

Background: Adult human corneal endothelial cells are G1-arrested, often necessitating transplantation in patients with endothelial dysfunction.
Results: WNT10B enhances proliferation in human corneal endothelial cells.
Conclusion: Modulation of the WNT10B pathway could be used to treat vision loss because of corneal endothelial dysfunction.
Significance: Modulation of the WNT10B pathway may provide a means of addressing the impending increase in donor cornea demand.
Keywords: β-catenin, cell proliferation, cornea, endothelium, Ras-related C3 botulinum toxin substrate 1 (Rac1)
Abstract
The cornea is the anterior, transparent tissue of the human eye that serves as its main refractive element. Corneal endothelial cells are arranged as a monolayer on the posterior surface of the cornea and function as a pump to counteract the leakiness of its basement membrane. Maintaining the cornea in a slightly dehydrated state is critical for the maintenance of corneal transparency. Adult human corneal endothelial cells are G1-arrested, even in response to injury, leading to an age-dependent decline in endothelial cell density. Corneal edema and subsequent vision loss ensues when endothelial cell density decreases below a critical threshold. Vision loss secondary to corneal endothelial dysfunction is a common indication for transplantation in developed nations. An impending increase in demand for and a current global shortage of donor corneas will necessitate the development of treatments for vision loss because of endothelial dysfunction that do not rely on donor corneas. Wnt ligands regulate many critical cellular functions, such as proliferation, making them attractive candidates for modulation in corneal endothelial dysfunction. We show that WNT10B causes nuclear transport and binding of RAC1 and β-catenin in human corneal endothelial cells, leading to the activation of Cyclin D1 expression and proliferation. Our findings indicate that WNT10B promotes proliferation in human corneal endothelial cells by simultaneously utilizing both β-catenin-dependent and -independent pathways and suggest that its modulation could be used to treat vision loss secondary to corneal endothelial dysfunction.
Introduction
The cornea consists of the epithelium, stroma, and endothelium and is the anterior, transparent tissue of the human eye that serves as its main refractive element. The corneal endothelium plays a critical role in maintaining corneal transparency, which is necessary for sharp vision by regulating corneal hydration through its pump function (1, 2). Adult human corneal endothelial cells (CECs)2 are arranged as a monolayer on the posterior surface and are G1-arrested (3, 4). As a result, there is an age-dependent decrease in CEC density that can be accelerated further by infection or injury. Corneal edema with subsequent vision loss results when the density decreases below a critical threshold of ∼500 cells/mm2. The current standard of care for severe vision loss secondary to endothelial dysfunction is corneal transplantation in the form of endothelial keratoplasty. Fortunately, there currently is no shortage of donor corneas in developed nations, but the demand is expected to increase because of the increasing age of the general population and an increasing number of patients undergoing intraocular surgery. Moreover, there are ∼8 million patients worldwide who have not been treated because of a local lack of donor corneas (5). Investigation of treatment strategies that do not rely on donor corneas, such as stimulating endogenous regeneration, may hold a key to addressing the current global shortage of donor corneas, which will likely be exacerbated further by the impending increase in donor cornea demand in developed nations.
A number of soluble factors have been reported to induce cell proliferation in the corneal endothelium, including IL-1β, a key mediator of inflammation in the cornea (6–8). In our previous studies, we reported that IL-1β stimulation led to FGF2 expression, resulting in enhanced proliferation in human and rabbit CECs (9–14). However, IL-1β and FGF2 have also been shown to be key mediators of endothelial-mesenchymal transition (EnMT) that can lead to retrocorneal membrane formation when lasting restoration of vision is no longer possible because of fibrosis, thereby making their use potentially dangerous (6, 10, 15). For a candidate to be clinically useful for treatment of vision loss secondary to endothelial dysfunction, it has to be able to promote proliferation in human CECs without inducing fibrosis.
The Wnt family of ligands regulates many processes that are critical for development and cellular homeostasis (16–20). Wnt signaling can be broadly classified into canonical (β-catenin-dependent) and non-canonical (β-catenin-independent) pathways. In the canonical pathway, binding of Wnt ligand to its receptor complex leads to nuclear transport of β-catenin, during which it interacts with the LEF-1/TCF family of transcription factors to activate transcription of target genes, including those critical for cell proliferation, such as Cyclin D1 and c-myc (16, 21–27). During eye development, the canonical Wnt signaling plays an important role in differentiation of the lens epithelium and lens fiber cells (28, 29). The non-canonical pathway does not signal through β-catenin and includes the planar cell polarity and Ca2+ pathways (30, 31). These pathways have been shown to regulate development, including extension movements during vertebrate gastrulation (32) and cell migration (33, 34). We have reported previously that WNT5A signals through the non-canonical pathway in human CECs and promotes migration through activation of CDC42 and inhibition of RHOA (35).
The RhoGTPase family members RAC1, RHOA, and CDC42 modulate various cellular functions through cycling between active GTP-bound and inactive GDP-bound forms (36, 37). It has been reported that these proteins regulate planar cell polarity in Drosophila (38) and convergent extension movement in Xenopus (32, 39). However, RAC1 has also been reported to be involved in the canonical pathway through modulation of β-catenin nuclear transport (40) and activation of β-catenin/TCF-mediated transcription of Cyclin D1 (41). The role of RAC1 and β-catenin signaling in proliferation of human CECs has not been reported previously.
In this study, we investigated the downstream targets of IL-1β capable of driving proliferation in human CECs. Our results show that IL-1β activates WNT10B expression and promotes proliferation in human CECs. WNT10B signals through Disheveled and induces independent nuclear transport of RAC1 and β-catenin, during which they form a complex to activate expression of Cyclin D1, leading to proliferation of human CECs. We provide evidence that WNT10B signals simultaneously through β-catenin-dependent and -independent pathways to enhance proliferation in human CECs, and our results suggest that modulation of WNT10B could be used to treat vision loss secondary to corneal endothelial dysfunction.
Experimental Procedures
Reagents
FGF2 was purchased from Cell Signaling Technology (Danvers, MA). Secreted Frizzled-related peptide (sFRP) 1 (42) was purchased from Fitzgerald Industries International (Acton, MA). Anti-JUN antibody, Disheveled-PDZ domain inhibitor II (43), Y27632, NSC23766, and SB203580 were obtained from Calbiochem. IL-1β, sulfasalazine, anti-β-actin, α-tubulin, p65 (RelA), and peroxidase-conjugated secondary antibodies were obtained from Sigma-Aldrich (St. Louis, MO). Recombinant human interleukin-1 receptor antagonist (IL-1ra), WNT10B, WNT3A, and WNT5A were obtained from R&D Systems (Minneapolis, MN). ML141 was purchased from Tocris Bioscience (Minneapolis, MN). AZD4547 (44) and XAV939 (45) were purchased from Selleckchem (Houston, TX). RHOA activator was obtained from Cytoskeleton Inc. (Denver, CO). Antibodies against WNT10B and β-catenin were obtained from GeneTex (Irvine, CA). Fascaplysin (46) and antibodies against CDC42 and lamin B were purchased from Santa Cruz Biotechnology (Dallas, TX).
Cell Culture
Immortalized human CEC line human CEC-B4G12 (DSMZ, Braunschweig, Germany) was cultured as described previously described (9, 35). Briefly, human CEC-B4G12 was cultured in human endothelial serum-free medium (SFM) supplemented with 10 ng/ml human recombinant FGF2 without antibiotics (SFM-F). Cells were grown in a humidified atmosphere containing 5% CO2 at 37 °C. For subculture, confluent cultures were treated with 0.05% trypsin and 5 mm EDTA in PBS for 5 min. Cells were plated in 100-mm tissue culture dishes coated with 10 mg/ml chondroitin-6-sulfate and 10 μg/ml laminin at a concentration of 1 × 106 cells. Second-passage human CECs maintained in SFM-F were used for all experiments. The culture medium was changed twice a week. In some experiments, pharmacologic inhibitors were used in the presence of IL-1β (5 ng/ml), WNT10B (200 ng/ml), WNT5A (300 ng/ml), or WNT3A (50 ng/ml) stimulation: IL-1ra (50 ng/ml), sFRP (3 μg/ml), SB203580 (a p38 inhibitor, 20 μm), sulfasalazine (an inhibitor of IκB degradation, 2 mm), Disheveled-PDZ domain inhibitor II (25 μm), XAV939 (20 μm), RHOA activator (1 μg/ml), Y27632 (a Rho-associated coil kinase inhibitor, 10 μm), ML141 (a CDC42 inhibitor, 10 μm), or NSC23766 (a RAC1 inhibitor, 100 μm). All experiments included a vehicle control (0.1 mm EDTA and 0.5% CHAPS in PBS or dimethyl sulfoxide) sample.
ChIP Assay
FGF2-starved human CECs were first pretreated with sulfasalazine or SB203580 for 2 h and then stimulated with IL-1β or vehicle control for 10 min. The cells were then maintained in SFM for 6 h prior to isolating protein-DNA complexes using anti-NF-κB (p65) and AP-1 (c-JUN) antibodies. Binding of NF-κB and AP-1 to the WNT10B promoter was evaluated by PCR using the following primer pairs: NF-κB site 1, 5′-CTGTCTAAGGTAATCCCATGG-3′ (forward) and 5′-TGACTCATCTGTTGCTGAGCC-3′ (reverse); NF-κB site 2, 5′-CTACTTGCCCTTGATGACACC-3′ (forward) and 5′-CACTTGCTCCACAACCTCGCC-3′ (reverse); NF-κB site 3, 5′-ACTCCCTGAGGTGTAAAGACC-3′ (forward) and 5′-CCTCCAGGGGTGAAGCTGTTT-3′ (reverse); AP-1 site 1, 5′-CAGCAGAAGGCGCATTAAGCG-3′ (forward) and 5′-GGACTAGGAGCAAGCTGGTTC-3′ (reverse); and AP-1 site 2, 5′-GCCAGCCGTGCCTTGGGCTGC-3′ (forward) and 5′-GAAAGAGCCTGGTCTTGGGGT-3′ (reverse). Standard PCR conditions were as follows: 5 min at 94 °C, followed by 33 cycles of 30 s at 94 °C, 30 s at 55 °C, 30 s at 72 °C, and a final extension for 2 min at 72 °C. Annealing temperature were adjusted depend on the PCR primer. PCR products were separated by 1.5% agarose gel electrophoresis and visualized by ethidium bromide staining.
