

Background: PLCϵ activation is sustained but the underlying regulatory mechanisms are unknown.
Results: PLCϵ gene deletion and rescue demonstrate that Rap1 activation by the CDC25 domain and regulation via the RA2 domain sustain thrombin-mediated PLCϵ and PKD activation and inflammatory gene expression.
Conclusion: Unique domains and compartmentalization of PLCϵ allow for sustained GPCR signaling.
Significance: Targeting these PLCϵ domains could ameliorate pathophysiological inflammation.
Keywords: astrocyte, cyclooxygenase (COX), diacylglycerol, G protein-coupled receptor (GPCR), guanine nucleotide exchange factor (GEF), Phospholipase C, protein kinase D (PKD), Ras-related protein 1 (Rap1), thrombin
Abstract
Phospholipase C-epsilon (PLCϵ) plays a critical role in G-protein-coupled receptor-mediated inflammation. In addition to its ability to generate the second messengers inositol 1,4,5-trisphosphate and diacylglycerol, PLCϵ, unlike the other phospholipase C family members, is activated in a sustained manner. We hypothesized that the ability of PLCϵ to function as a guanine nucleotide exchange factor (GEF) for Rap1 supports sustained downstream signaling via feedback of Rap1 to the enzyme Ras-associating (RA2) domain. Using gene deletion and adenoviral rescue, we demonstrate that both the GEF (CDC25 homology domain) and RA2 domains of PLCϵ are required for long term protein kinase D (PKD) activation and subsequent induction of inflammatory genes. PLCϵ localization is largely intracellular and its compartmentalization could contribute to its sustained activation. Here we show that localization of PLCϵ to the Golgi is required for activation of PKD in this compartment as well as for subsequent induction of inflammatory genes. These data provide a molecular mechanism by which PLCϵ mediates sustained signaling and by which astrocytes mediate pathophysiological inflammatory responses.
Introduction
Phospholipase C-epsilon (PLCϵ)3 has emerged as a novel signaling node through which G-protein coupled receptors (GPCRs) that activate small G-proteins can lead to biological responses (1–8). In addition to its ability to bind and respond to RhoA, PLCϵ mediates sustained phosphoinositide hydrolysis (9) and sustained activation of downstream kinases (2, 3, 10). Thus, PLCϵ may play an especially important role in chronic or dysregulated signaling involved in diseases such as cancer, heart failure, and central nervous system (CNS) injury (1–3, 10–23).
First identified in Caenorhabditis elegans as a Let-60 Ras-binding protein (24), PLCϵ was demonstrated to contain the X, Y, and C2 domains characteristic of enzymes in the phospholipase C (PLC) family (24). Like the other PLC family members, PLCϵ was found to function to hydrolyze phosphatidylinositol 4,5-bisphosphate to generate the second messengers inositol 1,4,5-trisphosphate (IP3)and diacylglycerol (DAG) (25). Additionally, PLCϵ has an extended N-terminal, which contains a CDC25 domain not found in the other PLCs and which functions as a GEF for the low molecular weight G-protein Rap1 (26–28). This domain has been shown to be important for PLCϵ localization to the Golgi, and its deletion leads to more transient PLCϵ localization to this compartment (26). Moreover, PLCϵ was found to be uniquely regulated by the small G-protein RhoA through a 65 amino acid sequence within the Y domain (4–8), as well as by other Ras family members through their interactions with the RA2 domain (5, 29).
Of particular interest, while both PLC-beta (PLCβ) and PLCϵ are regulated in response to endothelin-1 (ET-1), lysophosphatidic acid (LPA), and thrombin, knockdown of PLCβ inhibits inositol phosphate generation at short times (1–3 min) whereas knockdown of PLCϵ is required to inhibit inositol phosphate generation at longer times (10–60 min) (9). Using primary astrocytes from PLCϵ knock-out (KO) mice, we demonstrated that PLCϵ is needed for the sustained activation of protein kinase D (PKD) which occurs in response to ligands that activate Gα12/13/Rho whereas ligands that stimulate Gαq/PLCβ lead to a more transient activation of PKD (2). Our data also revealed that the sustained activation of PKD is necessary for induction of inflammatory gene expression (2).
We postulate and demonstrate here that the non-catalytic CDC25 and RA2 domains of PLCϵ are essential components required for sustained PKD activation and inflammatory gene expression. This conclusion is supported by studies using astrocytes from PLCϵ KO mice and rescue by adenoviral expression of wild-type (WT) and mutant PLCϵ. A role for compartmentalized PLCϵ signaling at the Golgi is also established. We conclude that PLCϵ signaling, initiated by GPCR stimulation and RhoA binding, is sustained by a feedback mechanism involving the CDC25 domain as a generator of active Rap1 and the RA2 domain of PLCϵ as its effector.
Experimental Procedures
Animals
All procedures were performed in accordance with NIH Guide and Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee at the University of California San Diego. Generation of homozygous C57BL6/Sv129 PLCϵ KO mice has been described previously (19). PLCϵ heterozygous KO mice were bred to generate homozygous KO animals.
