

Background: Niemann-Pick C2 (NPC2) protein is essential for intracellular cholesterol trafficking.
Results: Several regions on the surface of NPC2 are integral to its cholesterol transfer properties, which include the promotion of membrane-membrane interactions.
Conclusion: Rapid cholesterol transfer occurs via NPC2-mediated membrane interactions.
Significance: NPC2 may promote rapid efflux of late endosomal/lysosomal cholesterol by functioning at intra-lysosomal membrane contact sites.
Keywords: cholesterol, lipid transport, lipid-protein interaction, lysosomal storage disease, protein conformation, NPC2, Niemann-Pick C2, cholesterol-binding protein, membrane contact sites
Abstract
The cholesterol storage disorder Niemann-Pick type C (NPC) disease is caused by defects in either of two late endosomal/lysosomal proteins, NPC1 and NPC2. NPC2 is a 16-kDa soluble protein that binds cholesterol in a 1:1 stoichiometry and can transfer cholesterol between membranes by a mechanism that involves protein-membrane interactions. To examine the structural basis of NPC2 function in cholesterol trafficking, a series of point mutations were generated across the surface of the protein. Several NPC2 mutants exhibited deficient sterol transport properties in a set of fluorescence-based assays. Notably, these mutants were also unable to promote egress of accumulated intracellular cholesterol from npc2−/− fibroblasts. The mutations mapped to several regions on the protein surface, suggesting that NPC2 can bind to more than one membrane simultaneously. Indeed, we have previously demonstrated that WT NPC2 promotes vesicle-vesicle interactions. These interactions were abrogated, however, by mutations causing defective sterol transfer properties. Molecular modeling shows that NPC2 is highly plastic, with several intense positively charged regions across the surface that could interact favorably with negatively charged membrane phospholipids. The point mutations generated in this study caused changes in NPC2 surface charge distribution with minimal conformational changes. The plasticity, coupled with membrane flexibility, probably allows for multiple cholesterol transfer routes. Thus, we hypothesize that, in part, NPC2 rapidly traffics cholesterol between closely appositioned membranes within the multilamellar interior of late endosomal/lysosomal proteins, ultimately effecting cholesterol egress from this compartment.
Introduction
Niemann-Pick type C (NPC)2 disease is a lysosomal lipid storage disorder specifically characterized by the accumulation of unesterified cholesterol and glycolipids in the late endosome/lysosome (LE/LY) compartment. Over time, permanent damage to cells and tissues occurs as a result of this excessive lipid storage, particularly in the brain, peripheral nervous system, liver, spleen, and bone marrow (1, 2). The accumulation of cholesterol in this rare, autosomal recessive disorder is due to mutations in either of two lysosomal proteins, NPC1 or NPC2, which result in defective cholesterol trafficking. The mechanisms by which these proteins lead to cellular cholesterol egress are only beginning to be understood at the molecular level.
NPC1 is a polytopic transmembrane protein residing in the limiting lysosomal membrane. It contains a transmembrane sterol-sensing domain, binds cholesterol in its soluble N-terminal domain (3–5), and has recently been shown to bind oxysterol derivatives in a different, as yet unidentified, ligand binding site (6). NPC2, in contrast, is a small, 132-residue soluble intralysosomal protein. It has also been shown to bind cholesterol, with 1:1 stoichiometry (7, 8), and we have shown in vitro that it rapidly catalyzes cholesterol transfer between membranes via direct protein-membrane interactions (9, 10). Notably, sterol transport rates by NPC2 are dramatically enhanced by the unique LE/LY phospholipid lysobisphosphatidic acid (10), which is enriched in intralysosomal membranes (11, 12).
NPC2-deficient cells and mouse models accumulate free cholesterol in late edosomes/lysosomes, highlighting the essential role of NPC2 in normal cholesterol egress from this compartment. Although the structural basis for the cholesterol transport function of the protein remains largely unknown, one particular report has been informative in this regard. Prior to the availability of the NPC2 tertiary structure, Ko et al. (13) used site-directed mutagenesis of evolutionarily conserved residues and made the important observation that NPC2 point mutants unable to bind cholesterol could not reverse cholesterol accumulation in npc2−/− fibroblasts, thus demonstrating the functional requirement for cholesterol binding by NPC2 for the first time. Interestingly, they also described three point mutations in NPC2 that, despite normal cholesterol binding affinity, were nevertheless ineffective in npc2−/− cell cholesterol clearance (13). We hypothesized that these three residues were probably important in cholesterol transport, and the tertiary structure of NPC2 later revealed that all three were on the protein surface (7). More recently, it has been suggested that NPC2 directly transfers cholesterol to the luminal N-terminal domain (NTD) of NPC1 (3, 5, 14). Thus, the final step in cholesterol egress from the LE/LY compartment may involve sterol transfer between the two proteins, although direct interactions between NPC2 and the NPC1 NTD have not as yet been demonstrated (5).
In the present studies, we show that the three mutants identified by Ko et al. as being able to bind cholesterol normally but unable to “rescue” the cholesterol accumulation phenotype of NPC2-deficient cells (13) are markedly ineffective in transferring cholesterol between membranes. Several other point mutations on the surface of NPC2 also result in deficient cholesterol transfer properties and, interestingly, appear to form multiple regions on the surface of the protein. This observation suggests the potential for NPC2 to promote membrane-membrane interactions and to perhaps form bridges between membranes, as occurs at so-called membrane contact sites (MCSs) (15–17). Our laboratory and others have previously shown that WT NPC2 promotes interactions between membranes (18, 19). Computational modeling described here shows significant plasticity in NPC2, which may allow for multiple mechanisms of sterol transfer. It is thus possible that NPC2 stimulates rapid transfer of cholesterol at zones of close apposition between membranes, as exist in the interior of multivesicular endo/lysosomes (20), ultimately leading to cholesterol egress from the LE/LY compartment.
Experimental Procedures
Materials
Cholesterol was obtained from Sigma. Egg phosphatidylcholine (EPC), dehydroergosterol (DHE), dansyl phosphatidylethanolamine (dansyl-PE), nitrobenzoxadiozole (NBD)-phosphatidylcholine and lissamine rhodamine-PE were from Avanti (Alabaster, AL). Filipin III was obtained from Fisher. Human fibroblast cells from an apparently healthy donor (GM03652) and from an NPC2 patient (GM18455) were from Coriell Institute of Medical Research (Camden, NJ).
Generation and Purification of NPC2 Mutants
Point mutations were generated with the Stratagene QuikChange site-directed mutagenesis kit, using a Myc His6-tagged murine NPC2 plasmid (13). Wild type and mutant Myc His6-tagged NPC2 proteins were purified from transfected CHO KI cells as described previously (10, 13), using a 10 kDa cut-off flow filtration membrane to initially concentrate conditioned media (Millipore, Bedford, MA).
Cholesterol Binding by NPC2
NPC2 has two tryptophan residues at positions 109 and 122, and binding of cholesterol by the protein results in quenching of the endogenous tryptophan fluorescence. Thus, cholesterol binding by WT and mutant NPC2s was determined by incubating the proteins with increasing concentrations of cholesterol and monitoring the quenching. A 50 mm stock solution of cholesterol was prepared in DMSO. Proteins in citrate buffer (20 mm sodium citrate, 150 mm NaCl, pH 5.0) were incubated with increasing concentrations of cholesterol for 20 min at 25 °C. The final concentration of DMSO was ≤1% (v/v). Equilibrium binding constants were determined by a hyperbolic fit of the data using Sigma Plot software (San Jose, CA).
Membrane Vesicle Preparation
Small unilamellar vesicles were prepared by sonication and ultracentrifugation as described previously (9, 10, 21). Vesicles were maintained at temperatures above the phase transition temperatures of all constituent lipids. Standard vesicles were composed of 100 mol % EPC. 25 mol % DHE and 3 mol % dansyl-PE were substituted for EPC in the donor and acceptor vesicles, respectively, used in the intermembrane sterol transfer assays. All vesicles were prepared in citrate buffer. For the preparation of large unilamellar vesicles (LUVs), EPC dissolved in chloroform was dried under nitrogen for 1 h to create a lipid film and resuspended in citrate buffer, forming multilamellar structures. The lipid suspension was placed alternately on dry ice and in a water bath above 37 °C for seven freeze-thaw cycles, followed by extrusion through a 100-nm pore membrane (Avestin, Ottawa, Canada) using a miniextruder (Avestin) for 11 passes. The final phospholipid concentration of all vesicle preparations was determined by quantification of inorganic phosphate (21).