Isolation and Culture of Primary Human CECs
Isolation and culture of primary human CECs were performed according to previously published protocols (12, 47) with minor technical modifications. Briefly, corneas were removed from Optisol and washed several times with OptiMEM I (Gibco-BRL) containing 50 μg/ml gentamicin. The Descemet membrane and endothelium complex was stripped from corneas and treated with 0.2% collagenase type II and 0.05% hyaluronidase (Worthington Biochemical, Lakewood, NJ) for 3 h at 37 °C. After centrifugation, the primary cells were resuspended in culture medium (OptiMEM I supplemented with 5% fetal bovine serum, 10 μm Y27632, 0.5 μg/ml R-spondin, and 10 μm SB431542 (TGF-β receptor inhibitor kinase inhibitor VI) (48)) and plated on 24-well tissue culture plates precoated with FNC Coating Mix (Biological Research Faculty and Facility, Inc., Ijamsville, MD) and laminin (Sigma). For subculture, confluent primary cultures were treated with 0.05% trypsin and 5 mm EDTA in PBS for 10 min. Second- or third-passage human CECs were used for all experiments. For serum starvation, the culture medium was changed to OptiMEM I, and cultures were maintained for 24 h. In some experiments, pharmacologic inhibitors were used in the presence of IL-1β (5 ng/ml), FGF2 (10 ng/ml), WNT10B (200 ng/ml), WNT5A (300 ng/ml), or WNT3A (50 ng/ml) stimulation: IL-1ra (50 ng/ml), AZD4547 (an FGF receptor inhibitor, 0.1 μm), sFRP (3 μg/ml), Disheveled-PDZ domain inhibitor II (25 μm), XAV939 (20 μm), RHOA activator (1 μg/ml), Y27632 (10 μm), ML141 (10 μm), or NSC23766 (100 μm). Vehicle control (0.1 mm EDTA and 0.5% CHAPS in PBS or dimethyl sulfoxide) was included for all experiments.
In Vitro Proliferation Assays
Primary human CECs were used for all cell proliferation assays. The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay was used to assess proliferation as described previously (10). Briefly, cells were seeded in 96-well tissue culture plates at a concentration 4 × 103 cells/well. When cells reached ∼70% confluence, the medium was changed to OptiMEM I for serum starvation and maintained for 24 h. The serum-starved cells were then maintained for 24 h under each culture condition. Afterward, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (50 μg/ml) was added, and the culture was maintained for another 2 h at 37 °C. The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide-containing medium was discarded, and 100 μl of undiluted dimethyl sulfoxide was added to the cells. After 30 min of incubation at room temperature, the absorbance of the converted dye was measured at a wavelength of 570 nm with background subtraction at 650 nm using a spectrophotometric plate reader (model 680 microplate spectrophotometer, Bio-Rad). To examine the effect of Cyclin D1, β-catenin, and RAC1 siRNA transfection on cell proliferation, Accell delivery medium was used. 24 h after transfection, cells were stimulated and maintained with WNT10B for another 24 h. The cells were then harvested for the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide proliferation assay.
siRNA Knockdown
For gene knockdown by siRNA in human CECs, the Accell SMARTpool system was used as reported previously (35). Corneal endothelial cells were transfected at 70% confluence on a 6-well plate with 1.5 μm Accell SMARTpool of siRNA targeting p65 (RelA), JUN, Disheveled 2, RAC1, β-catenin, or Cyclin D1 (Dharmacon, Pittsburgh, PA) in Accell delivery medium according to the instructions of the manufacturer. 42 h after transfection, the medium was changed to each culture medium and maintained for another 6 h. Protein levels of p65, JUN, Disheveled 2, RAC1, β-catenin, and Cyclin D1 were analyzed by immunoblotting. Accell non-targeting (NT) control siRNA (Dharmacon) was used as a negative control. For RAC1, β-catenin, and Cyclin D1 gene knockdown, primary human CECs on 24-well plates were used.
Protein Preparation, Protein Assay, SDS-PAGE, Western Blot Analysis, Coimmunoprecipitation, and Nuclear Fractionation
All assays were performed following protocols reported previously (9, 10, 12, 13). The following gel concentrations were used: 15% polyacrylamide gel for CDC42 and RAC1; 12% polyacrylamide gel for WNT10B, JUN, and Cyclin D1; 10% polyacrylamide gel for p65, lamin B, α-tubulin, and β-actin; and 8% polyacrylamide gel for β-catenin and Disheveled 2. For coimmunoprecipitation of RAC1 and β-catenin, after purification of the nuclear fraction, the purified nuclei were suspended with 10 volumes of nucleus suspension medium (0.25 m sucrose, 5 mm MgCl2, and 50 mm Tris-Cl (pH 7.4)) using three to four gentle pipetting and then incubated for 1 h at 4 °C with treatment of DNase I and RNase to a final concentration of 0.25 μg/μl. Nuclear lysate protein concentrations were assessed with the Bradford protein assay system (Bio-Rad), and lysates were then used in coimmunoprecipitation experiments.
CDC42 and RAC1 GTPase Activation Assay
CDC42 and RAC1 activities were quantified as described previously (35) using CDC42 and RAC1 activation assay kits (EMD Millipore, Billerica, MA). After the pulldown reaction with GST-PAK-PBD (p21 binding domain of p21 activated kinase) beads, the precipitated proteins bound to the beads were subjected to immunoblot analysis with monoclonal antibody to CDC42 or to RAC1. Depletion with GDP and enrichment with GTPγS were used as negative and positive controls for pulldown, respectively. The total amount of each RhoGTPase was also determined by Western blot analysis using total cell lysates.
RHOA GTPase Activation Assay
RHOA activation was quantified as described previously (35) by measuring the amounts of RHOA-GTP using the ELISA-based G-LISATM RHOA activation biochem assay kit (Cytoskeleton Inc.). Active RHOA-GTP from lysates bound to the GTP-Rho binding domain of the Rho effector protein was detected by primary anti-RHOA antibody and HRP-conjugated secondary antibody. Results were normalized to the absorbance of lysate from SFM-maintained cells and expressed as a percentage of this value for the comparison of RHOA activity (level of GTP-bound RHOA) in different cell lysates.
Ex Vivo Proliferation Assay
Whole human corneas not suitable for transplantation were purchased from Vision Share (Ann Arbor, MI) and cut into three equal sections. The endothelium of each piece was injured with a small pipette tip measuring ∼0.5 mm in diameter, resulting in a linear wound ∼0.5 mm wide. The corneal pieces were then placed endothelial side up in individual wells of a 24-well tissue culture plate. The pieces were maintained with OptiMEM containing WNT10B with or without inhibitors for 72 h at 37 °C in a 5% carbon dioxide, humidified atmosphere. Pharmacologic inhibitors were used in the presence of WNT10B (200 ng/ml) stimulation: sFRP (3 μg/ml), Disheveled-PDZ domain inhibitor II (25 μm), XAV939 (20 μm), NSC23766 (100 μm), or fascaplysin (0.5 μm). For siRNA knockdown in ex vivo human corneal endothelium, the Accell SMARTpool system was used. 36 h after transfection with 1.5 μm Accell SMARTpool of siRNA targeting Disheveled 2 or Cyclin D1 (Dharmacon) in Accell Delivery media, the medium was changed to Opti-MEM with WNT10B and maintained for another 72 h. Accell non-targeting control siRNA (Dharmacon) was used as a negative control. After 72 h, the corneal pieces were fixed for 20 min in 1 ml of 4% paraformaldehyde and rinsed three times with PBS. They were then permeabilized using 0.5% Triton X-100 (Sigma-Aldrich) in PBS for 30 min at room temperature, followed by blocking with 4% bovine serum albumin (Sigma-Aldrich) in PBS for 1 h at room temperature. The tissues were incubated for 2 h at room temperature with both mouse anti-human proliferating cell nuclear antigen (PCNA) IgG (Sigma-Aldrich) or mouse anti-human phosphohistone H3 IgG (EMD Millipore) and rabbit anti-human ZO-1 IgG (Invitrogen) diluted 1:100 in blocking buffer. Each piece was rinsed in PBS three times for 10 min, followed by incubation with FITC-conjugated goat anti-mouse IgG (Sigma-Aldrich) and Rhodamine-conjugated donkey anti-rabbit IgG (Genetex) diluted 1:200 in blocking buffer for 2 h at room temperature. Incubation of primary or secondary antibody alone was used as a negative control. The corneal pieces were then washed three times for 10 min with PBS at room temperature. Excess sclera was removed to facilitate mounting (endothelial side down) with mounting solution with DAPI on a glass-bottom microwell dish (MatTek Co., Ashland, MA). A glass coverslip was placed on the epithelial side of the corneal sections, and a 20-g weight was placed on the coverslip for 15 min to flatten the sections for imaging. Images were captured with a BZ-X700 fluorescence microscope (Keyence, Itasca, IL) using a ×40 objective lens with an aperture of 0.95. All images were captured at room temperature on sections mounted with Vectashield with DAPI mounting medium (Vector Laboratories, Burlingame, CA) and analyzed with BZ-X analyzer software (Keyence). PCNA-positive nuclei were counted in an 85-μm2 field within the wound area. Five fields were counted per section, and three different sections were used for each culture condition. The proliferation index was calculated as the number of PCNA-positive nuclei divided by the number of all nuclei within each field, and the relative proliferation rate was calculated relative to the proliferation index of the vehicle-treated cornea.
Statistical Analysis
One-way analysis of variance (ANOVA) was performed to compare means within groups, and post-hoc Tukey's honest significant difference tests were done to perform pairwise comparisons between means within groups.
Results
IL-1β Stimulation Leads to Cell Proliferation and WNT10B Expression in Human CECs
We have reported previously that IL-1β stimulation led to FGF-2 expression in human (9) and rabbit (10, 11) CECs, resulting in enhanced proliferation (12–14). The FGF receptor 1–3 antagonist AZD4547 attenuated, but did not completely abolish, IL-1β-induced proliferation (Fig. 1A), suggesting that IL-1β-induced proliferation was mediated by a signaling pathway distinct from FGF2 in human CECs. Stimulation of human CECs with IL-1β resulted in activation of WNT10B expression, which, in turn, could be inhibited by cotreatment with the IL-1 receptor antagonist (Fig. 1B). IL-1β-dependent WNT10B expression in human CECs could be attenuated with pretreatment with the NF-κB inhibitor sulfasalazine or the AP-1 antagonist SB203580 (Fig. 1B). Pretreatment with both sulfasalazine and SB203580 resulted in complete inhibition of IL-1β-dependent WNT10B expression (Fig. 1B). Furthermore, IL-1β stimulation of human CECs transfected with either p65 siRNA targeting NF-κB or JUN siRNA targeting AP-1 resulted in attenuation of WNT10B expression, whereas transfection with both p65 and JUN siRNA completely inhibited IL-1β-induced WNT10B expression (Fig. 1B). Corresponding knockdowns of p65 and JUN proteins were observed in p65 and JUN siRNA-transfected human CECs, and neither use of small molecule inhibitors nor transfection with a non-targeting control siRNA affected p65 and JUN protein levels. The human WNT10B promoter contains several conserved and putative NF-κB and AP-1 binding sites (49). Binding of NF-κB and AP-1 to the WNT10B promoter in IL-1β-treated human CECs was shown by chromatin immunoprecipitation assays (Fig. 1C). NF-κB binding could be antagonized by pretreatment with sulfasalazine but not SB208530, and AP-1 binding could be antagonized by pretreatment with SB208530 but not sulfasalazine (Fig. 1C). Vehicle control treatment did not result in NF-κB and AP-1 binding. Treatment with IL-1β led to expression of WNT10B in human ex vivo corneal endothelium, and combining injury with IL-1β treatment resulted in greater WNT10B expression (Fig. 1D). Neither vehicle control nor injury alone resulted in expression of WNT10B in human ex vivo corneal endothelium.