Primary Culture of Astrocytes
Astrocytes were isolated from P1-P3 postnatal WT and KO mice as previously described (1). Purity of astrocytes was determined to be ∼95% based on GFAP staining. In all experiments, WT and PLCϵ KO astrocytes were used at passage 2.
Transduction of Astrocytes with Adenovirus
PLCϵ KO astrocytes were infected for 4–6 h in complete media with 200 multiplicity of infection (moi) of adenovirus expressing FLAG-tagged WT PLCϵ, CDC25-deleted mutant (CDC25Δ) PLCϵ, RA2 K2150E mutant, or enhanced yellow fluorescent protein (EYFP) as previously described (1, 16, 17). Following 4–6 h of infection, astrocytes were washed and serum starved for 18–24 h prior to agonist treatment.
Fluorescence Resonance Energy Transfer
Astrocytes were plated onto glass coverslips in 35-mm dishes and Golgi or plasma membrane-targeted DKAR constructs were transfected using Dharma-FECT 3 transfection reagent at a 1:3 DNA:Dharma-FECT3 ratio (Thermo Scientific). Cells were serum starved the next day for 18–24 h and then washed with HBSS (Gibco) prior to collecting DKAR images as described previously (30) on a Zeiss Axiovert microscope (Carl Zeiss MicroImaging, Inc.) with a cooled charge-couple device camera (Photometric) controlled by MetaFluor software (Universal Imaging Corp.). Images were collected at baseline for 4 min followed by treatment with thrombin for up to twelve minutes.
Immunofluorescence
Astrocytes were grown on glass slides and infected with 150 moi of mCherry-PLCϵ. Following serum starvation for 18–24 h, cells were fixed using 4% paraformaldehyde in PBS and then permeabilized with 0.1% Triton in PBS for 5 min before blocking with 5% BSA in PBS and 10% normal goat serum. Antibody GM-130 (BD Biosciences) was diluted in the blocking solution before addition of Alexa 488 mouse. Cells were then washed and mounted with coverslips using Vectashield with DAPI (Vector Labs). Pictures were acquired using the Olympus FV-1000 confocal microscope.
Quantitative-PCR (q-PCR)
Total RNA was extracted from agonist treated WT and PLCϵ KO astrocytes using an RNeasy kit (Invitrogen) as previously described (2). cDNA was amplified using the TaqMan Universal Master Mix in the presence of gene-specific primers for IL-6 and COX-2 with GAPDH used as an internal control (Applied Biosystems). Data were normalized to internal GAPDH and fold change determined according to published protocol (31).
Western Blotting
Astrocyte lysates were prepared in RIPA buffer. Western blot analysis was performed according to the previous described protocol (32). The antibodies used for immunoblotting were as follows: p-PKD (Ser-916), PKD, and GAPDH from Cell Signaling Technology, COX-2 from Cayman, and Rap1 from Santa Cruz. Immunoblots shown represent a single gel; images are split in cases where unnecessary lanes were removed.
Rap Pull-down
Serum-starved astrocytes were treated with thrombin for the indicated times and then lysed with buffer containing 50 mm Tris, 150 mm NaCl, 5 mm MgCl2, 0.1% Triton X-100, and 10% glycerol. Lysates were incubated with 20 μg of RalGDS RBD conjugated to glutathione beads for 45 min at 4 °C and then centrifuged to pellet the agarose beads. Agarose beads were washed with lysis buffer, and the pellets were resuspended in 2.5× Laemmli sample buffer containing DTT, boiled for 10 min, and centrifuged. Western blot analysis was then performed.
Statistical Analysis
Statistical differences were determined using Tukey's multicomparison analysis after one-way ANOVA with Prism software (GraphPad). p < 0.05 was considered significant.
Results
Rap1 Activation Is Sustained and Requires the CDC25 Domain of PLCϵ
Our earlier work demonstrated that endogenous PLCϵ functions as a Rap1GEF that is activated in response to GPCR stimulation (1, 16). Here we examined the kinetics of Rap1 activation in primary mouse astrocytes stimulated with thrombin, measuring activated Rap1 using a pull-down assay. Rap1 activation was significantly elevated at 15 min, further increased at 1 h, and sustained for up to 6 h (Fig. 1A). This response was absent in astrocytes from PLCϵ KO mice (Fig. 1A). To demonstrate that it is the CDC25 domain of PLCϵ that functions as the thrombin-regulated Rap1GEF, we used a previously generated mutant PLCϵ construct in which the CDC25 domain was deleted (CDC25Δ) (16). Adenoviral expression of WT PLCϵ in PLCϵ KO astrocytes lead to significant recovery of Rap1 activation (Fig. 1B). In contrast Rap1 activation was not recovered in the KO cells expressing the CDC25Δ mutant PLCϵ (Fig. 1B).
FIGURE 1.