Cholesterol Transfer from NPC2 to Membranes
The endogenous tryptophan fluorescence of NPC2 was used to monitor cholesterol transfer from NPC2 to membranes, as detailed previously, using a stopped-flow mixing chamber interfaced with a DX18-MV or SX20 spectrofluorometer (Applied Photophysics Ltd., Leatherhead, UK) (9, 10). Conditions to ensure the absence of photobleaching were established before each experiment. Data were analyzed with the software provided with the Applied Photophysics Instrument, and the cholesterol transfer rates were obtained by exponential fitting of the curves. All curves were fit well by a single exponential function.
Intermembrane Transfer of Sterol
As described by Xu et al. (10), the fluorescent cholesterol analog DHE was used in donor membranes, and its fluorescence resonance energy transfer (FRET) partner, dansyl-PE, was used in acceptor membranes. Intermembrane transfer of DHE was measured by DHE quenching or the sensitized emission of dansyl fluorescence and was examined in the absence of or upon the addition of apo-WT or mutant NPC2 protein (10). Conditions to ensure the absence of photobleaching were established before each experiment, and data were analyzed as described above.
Clearance of Cholesterol from npc2−/− Fibroblasts by WT and Mutant NPC2s
Wild type and npc2−/− fibroblasts were plated onto 8-well tissue culture slides (Nalgene) at a density of ∼6,000 cells/well. A single dose of purified WT or mutant NPC2 protein, at a final concentration of 0.4 nm, was added to the culture medium 24–36 h after cells were plated. Protein uptake, probably via bulk phase endocytosis, delivers the added NPC2 to the endosomal vesicle recycling system, and it has been shown that the addition of WT NPC2 protein can “rescue” the cholesterol accumulation phenotype in an NPC2 disease model (5, 13). Three days following administration of protein, cells were fixed and stained with 0.05 mg/ml filipin. Stained cells were imaged on a Nikon Eclipse E800 epifluorescence microscope using a ×60 oil objective. The filipin stain area was quantified with the accompanying NIS-Elements software (Nikon Inc.), as described previously (10, 18).
Membrane Aggregation and Fusion
LUVs were mixed with WT or mutant NPC2 proteins or bovine serum albumin (BSA), and absorbance at 350 nm was used to monitor membrane-membrane interaction (aggregation), as described by Schulz et al. (22). Membrane fusion was assessed in two ways: mixing of vesicle contents and mixing of membrane lipids. Monitoring fusion by mixing of contents was done as described previously (23) using 100 mol % EPC LUVs filled with >100 mm 5-carboxyfluorescein (5-CF), at which point the 5-CF is self-quenched. Release of self-quenching (i.e. increase in fluorescence) was monitored upon mixing of 50 μm 5-CF-filled EPC LUVs with 50 μm citrate buffer-filled EPC LUVs in the presence of 1 μm WT NPC2, using λex 493/λem 520 nm. As a positive control, 5-CF-filled EPC LUVs were mixed with 1% Triton X-100 lysis buffer, and absolute dequenching of the fluorophore was monitored. Membrane fusion was also determined using resonance energy transfer between NBD and rhodamine, as developed by Struck et al. (24). Briefly, 50 μm EPC small unilamellar vesicles containing 1 mol % NBD-phosphatidylcholine were mixed with 50 μm EPC small unilamellar vesicles containing 1 mol % lissamine rhodamine-PE, in the absence or presence of 1 μm WT NPC2. NBD is excited at 460 nm, and the decrease in NBD fluorescence (λ534) or the sensitized emission of rhodamine (λ571) upon mixing of the two vesicle populations, indicative of membrane fusion, was monitored over time. All experiments were conducted in citrate buffer at 25 °C. For each membrane fusion assay, conditions were established to verify the absence of photobleaching.
Computational Modeling of NPC2
The starting point for modeling was the crystallographic structure of bovine NPC2 (Protein Data Bank entry 1NEP) (7, 25). The bovine and murine proteins differ at several residue positions. Of these, only residues 29 and 32 were mutated in this study, both to alanine. To create a modeling “wild type,” we changed bovine lysine 29 to murine glutamine and bovine arginine 32 to murine lysine prior to minimizing the conformational energy. This procedure was used to maintain as many of the crystallographic coordinates as possible. The modeling is thus kept as close to known experimental data as possible. The resulting structure served as the starting point for modeling the mutant proteins.
All proteins were minimized to convergence using Insight II (Accelrys, San Diego, CA). In all cases, the structure was surrounded by a 5-Å shell of water molecules to simulate solution conditions. The models were all examined with Procheck (26, 27) and What_Check (28) to detect structural abnormalities. With minor exceptions, the results were within normal limits for crystallographic structures (26–28). The electrostatic surfaces of the wild type and all mutants were calculated with the APBS (adaptive Poisson-Boltzmann solver) plugin for PyMOL (29, 30). Solvent-accessible surface areas were calculated with a 1.4-Å probe using the programs ACCESS (31) and BINS (32). 100% accessibility was estimated from the areas of amino acids X in tripeptides of the form Gly-X-Gly in the extended conformation. Molecular images were created with PyMOL (PyMOL Molecular Graphics System, version 1.7.2.1 Schrödinger, LLC).
Results
Several Mutations on the Surface of NPC2 Prevent Efficient Sterol Transfer
Although the NPC2 protein binds cholesterol with high affinity, binding alone is not sufficient for egress of cholesterol from the LE/LY compartment (13). Therefore, to test the idea that surface residues on NPC2 might facilitate cholesterol transfer between membranes, we constructed point mutations of well conserved surface residues. All NPC2 point mutants generated in this study bound cholesterol similarly to WT protein (Table 1) with dissociation constants in the submicromolar range. Having verified that the point mutations had little or no effect on the cholesterol binding properties of the protein, the ability of each mutant to transport sterol to membranes or between membranes was assessed using the above described kinetic transfer assays. As shown in Table 1, the K97A, E99A, and K6A mutations had little effect on NPC2 sterol transfer rates in the two independent assays. The K16A and E70A mutations reduced sterol transfer rates by nearly half of WT values in both the protein to membrane and the intermembrane sterol transfer assays, indicating that they may play some role in cholesterol trafficking by NPC2. The most substantial attenuations in sterol transfer by NPC2, however, were observed with the remaining mutations shown in Table 1, which showed at least a 70% relative reduction in transfer rates. I62D, V64A, and D113A had the most dramatic effect of all of the mutations, with essentially undetectable cholesterol transfer.
TABLE 1.
Cholesterol binding affinity and relative in vitro sterol transfer rates of WT and mutant NPC2 proteins
The dissociation constants (Kd) for cholesterol were determined by tryptophan quenching using 5 μm protein as described under “Experimental Procedures.” S.E. values of 10–15% were found for all proteins; n ≥ 3 separate determinations. Cholesterol transfer from 2.5 μm NPC2 proteins to 250 μm membranes or DHE transfer from 50 μm donor to 250 μm acceptor membranes in the presence of NPC2 was determined as described under “Experimental Procedures.” Kinetic data are presented relative to WT NPC2 transfer rates ± S.E., and the mutants are listed in approximately decreasing order of effects on transfer rates and membrane interaction. Results are an average of n ≥ 3 separate determinations. The absolute rate of cholesterol transfer from WT NPC2 to membranes was 0.0367 ± 0.0031 s−1, whereas the intermembrane transfer rate of DHE in the presence of WT NPC2 was 0.0094 ± 0.0006 s−1. ND, not determined.