FIGURE 1.
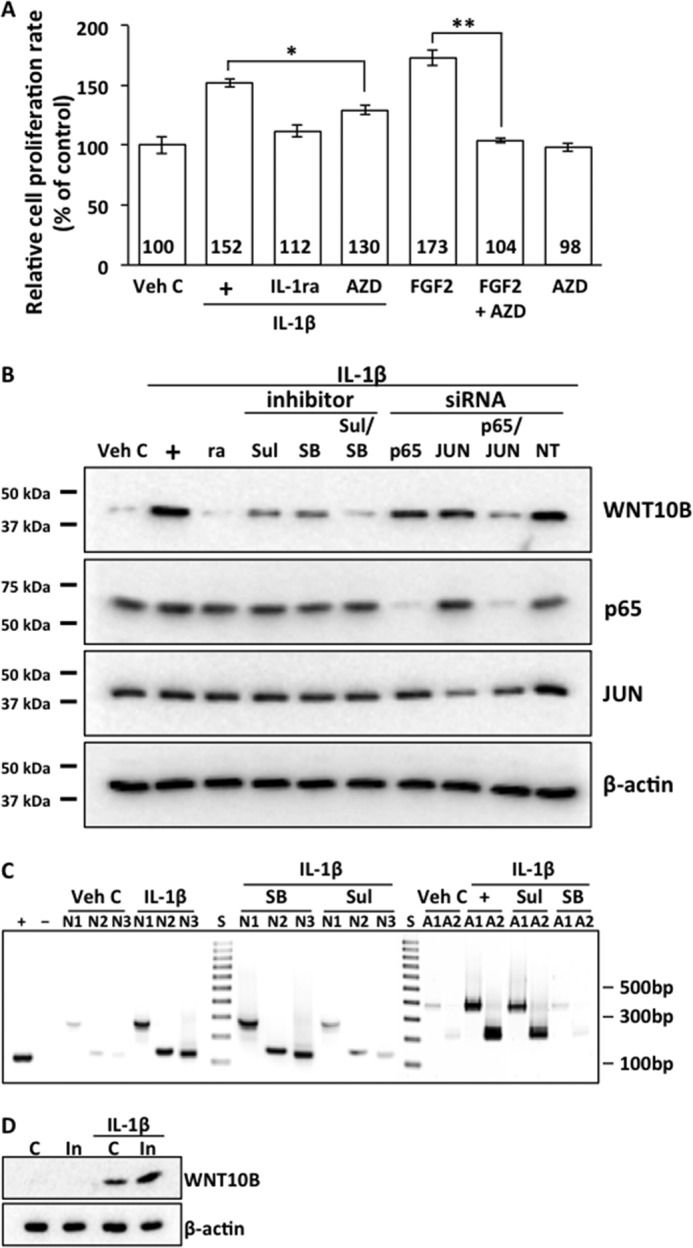
IL-1β stimulation enhances proliferation and activates expression of WNT10B in human CECs. A, treatment of primary human CECs with IL-1β (+) increased the relative proliferation rate compared with Veh C (152 ± 3.8 versus 100 ± 7.5, p < 0.01). IL-1β-dependent proliferation was abolished by cotreatment with IL-1ra (112 ± 5.4 versus 152 ± 3.8, p < 0.01). Cotreatment with AZD4547 (AZD), a FGF receptor 1–3 antagonist, attenuated IL-1β-enhanced proliferation (130 ± 4.3 versus 152 ± 3.8; *, p < 0.01), and it abolished FGF2-enhanced proliferation in human CECs (104 ± 2.4 versus 173 ± 7.1; **, p < 0.01). Treatment with AZD4547 alone did not change the proliferation rate compared with vehicle control (98 ± 3.7 versus 100 ± 7.5, p > 0.05). The relative proliferation rate was greater in IL-1β-stimulated CECs that were cotreated with AZD4547 compared with those cotreated with IL-1ra (130 ± 4.3 versus 112 ± 5.4; p < 0.01). One-way ANOVA, F(6, 35) = 186, p < 0.00001, n = 6/sample. Tukey's post-hoc test, HSD[0.05] = 9.4 and HSD[0.01] =11.3, where HSD is honestly significant difference. B, treatment of human CECs with IL-1β (+) activated expression of WNT10B, and IL-1β-dependent WNT10B expression could be abolished with IL-1ra (ra) cotreatment. IL-1β-dependent expression of WNT10B could be attenuated by cotreatment with either the NF-κB inhibitor sulfasalazine (Sul) or the AP-1 inhibitor SB203580 (SB). Cotreatment with both inhibitors abolished IL-1β-induced WNT10B expression. There was no effect on p65 and JUN protein levels by cotreatment with the inhibitors. Transfection with p65 siRNA and JUN siRNA resulted in corresponding knockdown of p65 and JUN protein levels in human CECs. Transfection with either p65 or JUN siRNA resulted in attenuation of IL-1β-dependent WNT10B expression, whereas transfection with both p65 and JUN siRNA severely decreased WNT10B expression. Transfection with NT control siRNA had no effect on p65, JUN, and WNT10B expression in human CECs. β-actin was used as a loading control. C, the expected PCR product sizes for NF-κB and AP-1 binding site were as follows: NF-κB site 1 (N1), 260 bp; NF-κB site 2 (N2), 147 bp; NF-κB site 3 (N3), 140 bp; AP-1 site 1 (A1), 380 bp; and AP-1 site 2 (A2), 204 bp. IL-1β, but not vehicle control treatment, resulted in binding of NF-κB and AP-1 to the WNT10B promoter in human CECs. NF-κB binding could be antagonized by pretreatment with sulfasalazine but not SB203580, and AP-1 binding could be antagonized with pretreatment with SB203580 but not sulfasalazine. Anti-histone H3 antibody was used as a positive control (+), and normal rabbit IgG was used as a negative control (−). D, WNT10B is not expressed in control (C) or injured (In) ex vivo human corneal endothelium. IL-1β treatment induced WNT10B expression in control ex vivo corneal endothelium, and injury induced greater WNT10B expression over the control in IL-1β-treated corneas.
WNT10B Signaling Proceeds through the Canonical Pathway in Human CECs
β-catenin was found in the cytoplasmic fraction but not in the nuclear fraction of human CECs treated with vehicle control (Fig. 2A). Treatment of human CECs with the positive control (WNT3A) resulted in nuclear transport of β-catenin whereas treatment with the negative control (WNT5A) did not. WNT3A was shown to signal through the canonical Wnt pathway, whereas WNT5A was shown to signal through the non-canonical Wnt pathway in human CECs.3 Treatment with WNT10B resulted in nuclear transport of β-catenin (Fig. 2A). WNT10B-dependent nuclear transport of β-catenin could be inhibited by cotreatment with sFRP, the β-catenin antagonist XAV, and Disheveled-PDZ domain inhibitor (DI) (Fig. 2B). Cotreatment with RHOA activator (RA), the Rho kinase inhibitor Y27632 (Y), and the RAC1 inhibitor NSC23766 (NSC) had no effect on WNT10B-dependent nuclear transport of β-catenin (Fig. 2B). Lamin B was used as a nuclear fraction marker, and α-tubulin was used as a cytoplasmic fraction marker.
FIGURE 2.
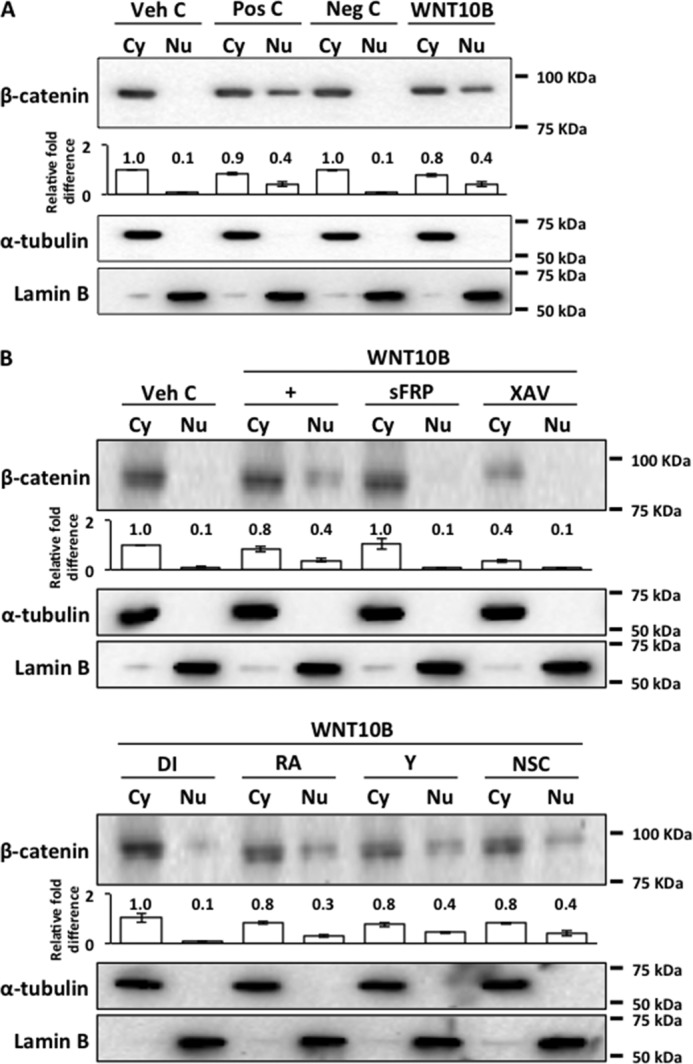
WNT10B stimulation leads to nuclear transport of β-catenin. A, a 4-fold increase in β-catenin was detected in nuclear (Nu) fractions of WNT10B and positive control (Pos C)-treated cells but not in Veh C and negative control (Neg C) cells. WNT3A was used as a positive control, and WNT5A was used as a negative control. The bar graph depicts the relative –fold difference in β-catenin levels. Cy, cytoplasmic fraction. B, WNT10B-dependent (+) nuclear transport of β-catenin was blocked by cotreatment with sFRP, the β-catenin antagonist XAV, or Disheveled-PDZ DI. As anticipated, total cytoplasmic β-catenin in XAV-treated cells was markedly lower compared with Veh C cells. Cotreatment with either RA, the Rho kinase inhibitor Y, or the Rac antagonist NSC, had no effect on WNT10B-dependent nuclear transport of β-catenin. The bar graph depicts the relative -fold difference in β-catenin levels. Lamin B and α-tubulin were used as nuclear and cytoplasmic fraction markers, respectively.