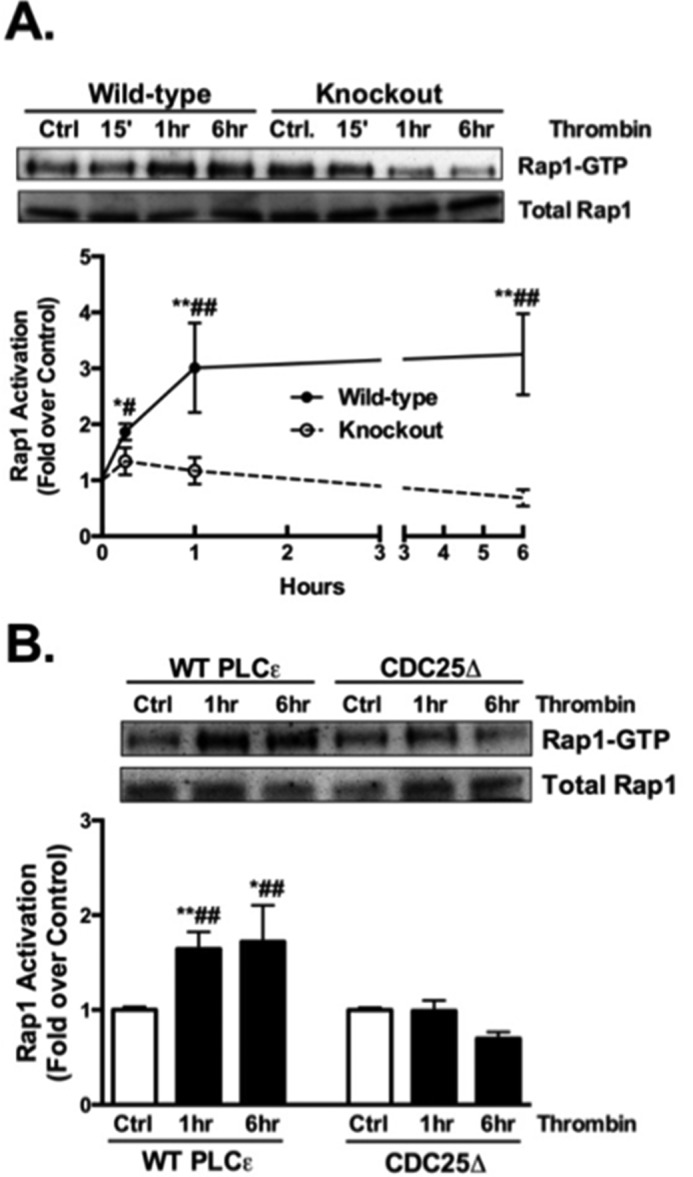
Rap1 activation is sustained and requires the CDC25 domain of PLCϵ. A, Rap1 activation was assessed by measuring levels of Rap1-GTP after Rap1 pull-down and Western blot in WT and PLCϵ KO astrocytes treated with vehicle or thrombin (5 nm) for 15 min, 1 h, and 6 h. Rap1-GTP was normalized to total Rap1 and data normalized to its own control. Representative Western blots are shown and data quantitated as the mean ± S.E. (n = 6) of three independent experiments. B, Rap1 activation was assessed in KO astrocytes that were infected with either WT PLCϵ adenovirus or mutant PLCϵ adenovirus that lacks the CDC25 domain (CDC25Δ) followed by vehicle or thrombin (5 nm) treatment for 1 h and 6 h. Rap1-GTP was normalized to total Rap1 and data normalized to each of its own control. Representative Western blots are shown and data quantitated as the mean ± S.E. (n = 9) of three independent experiments. *, p < .05 and **, p < .01 between control and thrombin treatment; #, p < .05 and ##, p < .01 between thrombin treatments, one-way ANOVA.
PKD Activation and COX-2 Expression Require the CDC25 Domain of PLCϵ
We previously demonstrated that sustained activation of PKD and subsequent COX-2 expression in astrocytes require PLCϵ (2). To determine whether Rap1 activation plays a role in these responses we compared the ability of the CDC25 domain mutant and WT PLCϵ to support activation of PKD and induction of COX-2 in PLCϵ KO astrocytes. The activation of PKD was assessed using an antibody directed at the PKD autophosphorylation site (Ser-916) and COX-2 expression was assessed by Western blotting. Thrombin activation of PKD was significantly attenuated in the CDC25Δ mutant compared with those expressing WT PLCϵ (Fig. 2A). COX-2 expression was also restored by WT PLCϵ, but not the CDC25Δ mutant (Fig. 2B).
FIGURE 2.

PKD activation and COX-2 expression require the CDC25 domain of PLCϵ. A, PKD phosphorylation (p-PKDS916) was measured in PLCϵ KO astrocytes that were infected with WT PLCϵ or CDC25Δ adenovirus followed by vehicle or thrombin (5 nm) treatment for 1 h. The p-PKDS916 protein levels were normalized to total PKD and expressed relative to its own averaged control. Representative Western blots are shown and data quantitated as the mean ± S.E. (n = 9) of four independent experiments (control error bars are small but present). B, COX-2 protein levels were measured in PLCϵ KO astrocytes infected with WT PLCϵ or CDC25Δ adenovirus followed by thrombin (5 nm) treatment for 1 h and 6 h. COX-2 protein levels were normalized to GAPDH and expressed relative to its own control. Representative Western blots are shown and data quantitated at the 6 h time point as the mean ± S.E. (n = 6) of three independent experiments. *, p < .01 between control and thrombin treatment; #, p < .01 between thrombin treatments, one-way ANOVA.
