

Background: MLIP (muscle enriched A-type lamin-interacting protein) is a unique protein of yet unknown function.
Results: MLIP impacts cardiac activity of Akt/mTOR pathways and is associated with and required for precocious cardiac adaptation to stress.
Conclusion: MLIP might be a new cardiac stress sensor.
Significance: These findings provide the first insight into the role of MLIP in vivo.
Keywords: Akt PKB, cardiac hypertrophy, cardiomyopathy, cardiovascular disease, heart failure, homeostasis, mammalian target of rapamycin (mTOR)
Abstract
Aging and diseases generally result from tissue inability to maintain homeostasis through adaptation. The adult heart is particularly vulnerable to disequilibrium in homeostasis because its regenerative abilities are limited. Here, we report that MLIP (muscle enriched A-type lamin-interacting protein), a unique protein of unknown function, is required for proper cardiac adaptation. Mlip−/− mice exhibited normal cardiac function despite myocardial metabolic abnormalities and cardiac-specific overactivation of Akt/mTOR pathways. Cardiac-specific MLIP overexpression led to an inhibition of Akt/mTOR, providing evidence of a direct impact of MLIP on these key signaling pathways. Mlip−/− hearts showed an impaired capacity to adapt to stress (isoproterenol-induced hypertrophy), likely because of deregulated Akt/mTOR activity. Genome-wide association studies showed a genetic association between Mlip and early response to cardiac stress, supporting the role of MLIP in cardiac adaptation. Together, these results revealed that MLIP is required for normal myocardial adaptation to stress through integrated regulation of the Akt/mTOR pathways.
Introduction
Cardiovascular diseases continue to be the leading cause of morbidity and mortality worldwide. Despite remarkable progress in the past decade, the fundamental mechanisms underlying cardiovascular pathophysiology remain poorly understood, which restricts effective identification of therapeutic targets for these diseases. Recent advances using system genetic approaches have highlighted novel genes and signaling pathways important for the cardiovascular system physiology and adaptation (1). Remodeling and adaptation are crucial properties of the myocardium and allow the heart to respond to changes in workload. Many molecules have been identified as cardiac mechanosensors and integrators of stress, all essential to preserve cardiac function (2). However, our understanding of the fundamental processes and early events that allow the myocardium to sense environmental cues and adapt accordingly is still limited.
We recently reported the discovery of MLIP (muscle enriched A-type lamin-interacting protein), encoded by a unique and conserved gene, ubiquitously expressed but enriched in the heart and muscles (3). MLIP was originally identified through its interaction with lamin A/C. Lamin A/C are ubiquitous proteins of the nuclear envelope. Mutations in lamin A/C give rise to a group of heterogeneous genetic disorders, collectively referred to as laminopathies, which display a large variety of complex clinical entities including skeletal and cardiac myopathies, lipodystrophy and metabolic abnormalities, neuropathy, leukodystrophy, and premature aging syndromes (4). The pathophysiologic mechanisms underlying the tissue-specific symptoms caused by mutant lamin A/C remain not fully understood. Among these A-type lamin-related disorders, cardiac disease is the most frequent form of laminopathies, and mutations in lamin A/C are also one of the most frequent causes of genetic dilated cardiomyopathy (5). Therefore, MLIP interaction with lamin might, in part, provide a solution to the complex manifestation of these specific mutations in lamin A/C. Yet the biological function of MLIP remains unknown.
Initial reports suggested that MLIP might play an important role in heart growth and disease (3, 6). To define the biological role of MLIP, we developed an Mlip knock-out mouse model (thereafter referred to as Mlip−/− mice). In the present study, we report that the loss of MLIP did not have an impact on cardiac function or structure but led to myocardial specific metabolic abnormalities and cardiac specific overactivation of Akt/mTOR6 pathways. In contrast, cardiac specific overexpression of MLIP led to an inhibition of Akt/mTOR signaling, providing evidence of a direct impact of MLIP on these key signaling pathways. Despite the absence of phenotype at baseline, Mlip−/− hearts showed a rapid increase in heart weight without cardiomyocyte hypertrophy and deregulated activity of Akt/mTOR pathways in response to isoproterenol-induced cardiac stress, thus unraveling an inadequate capacity to remodel and adapt. In addition, a systems genetic approach revealed a significant genetic association between Mlip and early cardiac response to isoproterenol-induced hypertrophy. Together, these data indicate that MLIP participates in the maintenance of cardiac homeostasis and the first response of the heart to workload changes. These findings provide the first insight into the role of MLIP in vivo and identify MLIP as a potential therapeutic target for cardiac diseases.
Experimental Procedures
Animals and Treatments
To generate the Mlip knock-out mouse model, a mutant Mlip allele was introduced into embryonic stem cells in which exon 1 and the putative proximal promoter was flanked by loxP sequences. Mice bearing this mutant Mlip allele (designated Mlipfl/+) in the inbred 129SvEv were mated to transgenic C57BL/6J CMV-Cre mice, which constitutively express Cre recombinase from the X-chromosome (7). In the presence of Cre, a new Mlip allele, designated Mlip−, was generated that lacks exon 1 and the putative proximal promoter. To remove the CMV-cre allele, male (CMV-Cre; Mlip+/−) mice were then mated with female 129SvEv. The male progeny from the CMV-Cre; Mlip+/− × Mlip+/+ were screened for the Mlip− allele and further backcrossed into the 129SvEv background.
To generate the cardiac-specific Mlip transgenic mouse model, the cDNA encoding the endogenous form of mouse Mlip was obtained by RT-PCR using total RNA isolated from the mouse cardiac ventricle. Full-length Mlip (∼0.97 kb) was subcloned, completely sequenced in both directions, and compared with GenBankTM cDNA database (accession number NM_027150.1). Full-length Mlip was subcloned into the SalI and HindIII restriction sites downstream of the mouse α-myosin heavy chain (α-MHC) promoter and the construct purified from the plasmid backbone after BamHI digestion. Microinjection of the linearized α-MHC promoter-Mlip construct transgene into fertilized eggs generated multiple lines of FVB/N transgenic mice (8). Four founders were obtained for this construct; the one chosen for experimentation (line 37) expressed the MLIP protein in the heart at a level that was ∼3.5-fold higher.
All the mice were studied according to protocols approved by the Canadian Council on Animal Care's Guide to the Care and Use of Experimental Animals and the Animals for Research Act. To determine the rate of cardiac global protein synthesis, puromycin (Sigma; 40 μmol·kg−1 of body weight) was intravenously injected to isofluorane-anesthetized animals (9). The heart was harvested 10 min after the injection.
For the isoproterenol-induced hypertrophy study, isoproterenol (40 mg·kg−1·day−1) was administered for 9 or 15 days to 12-week-old female and male Mlip+/+ and Mlip−/− mice through a dorsal implanted Alzet micro-osmotic pump (model 1004). Control mice were infused with saline.
Cardiac Function Measurement
Transthoracic echocardiography was performed at room temperature using an echocardiography-Doppler (Vevo 770 system; VisualSonics) with a probe RMV707B (15–45 MHz). The mice were slightly anesthetized with 0.5–1% isoflurane in 100%O2. The two-dimensionally guided time motion mode recording of the left ventricle (LV) provided the following measurements: interventricular septal wall thickness in diastole (IVSd), posterior wall thickness in diastole, LV end-diastolic (LVDD), and LV end systolic (LVSD) diameters. Percentage of LV fractional shortening (FS) were calculated as follows: FS = (LVDD − LVSD)/LVDD × 100.
Cardiac Hemodynamic Measurement
In vivo pressure-volume analysis was performed as previously described (10). Briefly, after mice were deeply anesthetized with 2.5% isofluorane, right carotid artery and jugular vein were exposed, without damaging the vagus nerve. A 1.2F Scisense Pressure catheter (Transonic) was inserted into the carotid and advanced retrogradely across the aortic valve into the left ventricle. Hemodynamic measurements were recorded at baseline and after 2-min of isoproterenol infusion (20 pg·g−1·min−1) through the jugular vein.
Genome-wide Association Study
The hybrid mouse diversity panel used for this study consisted of 30 classical inbred and 75 recombinant inbred (AXB (9), BXA (10), BXD (44), BXH (5), and CXB (7)) strains. 8–10-week-old female mice were divided into control and treated groups. Isoproterenol (20 mg·kg−1·day−1) was administered for 21 days in 9-week-old female mice through an abdominally implanted Alzet micropump, in ∼4 mice per strain. Echocardiograms were performed at baseline and at weekly intervals up to 3 weeks. genome-wide association study of directly measured and calculated echocardiographic measures was performed using the efficient mixed model association algorithm to correct for population substructure (11, 12).
Micro-positron Emission Tomography (PET) Imaging
Mouse PET [18F]fluorodeoxyglucose (FDG) imaging was conducted in the InveonTM DPET small animal scanner (Siemens, Knoxsville, TN) as previously described (13, 14). A 60-min list mode acquisition was started together with a 10–20-s tail vein injection of FDG (18–72 MBq in 150 μl). List data were sorted into 26 dynamic frames (12 × 10 s, 3 × 60 s, and 11 × 300 s) and reconstructed using OSEM3D with 10 iterations, 16 subsets, zoom 2.5 with a 128 × 128 matrix, resulting in a 0.35-mm transaxial pixel size. Images were corrected for radioactive decay, random coincidences and dead time losses using the vendor software Inveon Acquisition Workplace (version 1.5). The standard uptake value to evaluate radiotracer uptake was calculated according to the following standard equation: activity concentration in a region of interest (Bq/cc)/injected activity (Bq) corrected for the weight of the animal (g).