WNT10B Activates RAC1 and Inhibits RHOA in Human CECs
RhoGTPases have been reported previously to regulate cell proliferation through modulation of β-catenin signaling (40, 41, 50, 51). GTP-bound CDC42 was detected in positive control (WNT5A)-treated cells but not in negative control- (WNT3A) or vehicle control-treated human CECs (Fig. 3A). WNT10B treatment of human CECs did not result in CDC42 activation (Fig. 3A). WNT10B stimulation led to activation of RAC1 in human CECs, as shown by the presence of GTP-RAC1 (Fig. 3B). WNT10B-dependent RAC1 activation could be inhibited with cotreatment with sFRP, Disheveled-PDZ DI, and the RAC1 inhibitor NSC (Fig. 3B). Cotreatment with the β-catenin antagonist XAV, RA, and the Rho kinase inhibitor Y had no effect on WNT10B-dependent RAC1 activation (Fig. 3B). Activated RAC1 was not present in human CECs treated with vehicle control, but it was present in cells treated with the positive control (WNT5A). In contrast to RAC1 activation, WNT10B treatment led to an inhibition of RHOA activity relative to vehicle control (Fig. 3C). WNT10B-dependent inhibition of RHOA activity could be attenuated by cotreatment with sFRP and Disheveled-PDZ DI, whereas cotreatment with XAV had no effect (Fig. 3C). Cotreatment with RA resulted in increased RHOA activity, whereas positive control (WNT5A) treatment resulted in decreased RHOA activity. Moreover, WNT10B stimulation led to an increased binding between Disheveled 2 and RAC1 compared with vehicle-treated cells, as shown by increased amounts of RAC1 protein detected in lysates precipitated with anti-Disheveled 2 antibody (Fig. 3D). This is consistent with previous reports describing binding between Disheveled with RAC1 in other cell types (52–54). WNT10B-dependent binding of Disheveled 2 and RAC1 could be inhibited by cotreatment with sFRP, Disheveled-PDZ DI, or NSC. Cotreatment with XAV, RA, or Y had no effect on WNT10B-dependent binding between Disheveled and RAC1 (Fig. 3D). Transfection of human CECs with Disheveled 2 siRNA resulted in the corresponding knockdown of Disheveled 2 protein and inhibited WNT10B-dependent binding between Disheveled 2 and RAC1 (Fig. 3D). Transfection with non-targeting control siRNA had no effect on Disheveled 2 protein levels or WNT10B-dependent binding between Disheveled and RAC1 (Fig. 3D).
FIGURE 3.

Regulation of CDC42, RAC1, and RHOA by WNT10B. A, GTP-CDC42 was not detected in WNT10B-treated (W10B) human CECs. It was detected in Pos C-treated (WNT5A-treated) but not in Veh C- or negative control (Neg C)-treated WNT3A cells. B, an ∼8-fold increase in GTP-RAC1 level was detected in WNT10B-treated (+) cells relative to vehicle control. WNT10B-dependent activation of RAC1 was inhibited by cotreatment with sFRP, the RAC1 inhibitor NSC, and Disheveled-PDZ DI. Cotreatment with the β-catenin antagonist XAV, RA, and the Rho kinase inhibitor Y had no effect on WNT10B-dependent RAC1 activation in human CECs. The bar graph depicts the relative -fold difference in GTP-RAC1 levels. GTP-RAC1 was detected in Pos C-treated (WNT5A-treated) but not in Veh C-treated cells. C, WNT10B treatment (+) inhibited RHOA activity relative to Veh C (100 ± 3.9 versus 65 ± 2.3, p < 0.01). WNT10B-dependent inhibition of RHOA activity was abolished by cotreatment with sFRP (93 ± 4.4 versus 65 ± 2.3; *, p < 0.01) and DI (89 ± 2.5 versus 65 ± 2.3; **, p < 0.01), but cotreatment with XAV (77 ± 14.2 versus 65 ± 2.3, p > 0.05) or NSC (72 ± 5.4 versus 65 ± 2.3, p > 0.05) had no effect. Cotreatment with RA in the absence of WNT10B stimulation resulted in higher RHOA activity (154 ± 6.1 versus 100 ± 3.9, p < 0.01), and treatment with the Pos C, WNT5A, resulted in lower RHOA activity (48 ± 2.0 versus 100 ± 3.9, p < 0.01) than Veh C. One-way ANOVA, F(7, 24) = 102, p < 0.00001, n = 4/sample. Tukey's post-hoc test, HSD[0.05] = 14.4 and HSD[0.01] = 17.3. D, treatment with WNT10B induced Disheveled 2 and RAC1 binding. A 3.5-fold increase in RAC1 levels could be detected in anti-Disheveled 2 antibody-precipitated lysates from WNT10B-treated (+) compared with vehicle control (C)-treated human CECs. Cotreatment with sFRP, DI, and NSC inhibited WNT10B-dependent Disheveled 2 and RAC1 binding, whereas cotreatment with XAV, Y, and RA had no effect on WNT10B-dependent Disheveled 2 and RAC1 binding. The bar graph depicts the relative -fold difference in RAC1 levels. Transfection with Disheveled 2 siRNA also blocked WNT10B (W10B)-dependent binding between Disheveled 2 and RAC1, whereas NT control siRNA had no effect. IP, immunoprecipitation; IB, immunoblot.
WNT10B Activates Nuclear Transport and Binding of RAC1 and β-Catenin
It has been reported previously that RAC1 plays a role in the nuclear transport of β-catenin (40, 41, 52). RAC1 and β-catenin bound to RAC1 were present in the nuclear fraction of WNT10B but not vehicle control-treated human CECs (Fig. 4A). WNT10B-dependent nuclear transport of RAC1 and its association with β-catenin were blocked by cotreatment with sFRP, Disheveled-PDZ DI, and the RAC1 inhibitor NSC, whereas cotreatment with the Rho kinase inhibitor Y and RA did not have an effect (Fig. 4A). Cotreatment with the β-catenin antagonist XAV did not interfere with nuclear transport of RAC1, but it was not complexed with β-catenin in the nucleus (Fig. 4A). Although RAC1 was present in the cytoplasmic fraction under all conditions, it was not bound to β-catenin in the cytoplasm (Fig. 4B). Lamin B was used as a nuclear fraction marker, and α-tubulin was used as a cytoplasmic fraction marker.
FIGURE 4.
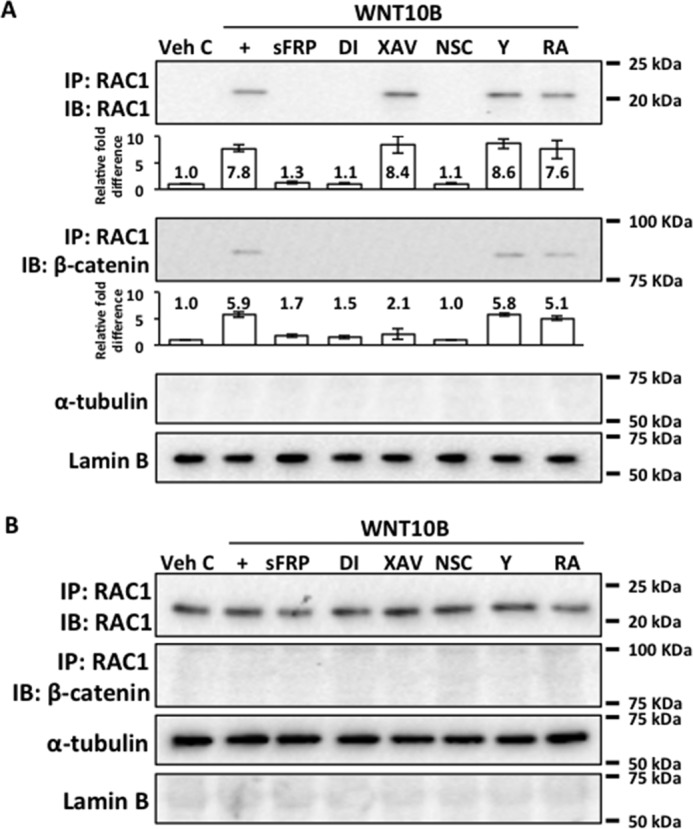
WNT10B induces nuclear transport and binding of RAC1 and β-catenin. A, an approximate 8-fold increase in RAC1 level along with an approximate 6-fold increase β-catenin level were detected in the nuclear fractions precipitated with RAC1 antibody in WNT10B-treated (+) but not in Veh C-treated human CECs. Cotreatment with sFRP, Disheveled-PDZ DI, and the RAC1 inhibitor NSC blocked WNT10B-dependent nuclear transport of RAC1 and its interaction with β-catenin. Inhibition of β-catenin with XAV treatment did not interfere with nuclear transport of RAC1, but β-catenin was not found in the nucleus complexed to RAC1. Cotreatment with RA or the Rho kinase inhibitor Y had no effect on WNT10B-dependent RAC1 nuclear transport and its interaction with β-catenin. The bar graphs depict the relative -fold difference in RAC1 and β-catenin levels. Lamin B and α-tubulin were used as nuclear and cytoplasmic markers, respectively. IP, immunoprecipitation; IB, immunoblot. B, RAC1 but not β-catenin complexed to RAC1 was detected in the cytoplasm under all treatment conditions.
WNT10B-stimulated Proliferation Is Dependent on β-Catenin and RAC1 in Human CECs
WNT10B treatment resulted in a significant increase in relative proliferation rates compared with vehicle control in human CECs (Fig. 5A). WNT10B-dependent proliferation could be inhibited by cotreatment with sFRP, Disheveled-PDZ DI, and the β-catenin antagonist XAV (Fig. 5A). Cotreatment with the negative control (WNT5A) had no effect, whereas cotreatment with the positive control (WNT3A) enhanced proliferation compared with vehicle control. Inhibition of RAC1 by cotreatment with NSC resulted in inhibition of WNT10B-dependent proliferation, whereas cotreatment with the Rho kinase inhibitor Y, RA, and the CDC42 antagonist ML141 did not have an effect (Fig. 5B). Transfection of primary human CECs with RAC1 and β-catenin siRNAs resulted in knockdown of RAC1 and β-catenin protein levels, whereas transfection with NT control siRNA did not alter RAC1 and β-catenin protein levels (Fig. 5C). RAC1 and β-catenin siRNA transfections significantly inhibited WNT10B-dependent proliferation in primary human CECs (Fig. 5D).
FIGURE 5.