PKD Activation and COX-2 Expression Require the RA2 Domain of PLCϵ
The data presented above provide evidence that the CDC25 domain of PLCϵ is important not only for sustained Rap1 activation but also for PKD activation and induction of COX-2 expression. PKD activation is mediated through second messengers generated by phosphoinositide hydrolysis, specifically DAG and its target protein kinase C. Accordingly, we hypothesized that continued PLCϵ-mediated phosphoinositide hydrolysis and DAG generation are facilitated by generation of activated Rap1 via the CDC25 domain. A mechanism through which this feedback could occur would be via Rap1 interaction with the RA2 domain. This domain was previously shown to regulate PLCϵ activity in response to heterologously expressed or recombinant Rap1 (5, 26). We utilized a RA2 mutant of PLCϵ (PLCϵ K2150E), shown to lack PLC activation by Ras family proteins (29), to test this hypothesis. In contrast to WT PLCϵ which supported thrombin stimulated PKD and COX-2 expression in PLCϵ KO cells (Fig. 3, A and B), the PLCϵ RA2 mutant was ineffective at supporting thrombin induced PKD activation and induction of COX-2 (Fig. 3, A and B).
FIGURE 3.

PKD activation and COX-2 expression require the RA2 domain of PLCϵ. A, PKD phosphorylation (p-PKDS916) was measured in PLCϵ KO astrocytes infected with WT PLCϵ or RA2 mutant (K2150E) adenovirus followed by thrombin (5 nm) treatment for 1 h. The p-PKDS916 protein levels were normalized to total PKD and expressed relative to their own averaged control. Representative Western blots and data quantitated as the mean ± S.E. (n = 6) of three independent experiments. B, COX-2 protein levels were measured in PLCϵ KO astrocytes infected with WT PLCϵ or RA2 mutant adenovirus followed by treatment with thrombin (5 nm) for 1 h and 6 h. COX-2 protein levels were normalized to GAPDH and expressed relative to its own control. Representative Western blots and data quantitated at the 6 h time point as the mean ± S.E. (n = 6) of three independent experiments. *, p < .05 and **, p < .01 between control and thrombin treatment; #, p < .05 and ##, p < .01 between thrombin treatments, one-way ANOVA.
The CDC25 and RA2 Domains Are Required for IL-6 mRNA Expression
To extend our findings on COX-2 regulation to another inflammatory gene, we assessed the induction of interleukin-6 (IL-6). Thrombin treatment increased IL-6 mRNA expression, and this response was also dependent on PLCϵ (Fig. 4A). To demonstrate the importance of the CDC25 and RA2 domains in thrombin stimulated IL-6 induction, we expressed WT PLCϵ, the CDC25Δ mutant, or the RA2 domain mutant in KO astrocytes. In contrast to what was observed in cells expressing WT PLCϵ, thrombin failed to induce IL-6 mRNA in cells expressing either the CDC25Δ or RA2 mutant PLCϵ (Fig. 4B).
FIGURE 4.

The CDC25 and RA2 domains are required for IL-6 mRNA expression. A, IL-6 mRNA levels in primary WT and PLCϵ KO astrocytes treated with thrombin (5 nm) or vehicle (control) for 1 h were assessed by q-PCR. Fold increase is expressed relative to the WT or KO averaged controls. Data shown are the mean ± S.E. of values (n = 6) from three independent experiments. B, IL-6 mRNA levels were measured by q-PCR in PLCϵ KO astrocytes infected with WT PLCϵ, CDC25Δ, or RA2 mutant (K2150E) adenovirus followed by thrombin (5 nm) treatment for 1 h. Fold increase is expressed relative to its own control. Data shown are the mean ± S.E. of values (n = 6) from three independent experiments. *, p < .05 and **, p < .01 between control and thrombin treatment; #, p < .05 between thrombin treatments, one-way ANOVA.
PLCϵ Is Localized to the Golgi
The observation that heterologously expressed PLCϵ localizes to an intracellular perinuclear compartment (26, 28, 33), confirmed by recent studies in cardiomyocytes (10), has important signaling implications. We hypothesized that this unique localization is critical for the feedback mechanism and sustained signaling proposed above. To examine PLCϵ localization in primary astrocytes, we expressed an adenoviral mCherry-PLCϵ construct in KO PLCϵ astrocytes. The mCherry fluorescence was most intense in an area surrounding the DAPI stained nucleus and was co-localized with the Golgi marker GM-130 (Fig. 5).
FIGURE 5.

PLCϵ is localized to the Golgi. KO astrocytes were infected with 150 moi of PLCϵ-mCherry adenovirus. The nucleus was stained with DAPI, and the Golgi was stained with GM-130.
PKD Is Activated at the Golgi in a PLCϵ-dependent Manner
To determine whether PKD is activated through PLCϵ signaling at the Golgi, we expressed a FRET reporter for PKD (Golgi-DKAR) that is targeted to this compartment (34, 35). Thrombin treatment significantly increased the FRET signal in astrocytes from WT mice (Fig. 6A). In contrast PKD activation was not evidenced by an increase in the FRET signal at the Golgi in cells lacking PLCϵ (Fig. 6B). Parallel studies using a plasma membrane targeted PKD activity reporter revealed minimal thrombin induced activation of PKD at the plasma membrane (Fig. 6C). Thus PKD activation in response to thrombin occurs through PLCϵ localized at the Golgi.
