers are too short. Rather, it is the hydrophobic nature of the third position residue that allows all three pockets to be filled. (In addition, the JNK docking groove structure may prevent spacer residues from binding to ΦL; see C). C, hypothesized non-cognate structure of the MKK6 D-site weakly bound to JNK. In this model, some currently unknown structural feature of the JNK docking groove impedes spacer residues from binding in the ΦL pocket (yellow X). Hence, the binding is weak because all three hydrophobic pockets cannot be filled. The presence of the second position isoleucine also contributes to the weakness of this interaction.
Published structures (42, 69) indicate that MKK6 and MEK2 are able to fill all three hydrophobic pockets in the docking groove of their cognate MAPKs by using a residue from the spacer that connects the basic and hydrophobic submotifs to fill the ΦL pocket (Fig. 9A). We hypothesize that the length/and or composition of the spacer makes this mode of binding untenable for the MKK4, MKK7, and Elk-1 D-sites. Rather, it is the extended hydrophobic submotif in these D-sites that allows them to occupy all three hydrophobic pockets in the docking groove (Fig. 9B). In addition, presumably the structure of the JNK docking groove hinders D-sites from using spacer residues to fill the ΦL pocket (Fig. 9C), thus explaining why the D-sites from MEK2, MKK6, etc. cannot bind to JNK (unless they are mutated to gain an extended hydrophobic residue). The possible structural basis by which JNK might discriminate against spacer residue binding is not readily apparent, however, although structures exist that allow a comparison of the docking grooves of JNK, ERK, and p38 (11).
Comparison with Other Models
In contrast to other published models of how specificity in MAPK docking interactions is determined, our results stress the importance of conserved residues in the hydrophobic submotif. Several studies have emphasized the potential role of residues outside the D-site in driving docking specificity (e.g. see Refs. 40 and 70–72; reviewed in Ref. 11). Clearly, because most of our experiments employed short D-site peptides, there is no possible involvement of regions outside the D-site in driving the selectivity differences we observed.
Another model of docking specificity was published recently by Garai et al. (42). These authors proposed that specificity is encoded in the spacer between the conserved basic and hydrophobic submotifs and that the conserved residues in the submotifs play the role of nonspecific anchors. In stark contrast, we found here that key hydrophobic submotif residues can drive specificity and that we could switch JNK binding specificity between two D-site peptides by switching only 1 or 2 residues in the hydrophobic submotif while leaving the spacers intact.
In attempting to accommodate these distinct models into a comprehensive picture of docking specificity, it is important to recall that published structural studies indicate that different D-sites can bind to the same docking groove in conformationally distinct modes (10, 11). Thus, in other MAPK/D-site interactions (including other JNK/D-site interactions), it is quite possible that different factors might play a predominant role in driving specific binding. For instance, as proposed by Garai et al. (42), the less charged and narrower docking groove of JNK may exclude certain D-sites (such as those found in MNK1 and RSK1) based on the size and composition of their spacer. At the same time, JNKs may discriminate against non-cognate D-sites in MKKs and Net based on differences in their hydrophobic submotifs, as shown here.
Although MAPKs may discriminate against different non-cognate D-sites in different ways, it is also likely that a D-site that is suboptimal for MAPK binding in one submotif can make up for this by being ideal in another binding motif. Thus, we should not be surprised to find MAPK substrates that do not “obey all the rules” (as defined here and elsewhere), particularly because substrates may not need to dock to their cognate MAPKs with the same high affinity with which MKKs do.
