

Background: The molecular mechanism of anti-allergic (−)-maackiain remains unknown.
Results: (−)-Maackiain and Hsp90 inhibitors inhibited PKCδ activation and suppressed H1R gene expression.
Conclusion: (−)-Maackiain is a novel Hsp90 pathway inhibitor, and its anti-allergic activity underlies the disruption of Hsp90-PKCδ interaction.
Significance: Hsp90 is involved in H1R gene up-regulation, and its inhibition could be a novel therapeutic strategy for allergic rhinitis.
Keywords: allergy, G protein-coupled receptor, gene expression, heat shock protein 90 (Hsp90), histamine, protein-protein interaction, (−)-maackiain, PKCdelta, allergic disease-sensitive gene, histamine H1 receptor gene
Abstract
The histamine H1 receptor (H1R) gene is an allergic disease sensitive gene, and its expression level is strongly correlated with the severity of allergic symptoms. (−)-Maackiain was identified as a Kujin-derived anti-allergic compound that suppresses the up-regulation of the H1R gene. However, the underlying mechanism of H1R gene suppression remains unknown. Here, we sought to identify a target protein of (−)-maackiain and investigate its mechanism of action. A fluorescence quenching assay and immunoblot analysis identified heat shock protein 90 (Hsp90) as a target protein of (−)-maackiain. A pull-down assay revealed that (−)-maackiain disrupted the interaction of Hsp90 with PKCδ, resulting in the suppression of phorbol 12-myristate 13-acetate (PMA)-induced up-regulation of H1R gene expression in HeLa cells. Additional Hsp90 inhibitors, including 17-(allylamino)-17-demethoxygeldanamycin, celastrol, and novobiocin also suppressed PMA-induced H1R gene up-regulation. 17-(Allylamino)-17-demethoxygeldanamycin inhibited PKCδ translocation to the Golgi and phosphorylation of Tyr311 on PKCδ. These data suggest that (−)-maackiain is a novel Hsp90 pathway inhibitor. The underlying mechanism of the suppression of PMA-induced up-regulation of H1R gene expression by (−)-maackiain and Hsp90 inhibitors is the inhibition of PKCδ activation through the disruption of Hsp90-PKCδ interaction. Involvement of Hsp90 in H1R gene up-regulation suggests that suppression of the Hsp90 pathway could be a novel therapeutic strategy for allergic rhinitis.
Introduction
Pollinosis is a seasonal allergic rhinitis caused by hypersensitivity to tree or grass pollens, and it affects more than 36 million people in the United States and about 30% of the Japanese population (1, 2). Histamine is a major chemical mediator of allergic reactions, and its action is mainly mediated through the activation of histamine H1 receptor (H1R).4 As a consequence, antihistamines are a widely employed first-line treatment for nasal symptoms of pollinosis. Recently, we reported that the H1R gene expression strongly correlated with the severity of allergic symptoms in toluene-2,4-diisocyanate (TDI)-sensitized rats, a commonly used allergy model, and patients with Japanese cedar pollinosis (3, 4). We also demonstrated that compounds that suppress up-regulation of H1R gene expression alleviate allergy symptoms (5–8). These findings strongly suggest that H1R signaling is very important for the development of pollinosis and that drugs targeting H1R signaling will be effective for allergic diseases. Nasal topical steroids are also used as first-line treatment for allergic rhinitis. They reduce the infiltration of inflammatory cells, such as mast cells, into the nasal mucosa. And, it is thought that because they do not inhibit mast cell degranulation or the effects of histamine, they do not improve symptoms immediately. However, it is shown that some symptoms are improved in <12 h (9). In our previous study, we demonstrated that treatment with dexamethasone suppressed TDI-induced up-regulation of H1R gene expression and alleviated nasal symptoms in TDI-sensitized rats (10). These findings suggest the existence of cross-talk between steroid signaling and H1R signaling.