FIGURE 6.

PKD is activated at the Golgi in a PLCϵ-dependent manner. Following transfection of Golgi-DKAR (1.5 μg) in WT astrocytes (A) or in KO astrocytes (B), the FRET response (CFP/FRET) was measured over time after the addition of thrombin (5 nm). Data quantitated as the mean ± S.E. (n = 8) of four independent experiments. C, plasma membrane-DKAR (1.5 μg) was transfected into WT astrocytes. The FRET response (CFP/FRET) was measured over time after the addition of thrombin (5 nm). Data are quantitated as the mean ± S.E. (n = 8) of four independent experiments.
Intact Golgi Is Necessary for Rap1 Activation, PKD Activation, and COX-2 Expression
We demonstrated in studies above that PLCϵ activation is required for thrombin stimulated activation of Rap1 and PKD and for induction of COX-2. To demonstrate the importance of compartmentalization at the Golgi, we disrupted the Golgi with brefeldin A (BFA) (Fig. 7A). Astrocytes pretreated with BFA showed significantly reduced Rap1 activation by thrombin (Fig. 7B). PKD activation (Fig. 7C) and COX-2 expression (Fig. 7D) were also markedly disrupted in BFA-treated cells compared with vehicle treated cells.
FIGURE 7.

Intact Golgi is necessary for Rap1 activation, PKD activation, and COX-2 expression. A, BFA (5.0 μg/ml) was used to disrupt the Golgi. B, following pretreatment with BFA (5.0 μg/ml), WT astrocytes were treated with vehicle and thrombin (5 nm) for 1 h and 6 h. Rap1 activation was assessed by measuring Rap1-GTP after Rap1 pull-down and Western blot. Rap1-GTP was normalized to total Rap1 and expressed relative to its own averaged control. Representative Western blots are shown and data quantitated as mean ± S.E. (n = 8) of four independent experiments. C, following pretreatment with BFA (5.0 μg/ml), WT astrocytes were treated with thrombin (5 nm) for 1 h, and PKD phosphorylation (p-PKDS916) was assessed via Western blot. The p-PKDS916 protein levels were normalized to total PKD and expressed relative to its own averaged control. Representative Western blots are shown and data quantitated as mean ± S.E. (n = 8) of four independent experiments. D, COX-2 mRNA levels were measured after pretreatment with BFA (5.0 μg/ml) and subsequent treatment with thrombin (5 nm) for 1 h. Fold increase is expressed relative to the averaged ± inhibitor controls. *, p < .05 and **, p < .01 between control and thrombin treatment; #, p < .05 and ##, p < .01 between thrombin treatments, one-way ANOVA.
Discussion
PLCϵ has been shown to be a critical mediator in a range of disorders including cancer, CNS injury, cardiac hypertrophy, and cardiac ischemia/reperfusion injury (1–3, 10–23). Our hypothesis is that the ability of PLCϵ to integrate signals from GPCRs to downstream changes in gene expression contributes to the pathophysiology of these diseases. PLCϵ has a unique structure and is compartmentalized within the cell. Data described here provide evidence that these features are central to its role in sustained inflammatory signaling in astrocytes.
Compared with other PLC family members, PLCϵ is unique in containing a CDC25 domain that functions as a GEF for Rap1 (26, 27). Previous work using heterologous expression of PLCϵ or a PLCϵ mutant lacking the CDC25 domain, demonstrated that the CDC25 domain is required for the Rap1 activation observed for up to 30 min following EGF stimulation (26). We previously reported that PLCϵ is required for the Rap1 activation observed 15 min following thrombin addition (1). The data presented here are the first to show that PLCϵ signaling is required for GPCR ligands to elicit long term activation of Rap1, persisting for at least 6 h. We demonstrate that the unique CDC25 domain is needed for sustained agonist-induced Rap1 activation, and further show that it is necessary for subsequent activation of chronic signaling resulting in PKD activation and induction of COX-2.
Ras family members including Ras, Rap1, Rap2, and TC21 have been shown to directly interact with PLCϵ to regulate its activity (5, 29). The findings presented here establish that the RA2 domain of the enzyme is responsible for sustained signaling and suggest that this occurs through feedback by active Rap1 generated through the CDC25 domain. Specifically studies using a mutant PLCϵ, which is unable to bind Ras or Rap (29), demonstrate the requirement for the RA2 domain in long term activation of PKD and subsequent COX-2 expression, responses that require continued generation of DAG.