Rapid Evolution of Docking Specificity
Our results show that a change of 1 or 2 residues in a MAPK-docking site can dramatically change specificity and, thus, that docking specificity can evolve rapidly by point mutation. Indeed, the TCF subfamily of transcription factors (also known as the ELK subfamily or ELK group) shows some evidence of this type of evolutionary plasticity. The divergence of the vertebrate TCF subfamily from a single ELK1-like ancestor into Elk-1, Net, and Sap-1 is thought to have occurred after the divergence of protostomes (which includes nematode worms and insects) from the deuterostomes (which includes vertebrates) but before the emergence of vertebrates (61). In the fruit fly Drosophila melanogaster, the proposed Elk ortholog Aop/Yan is a known a substrate of ERK (rolled in flies) but is not known to be phosphorylated by JNK (basket in flies) (73). Consistent with this, the hydrophobic submotif in the proposed D-site of Aop/Yan, ISLLR, is predicted to be a very poor JNK binder based on the results in this paper. In the nematode worm Caenorhabditis elegans, the ELK ortholog Lin-1 is a known substrate of ERK (Mpk-1 in worms) but is not known to be a substrate of any of the three JNK paralogs (74). The D-site of Lin-1 has the sequence LNLTA (73). Although it is not clear that this sequence would strongly discriminate against JNK, nor is it optimal for JNK binding. In vertebrate Net orthologs, the hydrophobic submotif, LEISA, is highly conserved, suggesting conservation of discrimination against JNK. The LELPL sequence of Elk-1 orthologs is also conserved but not as strongly. Notably, this sequence is LELPS in fishes. Collectively, these data are consistent with the following two hypotheses. First, the ancestral ELK group transcription factor was predominantly a substrate for ERK but not for JNK. Second, early in the evolution of vertebrates, there was positive selection pressure for Elk-1 to become a better JNK substrate, and this was accomplished, at least in part, by changes in the hydrophobic submotif of the Elk-1 D-site. At any rate, it appears that the D-sites of ELK group transcription factors display sequence variation between taxonomic classes that is consistent with corresponding variations in the fine-tuning of MAPK specificity.
Conclusions
We have investigated the sequence features of MAPK-docking sites that influence their ability to bind to cognate, within-pathway MAPKs over non-cognate MAPKs. Our findings provide insight into specificity in MAPK signaling networks and should be relevant to efforts to develop docking inhibitors (13–17) and to predict new MAPK substrates and regulators (75).
Author Contributions
A. J. B. and L. B. designed the experiments. A. J. B. performed the experiments. A. J. B. and L. B. analyzed the results and wrote the manuscript.
Acknowledgments
We thank David Ho for initial studies on the leucine/isoleucine difference, Erlynn Frankson and Ronak Zebarjedi for technical assistance, and Betsy Goldsmith for helpful discussions.
This work was supported, in whole or in part, by National Institutes of Health, NIGMS, Grants R01 GM60366, R01 GM84332, and P50 GM76516. The authors declare that they have no conflicts of interest with the contents of this article.
- CDK
- cyclin-dependent kinase
- TCF
- ternary complex factor
- MKK
- MAPK kinase.
References
- 1.Cohen P. (2002) Protein kinases: the major drug targets of the twenty-first century? Nat. Rev. Drug Discov. 1, 309–315 [DOI] [PubMed] [Google Scholar]
- 2.Papin J. A., Hunter T., Palsson B. O., and Subramaniam S. (2005) Reconstruction of cellular signalling networks and analysis of their properties. Nat. Rev. Mol. Cell Biol. 6, 99–111 [DOI] [PubMed] [Google Scholar]
- 3.Bhattacharyya R. P., Reményi A., Yeh B. J., and Lim W. A. (2006) Domains, motifs, and scaffolds: the role of modular interactions in the evolution and wiring of cell signaling circuits. Annu. Rev. Biochem. 75, 655–680 [DOI] [PubMed] [Google Scholar]