Metabolic Assessment
All analyses were performed on 12-week-old Mlip+/+ and Mlip−/− mice. Blood glucose was measured on animals fasted for 5 h using an Accu-Chek Aviva Nano glucometer as directed by the manufacturer. To perform glucose tolerance test, animals were fasted for 5 h, and blood glucose was measured, as described above, 15, 30, 60, and 120 min after intraperitoneal injection of 2 mg/g glucose solution. For the assessment of metabolic hormones levels, blood was collected at the saphene vein of animals fasted for 5 h. Plasma was extracted by centrifugation at 4000 rpm for 5 min and clarified by second centrifugation at 12,000 rpm for 20 min. Resistin, gastric inhibitory polypeptide, plasminogen activator inhibitor-1, glucagon-like petide-1, glucagon, ghrelin, leptin, and insulin concentrations were measured with Bio-PlexTM mouse diabetes 8-plex immunoassay (Bio-Rad) as recommended by the manufacturer. General metabolism was evaluated by indirect calorimetry using Oxymax/CLAMS monitoring system (Columbus Instruments). O2 consumption and CO2 production were recorded for 24 h and used to calculate the respiratory exchange rate.
Histology and Immunochemical Analysis
Fresh heart samples were fixed overnight in 1% formalin and paraffin-embedded. Cardiac sections (8 μm) were dewaxed and stained with hematein/eosin and Masson's trichrome for fibrosis visualization. Sections were analyzed by light microscopy. For wheat germ agglutinin (WGA) staining, cardiac sections were processed as described above, immersed in WGA solution for 20 min, and washed with PBS three times. For immunohistochemical analysis, fresh heart samples were snap frozen in liquid nitrogen-cooled isopentane, and stored at −80 °C until further processing. Frozen sections (8 μm) of cardiac muscle tissue were fixed for 10 min in 100% methanol at −20 °C, permeabilized 10 min at room temperature with PBS-Triton 0.3% and incubated for 30 min with blocking solution (5% bovine serum albumin in PBS-Triton 0.3%) at room temperature. Sections were incubated overnight at room temperature with primary mouse IgG1 anti-vinculin monoclonal antibody (1:250, V9131; Sigma-Aldrich), primary mouse IgG1 anti-α-actinin monoclonal antibody (1:250, A7811; Sigma-Aldrich), and a primary rabbit anti-MLIP polyclonal antibody (1:500, custom made) diluted in blocking solution. Sections were washed three times with PBS and incubated with secondary antibody (1:250, Alexa Fluor 488 goat anti-mouse IgG1, 1:500, Alexa Fluor 594 goat anti-rabbit IgG) for 1 h at room temperature. The nuclei were stained with DAPI and mounted with mounting medium (Invitrogen). Images were acquired with a Carl Zeiss fluorescence microscope.
Electron Microscopy
The ultrastructure of the myocardium of three mice per genotype was analyzed using electron microscope. Under deep anesthesia, mice were perfused with 0.3 m KOH to stop the heart in diastole. They were then perfused with fixative (2.5% glutaraldehyde, 2% paraformaldehyde, pH 7.4, in PBS). Freshly harvested left ventricle was cut in small pieces and kept in fixative solution overnight at 4 °C before processing. Ultrathin sections were cut, stained with uranyl acetate and lead citrate, and imaged on a JEOL 1230 transmission electron microscope using ATM software.
Microarray Expression Profiling and mRNA Analysis
The hearts of three mice per genotype were dissected and snap frozen in liquid nitrogen. Total RNA extraction was performed with TRIzol as previously described (3). For gene expression profiling, RNA were processed as recommended by the manufacturer (Affymetrix) and hybridized on Affymetrix Gene Chip Mouse Gene 1.0 ST Array. Quantitative PCR was performed with LightCycler 480 Sybr Green kit (Roche Diagnostic) following manufacturer's instructions. Individual expression values were normalized by comparison with Rplp0, Gapdh, Hprt, and Ubc mRNA, housekeeping genes. The sequences of oligonucleotides used for quantitative PCR analysis are listed in supplemental Table S2.
Protein Analysis
For Western blot analyses, proteins were extracted from frozen total heart (ventricles and atria) and Gastrocnemius muscle, as previously described (3). Proteins were separated by SDS-PAGE. A list of the antibodies used is provided in supplemental Table S3. Signals were developed using enhanced chemiluminescence reagent (SuperSignal West Femto, Thermo Scientific).
Statistical Analysis
Differences between groups were assessed using analysis of variance and Student's t test. Values of p < 0.05 were considered statistically significant.
Results
MLIP Is Not Required for Normal Cardiac Development and Function
To delineate the biological function of MLIP, we employed a Cre-mediated deletion strategy to generate an Mlip-deficient mouse model (Fig. 1A). Homozygote (Mlip−/−) Mlip knock-out mice were born with normal Mendelian ratios (χ2 test = 0.83). They were not overtly different from their control littermates (Fig. 1B), and the survival rate was similar in Mlip-deficient and control mice (oldest Mlip−/− mouse 16 months to date). Western blot analysis of Mlip-deficient hearts showed a complete loss of MLIP expression in Mlip−/− hearts with no major change in protein expression of A-type or B-type lamins (Fig. 1C). We previously reported that MLIP was found in the nucleus, interacting with A-type lamin, and in the cytoplasm of mouse C2C12 myoblasts (3). Using the Mlip−/− heart as negative control, we further characterized the localization of MLIP in the adult Mlip+/+ heart by immunostaining. MLIP was mainly located beneath the sarcolemma, partially colocalizing with vinculin staining in ventricular adult cardiomyocytes (Fig. 1D). In the atria, MLIP was found both beneath the sarcolemma, as in ventricular cardiomyocytes, and in the nucleus (data not shown).
FIGURE 1.

Mlip-deficient mice have normal cardiac structure and function. A, generation of Mlip KO mouse model by deletion of the first exon of the Mlip gene and its proximal promoter. B, genotyping and photographs of adult Mlip+/+ and Mlip−/− mice. C, Western blot of MLIP, A- and B-type lamins in Mlip+/+ and Mlip−/− hearts. D, Mlip+/+ cardiac sections stained with MLIP (red), vinculin, or α-actinin (green) antibodies. Nuclei were counterstained with DAPI (blue). Scale bars, 20 μm. E, HW:BW ratio of Mlip+/+ and Mlip−/− male mice (n = 3–8 per genotype and age, means ± S.D.). F, lung weight (LW):BW ratio of Mlip+/+ and Mlip−/− male mice (n = 3–8 per genotype and age, means ± S.D.). G, echocardiographic measurements values of 12-week-old Mlip+/+ and Mlip−/− male mice in diastole (d) and systole (s). IVS, interventricular septum; LVID, left ventricular internal diameter; FS, fractional shortening; EF, ejection fraction (n = 7 per genotype, means ± S.D.). Student's t test was used.
Detailed phenotypic analysis of the Mlip−/− mice did not reveal any remarkable cardiac abnormalities when compared with their control littermates. Heart weight to body weight ratio was similar in Mlip−/− and Mlip+/+ mice at all the ages examined (12–55 weeks of age; Fig. 1E), indicating no form of cardiac hypertrophy or atrophy. Lung morphological aspect and lungs weight to body weight ratio did not differ between Mlip−/− and Mlip+/+ mice (Fig. 1F), suggesting normal cardiac function in Mlip−/− mice. This result was confirmed by echocardiographic analysis and cardiac hemodynamic assessment of 12-week-old Mlip−/− mice, which revealed no contractile dysfunction or dilation of Mlip−/− hearts as compared with their control littermates (Fig. 1G and Table 1). In accordance with preserved cardiac function and dimensions, structural analysis of the myocardium revealed no myocardial disarray or fibrosis in Mlip−/− hearts (Fig. 2, A and B). Cardiomyocyte cross-section area was similar between Mlip−/− and Mlip+/+ mice at 12 weeks and 1 year of age (Fig. 2, C and D). Altogether, these results indicate that MLIP is not required to maintain normal cardiac structure and function.
TABLE 1.
Hemodynamic parameters of 12-week-old Mlip+/+ and Mlip−/− male mice
HR, heart rate; ESP, left ventricular end systolic pressure; EDP, end diastolic pressure; ESV, end systolic volume; EDV, end diastolic volume; SV, stroke volume; EF, ejection fraction; dP/dtmax, peak rate of pressure rise; dP/dtmin, peak rate of pressure decline; Tau (τ), relaxation time constant calculated by Glantz method (regression of dP/dt versus pressure). The values are means ± S.D.
| Mlip+/+ (n = 4) |
Mlip−/− (n = 4) |
|||
|---|---|---|---|---|
| Baseline | Isoproterenol | Baseline | Isoproterenol | |
| BW (g) | 24.9 ± 1.1 | 21.7 ± 0.6 | ||
| HR (bpm) | 569 ± 39 | 603 ± 29 | 601 ± 29 | 611 ± 36 |
| ESP (mm Hg) | 111.9 ± 16.7 | 106.5 ± 12.2 | 99.0 ± 19.4 | 95.2 ± 19.3 |
| EDP (mm Hg) | 13.6 ± 8.1 | 16.1 ± 9.3 | 9.2 ± 4.3 | 9.0 ± 4.0 |
| ESV (μl) | 27.4 ± 16.8 | 24.4 ± 14.3 | 21.6 ± 10.6 | 13.1 ± 6.1 |
| EDV (μl) | 42.5 ± 19.4 | 37.9 ± 15.8 | 43.0 ± 19.9 | 34.1 ± 15.5 |
| SV (μl) | 31.5 ± 12.7 | 28.3 ± 10.9 | 30.7 ± 12.8 | 27.6 ± 14.3 |
| Systolic function | ||||
| EF (%) | 68.62 ± 10.9 | 69.0 ± 12.5 | 72.0 ± 11.0 | 81.3 ± 20.1 |
| dP/dtmax (mm Hg/s) | 8437 ± 1339 | 8265 ± 1341 | 8225 ± 2145 | 9180 ± 1774 |
| Diastolic function | ||||
| dP/dtmin (−mm Hg/s) | 9326 ± 1651 | 8352 ± 1174 | 9215 ± 2655 | 9427 ± 1940 |
| Tau, τ (ms) | 9.1 ± 3.1 | 9.6 ± 3.9 | 7.4 ± 2.1 | 8.4 ± 4.4 |
FIGURE 2.