The CDC25 domain of PLCϵ has been shown to be important for its cellular localization. The Kataoka group demonstrated that Rap1 is required for PLCϵ localization to the Golgi and that this requires the CDC25 domain (26). Thus Rap1 activation occurs through the CDC25 domain and interaction of Rap1 with this domain contributes to PLCϵ localization. Not only does Rap1 retain PLCϵ at the Golgi but this organelle is also a rich source of PI4P that can serve as a substrate for PLCϵ and a source of DAG (10, 34, 36). Work shown here, as well as recent studies in cardiomyocytes (10), demonstrate that PLCϵ localized to the Golgi is important for PKD activation and subsequent induction of genes involved in inflammation and hypertrophy. Furthermore, localization of PLCϵ to the Golgi has a biological consequence. Disruption of the Golgi with BFA affects downstream PLCϵ signaling including Rap1 and PKD activation and subsequent induction of COX-2.
The protease activate receptor 1 (PAR1) has been implicated in CNS injury and disease and has been shown to induce astrocyte activation and proliferation in response to thrombin both in vitro and in vivo (37, 38). PAR1 is one of the most efficacious GPCRs in coupling to Gα12/13 and Rho (1, 39–44). We have previously demonstrated that thrombin stimulates phosphoinositide hydrolysis in astrocytes exclusively through PLCϵ and that thrombin signals through Rho and PLCϵ to mediate inflammatory gene expression in these cells (1, 2). Here we demonstrate that the molecular and cellular mechanism by which PLCϵ regulates astrocytic inflammatory gene expression requires its ability to function as a Rap1GEF, to generate activated Rap1, to further activate its RA2 domain, and to sustain PKD activation at the Golgi. As such, the domains that allow PLCϵ to function as a critical signaling node could represent possible therapeutic targets in diseases characterized by chronic inflammation.
Author Contributions
S. S. D. designed, performed, analyzed the experiments for the figures of the paper, and wrote the paper. M. T. K. provided technical assistance in particular for Fig. 6. A. V. S. provided technical assistance for Figs. 1, 2, 3, 4, and 5. J. H. B. conceived and coordinated the study and wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
This work was supported by National Institutes of Health Grants R01GM36927 (to J. H. B.), R01GM53536 (to A. V. S.), R03CA178524 (to M. T. K.). The authors declare that they have no conflicts of interest with the contents of this article.
- PLCϵ
- phospholipase C-epsilon
- BFA
- brefeldin A
- CNS
- central nervous system
- COX-2
- cyclooxygenase 2
- DAG
- diacylglycerol
- ET-1
- endothelin-1
- FRET
- fluorescence resonance energy transfer
- GEF
- guanine nucleotide exchange factor
- GPCR
- G-protein-coupled receptor
- IL-6
- interleukin-6
- IP3
- inositol 1,4,5-trisphosphate
- KO
- knockout
- LPA
- lysophosphatidic acid
- PAR1
- protease-activated receptor 1
- PKD
- protein kinase D
- q-PCR
- quantitative-PCR
- RA2
- Ras-associating domain 2.
References
- 1.Citro S., Malik S., Oestreich E. A., Radeff-Huang J., Kelley G. G., Smrcka A. V., and Brown J. H. (2007) Phospholipase Cϵ is a nexus for Rho and Rap-mediated G protein-coupled receptor-induced astrocyte proliferation. Proc. Natl. Acad. Sci. U.S.A. 104, 15543–15548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dusaban S. S., Purcell N. H., Rockenstein E., Masliah E., Cho M. K., Smrcka A. V., and Brown J. H. (2013) Phospholipase Cϵ links G protein-coupled receptor activation to inflammatory astrocytic responses. Proc. Natl. Acad. Sci. U.S.A. 110, 3609–3614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xiang S. Y., Ouyang K., Yung B. S., Miyamoto S., Smrcka A. V., Chen J., and Heller Brown J. (2013) PLCϵ, PKD1, and SSH1L transduce RhoA signaling to protect mitochondria from oxidative stress in the heart. Sci. Signal 6, ra108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hains M. D., Wing M. R., Maddileti S., Siderovski D. P., and Harden T. K. (2006) Galpha12/13- and rho-dependent activation of phospholipase Cϵ by lysophosphatidic acid and thrombin receptors. Mol. Pharmacol. 69, 2068–2075 [DOI] [PubMed] [Google Scholar]
- 5.Kelley G. G., Reks S. E., and Smrcka A. V. (2004) Hormonal regulation of phospholipase Cϵ through distinct and overlapping pathways involving G12 and Ras family G-proteins. Biochem. J. 378, 129–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seifert J. P., Wing M. R., Snyder J. T., Gershburg S., Sondek J., and Harden T. K. (2004) RhoA activates purified phospholipase Cϵ by a guanine nucleotide-dependent mechanism. J. Biol. Chem. 279, 47992–47997 [DOI] [PubMed] [Google Scholar]