Previously, we reported that histamine and phorbol 12-myristate 13-acetate (PMA) stimulation increased H1R at both mRNA and protein levels by the activation of H1R in HeLa cells endogenously expressing H1R (11). Recently, we demonstrated that the PKCδ/extracellular signal-regulated kinase (ERK)/poly(ADP-ribose) polymerase-1 signaling pathway was involved in histamine- and PMA-induced up-regulation of H1R gene expression in HeLa cells (12). In addition, we have reported the mechanism of up-regulation of H1R gene expression, in which two promoter regions, A and B1, were identified in the human H1R gene. Tandem binding of two molecules of AP-1 and one of Ets-1 to region A is crucial for H1R promoter activity. In region B1, dissociation of poly(ADP-ribosyl)ated Ku86 from the promoter caused activation of H1R gene transcription (13).
Kujin is the dried root of Sophorae flavescens AITON of the Leguminosae family. This Kampo herb has been used extensively in the treatment of allergic diseases and many other pathological conditions for many years in Asian countries. In a previous study, we showed that Kujin extract inhibited up-regulation of H1R and IL-4 gene expression in TDI-sensitized rats (6). We have identified (−)-maackiain as an anti-allergic component in Kujin (14). Treatment with synthetic maackiain alleviated nasal symptoms and suppressed up-regulation of H1R gene expression in TDI-sensitized rats. However, (−)-maackiain did not show antioxidant activity or inhibit PKCδ enzymatic activity. Studies using synthetic (−)-maackiain showed stereoselectivity for the suppression of IL-4 gene expression but not for H1R gene expression, suggesting the existence of distinct target proteins for each transcriptional signaling. However, the underlying mechanism of the suppressive activity of (−)-maackiain remains unknown.
In the present study, we investigated the molecular mechanism of anti-allergic activity of (−)-maackiain. Our data revealed that (−)-maackiain binds to Hsp90 and inhibits its interaction with PKCδ, resulting in the inhibition of Tyr311 phosphorylation on PKCδ and translocation of PKCδ to the Golgi and the suppression of H1R gene transcription. Additional Hsp90 inhibitors, including 17-(allylamino)-17-demethoxygeldanamycin (17-AAG), celastrol, and novobiocin, suppress PMA-induced up-regulation of H1R gene expression. These studies suggest that (−)-maackiain is a novel Hsp90 pathway inhibitor. The discovery of Hsp90 as a target protein of (−)-maackiain may shed light on a novel therapeutic strategy for allergic rhinitis.
Experimental Procedures
Identification of Hsp90 as (−)-Maackiain-binding Protein
HeLa cells were cultured at 37 °C under a humidified 5% CO2, 95% air atmosphere in minimal essential medium-α containing 8% fetal calf serum and 1% antibiotics-antimycotics (Invitrogen). HeLa cells were serum-starved for 24 h in 150-mm dishes. The cells from seven dishes were harvested in Tris-buffered saline (TBS) containing proteinase inhibitors (Complete Mini, Roche Applied Science) and phosphatase inhibitors (Phos STOP, Roche Applied Science), and whole cell extracts were prepared by sonication. The extracts were then applied to a HiTrapQ FF anion exchange column (GE Healthcare) pre-equilibrated with TBS, and proteins were eluted with a linear gradient of 0–0.5 m NaCl in TBS. The fractions were incubated with 1 μl of 100 mm (−)-maackiain, and then the tryptophan-derived fluorescence (λex = 285 nm and λem = 335 nm) was measured. For the control, 1 μl of DMSO was added to the fractions. Quenching activity was calculated by subtracting the fluorescence of the control from the fluorescence of the sample. The proteins in the fractions having high quenching activity were separated