WNT10B enhances proliferation through β-catenin and RAC1. A, WNT10B stimulation (+) increased the relative proliferation rate compared with Veh C in primary human CECs (137 ± 13.0 versus 100 ± 9.2; *, p < 0.01). WNT10B-dependent proliferation was blocked by treatment with sFRP (92 ± 9.1 versus 137 ± 13.0; **, p < 0.01), Disheveled-PDZ DI (104 ± 11.7 versus 137 ± 13.0; #, p < 0.01), and the β-catenin antagonist XAV (100 ± 7.2 versus 137 ± 13.0, ##, p < 0.01). Treatment with the negative control (Neg C), WNT5A, had no effect (104 ± 10.7 versus 100 ± 9.2, p > 0.05), whereas treatment with the Pos C, WNT3A, increased proliferation (141 ± 8.1 versus 100 ± 9.2, p < 0.01) in human CECs. One-way ANOVA, F(6, 35) = 22.8, p < 0.00001, n = 6/sample. Tukey's post-hoc test, HSD[0.05] = 18.2 and HSD[0.01] = 21.9. B, cotreatment with the RAC1 inhibitor NSC inhibited WNT10B-dependent proliferation in primary human CECs (105 ± 8.0 versus 137 ± 8.9; †, p < 0.05). Cotreatment with RA (131 ± 1.7 versus 137 ± 13.0, p > 0.05), the Rho kinase inhibitor Y (136 ± 4.8 versus 137 ± 13.0, p > 0.05), and the CDC42 inhibitor ML141 (ML, 134 ± 34.6 versus 137 ± 13.0, p > 0.05) had no effect on WNT10B-dependent proliferation. One-way ANOVA, F(5, 30) = 7.0, p = 0.0002, n = 6/sample. Tukey's post-hoc test, HSD[0.05] = 27.1 and HSD[0.01] = 32.9. C, transfection of primary human CECs with RAC1 and β-catenin (β-cat) siRNA resulted in knockdown of the respective protein levels. C, control. D, transfection with RAC1 siRNA resulted in inhibition of WNT10B-induced proliferation (106 ± 4.8 versus 142 ± 14.4; ††, p < 0.01). Transfection with β-catenin siRNA also resulted in inhibition of WNT10B-induced proliferation (105 ± 6.3 versus 142 ± 14.4; †††, p < 0.01). Transfection with NT control siRNA had no effect on WNT10B-dependent proliferation (140 ± 16.7 versus 142 ± 14.4, p > 0.05). One-way ANOVA, F(4, 40) = 34.2, p < 0.00001, n = 9/sample. Tukey's post-hoc test, HSD[0.05] = 14.3 and HSD[0.01] = 17.5.
Cyclin D1 Mediates WNT10B-dependent Cell Proliferation in Human CECs
Cyclin D1 has been reported previously as a transcriptional target of Wnt/β-catenin signaling (25, 55), and RAC1 has also been reported previously to activate Cyclin D1 expression through β-catenin in other cell types (41, 52). WNT10B stimulation increased Cyclin D1 expression compared with vehicle control in human CECs (Fig. 6A). WNT10B-dependent activation of Cyclin D1 expression could be inhibited by cotreatment with sFRP and Disheveled-PDZ DI and, to a lesser extent, by cotreatment with the β-catenin antagonist XAV and the RAC1 inhibitor NSC (Fig. 6A). Cotreatment with the Rho kinase inhibitor Y and RA did not have an effect on WNT10B-dependent activation of Cyclin D1 expression. Transfection of human CECs with Cyclin D1 siRNA resulted in a corresponding knockdown of Cyclin D1 protein levels in WNT10B-treated human CECs, whereas transfection with non-targeting control siRNA had no effect (Fig. 6B). Cyclin D1 (CCND1) siRNA transfection abolished WNT10B-dependent proliferation in human CECs, but transfection with non-targeting control siRNA had no effect compared with vehicle control (Fig. 6C). Transfection with NT control siRNA had no effect on relative proliferation rates.
FIGURE 6.
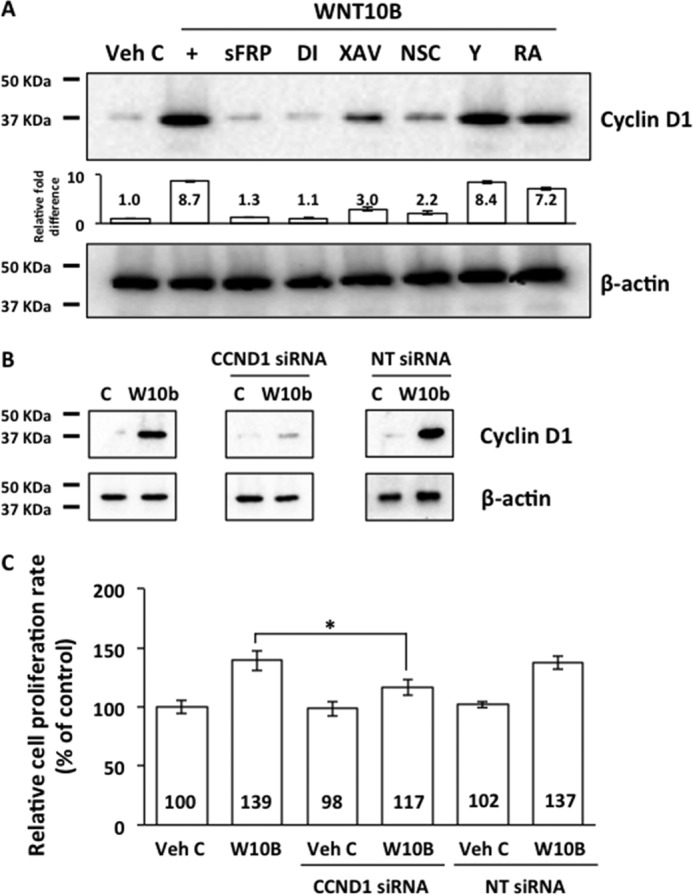
WNT10B induces Cyclin D1 expression through RAC1 and β-catenin and promotes cell proliferation. A, WNT10B stimulation (+) increased Cyclin D1 expression 8.7-fold compared with Veh C (C) in human CECs. Cotreatment with sFRP and Disheveled-PDZ DI abolished WNT10B-dependent Cyclin D1 expression, whereas cotreatment with the β-catenin antagonist XAV and the RAC1 inhibitor NSC attenuated expression of Cyclin D1 in WNT10B-treated cells. Rho kinase inhibitor Y and RA cotreatment had a minimal effect on WNT10B-dependent Cyclin D1 expression. The bar graph depicts the relative -fold difference in Cyclin D1 levels. β actin was used as a loading control. B, transfection with CCND1 siRNA knocked down WNT10B (W10B)-dependent Cyclin D1 expression, whereas transfection with NT siRNA had no effect. β actin was used as a loading control. C, Cyclin D1 siRNA transfection inhibited WNT10B-dependent proliferation in primary human CECs (117 ± 7.5 versus 139 ± 9.3; *, p < 0.01), whereas NT siRNA transfection had no effect (137 ± 6.1 versus 139 ± 9.3, p > 0.05). Transfection with CCND1 siRNA and NT siRNA had no effect on proliferation in vehicle control-treated cells (98 ± 6.8 versus 102 ± 2.5 versus 100 ± 6.5). One-way ANOVA, F(5, 30) = 46.4, p < 0.00001, n = 6/sample. Tukey's post-hoc test, HSD[0.05] = 11.8 and HSD[0.01] = 14.4.
WNT10B Promotes Proliferation in Human CECs ex Vivo
To further investigate the role of WNT10B signaling in human CEC proliferation, the proliferative response of endothelial cells to wounding in human ex vivo corneal endothelium was examined. PCNA and zonula occludens 1 (ZO-1) were used as markers for proliferation and CECs, respectively, in human ex vivo corneas. Exogenous WNT10B could not promote proliferation in CECs of human corneas ex vivo when the endothelium was not wounded (data not shown). WNT10B, but not vehicle control (Veh C), induced expression of PCNA in CECs in wounded endothelium of ex vivo human corneas (Fig. 7A). WNT10B-dependent expression of PCNA in human CECs ex vivo could be antagonized by cotreatment with sFRP, Disheveled-PDZ DI, the β-catenin antagonist XAV, NSC, and the CDK4-Cyclin D1 complex inhibitor fascaplysin (Fasc) (Fig. 7A). Quantification of PCNA expression in ex vivo human corneal endothelium showed significantly increased relative proliferation rates in response to WNT10B treatment (Fig. 7B). This, in turn, could be inhibited significantly by cotreatment with sFRP (p < 0.01), Disheveled-PDZ DI (p < 0.01), XAV (p < 0.01), NSC (p < 0.01), and Fasc (p < 0.01), and there was no statistically significant difference in PCNA expression between Veh C treatment and WNT10B + antagonist (sFRP, DI, XAV, NSC, and Fasc) treatment (p > 0.05, Fig. 7B). CCND1 siRNA transfection of ex vivo human corneal endothelium resulted in a knockdown of WNT10B-induced Cyclin D1 protein levels (Fig. 7C). Transfection with Disheveled 2 (DVL2) siRNA led to knockdown of DVL2 protein levels and inhibition of WNT10B-dependent Cyclin D1 expression (Fig. 7C). NT control siRNA transfection did not alter Cyclin D1 and Disheveled 2 protein levels. Transfection with Cyclin D1 and Disheveled 2 siRNA abolished PCNA expression in wounded ex vivo corneal endothelium treated with WNT10B, whereas non-targeting siRNA transfection did not affect PCNA expression (Fig. 7D). Cyclin D1 and Disheveled 2 siRNA transfection also significantly reduced relative proliferation rates in wounded ex vivo corneal endothelium treated with WNT10B (Fig. 7E). Transfection with non-targeting siRNA had no effect. To further evaluate cell cycle reentry of human ex vivo CECs, phosphohistone H3 was used as another marker. Treatment with WNT10B following injury led to phosphorylation of histone H3 whereas treatment with vehicle control did not (Fig. 7F).
FIGURE 7.