Normal myocardial structure of Mlip−/− hearts. A, hematein/eosin and Masson's trichrome staining of 12-week-old Mlip+/+ and Mlip−/− male hearts. Scale bars, 50 μm. B, electron microscopy pictures of 12-week-old Mlip+/+ and Mlip−/− male myocardium. Scale bars, 2 μm. C, cardiac section of 12-week-old and 1-year-old mice stained with WGA and vinculin antibody, respectively. Scale bar, 50 μm. D, measure of cardiomyocyte cross-section area (CSA) based on WGA and vinculin staining (means ± S.D.).
Loss of MLIP Results in Cardiac Metabolic Abnormalities
We previously reported that Mlip gene encodes a unique nucleotide sequence that is conserved among amniotes (3). Remarkably, MLIP shares no structural domain with any known protein. The positive evolutionary selection of Mlip, as well as its original structure, suggests that it might assume novel biological functions. Because MLIP is highly expressed in the heart, where its isoform expression pattern is complex (Ref. 3 and Fig. 1C), we hypothesized that MLIP would have specific functions in myocardial physiology. To gain more insight into the molecular functions of MLIP, gene expression profiling was performed in the heart and revealed 1093 genes differentially expressed in Mlip−/− versus Mlip+/+ hearts (p < 0.05) (supplemental Table S1). The microarray data were validated by quantitative PCR on a set of randomly selected genes with high/low fold change and high/low p values. A significant correlation was observed between the microarray and quantitative PCR results (r2 = 0.957, p = 1 × 10−8). Among the significantly deregulated genes, 121 genes displayed a fold change of ±1.3, with 78 genes being up-regulated and 43 genes being down-regulated in the Mlip−/− hearts (Fig. 3A and supplemental Table S1). The relatively small number of genes deregulated in Mlip−/− hearts was in accordance with the absence of phenotype observed in Mlip−/− hearts.
FIGURE 3.

Deregulation of metabolic and pro-hypertrophic pathways in Mlip-deficient hearts. A, gene expression array analysis (volcano lot) identified 121 genes deregulated in 12-week-old Mlip−/− male hearts (n = 3 per genotype). B, gene ontology biological process analysis of deregulated genes in Mlip−/− hearts (FunNet Transcriptional Networks Analysis). C, top deregulated pathways predicted by ingenuity pathway analysis in the heart of Mlip−/− mice.
To further explore and identify the affected pathways and transcriptional regulators impacted by the loss of MLIP in the heart, mRNA expression patterns were analyzed (FunNet Transcriptional Analysis Network) and revealed an enrichment of up-regulated transcripts associated with RNA processing and translation (up to 30% of the transcripts, p = 4.6 × 10−3 to 3.9 × 10−6) in Mlip−/− hearts. Down-regulated transcripts were predominantly associated with oxido-reduction processes (50% of the transcripts, p = 8.2 × 10−4) and transmembrane transport (33% of the transcripts, p = 0.039) (Fig. 3B). This analysis suggested that protein synthesis and general metabolism were affected in Mlip−/− hearts. In addition, in depth pathway analysis (ingenuity pathway analysis) of the deregulated genes in the Mlip−/− hearts revealed a significant enrichment of genes associated with the canonical pathways of EIF2 signaling, regulation of eIF4 and p70S6K signaling, mTOR signaling, and p53 signaling (Fig. 3C), pathways involved in the control of protein synthesis and metabolic adaptation. Taken together, these in silico analyses indicate that there may be alterations in the general metabolism of Mlip−/− hearts.
Metabolic remodeling is a classic adaptive feature of the heart that is observed during pathological stress conditions and hypertrophy. This adaptive mechanism allows the heart to cope with higher energy demand and involves an increased reliance on glucose to produce ATP through glucose oxidation. As a consequence, cardiac hypertrophy and remodeling are generally associated with an increase in cellular glucose uptake (15). To characterize the potential metabolic abnormalities of Mlip−/− hearts, we measured myocardial glucose uptake using FDG and PET imaging. A significant decrease in myocardial FDG uptake was observed in Mlip−/− mice compared with Mlip+/+ controls (Fig. 4, A and B). To determine whether the reduced cardiac glucose uptake resulted from overall systemic metabolic abnormalities because of the loss of MLIP, we performed general metabolic assessment of Mlip−/− mice. The general metabolic rate (based on the respiratory exchange rate) was investigated through indirect calorimetric chambers (Table 2). A glucose tolerance test was performed (Fig. 4F); serum levels of metabolic hormones (e.g. insulin, leptin, glucagon) and liver and adipose tissue weights were also measured (Table 2). For all the measured parameters, no difference was observed between Mlip+/+ and Mlip−/− mice. These results indicated no global metabolic defects in Mlip−/− mice and suggested that the decrease in cardiac FDG uptake was likely due to tissue intrinsic features. To test whether the loss of MLIP affected glucose uptake in a cardiac specific manner, we then analyzed the FDG uptake in skeletal muscle, tissue in which MLIP is highly expressed; in the liver; and in the brown adipose tissue. No difference in FDG uptake in the skeletal muscle (Quadriceps), liver and brown adipose tissue was observed in Mlip−/− mice when compared with Mlip+/+ mice (Fig. 4, C–E), indicating a cardiac specific decrease in glucose uptake in Mlip−/− mice. mRNA expression of glucose transporters (Glut) 1, 2, and 4 was similar in Mlip−/− and Mlip+/+ hearts (Fig. 4G), Glut4 being the major Glut expressed in the adult heart. However, total Glut1 protein levels were slightly lower (25% reduction, = 0.045) in Mlip−/− heart (Fig. 4, H and I), explaining in part the decreased glucose uptake observed in these hearts.
FIGURE 4.

Glucose uptake is altered in Mlip-deficient hearts. A, myocardial FDG uptake in 12-week-old Mlip+/+ and Mlip−/− male hearts detected by positron emission tomography imaging. B–E, quantification of the FDG uptake over time in the myocardium (B), skeletal muscle (Quadriceps, C), brown adipose tissue (BAT, D), and liver (E) (n = 6–7 per genotype, means ± S.E.). AUC, area under the curve. Student's t test was used. F, glucose tolerance test in 20-week-old Mlip+/+ and Mlip−/− male mice (n = 8 per genotype, means ± S.D.). Student's t test was used. G, cardiac Glut1, Glut2, and Glut4 mRNA levels in 12-week-old Mlip+/+ and Mlip−/− male mice (n = 3 per genotype, means ± S.D.). H and I, cardiac Glut1 and Glut4 protein levels in 12-week-old Mlip+/+ and Mlip−/− male hearts (n = 4 per genotype, means ± S.D.). *, p < 0.05. Student's t test was used. Lamin B was the loading control. J, cardiac mRNA levels of key genes involved in fatty acid transport (FAT), glycolysis, TCA cycle, fatty acid oxidation (FAO), and oxidative phosphorylation (OXPHOS) in the heart of 12-week-old Mlip+/+ and Mlip−/− male mice (n = 3 per genotype, means ± S.D.). *, p < 0.05. Student's t test was used. K, Western blot of the five mitochondrial oxidative phosphorylation complexes in 12-week-old Mlip+/+ and Mlip−/− male hearts.
TABLE 2.
Metabolic parameters of 12-week-old Mlip+/+ and Mlip−/− male mice
BW, body weight; WAT, abdominal white adipose tissue; BAT, brown adipose tissue; GIP, gastric inhibitory polypeptide; PAI-1, plasminogen activator inhibitor 1; GLP-1, glucagon-like peptide 1.