by 10% SDS-PAGE, digested with trypsin, and then subjected to tandem mass spectrometry (MS/MS) as described previously (15). Peptides were analyzed using a nanoflow-HPLC/nanospray ionization MS/MS on an Esquire 3000 ion trap mass spectrometer (Bruker-Daltonics, Bremen, Germany). MS/MS data were acquired using data analysis software (Bruker-Daltonics), converted to text files listing the mass values, and processed using the MASCOT algorithm (Matrix Science Ltd., London, UK) to assign peptides in the NCBI non-redundant sequence database. Human Hsp90α cDNA was PCR-amplified using a forward primer, 5′-AAATAAGTCGACATGCCTGAGGAAACCCAG-3′ and a reverse primer, 5′-CTTCATCTGCAGTTAGTCTACTTCTTCCAT-3′ (16). The fragment was cloned into the pGEM-T-Easy vector (Promega, Madison, WI), and the nucleotide sequence was confirmed. Hsp90 cDNA was then cloned into the expression vector pCold I (Takara Bio Inc., Kyoto, Japan) at the SalI and PstI sites. To overexpress Hsp90, BL21(DE3)pLys cells (Novagen) were transformed with the expression vector. After induction of Hsp90 protein by isopropyl 1-thio-β-d-galactopyranoside, protein expression was confirmed by immunoblot analysis using an anti-Hsp90 antibody (Santa Cruz Biotechnology). Recombinant Hsp90 protein was purified using TALON metal affinity resin (Clontech) followed by HisTrap HP (for HPLC; GE Healthcare).
Immunoblot Analysis
HeLa cells were serum-starved for 24 h and stimulated with 100 μm histamine for 1 min or with 100 nm of PMA for 10 min in 100-mm dishes. Cells were pretreated with (−)-maackiain or 17-AAG for 24 h before stimulation with histamine or PMA. The cells were harvested in TBS containing proteinase inhibitors (Complete Mini) and phosphatase inhibitors (Phos STOP), and whole cell extracts were prepared by sonication. For the immunoblot analysis, 30 μg of each protein sample was separated by 10% SDS-PAGE and then transferred onto a nitrocellulose membrane (Bio-Rad). The membrane was briefly rinsed in TBS containing 0.1% Tween 20 (TBS-T) and then incubated for 1 h at room temperature in TBS-T containing 5% skim milk (Difco) or 3% BSA (for detecting phosphoproteins; Sigma). The membrane was then incubated with a primary antibody (PKCδ (C-20), Santa Cruz Biotechnology; phospho-PKCδ (Tyr311), Hsp90, and β-actin, Cell Signaling) overnight at 4 °C. Goat anti-rabbit IgG (H±L)-HRP conjugate (Bio-Rad) or Immun-StarTM goat anti-mouse-HRP conjugate (Bio-Rad) was used as the secondary antibody, and proteins were visualized with an Immobilon Western Chemiluminescent HRP substrate (Millipore).
Immunoprecipitation Assay
HeLa cells were serum-starved for 24 h and treated with or without (−)-maackiain or 17-AAG in 100-mm dishes. The cells were harvested, washed with TBS containing proteinase inhibitors (Complete Mini) and phosphatase inhibitors (Phos STOP), and whole cell extracts were prepared in lysis buffer (20 mm Tris-HCl, pH 7.5, 100 mm NaCl, 0.5% Triton X-100) by sonication. rProtein A-Sepharose Fast Flow beads (GE Healthcare) and normal mouse IgG (Santa Cruz Biotechnology) were added to the whole cell extracts and incubated for 30 min at 4 °C. After centrifugation at 3,000 rpm for 2 min at 4 °C, aliquots were removed and immunoprecipitated with PKCδ antibody or rabbit control IgG. Proteins were separated by 10% SDS-PAGE and transferred to a nitrocellulose membrane (Bio-Rad). The membrane was briefly rinsed in TBS-T and incubated for 1 h at room temperature in TBS-T containing 5% skim milk (Difco). The membrane was incubated overnight at 4 °C with a primary antibody (Hsp90 (Cell Signaling) and PKCδ and Cdc37 (Santa Cruz Biotechnology)), and proteins were visualized using an Immobilon Western Chemiluminescent HRP substrate (Millipore).