- 7.Seifert J. P., Zhou Y., Hicks S. N., Sondek J., and Harden T. K. (2008) Dual activation of phospholipase Cϵ by Rho and Ras GTPases. J. Biol. Chem. 283, 29690–29698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wing M. R., Snyder J. T., Sondek J., and Harden T. K. (2003) Direct activation of phospholipase Cϵ by Rho. J. Biol. Chem. 278, 41253–41258 [DOI] [PubMed] [Google Scholar]
- 9.Kelley G. G., Kaproth-Joslin K. A., Reks S. E., Smrcka A. V., and Wojcikiewicz R. J. (2006) G-protein-coupled receptor agonists activate endogenous phospholipase Cϵ and phospholipase Cβ3 in a temporally distinct manner. J. Biol. Chem. 281, 2639–2648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang L., Malik S., Pang J., Wang H., Park K. M., Yule D. I., Blaxall B. C., and Smrcka A. V. (2013) Phospholipase Cϵ hydrolyzes perinuclear phosphatidylinositol 4-phosphate to regulate cardiac hypertrophy. Cell 153, 216–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bai Y., Edamatsu H., Maeda S., Saito H., Suzuki N., Satoh T., and Kataoka T. (2004) Crucial role of phospholipase Cϵ in chemical carcinogen-induced skin tumor development. Cancer Res. 64, 8808–8810 [DOI] [PubMed] [Google Scholar]
- 12.Harada Y., Edamatsu H., and Kataoka T. (2011) PLCϵ cooperates with the NF-κB pathway to augment TNFα-stimulated CCL2/MCP1 expression in human keratinocyte. Biochem. Biophys. Res. Commun. 414, 106–111 [DOI] [PubMed] [Google Scholar]
- 13.Ikuta S., Edamatsu H., Li M., Hu L., and Kataoka T. (2008) Crucial role of phospholipase Cϵ in skin inflammation induced by tumor-promoting phorbol ester. Cancer Res. 68, 64–72 [DOI] [PubMed] [Google Scholar]
- 14.Li M., Edamatsu H., Kitazawa R., Kitazawa S., and Kataoka T. (2009) Phospholipase Cϵ promotes intestinal tumorigenesis of Apc(Min/+) mice through augmentation of inflammation and angiogenesis. Carcinogenesis 30, 1424–1432 [DOI] [PubMed] [Google Scholar]
- 15.Martins M., McCarthy A., Baxendale R., Guichard S., Magno L., Kessaris N., El-Bahrawy M., Yu P., and Katan M. (2014) Tumor suppressor role of phospholipase Cϵ in Ras-triggered cancers. Proc. Natl. Acad. Sci. U.S.A. 111, 4239–4244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oestreich E. A., Malik S., Goonasekera S. A., Blaxall B. C., Kelley G. G., Dirksen R. T., and Smrcka A. V. (2009) Epac and phospholipase Cϵ regulate Ca2+ release in the heart by activation of protein kinase Cϵ and calcium-calmodulin kinase II. J. Biol. Chem. 284, 1514–1522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oestreich E. A., Wang H., Malik S., Kaproth-Joslin K. A., Blaxall B. C., Kelley G. G., Dirksen R. T., and Smrcka A. V. (2007) Epac-mediated activation of phospholipase C(ϵ) plays a critical role in β-adrenergic receptor-dependent enhancement of Ca2+ mobilization in cardiac myocytes. J. Biol. Chem. 282, 5488–5495 [DOI] [PubMed] [Google Scholar]
- 18.Oka M., Edamatsu H., Kunisada M., Hu L., Takenaka N., Dien S., Sakaguchi M., Kitazawa R., Norose K., Kataoka T., and Nishigori C. (2010) Enhancement of ultraviolet B-induced skin tumor development in phospholipase Cϵ-knockout mice is associated with decreased cell death. Carcinogenesis 31, 1897–1902 [DOI] [PubMed] [Google Scholar]
- 19.Wang H., Oestreich E. A., Maekawa N., Bullard T. A., Vikstrom K. L., Dirksen R. T., Kelley G. G., Blaxall B. C., and Smrcka A. V. (2005) Phospholipase Cϵ modulates β-adrenergic receptor-dependent cardiac contraction and inhibits cardiac hypertrophy. Circ. Res. 97, 1305–1313 [DOI] [PubMed] [Google Scholar]
- 20.Zhang L., Malik S., Kelley G. G., Kapiloff M. S., and Smrcka A. V. (2011) Phospholipase Cϵ scaffolds to muscle-specific A kinase anchoring protein (mAKAPβ) and integrates multiple hypertrophic stimuli in cardiac myocytes. J. Biol. Chem. 286, 23012–23021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dusaban S. S., and Brown J. H. (2014) PLCϵ mediated sustained signaling pathways. Adv. Biol. Regul. 57, 17–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smrcka A. V., Brown J. H., and Holz G. G. (2012) Role of phospholipase Cϵ in physiological phosphoinositide signaling networks. Cell Signal 24, 1333–1343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xiang S. Y., Dusaban S. S., and Brown J. H. (2013) Lysophospholipid receptor activation of RhoA and lipid signaling pathways. Biochim. Biophys. Acta 1831, 213–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shibatohge M., Kariya K., Liao Y., Hu C. D., Watari Y., Goshima M., Shima F., and Kataoka T. (1998) Identification of PLC210, a Caenorhabditis elegans phospholipase C, as a putative effector of Ras. J. Biol. Chem. 273, 6218–6222 [DOI] [PubMed] [Google Scholar]