| Kd, cholesterol | NPC2 to membranes | Intermembrane | |
|---|---|---|---|
| μm | |||
| WT NPC2 | 0.37 | 1.00 | 1.00 |
| K97A | 0.27 | 0.93 ± 0.038 | 0.70 ± 0.050 |
| E99A | 0.48 | 0.90 ± 0.048 | 0.75 ± 0.040 |
| K6A | 0.30 | 1.1 ± 0.027 | 0.56 ± 0.029 |
| K16A | 0.36 | 0.54 ± 0.012 | 0.40 ± 0.021 |
| E70A | 0.30 | 0.35 ± 0.019 | 0.43 ± 0.022 |
| E108A | 0.29 | 0.14 ± 0.003 | ND |
| H31A | 0.41 | 0.13 ± 0.007 | ND |
| Q29A | 0.13 | 0.10 ± 0.006 | ND |
| K32A | 0.19 | <0.01 | 0.32 ± 0.017 |
| N39A | 0.63 | <0.01 | 0.26 ± 0.014 |
| K115A | 0.33 | <0.01 | 0.22 ± 0.008 |
| D85A | 0.60 | <0.01 | 0.21 ± 0.012 |
| K75A | 0.27 | <0.01 | 0.21 ± 0.011 |
| D72A | 0.89 | <0.01 | 0.17 ± 0.009 |
| D113A | 0.31 | <0.01 | ND |
| I62D | 0.32 | <0.01 | ND |
| V64A | 0.31 | <0.01 | ND |
Sterol Transfer Kinetics Are Reflective of Cellular Events
The kinetic data obtained from the model membrane sterol transfer studies suggest that several point mutations on the surface of NPC2 result in very defective cholesterol trafficking properties by the protein. To determine whether the kinetic sterol transfer results reflect NPC2 function at the cellular level, npc2−/− fibroblasts were incubated with purified WT or mutant NPC2 proteins, and the effects on cellular cholesterol accumulation were observed using filipin staining. As shown in Fig. 1, A and B, and in agreement with prior literature (5, 13), cholesterol accumulation in the npc2−/− fibroblasts was essentially reversed following incubation with WT NPC2; the protein promoted efflux of greater than 90% of the cholesterol as compared with untreated controls. E99A and K97A, two mutants that had little effect on sterol transfer rates in the kinetic studies, were also found to stimulate cholesterol efflux from the npc2−/− fibroblasts; incubation with the mutants reduced filipin stain in the fibroblasts to nearly 15% of untreated controls. In contrast, E108A, H31A, Q29A, N39A, K115A, D85A, K75A, D72A, D113A, I62D, and V64A were all found to be ineffective at reversing the cellular cholesterol accumulation phenotype (Fig. 1B). Thus, the inability of these 11 mutants to reverse the cholesterol accumulation in npc2−/− fibroblasts is consistent with results from the sterol transfer assays, where the mutants exhibited considerably attenuated rates of cholesterol transfer (Table 2). Similarly, mutants exhibiting a moderate decline in relative sterol transfer rates (E70A and K16A) reduced filipin staining by ∼60–70% (Fig. 1B). Overall, the ability of an NPC2 form to clear cholesterol in human npc2−/− cells appears to be directly related to its rate of sterol transfer to and between model membranes (Fig. 1C).
FIGURE 1.
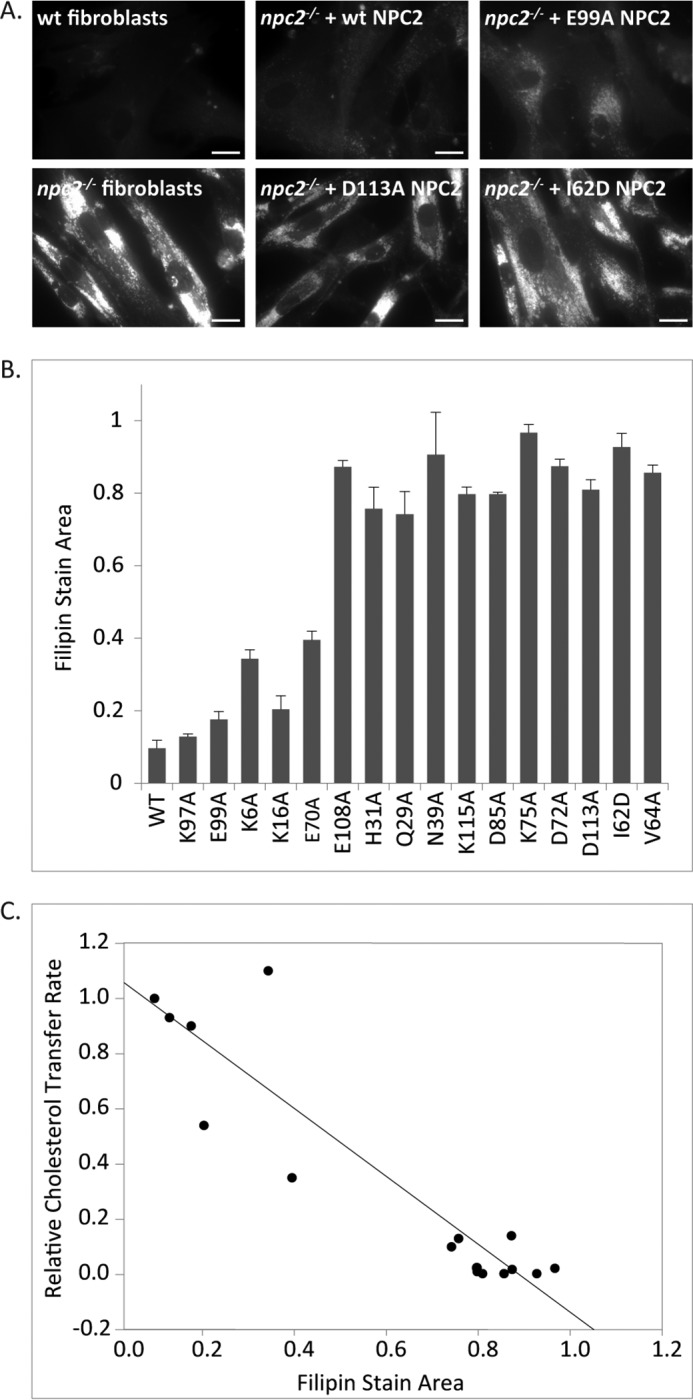
Effect of WT and mutant NPC2s on filipin accumulation in npc2−/− fibroblasts. Human NPC2-deficient fibroblasts were incubated with 0.4 nm purified WT or mutant NPC2 protein and were fixed and stained with filipin as described under “Experimental Procedures.” A, representative images of npc2−/− fibroblasts incubated with WT or mutant NPC2s. B, filipin accumulation was determined as the ratio of filipin stain area to total cell area. Results are expressed relative to control untreated cells and are represented as mean ± S.E. (error bars); n = 3 separate experiments. C, linear regression between cholesterol transport rates for WT or mutant proteins and filipin accumulation in npc2−/−-treated cells; r2 = 0.845.
Multiple Charged Regions on the Surface of NPC2 Promote Intermembrane Interactions
The wild type protein with each of the 17 mutated residues displayed as sticks (Fig. 2) shows that, as planned, all of the mutations are on the protein surface. For the purposes of discussion, the mutants fall into three groups: near the bottom (I62D, V64A, Q29A, K32A, H31A, E99A, and K97A), around the equator (K6A, E108A, Q29A, N39A, and E70A), and near the top (K16A, D113A, D85A, K115A, D72A, and K75A). Fig. 3 shows the structural forms as well as their electrostatic surfaces calculated as described above. Fig. 3A depicts the front of NPC2, similar to the orientation in Fig. 2. The electrostatic image of the back of the protein, shown in Fig. 3B, reveals a diagonal band of intense positive charge (dark blue). This region on the back of NPC2 extends, with some interruptions, from the upper left to the lower right and could interact favorably with negatively charged membrane phospholipid head groups. In contrast, there is a large, intensely negatively charged area (dark red) at the bottom of the protein (Fig. 3, B and C) and two negatively charged regions on the front (Fig. 3A), which are unlikely to interact with negatively charged membrane phospholipids. The front of the structure (Fig. 3A) additionally displays a mixture of relatively weaker positive and negative charged areas (light blue and light red, respectively), as does the top of the protein (Fig. 3D). Also on the front of the molecule is an area that is essentially white, indicating it is non-polar (Fig. 3A, within the oval). This region, where the rim of the entrance to the cholesterol binding cavity lies (8), is shown in magenta in the structural models, and the neutrality is consistent with its being the interaction point for the hydrophobic ligand.