Pull-down Assay
(−)-Maackiain-immobilized beads were prepared using nanomagnetic particle FG beads (epoxy beads, Tamagawa Seiki, Nagano, Japan). In brief, (−)-maackiain (final concentration 2 mm) was mixed with 0.5 mg of epoxy beads in N,N-dimethylformamide, and 2.8 mg of K2CO3 was added to the reaction mixture and incubated for 24 h at 60 °C. After the supernatant was removed by centrifugation at 15,000 rpm for 5 min at room temperature, the beads were washed three times with 50% N,N-dimethylformamide. Then, the beads were further washed three times with 50% methanol. The beads were then suspended in 50% methanol and stored at 4 °C until use. After being serum-starved for 24 h, HeLa cells were harvested and washed with TBS containing proteinase inhibitors and phosphatase inhibitors in 100-mm dishes. Whole cell extracts were prepared by sonication in lysis buffer (50 mm Tris-HCl, pH 8, 150 mm NaCl, 1% Nonidet P-40, 1 mm DTT, 0.5 mm PMSF). For the pull-down assay, the extract was incubated with (−)-maackiain-immobilized beads for 1 h at room temperature. After magnetic separation of the beads from the extracts, the beads were subjected to SDS-PAGE analysis. For the maackiain competition assay, the extracts were preincubated on ice with 1 mm (−)-maackiain for 30 min. To assess the effect of (−)-maackiain on ATP binding by Hsp90, γ-aminophenyl ATP-immobilized agarose (Jena Bioscience, Jena, Germany) was used. HeLa cells were serum-starved for 24 h and treated with (−)-maackiain (30 μm) or 17-AAG (5 μm) for 24 h in 100-mm dishes before harvesting. Cells were incubated on ice with TNESV buffer (50 mm Tris-HCl, pH 7.5, 2 mm EDTA, 100 nm NaCl, 1 mm sodium orthovanadate, 25 mm NaF, and 1% Triton X-100) for 30 min, sonicated, and centrifuged at 12,000 rpm for 30 min at 4 °C to obtain total cell extracts. The extract was incubated with ATP-immobilized agarose for 1 h at room temperature. After separation of the ATP-immobilized agarose from the extracts by centrifugation, Hsp90 bound to the column was detected by immunoblot analysis.
Geldanamycin Competition Assay
The geldanamycin competition assay was performed using the Hsp90α Assay Kit (BPS Bioscience San Diego, CA), according to the manufacturer's instructions. Briefly, recombinant Hsp90α (0.7 μg) and FITC-labeled geldanamycin (5 nm) were incubated with or without various concentrations of (−)-maackiain or 17-AAG for 2 h at 25 °C. Then fluorescence was measured at λex = 485 nm and λem = 530 nm using the PerkinElmer Enspire multimode plate reader (PerkinElmer Life Sciences).
Hsp90 ATPase Assay
The Hsp90 ATPase assay was performed by the methods described by Rowlands et al. (17). In brief, recombinant Hsp90α (2 μg; Abcam) and 1 mm ATP in the assay buffer (100 mm Tris-HCl, pH 7.4, 20 mm KCl, 6 mm MgCl2) were incubated with or without various concentrations of (−)-maackiain or 17-AAG in a final volume of 25 μl for 16 h at 37 °C. The reaction was stopped by the addition of 80 μl of malachite green reagent (0.0812% (w/v) malachite green, 2.32% (w/v) polyvinyl alcohol, 5.72% (w/v) ammonium molybdate, and MilliQ in a 2:1:1:2 ratio). Then 10 μl of 34% sodium citrate was added to the reaction mixture and left to stand for 15 min at room temperature, and the absorbance was measured at 620 nm using the PerkinElmer Enspire multimode plate reader.