- 4.Goldsmith E. J., Akella R., Min X., Zhou T., and Humphreys J. M. (2007) Substrate and docking interactions in serine/threonine protein kinases. Chem. Rev. 107, 5065–5081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ubersax J. A., and Ferrell J. E. Jr. (2007) Mechanisms of specificity in protein phosphorylation. Nat. Rev. Mol. Cell Biol. 8, 530–541 [DOI] [PubMed] [Google Scholar]
- 6.Bhaduri S., and Pryciak P. M. (2011) Cyclin-specific docking motifs promote phosphorylation of yeast signaling proteins by G1/S Cdk complexes. Curr. Biol. 21, 1615–1623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kõivomägi M., Valk E., Venta R., Iofik A., Lepiku M., Morgan D. O., and Loog M. (2011) Dynamics of Cdk1 substrate specificity during the cell cycle. Mol Cell 42, 610–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dhanasekaran D. N., Kashef K., Lee C. M., Xu H., and Reddy E. P. (2007) Scaffold proteins of MAP-kinase modules. Oncogene 26, 3185–3202 [DOI] [PubMed] [Google Scholar]
- 9.Bardwell L. (2006) Mechanisms of MAPK signalling specificity. Biochem. Soc. Trans. 34, 837–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reményi A., Good M. C., and Lim W. A. (2006) Docking interactions in protein kinase and phosphatase networks. Curr. Opin. Struct. Biol. 16, 676–685 [DOI] [PubMed] [Google Scholar]
- 11.Peti W., and Page R. (2013) Molecular basis of MAP kinase regulation. Protein Sci. 22, 1698–1710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grewal S., Molina D. M., and Bardwell L. (2006) Mitogen-activated protein kinase (MAPK)-docking sites in MAPK kinases function as tethers that are crucial for MAPK regulation in vivo. Cell. Signal. 18, 123–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hancock C. N., Macias A. T., Mackerell A. D. Jr., and Shapiro P. (2006) Mitogen activated protein (MAP) kinases: development of ATP and non-ATP dependent inhibitors. Med. Chem. 2, 213–222 [DOI] [PubMed] [Google Scholar]
- 14.Akella R., Moon T. M., and Goldsmith E. J. (2008) Unique MAP kinase binding sites. Biochim. Biophys. Acta 1784, 48–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stebbins J. L., De S. K., Machleidt T., Becattini B., Vazquez J., Kuntzen C., Chen L. H., Cellitti J. F., Riel-Mehan M., Emdadi A., Solinas G., Karin M., and Pellecchia M. (2008) Identification of a new JNK inhibitor targeting the JNK-JIP interaction site. Proc. Natl. Acad. Sci. U.S.A. 105, 16809–16813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gaestel M., and Kracht M. (2009) Peptides as signaling inhibitors for mammalian MAP kinase cascades. Curr. Pharm. Des. 15, 2471–2480 [DOI] [PubMed] [Google Scholar]
- 17.Bubici C., and Papa S. (2014) JNK signalling in cancer: in need of new, smarter therapeutic targets. Br. J. Pharmacol. 171, 24–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kyriakis J. M., and Avruch J. (2012) Mammalian MAPK signal transduction pathways activated by stress and inflammation: a 10-year update. Physiol. Rev. 92, 689–737 [DOI] [PubMed] [Google Scholar]
- 19.Morrison D. K. (2012) MAP kinase pathways. Cold Spring Harb. Perspect. Biol. 4, a011254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Plotnikov A., Zehorai E., Procaccia S., and Seger R. (2011) The MAPK cascades: signaling components, nuclear roles and mechanisms of nuclear translocation. Biochim. Biophys. Acta 1813, 1619–1633 [DOI] [PubMed] [Google Scholar]
- 21.Yang S. H., Sharrocks A. D., and Whitmarsh A. J. (2013) MAP kinase signalling cascades and transcriptional regulation. Gene 513, 1–13 [DOI] [PubMed] [Google Scholar]
- 22.Caunt C. J., and Keyse S. M. (2013) Dual-specificity MAP kinase phosphatases (MKPs): shaping the outcome of MAP kinase signalling. FEBS J. 280, 489–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kusari A. B., Molina D. M., Sabbagh W. Jr., Lau C. S., and Bardwell L. (2004) A conserved protein interaction network involving the yeast MAP kinases Fus3 and Kss1. J. Cell Biol. 164, 267–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martín H., Flández M., Nombela C., and Molina M. (2005) Protein phosphatases in MAPK signalling: we keep learning from yeast. Mol. Microbiol. 58, 6–16 [DOI] [PubMed] [Google Scholar]