FIGURE 2.
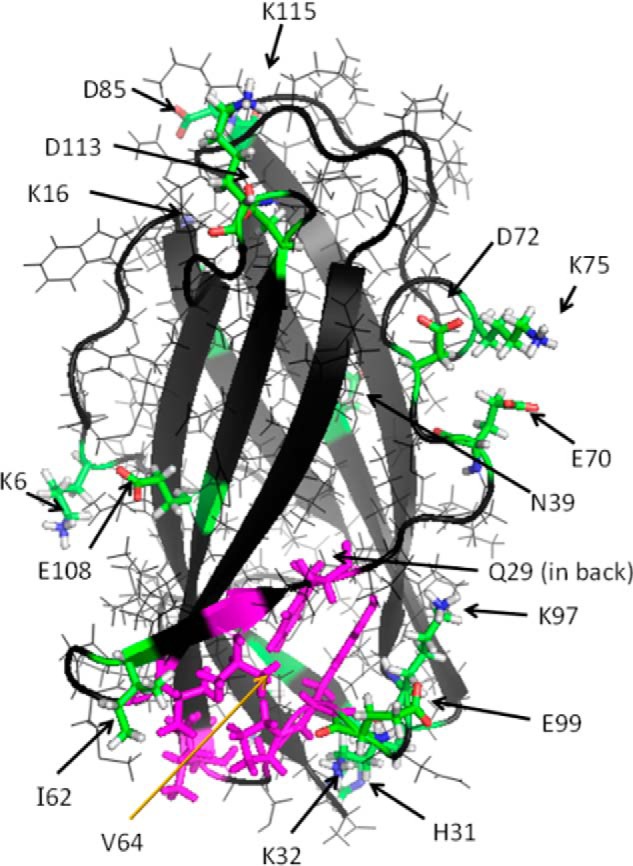
Wild type NPC2 with bovine Lys-29 and Arg-32 replaced by mouse Asn-29 and Lys-32, respectively. The side chains of all mutants made in this study are shown as sticks with functional coloring (green, carbon; white, hydrogen; blue, nitrogen; red, oxygen) except for Val-64. Val-64 along with Tyr-100, Pro-101, Ile-103, Val-59, and Phe-66 form the outer rim of the cholesterol binding pocket as described in Fig. 6 of Xu et al. (8). The rim, with the full residues shown as sticks, is shown in magenta. This orientation shows the “front” of the protein.
FIGURE 3.

Wild type NPC2 rotated to show electrostatic features of the molecular surface. The “front” (A) and “back” (B) of NPC2 are shown, with an oval enclosing the residues constituting the rim of the entry to the cholesterol binding cavity as given in Fig. 6 of Xu et al. (8). The molecule has also been rotated to display the “bottom” (C) and “top” (D) surfaces. The rim of the binding site for cholesterol is shown in magenta sticks in the left images of each panel.
The extensive positive zones on the NPC2 surface are potential interaction sites with negatively charged membrane surfaces. Considering that NPC2 directly interacts with membranes as a mechanism for transferring cholesterol (9, 10), the presence of several possible opposing cholesterol transport regions on the surface of NPC2 suggests that the protein may be able to interact with more than one membrane simultaneously. To test this hypothesis, a light scattering assay that monitors membrane-membrane interactions was conducted.
As shown in Fig. 4 and in confirmation of our previous findings (18), when EPC LUVs are mixed with WT NPC2, an increase in A350 is observed over time. These results indicate the occurrence of membrane-membrane interactions. Two mutants found to have cholesterol transport properties similar to WT, K6A and K97A, also cause an increase in light scattering upon their addition to EPC LUVs. In contrast, no change in A350 is observed when BSA, a protein that binds cholesterol but does not cause membrane-membrane interactions (18, 22), is mixed with vesicles. Similarly, mutants with deficient cholesterol transfer properties (D85A, K75A, V64A, I62D, and Q29A) do not cause changes in A350, indicating that they are unable to promote membrane-membrane interactions. Thus, there are distinct regions on the surface of NPC2 that facilitate the interactions of membranes.
FIGURE 4.
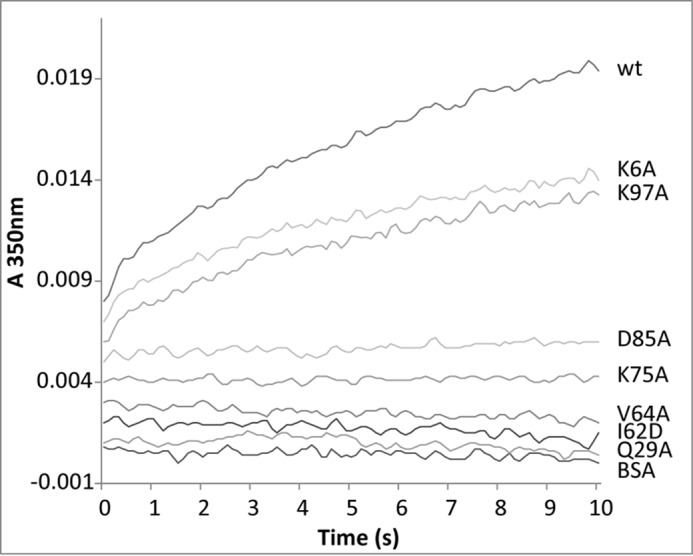
Promotion of membrane-membrane interactions by NPC2. Time-dependent changes in the absorbance of 50 μm LUVs mixed with 1 μm bovine serum albumin (BSA), WT, or mutant NPC2 proteins were observed at 350 nm using an SX20 stopped-flow spectrofluorometer. Results are representative of three experiments.
NPC2 Promotes Membrane Aggregation, Not Fusion
Although the above turbidity assay allows for identification of membrane-membrane interactions facilitated by NPC2, it cannot distinguish between vesicle aggregation and fusion. Thus, in order to determine whether NPC2 promotes membrane fusion, an assay that monitors the mixing of vesicle contents was performed. Mixing of 5-CF-filled LUVs with buffer-filled EPC LUVs, in the presence of WT NPC2, resulted in a small increase in the relative fluorescence of 5-CF (Fig. 5A), indicating a possible membrane fusion event. The relative fluorescence of 5-CF continued to increase up to 15 min after being mixed; however, the degree of 5-CF dequenching was quite low compared with that caused by a cell lysis buffer (Fig. 5A). Conversely, upon mixing of the two vesicle populations in the presence of NPC2 using a stopped flow spectrofluorometer, a moderate decrease in 5-CF emission was observed, probably due to slight photobleaching of the CF signal (Fig. 5A, inset). Considering these conflicting results, in addition to the possibility that the relatively slow increase in fluorescence may reflect CF leakage due to membrane interactions by NPC2, a second membrane fusion assay involving mixing of vesicles containing NBD- and rhodamine-labeled phospholipids was undertaken. Upon mixing of the two vesicle populations in the presence of WT NPC2, no appreciable changes in the emission of either fluorophore were observed over a period of 12 min (Fig. 5B), indicating the absence of membrane fusion. Additionally, no changes in the relative fluorescence of NBD were observed over a period of 100 s following stopped-flow mixing (Fig. 5B, inset), again suggesting that membrane fusion is not occurring. Taken together, these results indicate that NPC2 interacts with membranes but does not promote membrane fusion.
FIGURE 5.