Real-time Quantitative RT-PCR
HeLa cells cultured to 70% confluence in 6-well dishes were serum-starved for 24 h and then treated with reagents 24 h before PMA stimulation. After a 3-h treatment with PMA, the cells were harvested with 700 μl of RNAiso Plus (Takara Bio Inc.), mixed with 140 μl of chloroform, and centrifuged at 15,000 rpm for 15 min at 4 °C. The aqueous phase was collected, and RNA was precipitated by the addition of isopropyl alcohol. After centrifugation at 15,000 rpm for 15 min at 4 °C, the resulting RNA pellet was washed with ice-cold 70% ethanol. Total RNA was resuspended in 10 μl of diethylpyrocarbonate-treated water, and 5 μg of each RNA sample was used for the reverse transcription reaction. For the animal study, rat nasal mucosa samples were collected in RNAlater (Applied Biosystems) 4 h after provocation. Nasal mucosa samples were homogenized using a Polytron homogenizer (model PT-K; Kinematica AG, Littau/Luzern, Switzerland) in 10 volumes of ice-cold RNAiso Plus reagent. The homogenates were mixed with chloroform and centrifuged at 15,000 rpm for 15 min at 4 °C. The aqueous phase containing RNA was transferred to a new tube, and the RNA was precipitated by the addition of isopropyl alcohol and centrifugation at 15,000 rpm for 15 min at 4 °C. The RNA samples were reverse-transcribed to cDNA using a high capacity cDNA reverse transcription kit (Applied Biosystems). TaqMan primers and the probe were designed using Primer Express (Applied Biosystems). Real-time PCR was conducted using a GeneAmp 7300 sequence detection system (Applied Biosystems). The sequences of the primers and TaqMan probes are listed in Table 1. To standardize the starting material, the human GAPDH gene and rodent GAPDH control reagents (VICTM probe, Applied Biosystems) were used, and data were expressed as the ratio of H1R and IL-4 mRNA to GAPDH mRNA.
TABLE 1.
Nucleotide sequences for primers and probes used in this study
| Primer/probe name | Sequence |
|---|---|
| Human H1R mRNA | |
| Sense primer | 5′-CAGAGGATCAGATGTTAGGTGATAGC-3′ |
| Antisense primer | 5′-AGCGGAGCCTCTTCCAAGTAA-3′ |
| Probe | FAMa-CTTCTCTCGAACGGACTCAGATACCACC-TAMRAb |
| Rat H1R mRNA | |
| Sense primer | 5′-TATGTGTCCGGGCTGCACT-3′ |
| Antisense primer | 5′-CGCCATGATAAAACCCAACTG-3′ |
| Probe | FAM-CCGAGAGCGGAAGGCAGCCA-TAMRA |
| Rat IL-4 mRNA | |
| Sense primer | 5′-CAGGGTGCTTCGCAAATTTTAC-3′ |
| Antisense primer | 5′-CACCGAGAACCCCAGACTTG-3′ |
| Probe | FAM-CCCACGTGATGTACCTCCGTGCTTG-TAMRA |
a FAM, 6-carboxyfluorescein.
b TAMRA, tetramethylrhodamine.
Subcellular Localization of PKCδ
To determine the subcellular localization of PKCδ, HeLa cells were plated onto 35-mm glass-bottomed dishes (Asahi Techno Glass, Chiba, Japan). HeLa cells were serum-starved for 24 h. The cells were then stimulated with 100 nm PMA for 5 min. The cells were treated with 17-AAG (1 μm) for 24 h before PMA stimulation. After stimulation, the cells were washed once with PBS and fixed with ice-cold methanol, and the PBS was then replaced. The subcellular localization of the PKCδ was determined with anti-PKCδ antibody as the primary antibody and Cy3-conjugated donkey anti-rabbit IgG as the secondary antibody (Jackson ImmunoResearch) using a confocal laser microscope (LSM510; Carl Zeiss, Oberkochen, Germany). Localization of the Golgi was determined with anti-58K Golgi marker protein antibody (as the primary antibody; Abcam) and DyLight488-conjugated donkey anti-rabbit IgG (as the secondary antibody; Jackson ImmunoResearch).