| Mlip+/+ | Mlip−/− | p value | n | |
|---|---|---|---|---|
| BW (g) | 24.8 ± 2.3 | 25.9 ± 2.6 | 0.181 | 17/19 |
| WAT weight (mg) | 348.7 ± 149.2 | 458.0 ± 266.7 | 0.387 | 6/8 |
| WAT weight to BW (mg/g) | 14.8 ± 6.9 | 17.1 ± 8.1 | 0.573 | 6/8 |
| BAT weight (mg) | 94.0 ± 18.0 | 125.6 ± 63.0 | 0.259 | 6/8 |
| BAT weight to BW (mg/g) | 3.91 ± 0.75 | 4.70 ± 1.76 | 0.326 | 6/8 |
| Liver weight (mg) | 931.7 ± 121.6 | 980.9 ± 103.7 | 0.430 | 6/8 |
| Liver weight to BW (mg/g) | 38.6 ± 3.7 | 37.6 ± 4.0 | 0.713 | 6/8 |
| Fasted blood glucose (mmol/liter) | 6.76 ± 0.84 | 6.81 ± 1.01 | 0.869 | 17/18 |
| Hormones levels (pg/ml) | ||||
| Resistin | 106,632 ± 25,988 | 116,911 ± 34,038 | 0.466 | 11/9 |
| GIP | 553 ± 159 | 575 ± 145 | 0.743 | 11/9 |
| PAI-1 | 4225 ± 1468 | 3744 ± 1381 | 0.461 | 11/9 |
| GLP-1 | 58 ± 25 | 56 ± 16 | 0.756 | 11/9 |
| Glucagon | 298 ± 94 | 283 ± 35 | 0.637 | 11/9 |
| Ghrelin | 31,634 ± 7469 | 24,031 ± 11,723 | 0.110 | 11/9 |
| Leptin | 2939 ± 1924 | 3794 ± 2505 | 0.412 | 11/9 |
| Insulin | 2212 ± 639 | 2585 ± 729 | 0.244 | 11/9 |
| Respiratory exchange rate | ||||
| Light | 0.889 ± 0.030 | 0.870 ± 0.046 | 0.466 | 5/6 |
| Dark | 0.921 ± 0.025 | 0.909 ± 0.045 | 0.602 | 5/6 |
We next measured the expression of genes involved in fatty acid transport, glycolysis, TCA cycle, fatty acid oxidation, and oxidative phosphorylation in the heart to determine whether the loss of MLIP affected general cellular metabolic processes in addition to glucose uptake. Despite higher expression of Hk1 (encoding the Hexokinase 1, a key regulator of glycolytic flux) in Mlip−/− hearts, the expression of fatty acid transport, glycolysis, TCA cycle, fatty acid oxidation, and oxidative phosphorylation genes was similar in Mlip−/− and Mlip+/+ hearts (Fig. 4J), suggesting no general metabolic remodeling at the gene level in Mlip−/− hearts. In addition, mitochondrial structure and expression of the respiratory chain complex proteins were similar in Mlip−/− and Mlip+/+ hearts (Figs. 2B and 4K).
MLIP Regulates the Cardiac Activity of Akt/mTOR Signaling Pathways
To gain more insights into the molecular basis of the observed decreased glucose uptake in MLIP-deficient hearts, we then examined the upstream regulators involved in this process. One of the major regulators of “beat to beat” glucose uptake in the myocardium is the AMP-activated protein kinase (AMPK) (16), which plays a central role in cellular energy sensing and homeostasis (17). Phosphorylation of Thr-172 of the AMPKα catalytic subunit is crucial for the activation of the AMPK complex (18), subsequently resulting in an increase in cellular glucose uptake (16, 19). In Mlip−/− hearts, phosphorylation of AMPKα-Thr-172 was lower than in Mlip+/+ hearts, despite similar expression and phosphorylation of the regulatory AMPKβ subunits (Fig. 5A). The activity of LKB1, the upstream activating kinase of AMPK (20), was similar in Mlip+/+ and Mlip−/− hearts, as reflected by identical phosphorylation levels (Fig. 5A). These results indicated an LKB1-independent inactivation of AMPK, supporting the decreased glucose uptake observed in Mlip−/− hearts.
FIGURE 5.

MLIP impacts the activity of AMPK, Akt, and mTOR pathways in the heart. A–C, representative Western blot and quantification of pan- and phosphorylated AMPK, Akt, mTOR, and their upstream regulators LKB1, PDK1, and PTEN and downstream targets p70S6 kinase and 4E-BP1 in 12-week-old Mlip+/+ and Mlip−/− male hearts (n = 4 per genotype, means ± S.D.). *, p < 0.05; **, p < 0.01. Student's t test was used. Lamin B was the loading control. D, representative Western blot showing puromycin incorporation in polypeptide chains reflecting the global rate of protein synthesis in 12-week-old Mlip+/+ and Mlip−/− male hearts (n = 4 per genotype, means ± S.D.). Actin was the loading control. E and F, representative Western blot and quantification of MLIP, pan- and phosphorylated Akt, mTOR, and their downstream target 4E-BP1 in 12-week-old Mlip+/+ and Mlip−/− male skeletal muscle (Gastrocnemius) (n = 4 per genotype, means ± S.D.). Lamin B was the loading control. G, generation of cardiac-specific Mlip transgenic (Mlip-Tg) mouse model by insertion of Mlip full-length cDNA downstream from the α-MHC promoter. H, representative Western blot of MLIP in 20-week-old FVB WT and Mlip transgenic male hearts. Short and long exposure of the blot are shown. I, representative Western blot and quantification of pan- and phosphorylated Akt, mTOR and their downstream target 4E-BP1 in 20-week-old Wt and Mlip transgenic male hearts (n = 4 per genotype, means ± S.D.). *, p < 0.05; **, p < 0.01. Student's t test was used. Lamin B was the loading control.
Cardiac metabolic balance is finely tuned by two major signaling pathways that cross-regulate each other: AMPK signaling and Akt/mTOR signaling (21, 22). The Akt pathway represents one of the most well characterized positive regulators of mTOR activity and negative regulator of AMPK activity. Akt is activated by phospholipid binding (phosphatidylinositol 3,4,5-triphoshate, PIP3) at the plasma membrane and phosphorylation on Thr-308 by PDK1 (23). This process is negatively regulated through a PTEN-dependent mechanism, a lipid phosphatase (24). Dephosphorylation of PTEN-Ser-380 leads to its recruitment to the plasma membrane, where it dephosphorylates PIP3 (25), leading to the inactivation of Akt. In Mlip−/− hearts, Akt was activated as reflected by increased phosphorylation of Thr-308, without alteration of PDK1 phosphorylation/activity (Fig. 5B). However, phosphorylation of PTEN-Ser-380 was reduced, indicating an increased activity of PTEN in Mlip−/− hearts (Fig. 5B). This result suggested an uncoupling of the regulation of Akt activation, likely occurring at the sarcolemma in Mlip−/− hearts. mTOR activity was assessed as a downstream target of Akt and AMPK cascades (21). Phosphorylation of Thr-389 of p70S6 kinase and Thr-37/46 of 4E-BP1, two mTOR classical targets, was significantly increased in Mlip−/− hearts (Fig. 5C), indicating activation of the mTOR pathway in these hearts. To investigate whether this alteration in mTOR signaling could result in changes in the protein synthesis process, we measured the rate of protein synthesis in vivo using the puromycin incorporation method (9). Despite the activation of mTOR pathway in Mlip−/− hearts, the steady state of protein synthesis was similar in Mlip−/− and Mlip+/+ hearts (Fig. 5D). A discrepancy between mTOR activation and increased steady state of protein synthesis has been reported by others (26). This result was in accordance with the absence of cardiac morphological changes observed in the absence of MLIP (Fig. 2, C and D).
Collectively, the Western blot analyses of the AMPK/Akt/mTOR pathways revealed a deregulated activity of the key stress/nutrient sensors, AMPK and Akt, resulting in a hyperactivation of the mTOR pathway in Mlip−/− hearts. We then investigated whether the hyperactivation of Akt/mTOR pathways was also observed in skeletal muscle, in which glucose uptake was not affected by the loss of MLIP (Fig. 4C). Phosphorylation of Akt, mTOR, and 4E-BP1 was similar in the skeletal muscle (Gastrocnemius) of Mlip+/+ and Mlip−/− mice (Fig. 5, E and F), indicating that the deregulation of Akt/mTOR pathways observed in Mlip−/− hearts was cardiac specific.
To rule out the possibility that the activation of Akt/mTOR pathways observed in Mlip−/− hearts occurred as a result of secondary changes, rather than direct effects of MLIP deletion, we examined a transgenic mouse model in which MLIP was overexpressed in a cardiac specific manner (α-myosin heavy chain promoter driven Mlip transgenic mice, referred to as Mlip transgenic mice; Fig. 5G). In this model, cardiac MLIP protein level was 3.5-fold higher when compared with control heart (Fig. 5H). Importantly, cardiac specific overexpression of MLIP had no deleterious effect on cardiac function or heart size (Table 3). Analysis of the phosphorylation status of Akt, mTOR, and 4E-BP1 showed that overexpression of MLIP led to a significant decrease of Akt and 4E-BP1 phosphorylation, indicating an inactivation of Akt and mTOR pathways in Mlip transgenic hearts (Fig. 5I). These results provided evidence for a direct effect of MLIP on the activity of Akt and mTOR pathways in the heart and support the notion that the hyperactivation of these pathways was primarily resulting from the loss of MLIP in Mlip−/− hearts.
TABLE 3.