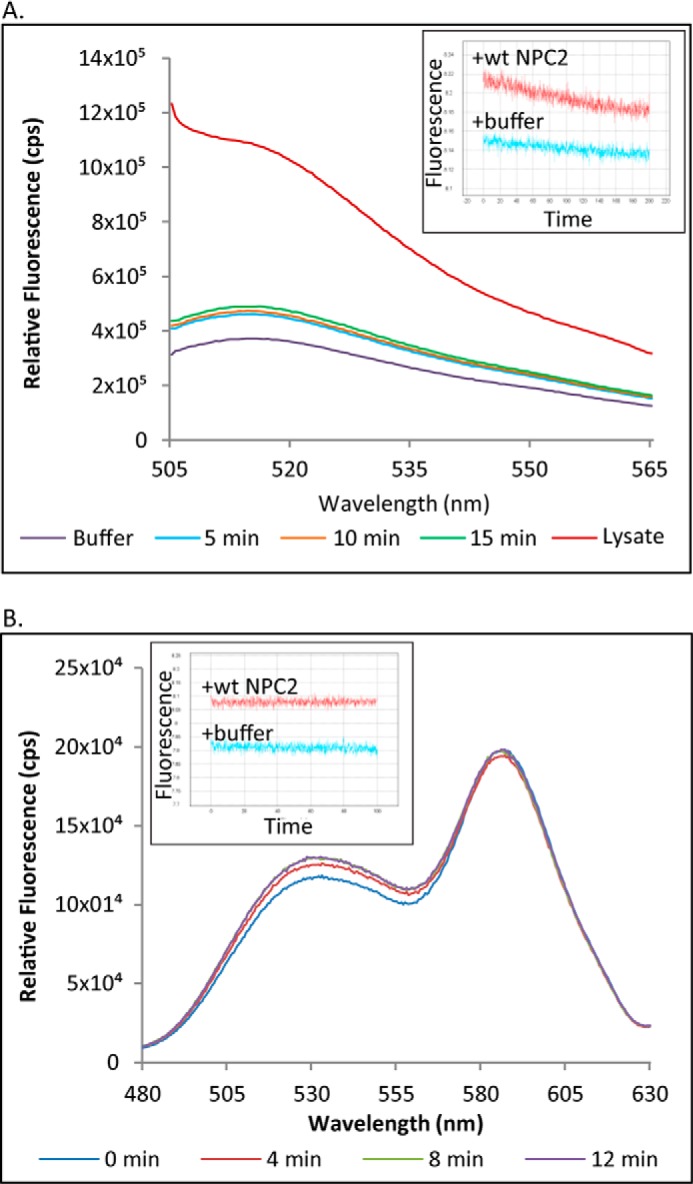
NPC2 does not promote membrane fusion. A, the ability of NPC2 to cause membrane fusion was tested by mixing 5-CF-filled vesicles with buffer-filled vesicles in the presence of WT NPC2 or cell lysis buffer. 5-CF fluorescence was also monitored over time on an SLM fluorometer or stopped flow spectrofluorometer using λex 493/λem 520 nm (inset). B, the ability of WT NPC2 to cause membrane fusion was also investigated by mixing NBD-PE vesicles with lissamine rhodamine-PE vesicles in the presence of WT NPC2. Changes in the fluorescence of either FRET partner using λex 460 were monitored over time on an SLM fluorometer. Change in NBD fluorescence (λ534) was also monitored using an SX20 stopped flow spectrofluorometer (inset).
Computational Modeling of NPC2 Mutant Proteins Reveals a Highly Plastic Structure
In an effort to pinpoint conformational differences between wild type NPC2 and the mutant proteins, the α-carbon coordinates of each mutant were superimposed onto those of wild type, and the displacements of the 130 carbons from wild type were calculated. The r.m.s. displacements for all 130 α-carbons range from 0.51 Å (I62D mutant) to 0.93 Å (E70A mutant) with a mean of 0.75 ± 0.11 Å. The r.m.s. displacement of the wild type NPC2 generated from the α-carbon coordinates of the 1NEP (7) crystal structure was comparable at 0.71 Å. By way of illustration, supplemental Fig. S1 shows the α-carbon superimpositions for wild type to 1NEP and for wild type to I62D (smallest r.m.s. displacement, 0.51 Å), Q29A (average r.m.s. displacement, 0.76 Å), and E70A (largest r.m.s. displacement 0.93 Å). In all cases, the displacements have little effect on the overall conformation of the protein.
For each of the 17 mutants, we rank-ordered the α-carbon distances of all 130 residues to the corresponding wild type α-carbons from largest to smallest, as shown in supplemental Table S1; the 10 α-carbons having the largest distances for each of the 17 mutants are listed. The locations in the three-dimensional structure where the distances between mutant and wild type α-carbons occur are shown in Fig. 6 for K97A and E99A (unimpaired for sterol transfer and npc2−/− cell rescue), K75A (severely impaired), and I62D (non-functional) as representative examples. Illustrative images, including those in Fig. 6, are shown for all 17 mutants in supplemental Fig. S2. Interestingly and without exception, the 10 largest displacements include residues distant from the site of the mutation. We averaged the 130 α-carbon displacements of the fully functional or moderately impaired mutants and separately averaged those of the severely impaired and of the non-functional structures. The three averages show no pattern that would distinguish the three classes of mutants (data not shown). Thus, NPC2 appears conformationally quite plastic, responding differently and subtly to the various mutations.
FIGURE 6.

Effects of representative missense mutations on the conformation and electrostatic surface of NPC2. Structural images and electrostatic surfaces of K97A (unimpaired) (A), E99A (unimpaired) (B), K75A (severely impaired) (C), and I62D (non-functional) (D) are shown in the same orientation. All are viewed from the front of the molecule as in Fig. 3A. In the structural models, most of the protein is shown as lines in normal functional colors: carbon in green, oxygen in red, nitrogen in blue, hydrogen in white, and sulfur in yellow. Yellow sticks identify the residues with the 10 largest displacements from wild type. Residues shown in magenta sticks form the rim to the cholesterol binding cavity as described by Xu et al. (8) except for Val-64 in the K97A and K75A mutants, which is cyan to indicate that it is both a rim residue and one of the 10 residues with the largest displacements for those mutants. For the mutations to alanine, the Ala methyl is shown as green spheres. The carboxylate of I62D is shown as red spheres.
Discussion
The NPC2 mutagenesis results for sterol transfer kinetics, as well as the ability of NPC2 to promote membrane interactions in vitro, suggest the presence of several regions on the surface that are necessary for normal functioning of the protein. The notable correspondence between these results and those of the npc2−/− cell rescue studies, in which transport-deficient mutants were unable to effect sterol clearance from NPC2-deficient cells, strongly supports the physiological relevance of the studies. Based on the present observations, we hypothesize that NPC2 may catalyze cholesterol transfer at zones of close apposition between membranes, as exist in the multilamellar interior of the LE/LY compartment. Computational modeling of NPC2, moreover, indicates the existence of several positively charged areas on the sides and back (Fig. 3) of the protein, which could bind to two closely situated membrane bilayers. Thus, NPC2 may bridge membranes at membrane contact sites, which have been proposed to be important for rapid lipid transfer between different organelles (16, 22).
Mutations with Dramatic Effects on NPC2 Function
By altering the surface properties of NPC2 beyond the immediate vicinity of a single amino acid, a number of the generated missense mutations had very dramatic functional effects. Interestingly, in many cases (E99A, K6A, E108A, N39A, and K75A) none of the biggest α-carbon displacements occurred at or near the mutated amino acid. This can only happen if the effects of the mutation are propagated through the protein by a cumulative series of small displacements that do not alter the overall conformation significantly. There is precedent for this in the study of a PDZ domain by McLaughlin et al. (33), in which residues of the third PDZ domain of the synaptic protein PDS-95 were mutated individually to each of the other 19 amino acids. It was observed that mutation at sites distant from the peptide binding site can affect function. Among the results shown here, for instance, mutating Lys-75 to alanine or Ile-62 to aspartate results in pronounced alteration in both surface charge distribution and function with little change in conformation. Replacement of lysine 75 by alanine allows negative charges from nearby Glu-70 and Asp-72 to have a strong effect on the surface. Consequently, the negatively charged zone around these residues intensifies and greatly expands, wrapping around the side of the protein and merging with the negative region on the top (Fig. 7A). Additionally, the back gains some negatively charged surface, whereas the region near the top gains positive intensity. The gains in negative surface charge on the side, especially, would reduce membrane interactions and thus the potential for NPC2 to transfer cholesterol. Indeed, the K75A mutant exhibits deficient transfer activity (Table 2). The side chain of Ile-62 in the wild type contributes heavily to the charge neutrality of the cholesterol binding pocket's surface. Replacement by an aspartate adds an electrostatic surface, which merges with a pre-existing negatively charged patch on the side nearby (Fig. 7C). Membrane interactions would be reduced as a result, interfering with the protein's ability to transfer cholesterol. Not surprisingly, then, this mutant is deficient in the cholesterol transfer assay (Table 2) and, importantly, is unable to reverse cholesterol accumulation in npc2−/− cells (Fig. 1).