Animal Study
Six-week-old male Brown Norway rats weighing 200–250 g (Japan SLC, Hamamatsu) were used for the present study. Rats were allowed free access to water and food and kept in a room maintained at 25 ± 2 °C and 55 ± 10% humidity with a 12-h light/dark cycle. Sensitization with TDI was performed by the method described by Dev et al. (6). In brief, 10 μl of a 10% solution of TDI in ethyl acetate (Wako Chemical, Tokyo, Japan) was applied bilaterally on the nasal vestibule of each rat once a day for 5 consecutive days. This sensitization procedure was then repeated after a 2-day interval. Nine days after the second sensitization, 10 μl of 10% TDI solution was again applied to the nasal vestibule to provoke nasal symptoms. The control group was sensitized and provoked with 10 μl of ethyl acetate using the same procedure. Celastrol (1 mg/kg) was administered orally once a day for 1 week, and nasal symptoms were measured during the 10-min period just after TDI provocation. Symptoms included the number of sneezes and the nasal score, which included the extent of watery rhinorrhea, swelling, and redness, measured on a scale ranging from 0 to 3 (Table 2). All experimental procedures were performed in accordance with the guidelines of the Animal Research Committee of Tokushima University.
TABLE 2.
Criteria for grading the severity of TDI-induced nasal responses in rats
| Nasal response | Score |
|||
|---|---|---|---|---|
| 0 | 1 | 2 | 3 | |
| Watery rhinorrhea | None | At nostril | Between 1 and 3 | Drops of discharge from nose |
| Swelling and redness | None | Slightly swollen | Between 1 and 3 | Strong swelling and redness |
Statistical Analysis
The results are shown as mean ± S.E. Statistical analyses were performed using analysis of variance with Dunnett's multiple comparison test using GraphPad Prism software (GraphPad Software Inc., La Jolla, CA). p < 0.05 was considered statistically significant.
Results
Identification of Hsp90 as a Target Protein for (−)-Maackiain
Quenching of intrinsic tryptophan fluorescence has been widely used to analyze the interaction of proteins and their ligands (18). We used this technique to identify the target protein of (−)-maackiain (Fig. 1). HeLa cell fractions, obtained through HiTrapQ FF anion exchange column chromatography, were incubated with (−)-maackiain or a DMSO control to detect quenching activity (Fig. 2A). Judging from the degree of the quenching activity and the elution profile obtained from SDS-PAGE, we selected some bands those are probably responsible for quenching activity and subjected to MS/MS. Hsp90 was identified as one of the candidates for (−)-maackiain-binding protein by MS/MS analysis (Fig. 2B). Next, to confirm the binding of Hsp90 with (−)-maackiain, we examined whether Hsp90 binds (−)-maackiain. Synthesized (−)-maackiain quenched the intrinsic tryptophan fluorescence of recombinant Hsp90 in a dose-dependent manner (Fig. 2C). Hsp90 bound to (−)-maackiain-immobilized resin, and free (−)-maackiain competed with binding of Hsp90 to the resin (Fig. 3A). Pull-down assays using ATP-immobilized beads showed that (−)-maackiain inhibited the binding of Hsp90 to ATP required for the binding of Hsp90 with its client proteins (Fig. 3B). Immunoprecipitation analysis revealed that PKCδ was a client protein of Hsp90, and pretreatment with (−)-maackiain disrupted the interaction of Hsp90 with PKCδ (Fig. 3C). Next, we investigated the effect of (−)-maackiain on Hsp90 enzymatic activity. First, we investigated whether (−)-maackiain could compete with FITC-geldanamycin for binding to the ATP-binding pocket of Hsp90α by a fluorescence polarization assay (19, 20). (−)-Maackiain competed with FITC-geldanamycin binding to the ATP binding pocket of Hsp90α with an IC50 value of 10.8 μm (Fig. 4A). This IC50 value is about 16 times higher than that of 17-AAG (0.64 μm; Fig. 4A), suggesting that (−)-maackiain binds to a site different from the ATP-binding pocket of Hsp90. Second, we investigated whether (−)-maackiain inhibits Hsp90 ATPase activity using the malachite green assay (17). As shown in Fig. 4B, inhibition of Hsp90 ATPase activity by (−)-maackiain was very weak compared with that of 17-AAG (52 μm; Fig. 4B). These data suggested that (−)-maackiain binds to the neighborhood of the ATP-binding pocket of Hsp90 and disrupts the Hsp90-PKCδ interaction. The cellular content of Hsp90 protein was increased by treatment with 17-AAG but not by treatment with (−)-maackiain (Fig. 3C, Input), indicating that (−)-maackiain does not induce the heat shock response.