Echocardiographic measures and heart weight of 20-week-old WT and Mlip transgenic male mice
d, diastole; s, systole; IVS, interventricular septum; LVID, left ventricular internal diameter; FS, fractional shortening; EF, ejection fraction; HW:BW, heart to body weight ratio; Mlip-Tg, Mlip transgenic. The values are means ± S.D. Student's t test was used.
| WT | Mlip-Tg | p value | n | |
|---|---|---|---|---|
| IVSd (mm) | 0.96 ± 0.10 | 0.94 ± 0.14 | 0.885 | 3/6 |
| LVIDd (mm) | 3.47 ± 0.45 | 3.40 ± 0.31 | 0.782 | 3/6 |
| LVIDs (mm) | 1.48 ± 0.25 | 1.54 ± 0.22 | 0.713 | 3/6 |
| FS (%) | 57.5 ± 1.83 | 54.2 ± 9.18 | 0.564 | 3/6 |
| EF (%) | 92.3 ± 1.00 | 89.4 ± 6.93 | 0.506 | 3/6 |
| Heart rate (bpm) | 393 ± 22 | 381 ± 56 | 0.727 | 3/6 |
| HW:BW (mg/g) | 5.27 ± 0.69 | 5.53 ± 0.60 | 0.464 | 5/10 |
MLIP Is Genetically Associated with and Required for Precocious Adaptation to Isoproterenol-induced Cardiac Hypertrophy
Given that the loss of MLIP resulted in deregulation of AMPK, Akt, and mTOR signaling, major pathways involved in homeostasis, we then tested whether Mlip−/− hearts were susceptible to stress. Using a well established isoproterenol (ISO)-induced hypertrophy model, female and male Mlip−/− mice were challenged with isoproterenol for 9 or 15 days. After 9 days of continuous ISO treatment, both female and male Mlip−/− mice displayed a robust increase in heart to body weight (HW:BW) ratio, whereas smaller or no change was observed in the Mlip+/+ mice compared with saline-treated mice (Fig. 6, A and B). Ventricular dimensions and function were assessed by echocardiography and showed that ISO-treated male Mlip−/− mice displayed thickened interventricular septum compared with saline-treated Mlip−/− mice (IVSd, 1.16 ± 0.16 versus 0.88 ± 0.06, p < 0.01; Table 4), explaining in part the increase in HW:BW ratio observed. In contrast, no wall thickening was observed in the ISO-treated male Mlip+/+ mice at that stage. In accordance with the inotropic effect of β-adrenergic receptor stimulation, ISO-treated Mlip+/+ mice demonstrated a temporary increase in contractility evidenced by higher ejection fraction and fractional shortening compared with their saline-treated controls (% ejection fraction, 91 ± 3 versus 62 ± 16; % FS, 61 ± 5 versus 34 ± 14; p < 0.05; Table 4). Conversely, ejection fraction and fractional shortening were not statistically different between ISO- and saline-treated Mlip−/− mice, indicating a blunted contractile response to ISO in these hearts.
FIGURE 6.

MLIP is required for proper cardiac response to stress. A, heart to body weight ratio of 12-week-old Mlip+/+ and Mlip−/− female mice treated with isoproterenol (40 mg·kg−1·day−1) for 9 days (n = 6 per group, means ± S.D.). **, p < 0.01; *, p < 0.05 versus genotype-matched saline control. Student's t test was used. B, heart to body weight ratio of 12-week-old Mlip+/+ and Mlip−/− male mice treated with isoproterenol (40 mg·kg−1·day−1) for 9 or 15 days (n = 3–8 per group, means ± S.D.). *, p < 0.05 versus genotype-matched saline control. Student's t test was used. C, hematein/eosin staining of Mlip+/+ and Mlip−/− male hearts after 15 days of isoproterenol infusion. Scale bars, 2.5 mm. D, Masson's Trichrome staining of Mlip+/+ and Mlip−/− male hearts after 15 days of isoproterenol infusion. Scale bars, 200 μm. E, cardiac section of Mlip+/+ and Mlip−/− male hearts after 15 days of isoproterenol infusion, stained with WGA. Scale bars, 50 μm. Measure of cardiomyocyte cross-section area (CSA) based on WGA staining (n = 3–4 per group, means ± S.D.). **, p < 0.01; *, p < 0.05. Student's t test was used. F, representative Western blot and quantification of total and phosphorylated Akt and mTOR in Mlip+/+ and Mlip−/− female hearts treated with isoproterenol for 9 days (n = 3–4 per group, means ± S.D.). **, p < 0.01; *, p < 0.05. Student's t test was used.
TABLE 4.
Echocardiographic measures of saline- and isoproterenol-treated Mlip+/+ and Mlip−/− male mice
d, diastole; s, systole; IVS, interventricular septum; LVID, left ventricular internal diameter; FS, fractional shortening; EF, ejection fraction; HW:BW, heart to body weight ratio. The values are means ± S.D. Student's t test was used.
| Saline |
ISO |
|||||
|---|---|---|---|---|---|---|
| 9 days |
15 days |
|||||
| Mlip+/+ | Mlip−/− | Mlip+/+ | Mlip−/− | Mlip+/+ | Mlip−/− | |
| IVSd (mm) | 1.02 ± 0.20 | 0.88 ± 0.06 | 1.12 ± 0.13 | 1.16 ± 0.16a | 1.42 ± 0.65a,b | 1.31 ± 0.08a |
| LVIDd (mm) | 3.65 ± 0.31 | 3.69 ± 0.42 | 3.31 ± 0.18 | 3.36 ± 0.76 | 3.60 ± 1.94 | 3.84 ± 0.25 |
| LVIDs (mm) | 2.41 ± 0.61 | 2.29 ± 0.47 | 1.29 ± 0.17c | 1.72 ± 0.68 | 1.95 ± 1.07 | 2.42 ± 0.56 |
| FS (%) | 34.4 ± 13.8 | 38.4 ± 7.52 | 61.2 ± 4.74c | 49.9 ± 10.7 | 46.4 ± 20.7b | 37.0 ± 10.7 |
| EF (%) | 62.0 ± 16.0 | 68.8 ± 9.55 | 90.7 ± 3.10c | 81.1 ± 11.3 | 77.7 ± 36.8 | 67.3 ± 15.0 |
a p < 0.01 versus genotype-matched saline-treated mice.
b p < 0.05 versus genotype-matched 9 days ISO-treated mice.
c p < 0.05 versus genotype-matched saline-treated mice.
By 15 days of ISO treatment, the increase in heart mass was similar in male Mlip+/+ and Mlip−/− ISO-treated mice and significantly higher when compared with their respective saline-treated controls (Fig. 6B). It is noteworthy that there was no further increase in the heart mass of Mlip−/− mice when compared with 9 days of treatment. Masson's Trichrome staining revealed a massive and similar deposition of extracellular matrix in both ISO-treated Mlip−/− and Mlip+/+ hearts (Fig. 6D), suggesting primary insult of similar severity in ISO-treated Mlip−/− and Mlip+/+ mice. However, despite Mlip−/− and Mlip+/+ mice having the same HW:BW ratios after 15 days of ISO, the cardiac morphology was different. ISO-treated Mlip+/+ hearts displayed robust hypertrophic phenotype as evidenced by the thickening of the ventricular walls (Fig. 6C and Table 4) and a massive increase in cardiomyocyte cross-section area (Fig. 6E). Conversely, ISO-treated Mlip−/− hearts were round-shaped (Fig. 6C), and cardiomyocyte cross-section area was not statistically different from the saline-treated Mlip−/− hearts (Fig. 6E). These results suggested that the rapid increase in heart mass of Mlip−/− mice under ISO reflected the incapacity of Mlip−/− hearts to buffer ISO-induced cardiac stress and adapt properly.
To rule out the possibility that the impaired adaptation to ISO observed in Mlip−/− hearts was due to altered β-adrenergic receptors sensitivity or function in the absence of MLIP, we assessed the cardiac response to acute ISO perfusion using hemodynamic measurement in Mlip−/− and Mlip+/+ mice. The results indicated that the response to acute inotropic stimulation was similar in Mlip−/− and Mlip+/+ mice (Table 1). Therefore, the effect observed under chronic ISO stimulation (Fig. 6) was not due to a defect in immediate β-adrenergic pathway but was likely related to the effect of MLIP on cardiac remodeling and long term adaptation.
Because Akt/mTOR pathways are major regulators of cardiac hypertrophy and were both deregulated in Mlip−/− hearts (Figs. 3C and 5), we investigated whether the rapid increase in HW:BW ratio observed in ISO-treated Mlip−/− hearts was associated with further deregulation of these pathways. As previously reported (27–29), ISO treatment resulted in increased activity of mTOR, as evidenced by the increase in phospho-mTOR to total mTOR ratio in ISO-treated Mlip+/+ when compared with saline-treated Mlip+/+ hearts (Fig. 6F). Unexpectedly, ISO-treated Mlip−/− hearts displayed a decreased phospho-mTOR to total mTOR ratio compared with saline-treated Mlip−/− hearts. Strikingly, the hyperactivation of mTOR observed in Mlip−/− hearts (Fig. 5C and saline-treated animals in Fig. 6F) was normalized under ISO infusion (Fig. 6F), possibly because of the inactivation of Akt in ISO-treated Mlip−/− hearts (Fig. 6F). Of note, the activation of Akt observed in Mlip−/− hearts (Fig. 5B) was absent in Mlip−/− mice after implantation with saline-delivering micro-osmotic pumps (saline-treated Mlip−/− mice in Fig. 6F). We hypothesized that this could be due to the stress of surgery, the inflammation resulting from the healing process, and/or the stress caused by the subcutaneous micro-osmotic pump. Altogether, these data indicated a blunted response to ISO-induced hypertrophy in Mlip−/− hearts and suggested that Mlip−/− hearts were unable to adapt properly, likely because of their inability to maintain adequate activity of Akt/mTOR pathways.