WNT10B enhances proliferation in human corneal endothelium ex vivo. A, WNT10B stimulation following injury of ex vivo human corneal endothelium induced PCNA expression in the nuclei (green) of endothelial cells immediately adjacent to the injury site. WNT10B-induced PCNA expression was severely attenuated by cotreatment with sFRP, Disheveled-PDZ DI, the β-catenin antagonist XAV, the Rac antagonist NSC, or the CDK4/Cyclin D1 antagonist Fasc. Corneal endothelial cell membranes were visualized with anti-ZO-1 (red) antibody, and nuclei were visualized with DAPI (blue). Scale bar = 50 μm. B, WNT10B stimulation (+) increased the relative proliferation rate compared with Veh C in human ex vivo corneal endothelium (13.7 ± 8.7 versus 1.0 ± 1.3; *, p < 0.01). WNT10B-dependent proliferation was blocked by treatment with sFRP (2.4 ± 2.2 versus 13.7 ± 8.7; **, p < 0.01), Disheveled-PDZ DI (2.4 ± 1.9 versus 12.8 ± 5.5; ***, p < 0.01), the β-catenin antagonist XAV (2.3 ± 2.0 versus 12.6 ± 6.4; #, p < 0.01), NSC (2.7 ± 2.1 versus 12.1 ± 4.6; ##, p < 0.01), and Fasc (2.3 ± 2.2 versus 14.3 ± 5.5; ###, p < 0.01). There was no significant difference in relative proliferation rates between vehicle treatment and WNT10B + inhibitor cotreatment (p > 0.05). One-way ANOVA, F(10, 154) = 24.2, p < 0.00001, n = 15/sample. Tukey's post-hoc test, HSD[0.05] = 5.4 and HSD[0.01] = 6.3. C, transfection of ex vivo human corneal endothelium with CCND1 siRNA resulted in knockdown of WNT10B-induced Cyclin D1 protein levels. Transfection with DVL2 siRNA resulted in knockdown of both DVL2 and WNT10B-induced CCND1 protein levels. Transfection with NT control siRNA had no effect on Cyclin D1 and Disheveled 2 protein levels. C, control. D, transfection of ex vivo human corneal endothelium with CCND1 siRNA and DVL2 siRNA resulted in inhibition of PCNA expression following injury and WNT10B (+) stimulation. Transfection with NT control siRNA had no effect on PCNA expression. Corneal endothelial cell membranes were visualized with anti-ZO-1 (red) antibody, and nuclei were visualized with DAPI (blue). Scale bar = 50 μm. E, transfection with CCND1 siRNA and DVL2 siRNA reduced relative proliferation rates compared with WNT10B treatment following injury in ex vivo human corneal endothelium (1.5 ± 1.7 versus 10.8 ± 2.4; †, p < 0.01 and 0.9 ± 1.4 versus 10.8 ± 2.4; ††, p < 0.01, respectively). Transection with NT control siRNA had no effect on WNT10B-induced proliferation (9.9 ± 2.6 versus 10.8 ± 2.4, p > 0.05). One-way ANOVA, F(4, 40) = 59.0, p < 0.00001, n = 9/sample. Tukey's post-hoc test, HSD[0.05] = 2.7 and HSD[0.01] = 3.2. F, WNT10B treatment following injury induced histone H3 phosphorylation (pH3, green) in ex vivo human corneal endothelium. Veh C treatment did not lead to histone H3 phosphorylation. Corneal endothelial cell membranes were visualized with anti-ZO-1 (red) antibody, and nuclei were visualized with DAPI (blue). Scale bar = 50 μm.
Discussion
Unlike other cells, in which proliferation plays a critical role in wound healing, endothelial cells in the adult human cornea are G1-arrested and do not proliferate even in response to injury (3, 4). This unusual feature of adult human CECs, in the setting of injury or infection, can lead to severe vision loss secondary to corneal edema. The current standard of care for severe vision loss secondary to endothelial dysfunction is corneal transplantation in the form of endothelial keratoplasty. There is currently a global shortage of donor corneas that will likely be exacerbated by an impending increase for donor corneas in developed nations. One method of addressing this issue is to develop strategies for treatment of vision loss because of endothelial dysfunction that do not rely on donor corneas. Tissue engineering approaches using human CECs derived from in vitro expansion methods are being investigated (56–61). However, these approaches have been hampered by fibroblastic transformation and uncertainty regarding the endothelial phenotype in cultured cells. Approaches targeting specific signaling pathways in human CECs or relying on alternative starting material hold the promise of generating a potentially unlimited supply of transplantable endothelium (62–65). All new approaches will have to ensure the endothelial phenotype and verify that the derived endothelial cells have not undergone mesenchymal transition to ensure safety and efficacy.
We have reported previously that IL-1β-induced FGF2 enhances proliferation and migration in rabbit and human CECs (9, 11–14). However, FGF2 has also been reported as a key mediator for EnMT, leading to retrocorneal fibrous membrane formation (13, 15, 66–68). This led us to investigate other downstream targets of IL-1β signaling in human CECs that could enhance cell proliferation without inducing fibrosis. Attenuation, but not complete inhibition, of cell proliferation by AZD4547 (Fig. 1A) strongly suggested the presence of another signaling pathway distinct from FGF2 that could drive proliferation in human CECs. Previous reports of a WNT10B promoter containing several conserved AP-1 and NF-κB sites (49) in conjunction with AP-1 and NF-κB being activated by IL-1β (9, 11, 35) led us to investigate the role of WNT10B in IL-1β-dependent proliferation in human CECs. As anticipated, IL-1β-dependent WNT10B expression proceeds through AP-1 and NF-κB activation in human CECs (Fig. 1, B and C). Also as anticipated, injury could not induce expression of WNT10B in human ex vivo CECs, but it could augment it in the presence of IL-1β (Fig. 1D). Nuclear transport of β-catenin indicated that WNT10B signals through the canonical pathway in human CECs (Fig. 2), similar to what has been reported for other cell types (16, 21, 69, 70). WNT3A was another potential candidate because it can induce nuclear transport of β-catenin in human CECs in culture, but it was not expressed in human CECs in response to IL-1β stimulation.3
Previous studies have reported that Disheveled 2 is required for the activation of RAC1 (32, 53) and that activated RAC1 regulates nuclear localization of β-catenin and β-catenin/TCF-dependent transcription (40, 52). Our results indicate that Disheveled 2 is critical for WNT10B signaling and occupies the nexus controlling both β-catenin nuclear transport and regulation of RhoGTPases. Antagonizing Disheveled 2 activity with Disheveled-PDZ DI resulted in inhibition of WNT10B-dependent nuclear transport of β-catenin (Fig. 2B), activation of RAC1 (Fig. 3B), and inhibition of RHOA (Fig. 3C). Inhibition of Disheveled 2 activity by either DI or siRNA knockdown led to an inhibition of WNT10B-induced binding between Disheveled 2 and RAC1 (Fig. 3D). In contrast to previous reports, activated RAC1 does not appear to be required for nuclear transport of β-catenin in human CECs, as evidenced by RAC1 inhibition with NSC23766 having no effect on WNT10B-dependent nuclear transport of β-catenin in human CECs (Fig. 2B). Moreover, β-catenin does not appear to play a role in RAC1 signaling in human CECs, as evidenced by inhibition of β-catenin with XAV939 having no effect on WNT10B-dependent activation (Fig. 3B) and nuclear transport (Fig. 4A) of RAC1. Although the nuclear transport of β-catenin and RAC1 are independent of one another, both are required for WNT10B-dependent proliferation in human CECs, as evidenced by XAV939 and NSC23766 independently being able to abolish WNT10B-dependent proliferation (Fig. 5), likely through attenuation of Cyclin D1 expression (Fig. 6A). It is interesting to note that, although WNT10B inhibited RHOA activity (Fig. 6C), neither modulation of RHOA by inhibition with Y27632 nor activation with RHOA activator had any effect on WNT10B-dependent proliferation in human CEC (Fig. 5B). This suggests that RHOA plays a very minor role in WNT10B-dependent proliferation in human CECs. Downstream of β-catenin and RAC1, WNT10B-dependent proliferation in human CECs proceeds through Cyclin D1. To address the concern over nonspecific effects of small molecule inhibitors, we confirmed the roles of RAC1 and β-catenin in WNT10B-induced CEC proliferation using siRNA-mediated knockdown of RAC1 and β-catenin (Fig. 5D).
WNT10B signals simultaneously through canonical and non-canonical Wnt pathways in human CEC proliferation. Treatment with either β-catenin antagonist XAV939 or NSC23766 substantially attenuated WNT10B-dependent expression of Cyclin D1 (Fig. 6A), and transfection of human CEC with Cyclin D1 siRNA resulted in knockdown of WNT10B-dependent Cyclin D1 expression (Fig. 6B) and inhibition of WNT10B-dependent proliferation (Fig. 6C) supporting the critical role of Cyclin D1.
To address the concern that the WNT10B-dependent proliferation could be the result of a culturing artifact, we used ex vivo human corneas to investigate the role of WNT10B signaling in CEC proliferation. The ability of WNT10B to stimulate proliferation in human CECs was comparable under ex vivo (Fig. 7) and in vitro (Figs. 5 and 6) conditions. Moreover, the results of vehicle control treatment of wounded ex vivo corneas showed a negligible number of CECs with nuclear expression of PCNA, consistent with previous reports (4, 71). Concerns over nonspecific effects of small molecule inhibitors were addressed by verifying the results from small molecule inhibitor experiments by utilizing siRNA knockdown of Cyclin D1 and Disheveled 2 in ex vivo corneal endothelium (Fig. 7, D and E). To address the concern that PCNA expression could be secondary to DNA damage rather than cell cycle reentry, we used phosphohistone H3 as an additional marker. Phosphorylation of histone H3 in wounded human ex vivo CECs treated with WNT10B indicates that they have entered mitosis (Fig. 7F). Taken together, these results suggest that modulating WNT10B signaling could play an important role in promoting endothelial cell proliferation in patients with vision loss because of endothelial dysfunction.
Although Cyclin D1 plays a crucial role in WNT10B-mediated proliferation, siRNA targeting of RAC1 and β-catenin compared with Cyclin D1 resulted in a greater inhibition of WNT10B-induced proliferation in primary human CECs (Figs. 5D and 6C). Although this strongly suggests that Cyclin D1 is not the only downstream target of WNT10B in promoting proliferation in primary human CECs, the results are slightly different in ex vivo human corneas. Targeting Disheveled 2, RAC1, and Cyclin D1 had similar effects on inhibition of proliferation in wounded ex vivo endothelial cells that were treated with WNT10B (Fig. 7). This highlights the notion that corneal endothelial cell behavior is, to some extent, dependent on their local environment.
The apparent minor role of RHOA in our results appears to contradict a previous report that showed inhibition of RHOA activity with the Rho kinase inhibitor Y27632 induced cyclin D expression and proliferation in monkey CECs (72). The differences in results could be rooted in differences in species and culture conditions causing different components, e.g. RAC1, RHOA, and β-catenin, to play different roles in mediating cell proliferation. This strongly suggests that the source and culturing conditions for corneal endothelial cells significantly influence their behavior.
With investigations of alternative approaches in very preliminary stages, a major concern that needs to be addressed is the potential induction of endothelial EnMT and its undesirable sequelae. Although both IL-1β and FGF2 can induce proliferation of human CECs, they also induce EnMT, making their use potentially dangerous because of fibrosis. Future studies investigating the potential induction of EnMT by WNT10B and other targets that can enhance proliferation are needed. Enhancing endothelial pump function through cell proliferation in vivo for patients with endothelial dysfunction not only avoids the postoperative morbidity associated with cornea transplants, but it also effectively enlarges the cornea donor pool by making more donor corneas available for other causes of corneal blindness. In summary, WNT10B activates RAC1 and β-catenin, resulting in induction of Cyclin D1 expression and subsequent proliferation in human CECs (Fig. 8).
FIGURE 8.
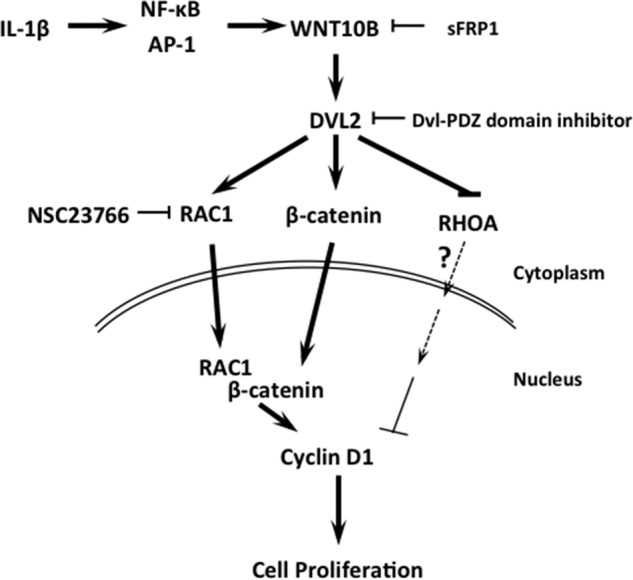
WNT10B signaling in human CECs. IL-1β-induced WNT10B expression activates of Disheveled 2. This leads to independent nuclear transport and binding of β-catenin and RAC1, leading to activation of Cyclin D1 expression and proliferation in human CECs.