FIGURE 1.
Structure of (−)-maackiain
FIGURE 2.
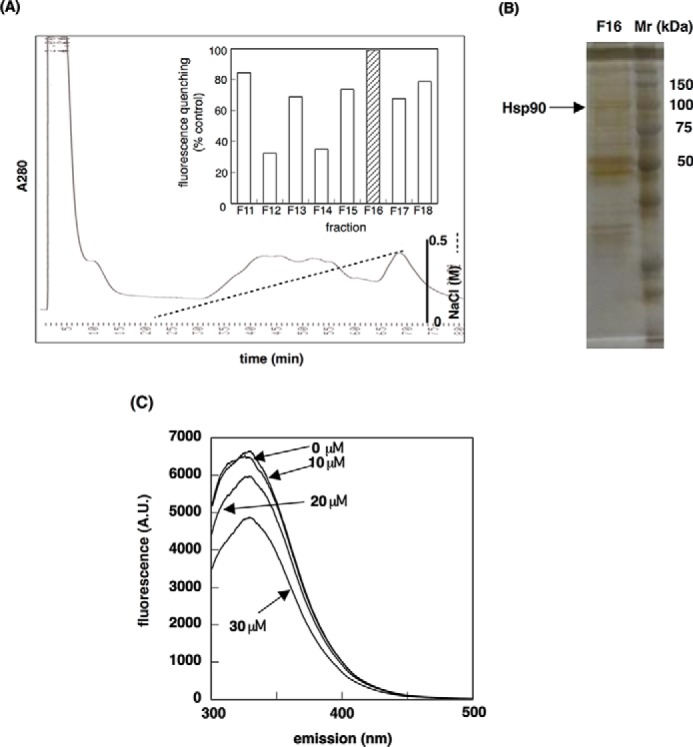
Identification of Hsp90 as a (−)-maackiain-binding protein. A, fluorescence quenching assay. The fractions obtained from the anion exchange column chromatography were incubated with 1 μl of 100 mm (−)-maackiain, and intrinsic tryptophan-derived fluorescence was measured (inset). For the blank, DMSO without (−)-maackiain was added to the fractions. B, silver-stained SDS-polyacrylamide gel of fraction 16 that showed high quenching activity. Hsp90 was identified from the MS/MS analysis. C, quenching of intrinsic tryptophan fluorescence of recombinant Hsp90 by (−)-maackiain. A.U., arbitrary units.
FIGURE 3.

(−)-Maackiain binds to Hsp90 and disrupts Hsp90-PKCδ interaction. A and B, pull-down assay. Whole cell extracts were incubated with (−)-maackiain-immobilized beads (A) or ATP-immobilized agarose (B), and Hsp90 was detected by immunoblot analysis (IB). C, immunoprecipitation assay (IP). The extracts were immunoprecipitated with a PKCδ antibody, and Hsp90 was detected by immunoblot analysis.