The implication of MLIP in the cardiac response to pro-hypertrophic stimulation was confirmed by an independent genome-wide association study that took advantage of the Hybrid Mouse Diversity Panel (11, 12). In this experiment, a system genetics approach was undertaken to identify, in an unbiased manner, key genes important for cardiac hypertrophy and remodeling, using a similar ISO-induced hypertrophy model. A panel of 105 strains of inbred female mice that have been densely genotyped and display natural phenotypic variation were induced to hypertrophy with continuous infusion of ISO. Echocardiography was performed prior to and every week during ISO infusion over a 3-week period (Fig. 7A). A genetic association with IVSd, an echocardiographic end point of response to ISO, was found after 1 week of continuous ISO infusion, as represented by Manhattan plot (Fig. 6B). The peak SNP rs13480288 (p = 4.1 × 10−9) and nearby SNPs (within correlation r2 > 0.8) spanned across three genes on chromosome 9 (Fig. 7D). Of these, Mlip gene alone harbored a missense variant rs30135861 (c.T568A, p.L154Q in exon 3; Fig. 7E) and a splice region variant rs30427253 near the 3′ acceptor site of exon 7 (Fig. 7E) in a conserved region of Mlip, which cosegregated with the peak SNP across our panel. The location of these specific variants next to exon-intron junctions might affect the alternative splicing of Mlip mRNA, favoring specific exon enrichment or skipping in the different mouse strains. After 2 weeks of continuous ISO infusion no genetic association was observed with ISO-induced IVSd wall thickening (Fig. 7C). mRNA expression measured by quantitative PCR revealed a significant down-regulation of Mlip expression with ISO treatment in all strains (p = 9.5 × 10−5) (Fig. 7F). These data provided compelling evidence that genetic variations in Mlip contribute to differential responses in early interventricular septum hypertrophy in response to ISO and supported the interaction between MLIP and precocious cardiac response to stress seen in the Mlip−/− mice (Fig. 6, A and B). Altogether these results strongly suggest that MLIP plays a cardioprotective role under stress by enabling appropriate remodeling and hypertrophic adaptation.
FIGURE 7.

Mlip is genetically associated with early cardiac hypertrophy. A, timeline of isoproterenol infusion (20 mg·kg−1·day−1) used in the genome-wide association study. B and C, Manhattan plot showing the association between genotype and IVSd at after 1 (B) and 2 (C) weeks of isoproterenol infusion. The red line represents the statistical threshold. D, regional association plot with interventricular septum thickness in diastole in response to isoproterenol (week 1), with linkage disequilibrium denoted by r2. E, characteristics of the two SNPs in Mlip gene associated with differential hypertrophy of the IVSd. F, Mlip mRNA level after 3 weeks of isoproterenol treatment in all mouse strains (means ± S.D.). ***, p < 0.001, paired t test.
Discussion
We recently reported the discovery of MLIP, a unique and conserved protein among the Amniotes (3). MLIP interacts with A-type lamin (3) and Islet1 transcription factor (6) and has been implicated in neonate cardiomyocyte hypertrophy in vitro (6). Despite these reported findings, the molecular and biological function(s) of MLIP remain to be defined.
In the present study, we show that in the heart (i) MLIP localized beneath the sarcolemma, a key cellular compartment in the transmission and integration of extracellular signals in adult ventricular cardiomyocytes; (ii) MLIP is critical for the maintenance of a balanced activity of the cardiac homeostatic pathways AMPK/Akt/mTOR; and (iii) MLIP is associated with and required for adequate precocious response to isoproterenol-induced cardiac hypertrophy. Taken together these results provide the first evidence that MLIP plays a pivotal role in the capacity of the heart to adapt, particularly in response to β-adrenergic agonist-induced stress.
The exact molecular function of MLIP remains unclear, but based on our study, we propose that in the heart, MLIP is essential for the integration of the AMPK/Akt/mTOR regulatory feedback loops, which are key traits of homeostasis (Fig. 8). In Mlip−/− hearts, the deregulated activity of Akt and AMPK is independent of their upstream activating kinases (PDK1 and LKB1, respectively; Fig. 5) and inactivating phosphatase (PTEN, which is activated in Mlip−/− hearts but uncoupled to downstream Akt; Fig. 5). These observations support the notion that MLIP functions by integrating and coupling signals transmitted by these upstream regulators (PDK1/PTEN), to translate them into appropriate Akt activity. The nature of the functional interaction between MLIP and Akt remains to be elucidated, but this process likely occurs beneath the sarcolemma in adult ventricular cardiomyocytes and contributes to the balanced regulation of Akt downstream targets: mTOR and AMPK (Fig. 8), thus preserving cardiac homeostasis. In the absence of MLIP, the aberrant activation of Akt is likely responsible for the inactivation of AMPK, through direct (30) and/or indirect mechanisms (31), and for the activation of mTOR signaling (21). mTOR activation is further sustained through the Akt-dependent inhibition of AMPK (Fig. 8). These feedback loops would contribute, in part, to the establishment of a displaced equilibrium between anabolic-hypertrophic signaling and energy producing pathways, which ultimately results in maladaptive remodeling and inadequate or limited adaptation to stress in Mlip−/− mice.
FIGURE 8.
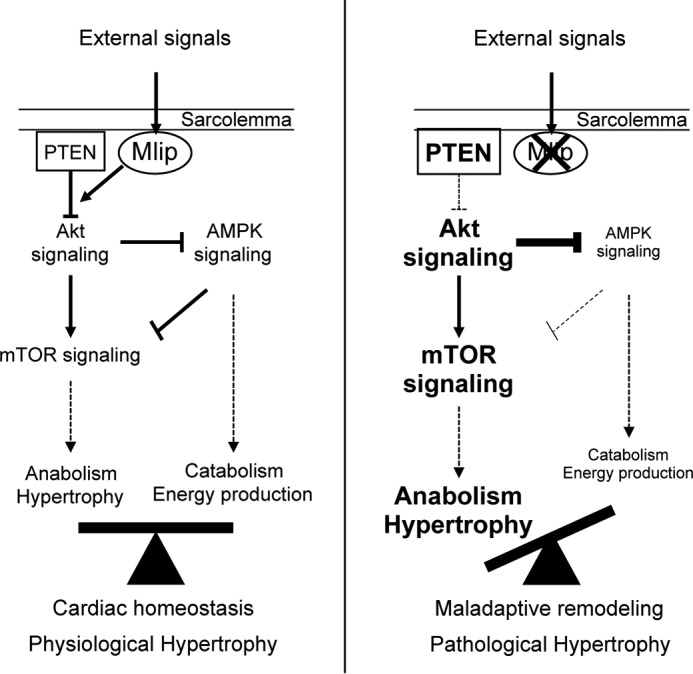
MLIP is required for maintenance of cardiac homeostasis in response to stress. In this model, MLIP integrates physiological signals (physiological growth, stress) and modulates the activation of AMPK/Akt/mTOR signaling pathways to maintain proper metabolic equilibrium in the myocardium, thereby promoting cardiac homeostasis and proper adaptation.
An imbalance or uncoupling of these key signaling pathways results in cell and tissue inadequacy to continually sense and adapt to environmental cues that often manifests as cellular and tissular dysfunction and disease. The heart adjusts to changes in its workload demands by promoting cellular hypertrophy. This phenomenon is initially beneficial and decreases ventricular wall stress to maintain cardiac output. However, if uncontrolled or persistent, cardiac hypertrophy ultimately leads to pathological remodeling and failure. A very large number of studies have been dedicated to understand the complex role of Akt and mTOR signaling pathways with regard to cardiac hypertrophy and adaptation (reviewed in Refs. 32 and 33). Most of the models used consisted in genetic modifications to either overexpress wild-type or mutant proteins or delete endogenous isoforms. More moderate strategies have used pharmacological inhibition, of mTOR notably. The conclusions of these studies revealed that both Akt and mTOR endorse very complex functions in the myocardium. Akt has been shown to be overall cardioprotective, through the promotion of physiological hypertrophy and pro-survival signaling. However, the biological outcome of Akt activation appears to be dependent on the duration, frequency, and intensity of this activation. For instance, short term activation of Akt, through cardiac overexpression of phosphomimetic constitutively active Akt1 or myristoylated Akt, promoted adaptive hypertrophy, whereas long term activation or high levels of expression of Akt induced pathological hypertrophy and heart failure (34–38). Similarly, mTOR plays crucial and complex roles in cardiac physiology and diseases. Indeed, mTOR is required for cardiac development and physiological hypertrophy in response to cardiac overload by promoting adaptive remodeling (39–41). However, mTOR also promotes pathological hypertrophy in pressure-overload and ischemia conditions (42–44). Conversely, the activation of mTOR has also been reported in several mouse models without hypertrophy at the organ or cellular level (45, 46). Noteworthy, the activation of mTOR alone is not sufficient to induce cardiac hypertrophy and morphological changes, as evidenced by the absence of myocardial structural changes in mice overexpressing mTOR in cardiomyocytes (47). It appears that the addition of stress (e.g. genetic defects, myocardial infarction, aortic banding, β-adrenergic receptor stimulation) is required to elicit the effect of mTOR activation on cardiac hypertrophy (reviewed in Ref. 33). In Mlip−/− mice, the increased in mTOR activity (2–3-fold increase in phosphorylation of its targets p70S6K and 4E-BP1; Fig. 5C) is comparable to the levels reported in mouse models that do not explicit cardiac hypertrophy (45–47). Interestingly, the α-MHC-mTOR transgenic mice have normal cardiac structure and function but a limited response to cardiac stress (aortic banding) (47), as observed in Mlip−/− mice under chronic isoproterenol infusion.
Taken together, these studies showed that cardiac homeostasis and adaptation are particularly sensitive to and dependent on proper integration and regulation of both Akt and mTOR signaling. In addition, Akt and mTOR pathways are tightly intricate in the regulation of hypertrophic processes, and mTOR has been shown to mediate the cardiac hypertrophy induced by Akt overexpression (34, 37).