FIGURE 7.
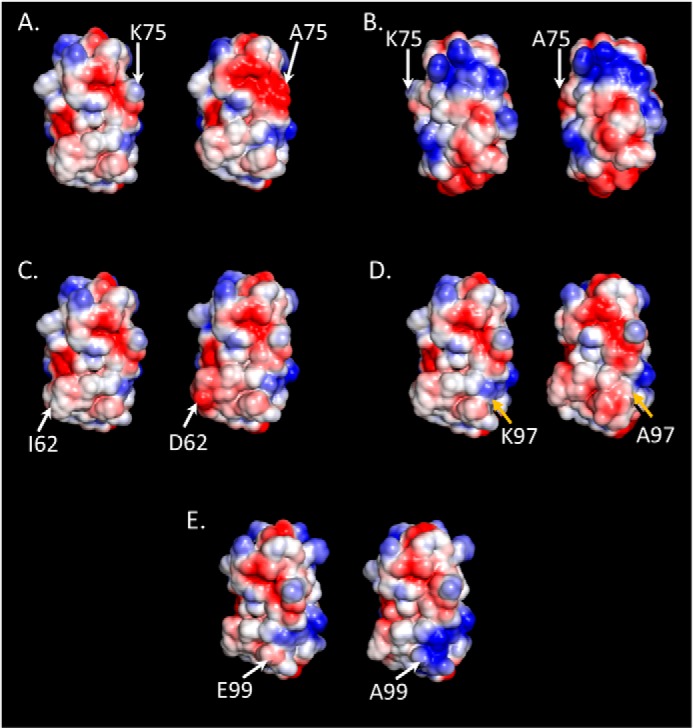
Comparison of WT and mutant electrostatic surfaces. Changes in the electrostatic surfaces of NPC2 are shown to the right of the WT protein for K75A (A and B), I62D (C), K97A (D), and E99A (E). The WT and mutant molecules are oriented to display the front of the protein, as in Fig. 3A, with the exception of the K75A mutant in B, which displays the back of the protein as orientated in Fig. 3B. Respective residues are indicated on the WT (left) and mutant (right) proteins. Blue, positively charged surface; red, negatively charged surface; white, neutral surface.
Similar surface electrostatic changes were observed upon neutralization of the Asp-72 surface residue, which resulted in an inactive protein. Consistent with these results, a missense mutation in Asp-72 (supplemental Fig. S2), which was identified in participants of the ClinSeq project (34), is considered to be disease-causing (35). A number of additional NPC2 missense mutations occurring at non-surface residues that are also not in the binding pocket, have also been shown to be damaging in individuals (36, 37). It will thus be interesting to investigate the effect of these additional mutations on NPC2 functional characteristics and on the conformation and electrostatic surfaces of the protein.
Mutations with Little or No Effect on NPC2 Function
Not all mutations had a substantial effect on the electrostatic surface of the protein, however. In the K97A mutant, the weak positive surface contribution of Lys-97 disappears, and the latent negative surface becomes weakly apparent. A strong negative area in the center of the front expands slightly (Fig. 7D), whereas the back, top, and bottom of the molecule show very little change (not shown). Replacement of the weak positive surface charge by a weak negative charge thus has little overall effect, and, accordingly, the K97A NPC2 mutant exhibits cholesterol transfer properties similar to WT (Table 2).
Another such example is the E99A mutant, where the weak negative charge, which is mostly canceled by Lys-97, is abolished and the positive charge of Lys-97 spreads down toward the bottom of the molecule, yielding a large positive region (Fig. 7E), which would certainly facilitate interactions with negatively charged membranes. Only minor changes in surface charge distribution are observed on the bottom, back, and top of the protein. Presumably due to these minimal changes, this mutation had little or no effect on sterol transfer kinetics (Table 2) and was able to effectively clear cholesterol when administered to NPC2-deficient fibroblasts (Fig. 1). Whereas a human Glu-99 mutation in NPC2 was lethal, it introduced a stop codon (36), and thus it is likely that no functional protein was generated in this patient. Future studies will investigate human mutations that are either benign or only slightly damaging, such as the missense mutation to the surface residue Asn-79, which is non-disease-causing, or the V20M mutation implicated in cases of adult onset NPC2 disease (36, 37).
Overall, considering the important role that charge-charge interactions probably play in the ability of NPC2 to transfer cholesterol to membranes, the effect on the electrostatic surface of the protein caused by a specific mutation may be more important than the precise location of the mutation itself.
Model of Cholesterol Transfer by NPC Proteins
Within the LE/LY compartment, two potential zones at which NPC2 could stimulate rapid transfer of cholesterol between membranes exist. The first is between inner lysosomal membranes, where cholesterol probably partitions (Fig. 8) (11, 12). This would allow NPC2 to transfer cholesterol from more interior membranes toward the limiting lysosomal membrane, which the sterol must ultimately cross in order to maintain cellular cholesterol homeostasis. The second is between the inner lysosomal membranes and the limiting lysosomal membrane, allowing for the deposition of cholesterol by NPC2 into the inner leaflet of the limiting lysosomal membrane (Fig. 8). In either case, transfer could occur following diffusion of NPC2 through the lysosomal lumen and direct interaction with acceptor membranes, or it could potentially occur at membrane contact sites. At the limiting lysosomal membrane, NPC2 could form a contact site by simply interacting with headgroups of the membrane phospholipids. Another possibility is that it may interact with the second luminal domain of NPC1, which has been shown to bind holo-NPC2 (14), or with the N-terminal domain of NPC1, as suggested by Wang et al. (5).
FIGURE 8.

Hypothesis for cholesterol transport in the LE/LY compartment. Free cholesterol is liberated by acid lipase and largely partitions to lysobisphosphatidic acid-rich inner LE/LY membranes. NPC2 extracts cholesterol from these membranes via direct interaction (a), potentially by facilitating sterol transfer at “membrane contact sites” between inner LE/LY membrane lamellae, which may involve NPC2-lysobisphosphatidic acid interaction (b). NPC2 may then transfer cholesterol directly to the N-terminal domain of NPC1 (c), which then facilitates cholesterol efflux from the LE/LY membrane by an unknown mechanism (d). It is also possible that NPC2 may deliver cholesterol directly to the limiting (outer) LE/LY membrane (e), in part, potentially, at membrane contact sites between the inner and outer LE/LY membranes (f). Cholesterol in the limiting LE/LY membrane may access NPC1 via rapid lateral diffusion in the plane of the membrane (g) or might also egress in an NPC1-independent manner (h).
NPC2 is a small protein, with its cholesterol binding pocket located near one end of the molecule. It is thus unlikely that a single NPC2 protein would be able to directly transfer cholesterol between membranes at contact sites while maintaining a stable bridge. In order for the cholesterol binding pocket to come in contact with an acceptor membrane, NPC2 would need to turn, potentially resulting in loss of a stable membrane contact site. Because both the protein and the membrane are deformable, however, the interaction could allow cholesterol that is not necessarily within the NPC2 pocket to move from one bilayer to another. This suggestion is supported by the observation that all 17 mutants bind the ligand with approximately equal affinity, although many of them are unable to catalyze transport. Because cholesterol binding is necessary for NPC2 to clear sterol accumulation in deficient cells (13), the present results indicate that ligand binding may be a necessary but insufficient condition for transport. Stabilization of membrane contact sites by NPC2 could thus lower the activation energy for desorption of cholesterol from the donor membrane, allowing for diffusion of cholesterol through the small interbilayer space to the acceptor membrane at rates that greatly exceed spontaneous transfer. Conformational plasticity coupled with the presence of several strongly positively charged zones distributed over the back and sides of the protein also raises the possibility that there may be more than one route for cholesterol movement. Membrane to membrane transfer may occur with several side-to-side orientations of the protein, including, perhaps, to the limiting lysosomal membrane. Direct transfer to NPC1 in the limiting bilayer is also possible.