Author Contributions
J. G. L. and M. H. conceived and designed the experiments. J. G. L. performed the experiments. J. G. L. and M. H. analyzed and interpreted the data, and M. H. performed the statistical analysis. J. G. L. and M. H. wrote the manuscript and approved the final version of the manuscript.
Acknowledgments
We thank the Kehle family for their generous support.
This study was supported by Grant K08 EY021485 from the NEI/National Institutes of Health (to M. H.) and a career development award from Research to Prevent Blindness (to M. H.). The authors declare that they have no conflicts of interest with the contents of this article.
J. G. Lee and M. Heur, unpublished data.
- CEC
- corneal endothelial cell
- EnMT
- endothelial-mesenchymal transition
- IL-1ra
- interleukin-1 receptor antagonist
- SFM
- serum-free medium
- GTPγS
- guanosine 5′-3-O-(thio)triphosphate
- PCNA
- proliferating cell nuclear antigen
- ANOVA
- analysis of variance
- XAV
- XAV939
- DI
- domain inhibitor
- RA
- RHOA activator
- Y
- Y27632
- NSC
- NSC23766
- sFRP
- secreted Frizzled-related peptide
- NT
- non-targeting
- Veh C
- vehicle control
- Fasc
- fascaplysin
- Pos C
- positive control.
References
- 1.Kreutziger G. O. (1976) Lateral membrane morphology and gap JUNction structure in rabbit corneal endothelium. Exp. Eye Res. 23, 285–293 [DOI] [PubMed] [Google Scholar]
- 2.Geroski D. H., and Edelhauser H. F. (1984) Quantitation of Na/K ATPase pump sites in the rabbit corneal endothelium. Invest. Ophthalmol. Vis. Sci. 25, 1056–1060 [PubMed] [Google Scholar]
- 3.Joyce N. C., Navon S. E., Roy S., and Zieske J. D. (1996) Expression of cell cycle-associated proteins in human and rabbit corneal endothelium in situ. Invest. Ophthalmol. Vis. Sci. 37, 1566–1575 [PubMed] [Google Scholar]
- 4.Senoo T., and Joyce N. C. (2000) Cell cycle kinetics in corneal endothelium from old and young donors. Invest. Ophthalmol. Vis. Sci. 41, 660–667 [PubMed] [Google Scholar]
- 5.Whitcher J. P., Srinivasan M., and Upadhyay M. P. (2001) Corneal blindness: a global perspective. Bull. World Health Organ. 79, 214–221 [PMC free article] [PubMed] [Google Scholar]
- 6.Song J. S., Lee J. G., and Kay E. P. (2010) Induction of FGF-2 synthesis by IL-1β in aqueous humor through P13-kinase and p38 in rabbit corneal endothelium. Invest. Ophthalmol. Vis. Sci. 51, 822–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Djalilian A. R., Nagineni C. N., Mahesh S. P., Smith J. A., Nussenblatt R. B., and Hooks J. J. (2006) Inhibition of inflammatory cytokine production in human corneal cells by dexamethasone, but not cyclosporin. Cornea 25, 709–714 [DOI] [PubMed] [Google Scholar]
- 8.Moore J. E., McMullen T. C., Campbell I. L., Rohan R., Kaji Y., Afshari N. A., Usui T., Archer D. B., and Adamis A. P. (2002) The inflammatory milieu associated with conJUNctivalized cornea and its alteration with IL-1 RA gene therapy. Invest. Ophthalmol. Vis. Sci. 43, 2905–2915 [PubMed] [Google Scholar]
- 9.Lee J. G., and Heur M. (2013) Interleukin-1β enhances cell migration through AP-1 and NF-κB pathway-dependent FGF2 expression in human corneal endothelial cells. Biol. Cell 105, 175–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee J. G., and Kay E. P. (2009) Common and distinct pathways for cellular activities in FGF-2 signaling induced by IL-1β in corneal endothelial cells. Invest. Ophthalmol. Vis. Sci. 50, 2067–2076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee J. G., and Kay E. P. (2012) NF-κB is the transcription factor for FGF-2 that causes endothelial mesenchymal transformation in cornea. Invest Ophthalmol. Vis. Sci. 53, 1530–1538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee J. G., Song J. S., Smith R. E., and Kay E. P. (2011) Human corneal endothelial cells employ phosphorylation of p27(Kip1) at both Ser10 and Thr187 sites for FGF-2-mediated cell proliferation via PI 3-kinase. Invest. Ophthalmol. Vis. Sci. 52, 8216–8223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee J. G., and Kay E. P. (2007) Two populations of p27 use differential kinetics to phosphorylate Ser-10 and Thr-187 via phosphatidylinositol 3-kinase in response to fibroblast growth factor-2 stimulation. J. Biol. Chem. 282, 6444–6454 [DOI] [PubMed] [Google Scholar]
- 14.Lee J. G., and Kay E. P. (2011) PI 3-kinase/RAC1 and ERK1/2 regulate FGF-2-mediated cell proliferation through phosphorylation of p27 at Ser10 by KIS and at Thr187 by Cdc25A/Cdk2. Invest. Ophthalmol. Vis. Sci. 52, 417–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee H. T., Lee J. G., Na M., and Kay E. P. (2004) FGF-2 induced by interleukin-1β through the action of phosphatidylinositol 3-kinase mediates endothelial mesenchymal transformation in corneal endothelial cells. J. Biol. Chem. 279, 32325–32332 [DOI] [PubMed] [Google Scholar]
- 16.Angers S., and Moon R. T. (2009) Proximal events in Wnt signal transduction. Nat. Rev. Mol. Cell Biol. 10, 468–477 [DOI] [PubMed] [Google Scholar]
- 17.Moon R. T., Bowerman B., Boutros M., and Perrimon N. (2002) The promise and perils of Wnt signaling through β-catenin. Science 296, 1644–1646 [DOI] [PubMed] [Google Scholar]
- 18.Nusse R. (2005) Wnt signaling in disease and in development. Cell Res. 15, 28–32 [DOI] [PubMed] [Google Scholar]
- 19.Nusse R., and Varmus H. E. (1982) Many tumors induced by the mouse mammary tumor virus contain a provirus integrated in the same region of the host genome. Cell 31, 99–109 [DOI] [PubMed] [Google Scholar]
- 20.Polakis P. (2000) Wnt signaling and cancer. Genes Dev. 14, 1837–1851 [PubMed] [Google Scholar]
- 21.MacDonald B. T., Tamai K., and He X. (2009) Wnt/β-catenin signaling: components, mechanisms, and diseases. Dev. Cell 17, 9–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goel S., Chin E. N., Fakhraldeen S. A., Berry S. M., Beebe D. J., and Alexander C. M. (2012) Both LRP5 and LRP6 receptors are required to respond to physiological Wnt ligands in mammary epithelial cells and fibroblasts. J. Biol. Chem. 287, 16454–16466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zeng X., Huang H., Tamai K., Zhang X., Harada Y., Yokota C., Almeida K., Wang J., Doble B., Woodgett J., Wynshaw-Boris A., Hsieh J. C., and He X. (2008) Initiation of Wnt signaling: control of Wnt coreceptor Lrp6 phosphorylation/activation via frizzled, dishevelled and axin functions. Development 135, 367–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tamai K., Zeng X., Liu C., Zhang X., Harada Y., Chang Z., and He X. (2004) A mechanism for Wnt coreceptor activation. Mol. Cell 13, 149–156 [DOI] [PubMed] [Google Scholar]
- 25.Tetsu O., and McCormick F. (1999) β-Catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 398, 422–426 [DOI] [PubMed] [Google Scholar]
- 26.Zhang J., Gill A. J., Issacs J. D., Atmore B., Johns A., Delbridge L. W., Lai R., and McMullen T. P. (2012) The Wnt/β-catenin pathway drives increased cyclin D1 levels in lymph node metastasis in papillary thyroid cancer. Hum. Pathol. 43, 1044–1050 [DOI] [PubMed] [Google Scholar]
- 27.Roose J., and Clevers H. (1999) TCF transcription factors: molecular switches in carcinogenesis. Biochim. Biophys. Acta 1424, M23–37 [DOI] [PubMed] [Google Scholar]
- 28.Stump R. J., Ang S., Chen Y., von Bahr T., Lovicu F. J., Pinson K., de Iongh R. U., Yamaguchi T. P., Sassoon D. A., and McAvoy J. W. (2003) A role for Wnt/β-catenin signaling in lens epithelial differentiation. Dev. Biol. 259, 48–61 [DOI] [PubMed] [Google Scholar]
- 29.Chen Y., Stump R. J., Lovicu F. J., and McAvoy J. W. (2006) A role for Wnt/planar cell polarity signaling during lens fiber cell differentiation? Semin. Cell Dev. Biol. 17, 712–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Randall R. M., Shao Y. Y., Wang L., and Ballock R. T. (2012) Activation of Wnt planar cell polarity (PCP) signaling promotes growth plate column formation in vitro. J. Orthop. Res. 30, 1906–1914 [DOI] [PubMed] [Google Scholar]
- 31.Dejmek J., Säfholm A., Kamp Nielsen C., Andersson T., and Leandersson K. (2006) Wnt-5a/Ca2+-induced NFAT activity is counteracted by Wnt-5a/Yes-Cdc42-casein kinase 1α signaling in human mammary epithelial cells. Mol. Cell. Biol. 26, 6024–6036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Habas R., Dawid I. B., and He X. (2003) Coactivation of Rac and Rho by Wnt/Frizzled signaling is required for vertebrate gastrulation. Genes Dev. 17, 295–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kikuchi A., Yamamoto H., and Sato A. (2009) Selective activation mechanisms of Wnt signaling pathways. Trends Cell Biol. 19, 119–129 [DOI] [PubMed] [Google Scholar]
- 34.Semenov M. V., Habas R., Macdonald B. T., and He X. (2007) SnapShot: noncanonical Wnt signaling pathways. Cell 131, 1378. [DOI] [PubMed] [Google Scholar]
- 35.Lee J. G., and Heur M. (2014) Interleukin-1β-induced Wnt5a enhances human corneal endothelial cell migration through regulation of Cdc42 and RHOA. Mol. Cell. Biol. 34, 3535–3545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hall A. (1998) Rho GTPases and the actin cytoskeleton. Science 279, 509–514 [DOI] [PubMed] [Google Scholar]
- 37.Schmitz A. A., Govek E. E., Böttner B., and Van Aelst L. (2000) Rho GTPases: signaling, migration, and invasion. Exp. Cell Res. 261, 1–12 [DOI] [PubMed] [Google Scholar]