Aberrant activation of Akt-mTOR has been documented in cardiomyopathies of different etiologies and notably in A-type lamin-related dilated cardiomyopathy models in which it contributes to the pathological process of the disease (46, 48). The exact mechanisms driving this activation in a context of A-type lamin alterations are yet to be elucidated. Whether MLIP plays a role in this aberrant activation in context of lamin A/C mutations would need further exploration. More generally, sustained activation of mTOR is associated with a wide spectrum of diseases (49, 50) and accelerating aging in many species, from yeast to mammals (51), whereas mTOR inhibition has been shown to be beneficial in curbing the aging process and prolonging survival (49). In Mlip−/− hearts, the global gene expression profiling revealed an overall activation of p53 signaling (Fig. 3C), which together with the hyperactivation of Akt and mTOR, may reflect an accelerated aging of the MLIP-deficient hearts.
Genome-wide association studies represent a powerful genetic tool to identify new candidate genes responsible for complex traits and diseases. To uncover the genetic causes underlying heart failure, several genome-wide association studies have been performed in different populations of patients suffering from heart failure (1). However, only limited number of candidate genes have been reported, which may in part be due to the heterogeneous etiologies of heart failure and stage of the disease. To overcome this issue, animal models have been develop to study the influence of genetic factors in the setting of cardiac hypertrophy and heart failure. Chronic stimulation of β-adrenergic receptors, a common feature of many cardiovascular diseases and a determinant contributor to cardiac hypertrophy and ultimately heart failure (52), was used in the present study to identify initiating genetic interactions with the progression of the disease. As evidenced by our results presented in Fig. 7 (B and C), genetic associations were found only at early stage of the pathological process (week 1) but failed to reach the statistical threshold when the phenotype was more pronounced (week 2). This is in accordance with the genome-wide association studies on heart failure patients (1) and is likely due to the masking of discrete and transient genetic associations by the overall cardiac remodeling process and progression of the disease. The early association of Mlip with interventricular septum thickening, a hallmark of cardiac hypertrophy, strongly suggests that MLIP is part of the first responders to pro-hypertrophic stress, reinforcing its role in sensing and maintenance of homeostasis in the heart.
Importantly, in accordance with the notion of MLIP being a homeostasis “gatekeeper,” our results show that MLIP expression levels need to be finely regulated to ensure proper adaptation to myocardial stress. Mlip expression is significantly down-regulated under ISO-induced hypertrophic stimulation to promote adequate remodeling of the heart (Fig. 7F). This finding is in accordance with a previous report that MLIP overexpression inhibits neonate cardiomyocyte hypertrophy in vitro (6). However, the complete loss of MLIP in Mlip−/− hearts leads to inadequate adaptation, especially under stress conditions, and results in maladaptive remodeling (i.e. decreased glucose uptake, absence of cardiomyocyte hypertrophy under ISO) and blunted adaptive response. In addition, the respective roles of the different MLIP spliced isoforms remain to be elucidated but will likely increase the complexity of the biological functions of MLIP in the heart. Therefore, the therapeutic targeting of MLIP may represent a new strategy for the treatment of cardiomyopathies and heart failure.
Author Contributions
P. G. B. developed the research program; P. G. B. and M.-E. C. designed the study; P. G. B., M.-E. C., C. L. R., J. J. W., and E. M.-W. carried out the experiments and analyzed data; S. L. T. and J. N. D. performed and analyzed micro-PET studies; J. W., Y. W., and A. J. L. designed and executed genome-wide association study experiments and analyzed data. P. G. B. and M.-E. C. wrote the paper with editorial input from all listed authors.
Supplementary Material
Acknowledgments
We thank Rob Beanlands, Rob deKemp, and the Small Animal Imaging Facility at the University of Ottawa Heart Institute for technical advice. We thank Julia Petryk, Richard Seymour, and the staff of the Animal Care and Veterinarian Services of the University of Ottawa Heart Institute for technical assistance.
This work was supported, in whole or in part, by Canadian Institutes of Health Research Grant MOP119470 (to P. G. B.), Heart and Stroke Foundation of Canada Grant 000007 (to P. G. B.), and National Institutes of Health Grants HL123295 and HL114437 (to A. J. L.). The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Tables S1–S3.
- mTOR
- mammalian target of rapamycin
- ISO
- isoproterenol
- LV
- left ventricle
- IVSd
- interventricular septal wall thickness in diastole
- LVDD
- LV end diastolic
- LVSD
- LV end systolic
- FS
- fractional shortening
- PET
- positron emission tomography
- FDG
- [18F]fluorodeoxyglucose
- WGA
- wheat germ agglutinin
- Glut
- glucose transporter
- AMPK
- AMP-activated protein kinase
- HW
- heart weight
- BW
- body weight
- MLIP
- muscle enriched A-type lamin-interacting protein.
References
- 1.Rau C. D., Lusis A. J., and Wang Y. (2015) Genetics of common forms of heart failure: challenges and potential solutions. Curr. Opin. Cardiol. 30, 222–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lyon R. C., Zanella F., Omens J. H., and Sheikh F. (2015) Mechanotransduction in cardiac hypertrophy and failure. Circ. Res. 116, 1462–1476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ahmady E., Deeke S. A., Rabaa S., Kouri L., Kenney L., Stewart A. F., and Burgon P. G. (2011) Identification of a novel muscle A-type lamin-interacting protein (MLIP). J. Biol. Chem. 286, 19702–19713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bertrand A. T., Chikhaoui K., Yaou R. B., and Bonne G. (2011) Clinical and genetic heterogeneity in laminopathies. Biochem. Soc. Trans. 39, 1687–1692 [DOI] [PubMed] [Google Scholar]
- 5.Hershberger R. E., Morales A., and Siegfried J. D. (2010) Clinical and genetic issues in dilated cardiomyopathy: a review for genetics professionals. Genet. Med. 12, 655–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang Z. P., Young Seok H., Zhou B., Chen J., Chen J. F., Tao Y., Pu W. T., and Wang D. Z. (2012) CIP, a cardiac Isl1-interacting protein, represses cardiomyocyte hypertrophy. Circ. Res. 110, 818–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schwenk F., Baron U., and Rajewsky K. (1995) A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucleic Acids Res. 23, 5080–5081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Palermo J., Gulick J., Colbert M., Fewell J., and Robbins J. (1996) Transgenic remodeling of the contractile apparatus in the mammalian heart. Circ. Res. 78, 504–509 [DOI] [PubMed] [Google Scholar]
- 9.Kelleher A. R., Kimball S. R., Dennis M. D., Schilder R. J., and Jefferson L. S. (2013) The mTORC1 signaling repressors REDD1/2 are rapidly induced and activation of p70S6K1 by leucine is defective in skeletal muscle of an immobilized rat hindlimb. Am. J. Physiol. Endocrinol. Metab. 304, E229–E236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pacher P., Nagayama T., Mukhopadhyay P., Bátkai S., and Kass D. A. (2008) Measurement of cardiac function using pressure-volume conductance catheter technique in mice and rats. Nat. Protoc. 3, 1422–1434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rau C. D., Wang J., Avetisyan R., Romay M. C., Martin L., Ren S., Wang Y., and Lusis A. J. (2015) Mapping genetic contributions to cardiac pathology induced by β-adrenergic stimulation in mice. Circ. Cardiovasc. Genet. 8, 40–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Farber C. R., Bennett B. J., Orozco L., Zou W., Lira A., Kostem E., Kang H. M., Furlotte N., Berberyan A., Ghazalpour A., Suwanwela J., Drake T. A., Eskin E., Wang Q. T., Teitelbaum S. L., and Lusis A. J. (2011) Mouse genome-wide association and systems genetics identify Asxl2 as a regulator of bone mineral density and osteoclastogenesis. PLoS Genet. 7, e1002038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thorn S. L., deKem R. A., Dumouchel T., Klein R., Renaud J. M., Wells R. G., Gollob M. H., Beanlands R. S., and DaSilva J. N. (2013) Repeatable noninvasive measurement of mouse myocardial glucose uptake with 18F-FDG: evaluation of tracer kinetics in a type 1 diabetes model. J. Nucl. Med. 54, 1637–1644 [DOI] [PubMed] [Google Scholar]
- 14.Thorn S. L., Gollob M. H., Harper M. E., Beanlands R. S., Dekemp R. A., and Dasilva J. N. (2013) Chronic AMPK activity dysregulation produces myocardial insulin resistance in the human Arg302Gln-PRKAG2 glycogen storage disease mouse model. EJNMMI Res. 3, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Neubauer S. (2007) The failing heart: an engine out of fuel. N. Engl. J. Med. 356, 1140–1151 [DOI] [PubMed] [Google Scholar]
- 16.Steinbusch L. K., Schwenk R. W., Ouwens D. M., Diamant M., Glatz J. F., and Luiken J. J. (2011) Subcellular trafficking of the substrate transporters GLUT4 and CD36 in cardiomyocytes. Cell Mol. Life Sci. 68, 2525–2538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim A. S., Miller E. J., and Young L. H. (2009) AMP-activated protein kinase: a core signalling pathway in the heart. Acta Physiol. (Oxf.) 196, 37–53 [DOI] [PubMed] [Google Scholar]