Although the mechanism by which NPC1 transfers cholesterol out of the LE/LY compartment was not directly investigated in the present study, the roles of both NPC2 and NPC1 must be considered in elucidating the normal path of cholesterol efflux. The N-terminal domain of NPC1, which projects into the lysosomal lumen, has been shown to bind cholesterol (3), and other transmembrane sterol-binding domains have been identified in NPC1 (6, 38), yet little is known regarding the molecular basis of NPC1 function in cholesterol efflux from the LE/LY compartment. It has been hypothesized that normal trafficking of cholesterol in the LE/LY compartment involves a “handoff” of cholesterol from NPC2 to the NPC1 NTD (5), although direct interactions have yet to be demonstrated. Our sterol transfer studies (9, 10) have suggested that NPC2 could also deliver cholesterol directly to the limiting lysosomal membrane, potentially at MCSs.
Studies identifying MCSs between the endoplasmic reticulum and other organelles, where hydrophobic species may rapidly move through a narrow aqueous phase, indicate distances between organellar membranes of ∼10 nm. The 8-nm thickness of the limiting lysosomal membrane glycocalyx, which contrasts sharply with cell membrane glycocalyces of several hundred nm (39), may allow NPC2 to bring inner and limiting lysosomal membranes within a distance reported for other MCSs. Regardless of the mechanism, once the cholesterol is deposited in the limiting membrane, it can diffuse laterally in the plane of the membrane to NPC1. NPC1 could then facilitate egress from the LE/LY compartment by an unknown mechanism (Fig. 8).
Recent evidence has suggested, however, that egress of at least some cholesterol from the LE/LY compartment could occur in an NPC1-independent manner. Kennedy et al. (40) showed that NPC2 mutants unable to transfer cholesterol to NPC1 were still able to promote the efflux of LE/LY cholesterol to mitochondria. It has also been recently suggested that the two proteins work independently of each other in the egress of lysosomal cargo more generally (41). Thus, transfer of cholesterol across the limiting lysosomal membrane could occur directly following deposition of the sterol into the inner leaflet by NPC2, without interaction with NPC1 (Fig. 8). The two routes are not mutually exclusive. The fact that Kennedy et al. (40) further demonstrated that the transfer of LE/LY cholesterol to mitochondria occurred in a non-vesicular manner suggests that the efflux may involve either a cytosolic cholesterol transport protein or perhaps the formation of contact sites between the limiting lysosomal membrane and other organelles, such as mitochondria or the endoplasmic reticulum. Interestingly, MCSs between lysosomes and peroxisomes were recently suggested to be important for normal cholesterol trafficking to that organelle (42). Although an NPC1-independent manner of LE/LY cholesterol efflux may likely exist, the debilitating effects of NPC1 mutations indicate that that the primary mechanism of efflux requires NPC1 participation. Further understanding of potential NPC1-independent mechanisms of cholesterol trafficking could allow for therapeutic exploitation of such a pathway, however, with the potential of ameliorating NPC1 deficiencies, which account for ∼95% of NPC disease cases.
Although further investigation is necessary to determine the mechanisms by which NPC2, and ultimately NPC1, transfer cholesterol out of the LE/LY compartment, the present studies indicate that NPC2 acts to traffic cholesterol rapidly between membranes. As depicted in Fig. 8, we hypothesize that NPC2 functions, at least in part, at membrane contact sites within the multilamellar interior of late endosomes and lysosomes, thereby promoting rapid cholesterol transfer between closely appositioned membranes. Ultimately, in conjunction with NPC1 and/or in an NPC1-independent manner, sterol transport by NPC2 leads to the egress of cholesterol from the LE/LY compartment, a requisite step for normal intracellular cholesterol homeostasis.
Author Contributions
L. A. M., Z. X., and J. S. designed the study. L. A. M., J. S., and P. C. K. composed the manuscript. L. A. M. and Z. X. performed the cholesterol binding and transfer assays and designed the NPC2 mutant primers. All NPC2 proteins were prepared and purified by L. A. M., Z. X., D. C. K., and S. K. L. A. M. additionally completed the cell rescue assays, including all microscopic work and filipin stain measurements, and conducted the membrane aggregation and fusion studies. Protein modeling studies were conducted by P. C. K., and R. L. aided with the collection and analysis of accompanying data. D. C. K. and M. P. S. generated reagents for initial studies and provided ongoing insights. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
The structure of NPC2 was acquired from the Protein Data Bank, entry 1NEP. We thank Thomas Holder for assistance with PyMOL.
This work was supported by funds from the Ara Parseghian Medical Research Foundation (to J. S.), the American Heart Association (predoctoral fellowship 11PRE7330012 (to L. A. M.), postdoctoral fellowship 0625994T (to Z. X.), and Grant-in-Aid 14GRNT19990014 (to J. S.)), and an anonymous private source (to P. C. K.). The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Table S1 and Figs. S1 and S2.
- NPC
- Niemann-Pick type C
- NPC1
- Niemann-Pick C 1 protein
- NPC2
- Niemann-Pick C 2 protein
- LE/LY
- late endosomal/lysosomal
- EPC
- egg phosphatidylcholine
- LUV
- large unilamellar vesicle
- DHE
- dehydroergosterol
- MCS
- membrane contact site
- PE
- phosphatidylethanolamine
- NBD
- nitrobenzoxadiozole
- 5-CF
- 5-carboxyfluorescein
- r.m.s.
- root mean square.
References
- 1.Pentchev P. G., Brady R. O., Blanchette-Mackie E. J., Vanier M. T., Carstea E. D., Parker C. C., Goldin E., and Roff C. F. (1994) The Niemann-Pick C lesion and its relationship to the intracellular distribution and utilization of LDL cholesterol. Biochim. Biophys. Acta 1225, 235–243 [DOI] [PubMed] [Google Scholar]
- 2.Runz H., Dolle D., Schlitter A. M., and Zschocke J. (2008) NPC-db, a Niemann-Pick type C disease gene variation database. Hum. Mutat. 29, 345–350 [DOI] [PubMed] [Google Scholar]
- 3.Kwon H. J., Abi-Mosleh L., Wang M. L., Deisenhofer J., Goldstein J. L., Brown M. S., and Infante R. E. (2009) Structure of N-terminal domain of NPC1 reveals distinct subdomains for binding and transfer of cholesterol. Cell 137, 1213–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Infante R. E., Wang M. L., Radhakrishnan A., Kwon H. J., Brown M. S., and Goldstein J. L. (2008) NPC2 facilitates bidirectional transfer of cholesterol between NPC1 and lipid bilayers, a step in cholesterol egress from lysosomes. Proc. Natl. Acad. Sci. U.S.A. 105, 15287–15292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang M. L., Motamed M., Infante R. E., Abi-Mosleh L., Kwon H. J., Brown M. S., and Goldstein J. L. (2010) Identification of surface residues on Niemann-Pick C2 essential for hydrophobic handoff of cholesterol to NPC1 in lysosomes. Cell Metab. 12, 166–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ohgane K., Karaki F., Dodo K., and Hashimoto Y. (2013) Discovery of oxysterol-derived pharmacological chaperones for NPC1: implication for the existence of second sterol-binding site. Chem. Biol. 20, 391–402 [DOI] [PubMed] [Google Scholar]
- 7.Friedland N., Liou H. L., Lobel P., and Stock A. M. (2003) Structure of a cholesterol-binding protein deficient in Niemann-Pick type C2 disease. Proc. Natl. Acad. Sci. U.S.A. 100, 2512–2517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu S., Benoff B., Liou H. L., Lobel P., and Stock A. M. (2007) Structural basis of sterol binding by NPC2, a lysosomal protein deficient in Niemann-Pick type C2 disease. J. Biol. Chem. 282, 23525–23531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheruku S. R., Xu Z., Dutia R., Lobel P., and Storch J. (2006) Mechanism of cholesterol transfer from the Niemann-Pick type C2 protein to model membranes supports a role in lysosomal cholesterol transport. J. Biol. Chem. 281, 31594–31604 [DOI] [PubMed] [Google Scholar]