- 25.Futran A. S., Link A. J., Seger R., and Shvartsman S. Y. (2013) ERK as a model for systems biology of enzyme kinetics in cells. Curr. Biol. 23, R972–R979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bardwell A. J., Flatauer L. J., Matsukuma K., Thorner J., and Bardwell L. (2001) A conserved docking site in MEKs mediates high-affinity binding to MAP kinases and cooperates with a scaffold protein to enhance signal transmission. J. Biol. Chem. 276, 10374–10386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bardwell L., and Thorner J. (1996) A conserved motif at the amino termini of MEKs might mediate high-affinity interaction with the cognate MAPKs. Trends Biochem. Sci. 21, 373–374 [PubMed] [Google Scholar]
- 28.Lawrence M. C., Jivan A., Shao C., Duan L., Goad D., Zaganjor E., Osborne J., McGlynn K., Stippec S., Earnest S., Chen W., and Cobb M. H. (2008) The roles of MAPKs in disease. Cell Res. 18, 436–442 [DOI] [PubMed] [Google Scholar]
- 29.Tidyman W. E., and Rauen K. A. (2009) The RASopathies: developmental syndromes of Ras/MAPK pathway dysregulation. Curr. Opin. Genet. Dev. 19, 230–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dhillon A. S., Hagan S., Rath O., and Kolch W. (2007) MAP kinase signalling pathways in cancer. Oncogene 26, 3279–3290 [DOI] [PubMed] [Google Scholar]
- 31.Deschênes-Simard X., Gaumont-Leclerc M. F., Bourdeau V., Lessard F., Moiseeva O., Forest V., Igelmann S., Mallette F. A., Saba-El-Leil M. K., Meloche S., Saad F., Mes-Masson A. M., and Ferbeyre G. (2013) Tumor suppressor activity of the ERK/MAPK pathway by promoting selective protein degradation. Genes Dev. 27, 900–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cuadrado A., and Nebreda A. R. (2010) Mechanisms and functions of p38 MAPK signalling. Biochem. J. 429, 403–417 [DOI] [PubMed] [Google Scholar]
- 33.de Nadal E., Ammerer G., and Posas F. (2011) Controlling gene expression in response to stress. Nat. Rev. Genet. 12, 833–845 [DOI] [PubMed] [Google Scholar]
- 34.Johnson G. L., and Nakamura K. (2007) The c-jun kinase/stress-activated pathway: regulation, function and role in human disease. Biochim. Biophys. Acta 1773, 1341–1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sabio G., and Davis R. J. (2010) cJun NH2-terminal kinase 1 (JNK1): roles in metabolic regulation of insulin resistance. Trends Biochem. Sci. 35, 490–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bardwell A. J., Frankson E., and Bardwell L. (2009) Selectivity of docking sites in MAPK kinases. J. Biol. Chem. 284, 13165–13173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bogoyevitch M. A., and Kobe B. (2006) Uses for JNK: the many and varied substrates of the c-Jun N-terminal kinases. Microbiol. Mol. Biol. Rev. 70, 1061–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yoon S., and Seger R. (2006) The extracellular signal-regulated kinase: multiple substrates regulate diverse cellular functions. Growth Factors 24, 21–44 [DOI] [PubMed] [Google Scholar]
- 39.Yang S. H., Whitmarsh A. J., Davis R. J., and Sharrocks A. D. (1998) Differential targeting of MAP kinases to the ETS-domain transcription factor Elk-1. EMBO J. 17, 1740–1749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barsyte-Lovejoy D., Galanis A., and Sharrocks A. D. (2002) Specificity determinants in MAPK signaling to transcription factors. J. Biol. Chem. 277, 9896–9903 [DOI] [PubMed] [Google Scholar]
- 41.Galanis A., Yang S. H., and Sharrocks A. D. (2001) Selective targeting of MAPKs to the ETS domain transcription factor SAP-1. J. Biol. Chem. 276, 965–973 [DOI] [PubMed] [Google Scholar]