- 38.Fanto M., Weber U., Strutt D. I., and Mlodzik M. (2000) Nuclear signaling by Rac and Rho GTPases is required in the establishment of epithelial planar polarity in the Drosophila eye. Curr. Biol. 10, 979–988 [DOI] [PubMed] [Google Scholar]
- 39.Habas R., Kato Y., and He X. (2001) Wnt/Frizzled activation of Rho regulates vertebrate gastrulation and requires a novel Formin homology protein Daam1. Cell 107, 843–854 [DOI] [PubMed] [Google Scholar]
- 40.Wu X., Tu X., Joeng K. S., Hilton M. J., Williams D. A., and Long F. (2008) RAC1 activation controls nuclear localization of β-catenin during canonical Wnt signaling. Cell 133, 340–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Esufali S., and Bapat B. (2004) Cross-talk between RAC1 GTPase and dysregulated Wnt signaling pathway leads to cellular redistribution of β-catenin and TCF/LEF-mediated transcriptional activation. Oncogene 23, 8260–8271 [DOI] [PubMed] [Google Scholar]
- 42.Chim C. S., Pang R., Fung T. K., Choi C. L., and Liang R. (2007) Epigenetic dysregulation of Wnt signaling pathway in multiple myeloma. Leukemia 21, 2527–2536 [DOI] [PubMed] [Google Scholar]
- 43.Grandy D., Shan J., Zhang X., Rao S., Akunuru S., Li H., Zhang Y., Alpatov I., Zhang X. A., Lang R. A., Shi D. L., and Zheng J. J. (2009) Discovery and characterization of a small molecule inhibitor of the PDZ domain of dishevelled. J. Biol. Chem. 284, 16256–16263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gavine P. R., Mooney L., Kilgour E., Thomas A. P., Al-Kadhimi K., Beck S., Rooney C., Coleman T., Baker D., Mellor M. J., Brooks A. N., and Klinowska T. (2012) AZD4547: an orally bioavailable, potent, and selective inhibitor of the fibroblast growth factor receptor tyrosine kinase family. Cancer Res. 72, 2045–2056 [DOI] [PubMed] [Google Scholar]
- 45.Huang S. M., Mishina Y. M., Liu S., Cheung A., Stegmeier F., Michaud G. A., Charlat O., Wiellette E., Zhang Y., Wiessner S., Hild M., Shi X., Wilson C. J., Mickanin C., Myer V., Fazal A., Tomlinson R., Serluca F., Shao W., Cheng H., Shultz M., Rau C., Schirle M., Schlegl J., Ghidelli S., Fawell S., Lu C., Curtis D., Kirschner M. W., Lengauer C., Finan P. M., Tallarico J. A., Bouwmeester T., Porter J. A., Bauer A., and Cong F. (2009) Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 461, 614–620 [DOI] [PubMed] [Google Scholar]
- 46.Hamilton G. (2014) Cytotoxic effects of fascaplysin against small cell lung cancer cell lines. Mar. Drugs 12, 1377–1389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hirata-Tominaga K., Nakamura T., Okumura N., Kawasaki S., Kay E. P., Barrandon Y., Koizumi N., and Kinoshita S. (2013) Corneal endothelial cell fate is maintained by LGR5 through the regulation of hedgehog and Wnt pathway. Stem Cells 31, 1396–1407 [DOI] [PubMed] [Google Scholar]
- 48.Okumura N., Kay E. P., Nakahara M., Hamuro J., Kinoshita S., and Koizumi N. (2013) Inhibition of TGF-β signaling enables human corneal endothelial cell expansion in vitro for use in regenerative medicine. PLoS ONE 8, e58000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Katoh M., and Katoh M. (2007) AP1- and NF-κB-binding sites conserved among mammalian WNT10B orthologs elucidate the TNFα-WNT10B signaling loop implicated in carcinogenesis and adipogenesis. Int. J. Mol. Med. 19, 699–703 [PubMed] [Google Scholar]
- 50.Chahdi A., and Raufman J. P. (2013) The Cdc42/Rac nucleotide exchange factor protein β1Pix (Pak-interacting exchange factor) modulates β-catenin transcriptional activity in colon cancer cells: evidence for direct interaction of β1PIX with β-catenin. J. Biol. Chem. 288, 34019–34029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rossol-Allison J., Stemmle L. N., Swenson-Fields K. I., Kelly P., Fields P. E., McCall S. J., Casey P. J., and Fields T. A. (2009) Rho GTPase activity modulates Wnt3a/β-catenin signaling. Cell. Signal. 21, 1559–1568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Buongiorno P., Pethe V. V., Charames G. S., Esufali S., and Bapat B. (2008) RAC1 GTPase and the RAC1 exchange factor Tiam1 associate with Wnt-responsive promoters to enhance β-catenin/TCF-dependent transcription in colorectal cancer cells. Mol. Cancer 7, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cajanek L., Ganji R. S., Henriques-Oliveira C., Theofilopoulos S., Konik P., Bryja V., and Arenas E. (2013) Tiam1 regulates the Wnt/Dvl/RAC1 signaling pathway and the differentiation of midbrain dopaminergic neurons. Mol. Cell. Biol. 33, 59–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rosso S. B., Sussman D., Wynshaw-Boris A., and Salinas P. C. (2005) Wnt signaling through Dishevelled, Rac and JNK regulates dendritic development. Nat. Neurosci. 8, 34–42 [DOI] [PubMed] [Google Scholar]
- 55.Shtutman M., Zhurinsky J., Simcha I., Albanese C., D'Amico M., Pestell R., and Ben-Ze'ev A. (1999) The cyclin D1 gene is a target of the β-catenin/LEF-1 pathway. Proc. Natl. Acad. Sci. U.S.A. 96, 5522–5527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hatou S., Yoshida S., Higa K., Miyashita H., Inagaki E., Okano H., Tsubota K., and Shimmura S. (2013) Functional corneal endothelium derived from corneal stroma stem cells of neural crest origin by retinoic acid and Wnt/β-catenin signaling. Stem Cells Dev. 22, 828–839 [DOI] [PubMed] [Google Scholar]
- 57.Hsiue G. H., Lai J. Y., Chen K. H., and Hsu W. M. (2006) A novel strategy for corneal endothelial reconstruction with a bioengineered cell sheet. Transplantation 81, 473–476 [DOI] [PubMed] [Google Scholar]
- 58.Li W., Sabater A. L., Chen Y. T., Hayashida Y., Chen S. Y., He H., and Tseng S. C. (2007) A novel method of isolation, preservation, and expansion of human corneal endothelial cells. Invest. Ophthalmol. Vis. Sci. 48, 614–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sumide T., Nishida K., Yamato M., Ide T., Hayashida Y., Watanabe K., Yang J., Kohno C., Kikuchi A., Maeda N., Watanabe H., Okano T., and Tano Y. (2006) Functional human corneal endothelial cell sheets harvested from temperature-responsive culture surfaces. FASEB J. 20, 392–394 [DOI] [PubMed] [Google Scholar]
- 60.Honda N., Mimura T., Usui T., and Amano S. (2009) Descemet stripping automated endothelial keratoplasty using cultured corneal endothelial cells in a rabbit model. Arch. Ophthalmol. 127, 1321–1326 [DOI] [PubMed] [Google Scholar]
- 61.Okumura N., Kinoshita S., and Koizumi N. (2014) Cell-based approach for treatment of corneal endothelial dysfunction. Cornea 33, S37–41 [DOI] [PubMed] [Google Scholar]
- 62.Numata R., Okumura N., Nakahara M., Ueno M., Kinoshita S., Kanematsu D., Kanemura Y., Sasai Y., and Koizumi N. (2014) Cultivation of corneal endothelial cells on a pericellular matrix prepared from human decidua-derived mesenchymal cells. PLoS ONE 9, e88169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhu Y. T., Li F., Han B., Tighe S., Zhang S., Chen S. Y., Liu X., and Tseng S. C. (2014) Activation of RHOA-ROCK-BMP signaling reprograms adult human corneal endothelial cells. J. Cell Biol. 206, 799–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nakano Y., Oyamada M., Dai P., Nakagami T., Kinoshita S., and Takamatsu T. (2008) Connexin43 knockdown accelerates wound healing but inhibits mesenchymal transition after corneal endothelial injury in vivo. Invest. Ophthalmol. Vis. Sci. 49, 93–104 [DOI] [PubMed] [Google Scholar]
- 65.Okumura N., Koizumi N., Ueno M., Sakamoto Y., Takahashi H., Tsuchiya H., Hamuro J., and Kinoshita S. (2012) ROCK inhibitor converts corneal endothelial cells into a phenotype capable of regenerating in vivo endothelial tissue. Am. J. Pathol. 181, 268–277 [DOI] [PubMed] [Google Scholar]
- 66.Lee J. G., and Kay E. P. (2006) FGF-2-induced wound healing in corneal endothelial cells requires Cdc42 activation and Rho inactivation through the phosphatidylinositol 3-kinase pathway. Invest. Ophthalmol. Vis. Sci. 47, 1376–1386 [DOI] [PubMed] [Google Scholar]
- 67.Ko M. K., and Kay E. P. (2005) Regulatory role of FGF-2 on type I collagen expression during endothelial mesenchymal transformation. Invest. Ophthalmol. Vis. Sci. 46, 4495–4503 [DOI] [PubMed] [Google Scholar]
- 68.Lee J. G., and Kay E. P. (2006) Cross-talk among Rho GTPases acting downstream of PI 3-kinase induces mesenchymal transformation of corneal endothelial cells mediated by FGF-2. Invest. Ophthalmol. Vis. Sci. 47, 2358–2368 [DOI] [PubMed] [Google Scholar]
- 69.Seidensticker M. J., and Behrens J. (2000) Biochemical interactions in the Wnt pathway. Biochim. Biophys. Acta 1495, 168–182 [DOI] [PubMed] [Google Scholar]
- 70.Cadigan K. M., and Nusse R. (1997) Wnt signaling: a common theme in animal development. Genes Dev. 11, 3286–3305 [DOI] [PubMed] [Google Scholar]
- 71.Joyce N. C., Meklir B., Joyce S. J., and Zieske J. D. (1996) Cell cycle protein expression and proliferative status in human corneal cells. Invest. Ophthalmol. Vis. Sci. 37, 645–655 [PubMed] [Google Scholar]
- 72.Okumura N., Nakano S., Kay E. P., Numata R., Ota A., Sowa Y., Sakai T., Ueno M., Kinoshita S., and Koizumi N. (2014) Involvement of cyclin D and p27 in cell proliferation mediated by ROCK inhibitors Y-27632 and Y-39983 during corneal endothelium wound healing. Invest. Ophthalmol. Vis. Sci. 55, 318–329 [DOI] [PubMed] [Google Scholar]