- 18.Hawley S. A., Davison M., Woods A., Davies S. P., Beri R. K., Carling D., and Hardie D. G. (1996) Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J. Biol. Chem. 271, 27879–27887 [DOI] [PubMed] [Google Scholar]
- 19.Lee C. T., Ussher J. R., Mohammad A., Lam A., and Lopaschuk G. D. (2014) 5′-AMP-activated protein kinase increases glucose uptake independent of GLUT4 translocation in cardiac myocytes. Can. J. Physiol. Pharmacol. 92, 307–314 [DOI] [PubMed] [Google Scholar]
- 20.Woods A., Johnstone S. R., Dickerson K., Leiper F. C., Fryer L. G., Neumann D., Schlattner U., Wallimann T., Carlson M., and Carling D. (2003) LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr. Biol. 13, 2004–2008 [DOI] [PubMed] [Google Scholar]
- 21.Shimobayashi M., and Hall M. N. (2014) Making new contacts: the mTOR network in metabolism and signalling crosstalk. Nat. Rev. Mol. Cell Biol. 15, 155–162 [DOI] [PubMed] [Google Scholar]
- 22.Zoncu R., Efeyan A., and Sabatini D. M. (2011) mTOR: from growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 12, 21–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alessi D. R., James S. R., Downes C. P., Holmes A. B., Gaffney P. R., Reese C. B., and Cohen P. (1997) Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Bα. Curr. Biol. 7, 261–269 [DOI] [PubMed] [Google Scholar]
- 24.Stambolic V., Suzuki A., de la Pompa J. L., Brothers G. M., Mirtsos C., Sasaki T., Ruland J., Penninger J. M., Siderovski D. P., and Mak T. W. (1998) Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell 95, 29–39 [DOI] [PubMed] [Google Scholar]
- 25.Rahdar M., Inoue T., Meyer T., Zhang J., Vazquez F., and Devreotes P. N. (2009) A phosphorylation-dependent intramolecular interaction regulates the membrane association and activity of the tumor suppressor PTEN. Proc. Natl. Acad. Sci. U.S.A. 106, 480–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hayasaka M., Tsunekawa H., Yoshinaga M., and Murakami T. (2014) Endurance exercise induces REDD1 expression and transiently decreases mTORC1 signaling in rat skeletal muscle. Physiol. Rep. 2, e12254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang W., Yano N., Deng M., Mao Q., Shaw S. K., and Tseng Y. T. (2011) β-Adrenergic receptor-PI3K signaling crosstalk in mouse heart: elucidation of immediate downstream signaling cascades. PLoS One 6, e26581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lundby A., Andersen M. N., Steffensen A. B., Horn H., Kelstrup C. D., Francavilla C., Jensen L. J., Schmitt N., Thomsen M. B., and Olsen J. V. (2013) In vivo phosphoproteomics analysis reveals the cardiac targets of β-adrenergic receptor signaling. Sci. Signal 6, rs11. [DOI] [PubMed] [Google Scholar]
- 29.Chen X., Zeng S., Zou J., Chen Y., Yue Z., Gao Y., Zhang L., Cao W., and Liu P. (2014) Rapamycin attenuated cardiac hypertrophy induced by isoproterenol and maintained energy homeostasis via inhibiting NF-κB activation. Mediators Inflamm. 2014, 868753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hawley S. A., Ross F. A., Gowans G. J., Tibarewal P., Leslie N. R., and Hardie D. G. (2014) Phosphorylation by Akt within the ST loop of AMPK-α1 down-regulates its activation in tumour cells. Biochem. J. 459, 275–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu L., Siu F. M., Che C. M., Xu A., and Wang Y. (2012) Akt blocks the tumor suppressor activity of LKB1 by promoting phosphorylation-dependent nuclear retention through 14–3-3 proteins. Am. J. Transl. Res. 4, 175–186 [PMC free article] [PubMed] [Google Scholar]
- 32.Sussman M. A., Völkers M., Fischer K., Bailey B., Cottage C. T., Din S., Gude N., Avitabile D., Alvarez R., Sundararaman B., Quijada P., Mason M., Konstandin M. H., Malhowski A., Cheng Z., Khan M., and McGregor M. (2011) Myocardial AKT: the omnipresent nexus. Physiol. Rev. 91, 1023–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sciarretta S., Volpe M., and Sadoshima J. (2014) Mammalian target of rapamycin signaling in cardiac physiology and disease. Circ. Res. 114, 549–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shioi T., McMullen J. R., Kang P. M., Douglas P. S., Obata T., Franke T. F., Cantley L. C., and Izumo S. (2002) Akt/protein kinase B promotes organ growth in transgenic mice. Mol. Cell Biol. 22, 2799–2809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matsui T., Li L., Wu J. C., Cook S. A., Nagoshi T., Picard M. H., Liao R., and Rosenzweig A. (2002) Phenotypic spectrum caused by transgenic overexpression of activated Akt in the heart. J. Biol. Chem. 277, 22896–22901 [DOI] [PubMed] [Google Scholar]
- 36.Condorelli G., Drusco A., Stassi G., Bellacosa A., Roncarati R., Iaccarino G., Russo M. A., Gu Y., Dalton N., Chung C., Latronico M. V., Napoli C., Sadoshima J., Croce C. M., and Ross J. Jr. (2002) Akt induces enhanced myocardial contractility and cell size in vivo in transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 99, 12333–12338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shiojima I., Sato K., Izumiya Y., Schiekofer S., Ito M., Liao R., Colucci W. S., and Walsh K. (2005) Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J. Clin. Invest. 115, 2108–2118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schiekofer S., Shiojima I., Sato K., Galasso G., Oshima Y., and Walsh K. (2006) Microarray analysis of Akt1 activation in transgenic mouse hearts reveals transcript expression profiles associated with compensatory hypertrophy and failure. Physiol. Genomics 27, 156–170 [DOI] [PubMed] [Google Scholar]
- 39.Zhu Y., Pires K. M., Whitehead K. J., Olsen C. D., Wayment B., Zhang Y. C., Bugger H., Ilkun O., Litwin S. E., Thomas G., Kozma S. C., and Abel E. D. (2013) Mechanistic target of rapamycin (mTOR) is essential for murine embryonic heart development and growth. PLoS One 8, e54221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang D., Contu R., Latronico M. V., Zhang J. L., Rizzi R., Catalucci D., Miyamoto S., Huang K., Ceci M., Gu Y., Dalton N. D., Peterson K. L., Guan K. L., Brown J. H., Chen J., Sonenberg N., and Condorelli G. (2010) MTORC1 regulates cardiac function and myocyte survival through 4E-BP1 inhibition in mice. J. Clin. Invest. 120, 2805–2816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shende P., Plaisance I., Morandi C., Pellieux C., Berthonneche C., Zorzato F., Krishnan J., Lerch R., Hall M. N., Rüegg M. A., Pedrazzini T., and Brink M. (2011) Cardiac raptor ablation impairs adaptive hypertrophy, alters metabolic gene expression, and causes heart failure in mice. Circulation 123, 1073–1082 [DOI] [PubMed] [Google Scholar]
- 42.Shioi T., McMullen J. R., Tarnavski O., Converso K., Sherwood M. C., Manning W. J., and Izumo S. (2003) Rapamycin attenuates load-induced cardiac hypertrophy in mice. Circulation 107, 1664–1670 [DOI] [PubMed] [Google Scholar]
- 43.McMullen J. R., Sherwood M. C., Tarnavski O., Zhang L., Dorfman A. L., Shioi T., and Izumo S. (2004) Inhibition of mTOR signaling with rapamycin regresses established cardiac hypertrophy induced by pressure overload. Circulation 109, 3050–3055 [DOI] [PubMed] [Google Scholar]
- 44.Buss S. J., Muenz S., Riffel J. H., Malekar P., Hagenmueller M., Weiss C. S., Bea F., Bekeredjian R., Schinke-Braun M., Izumo S., Katus H. A., and Hardt S. E. (2009) Beneficial effects of mammalian target of rapamycin inhibition on left ventricular remodeling after myocardial infarction. J. Am. Coll. Cardiol. 54, 2435–2446 [DOI] [PubMed] [Google Scholar]
- 45.Choi J. C., Wu W., Muchir A., Iwata S., Homma S., and Worman H. J. (2012) Dual specificity phosphatase 4 mediates cardiomyopathy caused by lamin A/C (LMNA) gene mutation. J. Biol. Chem. 287, 40513–40524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ramos F. J., Chen S. C., Garelick M. G., Dai D. F., Liao C. Y., Schreiber K. H., MacKay V. L., An E. H., Strong R., Ladiges W. C., Rabinovitch P. S., Kaeberlein M., and Kennedy B. K. (2012) Rapamycin reverses elevated mTORC1 signaling in lamin A/C-deficient mice, rescues cardiac and skeletal muscle function, and extends survival. Sci. Transl. Med. 4, 144ra103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Song X., Kusakari Y., Xiao C. Y., Kinsella S. D., Rosenberg M. A., Scherrer-Crosbie M., Hara K., Rosenzweig A., and Matsui T. (2010) mTOR attenuates the inflammatory response in cardiomyocytes and prevents cardiac dysfunction in pathological hypertrophy. Am. J. Physiol. Cell Physiol. 299, C1256–C1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Choi J. C., Muchir A., Wu W., Iwata S., Homma S., Morrow J. P., and Worman H. J. (2012) Temsirolimus activates autophagy and ameliorates cardiomyopathy caused by lamin A/C gene mutation. Sci. Transl. Med. 4, 144ra102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Johnson S. C., Rabinovitch P. S., and Kaeberlein M. (2013) mTOR is a key modulator of ageing and age-related disease. Nature 493, 338–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li J., Kim S. G., and Blenis J. (2014) Rapamycin: one drug, many effects. Cell Metab. 19, 373–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu S., Cai Y., and Wei Y. (2014) mTOR signaling from cellular senescence to organismal aging. Aging Dis. 5, 263–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dorn G. W., 2nd. (2010) Adrenergic signaling polymorphisms and their impact on cardiovascular disease. Physiol. Rev. 90, 1013–1062 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.