- 42.Garai Á., Zeke A., Gógl G., Törő I., Fördős F., Blankenburg H., Bárkai T., Varga J., Alexa A., Emig D., Albrecht M., and Rémenyi A. (2012) Specificity of linear motifs that bind to a common mitogen-activated protein kinase docking groove. Sci. Signal. 5, ra74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bardwell A. J., Abdollahi M., and Bardwell L. (2004) Anthrax lethal factor-cleavage products of MAPK (mitogen-activated protein kinase) kinases exhibit reduced binding to their cognate MAPKs. Biochem. J. 378, 569–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bardwell A. J., Abdollahi M., and Bardwell L. (2003) Docking sites on mitogen-activated protein kinase (MAPK) kinases, MAPK phosphatases and the Elk-1 transcription factor compete for MAPK binding and are crucial for enzymic activity. Biochem. J. 370, 1077–1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ho D. T., Bardwell A. J., Abdollahi M., and Bardwell L. (2003) A docking site in MKK4 mediates high affinity binding to JNK MAPKs and competes with similar docking sites in JNK substrates. J. Biol. Chem. 278, 32662–32672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ho D. T., Bardwell A. J., Grewal S., Iverson C., and Bardwell L. (2006) Interacting JNK-docking sites in MKK7 promote binding and activation of JNK mitogen-activated protein kinases. J. Biol. Chem. 281, 13169–13179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Callaway K., Abramczyk O., Martin L., and Dalby K. N. (2007) The anti-apoptotic protein PEA-15 is a tight binding inhibitor of ERK1 and ERK2, which blocks docking interactions at the D-recruitment site. Biochemistry 46, 9187–9198 [DOI] [PubMed] [Google Scholar]
- 48.Ember B., Kamenecka T., and LoGrasso P. (2008) Kinetic mechanism and inhibitor characterization for c-jun-N-terminal kinase 3α1. Biochemistry 47, 3076–3084 [DOI] [PubMed] [Google Scholar]
- 49.Niu L., Chang K. C., Wilson S., Tran P., Zuo F., and Swinney D. C. (2007) Kinetic characterization of human JNK2α2 reaction mechanism using substrate competitive inhibitors. Biochemistry 46, 4775–4784 [DOI] [PubMed] [Google Scholar]
- 50.Brandt R. B., Laux J. E., and Yates S. W. (1987) Calculation of inhibitor Ki and inhibitor type from the concentration of inhibitor for 50% inhibition for Michaelis-Menten enzymes. Biochem. Med. Metab. Biol. 37, 344–349 [DOI] [PubMed] [Google Scholar]
- 51.Cer R. Z., Mudunuri U., Stephens R., and Lebeda F. J. (2009) IC50-to-Ki: a web-based tool for converting IC50 to Ki values for inhibitors of enzyme activity and ligand binding. Nucleic Acids Res. 37, W441–W445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dérijard B., Raingeaud J., Barrett T., Wu I. H., Han J., Ulevitch R. J., and Davis R. J. (1995) Independent human MAP-kinase signal transduction pathways defined by MEK and MKK isoforms. Science 267, 682–685 [DOI] [PubMed] [Google Scholar]
- 53.Han J., Lee J. D., Jiang Y., Li Z., Feng L., and Ulevitch R. J. (1996) Characterization of the structure and function of a novel MAP kinase kinase (MKK6). J. Biol. Chem. 271, 2886–2891 [DOI] [PubMed] [Google Scholar]
- 54.Tournier C., Whitmarsh A. J., Cavanagh J., Barrett T., and Davis R. J. (1997) Mitogen-activated protein kinase kinase 7 is an activator of the c-Jun NH2-terminal kinase. Proc. Natl. Acad. Sci. U.S.A. 94, 7337–7342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu Z., Wu J., Jacinto E., and Karin M. (1997) Molecular cloning and characterization of human JNKK2, a novel Jun NH2-terminal kinase-specific kinase. Mol. Cell. Biol. 17, 7407–7416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lin A., Minden A., Martinetto H., Claret F. X., Lange-Carter C., Mercurio F., Johnson G. L., and Karin M. (1995) Identification of a dual specificity kinase that activates the Jun kinases and p38-Mpk2. Science 268, 286–290 [DOI] [PubMed] [Google Scholar]
- 57.Chang C. I., Xu B. E., Akella R., Cobb M. H., and Goldsmith E. J. (2002) Crystal structure of MAP kinase p38 complexed to the docking sites on its nuclear substrate MEF2A and activator MKK3b. Mol. Cell 9, 1241–1249 [DOI] [PubMed] [Google Scholar]
- 58.Reményi A., Good M. C., Bhattacharyya R. P., and Lim W. A. (2005) The role of docking interactions in mediating signaling input, output, and discrimination in the yeast MAPK network. Mol. Cell 20, 951–962 [DOI] [PubMed] [Google Scholar]