- 25.Bunney T. D., and Katan M. (2006) Phospholipase Cϵ: linking second messengers and small GTPases. Trends Cell Biol. 16, 640–648 [DOI] [PubMed] [Google Scholar]
- 26.Jin T. G., Satoh T., Liao Y., Song C., Gao X., Kariya K., Hu C. D., and Kataoka T. (2001) Role of the CDC25 homology domain of phospholipase Cϵ in amplification of Rap1-dependent signaling. J. Biol. Chem. 276, 30301–30307 [DOI] [PubMed] [Google Scholar]
- 27.Song C., Satoh T., Edamatsu H., Wu D., Tadano M., Gao X., and Kataoka T. (2002) Differential roles of Ras and Rap1 in growth factor-dependent activation of phospholipase Cϵ. Oncogene 21, 8105–8113 [DOI] [PubMed] [Google Scholar]
- 28.Satoh T., Edamatsu H., and Kataoka T. (2006) Phospholipase Cϵ guanine nucleotide exchange factor activity and activation of Rap1. Methods Enzymol. 407, 281–290 [DOI] [PubMed] [Google Scholar]
- 29.Kelley G. G., Reks S. E., Ondrako J. M., and Smrcka A. V. (2001) Phospholipase C(ϵ): a novel Ras effector. EMBO J. 20, 743–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Violin J. D., Zhang J., Tsien R. Y., and Newton A. C. (2003) A genetically encoded fluorescent reporter reveals oscillatory phosphorylation by protein kinase C. J. Cell Biol. 161, 899–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schmittgen T. D., and Livak K. J. (2008) Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc 3, 1101–1108 [DOI] [PubMed] [Google Scholar]
- 32.Del Re D. P., Miyamoto S., and Brown J. H. (2007) RhoA/Rho kinase up-regulate Bax to activate a mitochondrial death pathway and induce cardiomyocyte apoptosis. J. Biol. Chem. 282, 8069–8078 [DOI] [PubMed] [Google Scholar]
- 33.Song C., Hu C. D., Masago M., Kariyai K., Yamawaki-Kataoka Y., Shibatohge M., Wu D., Satoh T., and Kataoka T. (2001) Regulation of a novel human phospholipase C, PLCϵ, through membrane targeting by Ras. J. Biol. Chem. 276, 2752–2757 [DOI] [PubMed] [Google Scholar]
- 34.Kunkel M. T., and Newton A. C. (2010) Calcium transduces plasma membrane receptor signals to produce diacylglycerol at Golgi membranes. J. Biol. Chem. 285, 22748–22752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kunkel M. T., Toker A., Tsien R. Y., and Newton A. C. (2007) Calcium-dependent regulation of protein kinase D revealed by a genetically encoded kinase activity reporter. J. Biol. Chem. 282, 6733–6742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gallegos L. L., Kunkel M. T., and Newton A. C. (2006) Targeting protein kinase C activity reporter to discrete intracellular regions reveals spatiotemporal differences in agonist-dependent signaling. J. Biol. Chem. 281, 30947–30956 [DOI] [PubMed] [Google Scholar]
- 37.Nicole O., Goldshmidt A., Hamill C. E., Sorensen S. D., Sastre A., Lyuboslavsky P., Hepler J. R., McKeon R. J., and Traynelis S. F. (2005) Activation of protease-activated receptor-1 triggers astrogliosis after brain injury. J. Neurosci. 25, 4319–4329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sorensen S. D., Nicole O., Peavy R. D., Montoya L. M., Lee C. J., Murphy T. J., Traynelis S. F., and Hepler J. R. (2003) Common signaling pathways link activation of murine PAR-1, LPA, and S1P receptors to proliferation of astrocytes. Mol. Pharmacol. 64, 1199–1209 [DOI] [PubMed] [Google Scholar]
- 39.Coughlin S. R. (2000) Thrombin signalling and protease-activated receptors. Nature 407, 258–264 [DOI] [PubMed] [Google Scholar]
- 40.Martin C. B., Mahon G. M., Klinger M. B., Kay R. J., Symons M., Der C. J., and Whitehead I. P. (2001) The thrombin receptor, PAR-1, causes transformation by activation of Rho-mediated signaling pathways. Oncogene 20, 1953–1963 [DOI] [PubMed] [Google Scholar]
- 41.Aragay A. M., Collins L. R., Post G. R., Watson A. J., Feramisco J. R., Brown J. H., and Simon M. I. (1995) G12 requirement for thrombin-stimulated gene expression and DNA synthesis in 1321N1 astrocytoma cells. J. Biol. Chem. 270, 20073–20077 [DOI] [PubMed] [Google Scholar]
- 42.Sah V. P., Seasholtz T. M., Sagi S. A., and Brown J. H. (2000) The role of Rho in G protein-coupled receptor signal transduction. Annu. Rev. Pharmacol. Toxicol. 40, 459–489 [DOI] [PubMed] [Google Scholar]
- 43.Siehler S. (2009) Regulation of RhoGEF proteins by G12/13-coupled receptors. Br. J. Pharmacol. 158, 41–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sternweis P. C., Carter A. M., Chen Z., Danesh S. M., Hsiung Y. F., and Singer W. D. (2007) Regulation of Rho guanine nucleotide exchange factors by G proteins. Adv. Protein Chem. 74, 189–228 [DOI] [PubMed] [Google Scholar]