- 10.Xu Z., Farver W., Kodukula S., and Storch J. (2008) Regulation of sterol transport between membranes and NPC2. Biochemistry 47, 11134–11143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Möbius W., van Donselaar E., Ohno-Iwashita Y., Shimada Y., Heijnen H. F., Slot J. W., and Geuze H. J. (2003) Recycling compartments and the internal vesicles of multivesicular bodies harbor most of the cholesterol found in the endocytic pathway. Traffic 4, 222–231 [DOI] [PubMed] [Google Scholar]
- 12.Kobayashi T., Beuchat M. H., Chevallier J., Makino A., Mayran N., Escola J. M., Lebrand C., Cosson P., Kobayashi T., and Gruenberg J. (2002) Separation and characterization of late endosomal membrane domains. J. Biol. Chem. 277, 32157–32164 [DOI] [PubMed] [Google Scholar]
- 13.Ko D. C., Binkley J., Sidow A., and Scott M. P. (2003) The integrity of a cholesterol-binding pocket in Niemann-Pick C2 protein is necessary to control lysosome cholesterol levels. Proc. Natl. Acad. Sci. U.S.A. 100, 2518–2525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deffieu M. S., and Pfeffer S. R. (2011) Niemann-Pick type C 1 function requires lumenal domain residues that mediate cholesterol-dependent NPC2 binding. Proc. Natl. Acad. Sci. U.S.A. 108, 18932–18936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Levine T. (2004) Short-range intracellular trafficking of small molecules across endoplasmic reticulum junctions. Trends Cell Biol. 14, 483–490 [DOI] [PubMed] [Google Scholar]
- 16.Holthuis J. C., and Levine T. P. (2005) Lipid traffic: floppy drives and a superhighway. Nat. Rev. Mol. Cell Biol. 6, 209–220 [DOI] [PubMed] [Google Scholar]
- 17.Levine T., and Loewen C. (2006) Inter-organelle membrane contact sites: through a glass, darkly. Curr. Opin. Cell Biol. 18, 371–378 [DOI] [PubMed] [Google Scholar]
- 18.McCauliff L. A., Xu Z., and Storch J. (2011) Sterol transfer between cyclodextrin and membranes: similar but not identical mechanism to NPC2-mediated cholesterol transfer. Biochemistry 50, 7341–7349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abdul-Hammed M., Breiden B., Adebayo M. A., Babalola J. O., Schwarzmann G., and Sandhoff K. (2010) Role of endosomal membrane lipids and NPC2 in cholesterol transfer and membrane fusion. J. Lipid Res. 51, 1747–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gruenberg J. (2001) The endocytic pathway: a mosaic of domains. Nat. Rev. Mol. Cell Biol. 2, 721–730 [DOI] [PubMed] [Google Scholar]
- 21.Storch J., and Kleinfeld A. M. (1986) Transfer of long-chain fluorescent free fatty acids between unilamellar vesicles. Biochemistry 25, 1717–1726 [DOI] [PubMed] [Google Scholar]
- 22.Schulz T. A., Choi M. G., Raychaudhuri S., Mears J. A., Ghirlando R., Hinshaw J. E., and Prinz W. A. (2009) Lipid-regulated sterol transfer between closely apposed membranes by oxysterol-binding protein homologues. J. Cell Biol. 187, 889–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kleinfeld A. M., Chu P., and Storch J. (1997) Flip-flop is slow and rate limiting for the movement of long chain anthroyloxy fatty acids across lipid vesicles. Biochemistry 36, 5702–5711 [DOI] [PubMed] [Google Scholar]
- 24.Struck D. K., Hoekstra D., and Pagano R. E. (1981) Use of resonance energy transfer to monitor membrane fusion. Biochemistry 20, 4093–4099 [DOI] [PubMed] [Google Scholar]
- 25.Berman H., Henrick K., and Nakamura H. (2003) Announcing the worldwide Protein Data Bank. Nat. Struct. Biol. 10, 980. [DOI] [PubMed] [Google Scholar]
- 26.Laskowski R. A., MacArthur M. W., Moss D. S., and Thornton J. M. (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Cryst. 26, 283–291 [Google Scholar]
- 27.Laskowski R. A., MacArthur M. W., and Thornton J. M. (2001) in International Tables of Crystallography, Vol. F (Rossmann M. G., and Arnold E., eds) pp. 722–725, Kluwer Academic Publishers, Dordrecht, The Netherlands [Google Scholar]
- 28.Hooft R. W., Vriend G., Sander C., and Abola E. E. (1996) Errors in protein structures. Nature 381, 272. [DOI] [PubMed] [Google Scholar]
- 29.Holst M., and Saied F. (1995) Numerical solution of the nonlinear Poisson-Boltzmann equation: developing more robust and efficient methods. J. Comput. Chem. 16, 337–364 [Google Scholar]
- 30.Baker N. A., Sept D., Joseph S., Holst M. J., and McCammon J. A. (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Richards F. M. (1977) Areas, volumes, packing and protein structure. Annu. Rev. Biophys. Bioeng. 6, 151–176 [DOI] [PubMed] [Google Scholar]
- 32.Kajander T., Kahn P. C., Passila S. H., Cohen D. C., Lehtiö L., Adolfsen W., Warwicker J., Schell U., and Goldman A. (2000) Buried charged surface in proteins. Structure 8, 1203–1214 [DOI] [PubMed] [Google Scholar]
- 33.McLaughlin R. N. Jr., Poelwijk F. J., Raman A., Gosal W. S., and Ranganathan R. (2012) The spatial architecture of protein function and adaptation. Nature 491, 138–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Biesecker L. G., Mullikin J. C., Facio F. M., Turner C., Cherukuri P. F., Blakesley R. W., Bouffard G. G., Chines P. S., Cruz P., Hansen N. F., Teer J. K., Maskeri B., Young A. C., NISC Comparative Sequencing Program, Manolio T. A., Wilson A. F., Finkel T., Hwang P., Arai A., Remaley A. T., Sachdev V., Shamburek R., Cannon R. O., and Green E. D. (2009) The ClinSeq Project: piloting large-scale genome sequencing for research in genomic medicine. Genome Res. 19, 1665–1674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wassif C. A., Cross J. L., Iben J., Sanchez-Pulido L., Cougnoux A., Platt F. M., Ory D. S., Ponting C. P., Bailey-Wilson J. E., Biesecker L. G., and Porter F. D. (2015) High incidence of unrecognized visceral/neurological late-onset Niemann-Pick disease, type C1, predicted by analysis of massively parallel sequencing data sets. Genet. Med. 10.1038/gim.2015.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vanier M. T., and Millat G. (2004) Structure and function of the NPC2 protein. Biochim. Biophys. Acta 1685, 14–21 [DOI] [PubMed] [Google Scholar]
- 37.Park W. D., O'Brien J. F., Lundquist P. A., Kraft D. L., Vockley C. W., Karnes P. S., Patterson M. C., and Snow K. (2003) Identification of 58 novel mutations in Niemann-Pick disease type C: correlation with biochemical phenotype and importance of PTC1-like domains in NPC1. Hum. Mutat. 22, 313–325 [DOI] [PubMed] [Google Scholar]
- 38.Ohgami N., Ko D. C., Thomas M., Scott M. P., Chang C. C., and Chang T. Y. (2004) Binding between the Niemann-Pick C1 protein and a photoactivatable cholesterol analog requires a functional sterol-sensing domain. Proc. Natl. Acad. Sci. U.S.A. 101, 12473–12478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wilke S., Krausze J., and Büssow K. (2012) Crystal structure of the conserved domain of the DC lysosomal associated membrane protein: implications for the lysosomal glycocalyx. BMC Biol. 10, 62-7007-10-62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kennedy B. E., Charman M., and Karten B. (2012) Niemann-Pick Type C2 protein contributes to the transport of endosomal cholesterol to mitochondria without interacting with NPC1. J. Lipid Res. 53, 2632–2642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goldman S. D., and Krise J. P. (2010) Niemann-Pick C1 functions independently of Niemann-Pick C2 in the initial stage of retrograde transport of membrane-impermeable lysosomal cargo. J. Biol. Chem. 285, 4983–4994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chu B. B., Liao Y. C., Qi W., Xie C., Du X., Wang J., Yang H., Miao H. H., Li B. L., and Song B. L. (2015) Cholesterol transport through lysosome-peroxisome membrane contacts. Cell 161, 291–306 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.