- 59.Laughlin J. D., Nwachukwu J. C., Figuera-Losada M., Cherry L., Nettles K. W., and LoGrasso P. V. (2012) Structural mechanisms of allostery and autoinhibition in JNK family kinases. Structure 20, 2174–2184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pearson W. R. (1990) Rapid and sensitive sequence comparison with FASTP and FASTA. Methods Enzymol. 183, 63–98 [DOI] [PubMed] [Google Scholar]
- 61.Laudet V., Hänni C., Stéhelin D., and Duterque-Coquillaud M. (1999) Molecular phylogeny of the ETS gene family. Oncogene 18, 1351–1359 [DOI] [PubMed] [Google Scholar]
- 62.Buchwalter G., Gross C., and Wasylyk B. (2004) Ets ternary complex transcription factors. Gene 324, 1–14 [DOI] [PubMed] [Google Scholar]
- 63.Hollenhorst P. C., McIntosh L. P., and Graves B. J. (2011) Genomic and biochemical insights into the specificity of ETS transcription factors. Annu. Rev. Biochem. 80, 437–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ducret C., Maira S. M., Lutz Y., and Wasylyk B. (2000) The ternary complex factor Net contains two distinct elements that mediate different responses to MAP kinase signalling cascades. Oncogene 19, 5063–5072 [DOI] [PubMed] [Google Scholar]
- 65.Wozniak M. A., Cheng C. Q., Shen C. J., Gao L., Olarerin-George A. O., Won K. J., Hogenesch J. B., and Chen C. S. (2012) Adhesion regulates MAP kinase/ternary complex factor exchange to control a proliferative transcriptional switch. Curr. Biol. 22, 2017–2026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fantz D. A., Jacobs D., Glossip D., and Kornfeld K. (2001) Docking sites on substrate proteins direct extracellular signal-regulated kinase to phosphorylate specific residues. J. Biol. Chem. 276, 27256–27265 [DOI] [PubMed] [Google Scholar]
- 67.Lee T., Hoofnagle A. N., Kabuyama Y., Stroud J., Min X., Goldsmith E. J., Chen L., Resing K. A., and Ahn N. G. (2004) Docking motif interactions in MAP kinases revealed by hydrogen exchange mass spectrometry. Mol. Cell 14, 43–55 [DOI] [PubMed] [Google Scholar]
- 68.Yang S. H., Yates P. R., Whitmarsh A. J., Davis R. J., and Sharrocks A. D. (1998) The Elk-1 ETS-domain transcription factor contains a mitogen-activated protein kinase targeting motif. Mol. Cell. Biol. 18, 710–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gógl G., Törő I., and Reményi A. (2013) Protein-peptide complex crystallization: a case study on the ERK2 mitogen-activated protein kinase. Acta Crystallogr. D Biol. Crystallogr. 69, 486–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tanoue T., Yamamoto T., and Nishida E. (2002) Modular structure of a docking surface on MAPK phosphatases. J. Biol. Chem. 277, 22942–22949 [DOI] [PubMed] [Google Scholar]
- 71.Muñoz J. J., Tárrega C., Blanco-Aparicio C., and Pulido R. (2003) Differential interaction of the tyrosine phosphatases PTP-SL, STEP and HePTP with the mitogen-activated protein kinases ERK1/2 and p38α is determined by a kinase specificity sequence and influenced by reducing agents. Biochem. J. 372, 193–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Francis D. M., Różycki B., Koveal D., Hummer G., Page R., and Peti W. (2011) Structural basis of p38α regulation by hematopoietic tyrosine phosphatase. Nat. Chem. Biol. 7, 916–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jacobs D., Glossip D., Xing H., Muslin A. J., and Kornfeld K. (1999) Multiple docking sites on substrate proteins form a modular system that mediates recognition by ERK MAP kinase. Genes Dev. 13, 163–175 [PMC free article] [PubMed] [Google Scholar]
- 74.Sakaguchi A., Matsumoto K., and Hisamoto N. (2004) Roles of MAP kinase cascades in Caenorhabditis elegans. J. Biochem. 136, 7–11 [DOI] [PubMed] [Google Scholar]
- 75.Whisenant T. C., Ho D. T., Benz R. W., Rogers J. S., Kaake R. M., Gordon E. A., Huang L., Baldi P., and Bardwell L. (2010) Computational prediction and experimental verification of new MAP kinase docking sites and substrates including Gli transcription factors. PLoS Comput. Biol. 10.1371/journal.pcbi.1000908 [DOI] [PMC free article] [PubMed] [Google Scholar]