FIGURE 4.

Effect of (−)-maackiain on Hsp90 enzymatic activity. A, geldanamycin competition assay. Recombinant Hsp90α (0.7 μg) and FITC-labeled geldanamycin (5 nm) were incubated with or without various concentrations of (−)-maackiain (●) or 17-AAG (■) for 2 h at 25 °C. Then fluorescence was measured at λex = 485 nm and λem = 530 nm using a microtiter plate reader. B, Hsp90 ATPase assay. Recombinant Hsp90α (2 ng) and 1 mm ATP were incubated with or without various concentrations of (−)-maackiain (●) or 17-AAG (■) for 16 h at 37 °C. The reaction was stopped by the addition of malachite green reagent. 15 min after the addition of sodium citrate, the absorbance at 620 nm was measured using a microtiter plate reader.
Effect of Hsp90 Inhibitors on PMA-induced Up-regulation of H1R Gene Expression in HeLa Cells
Our data indicated that (−)-maackiain inhibited the Hsp90 pathway. Therefore, we examined the effect of the commercially available Hsp90 inhibitors 17-AAG, novobiocin, and celastrol on PMA-induced up-regulation of H1R gene expression in HeLa cells. 17-AAG suppressed PMA-induced up-regulation of H1R gene expression in a dose-dependent manner (Fig. 5A). In addition, pretreatment with 17-AAG inhibited PMA-induced phosphorylation of Tyr311 on PKCδ (Fig. 5B), inhibited translocation of PKCδ to the Golgi (Fig. 5C), and disrupted the interaction of Hsp90 with PKCδ (Fig. 3C). Celastrol and novobiocin also suppressed PMA-induced up-regulation of H1R gene expression in HeLa cells in a dose-dependent manner (Fig. 5, D and E). These results suggest that Hsp90 is involved in PMA-induced up-regulation of H1R gene expression in HeLa cells, and Hsp90 inhibitors suppressed H1R gene up-regulation through the inhibition of PKCδ activation.
FIGURE 5.

Effect of Hsp90 inhibitors on H1R signaling. A, effect of 17-AAG on PMA-induced up-regulation of H1R gene expression in HeLa cells. B, effect of 17-AAG on PMA-induced phosphorylation of PKCδ on Tyr311. C, effect of 17-AAG on translocation of PKCδ in response to PMA stimulation. D and E, effect of celastrol (D) or novobiocin (E) on PMA-induced up-regulation of H1R gene expression in HeLa cells. HeLa cells were serum-starved for 24 h and treated with varying concentrations of 17-AAG (A–C), celastrol (D), or novobiocin (E) for 24 h before stimulation with PMA for 3 h (A, D, and E), 10 min (B), or 5 min (C). In B and C, 1 μm 17-AAG was used. In A, D, and E, total RNA was isolated, and the H1R mRNA levels were determined by real-time RT-PCR. Data are presented as the mean ± S.E. (error bars) (n = 3). **, p < 0.01; *, p < 0.05 versus PMA. In B, total cell lysates were prepared and subjected to immunoblot analysis. In C, the subcellular localization of PKCδ was determined using a confocal laser microscope. The images of control and PMA stimulation were taken from Ref. 14. Scale bars, 20 μm.
Cdc37 Does Not Bind Hsp90-PKCδ Complex
It is well known that Cdc37 associates with many kinases (21, 22). Recent work also demonstrated that Cdc37 is a highly specialized co-chaperon adaptor for kinases and Hsp90 and Cdc37 act in concert in chaperoning client kinases (23). Thus, we investigated whether Cdc37 acts as co-chaperon for Hsp90-PKCδ interaction. Immunoprecipitation studies revealed that Cdc37 did not bind Hsp90-PKCδ complex (Fig. 6).
FIGURE 6.

Cdc37 does not bind to PKCδ-Hsp90 complex. HeLa cells were serum-starved for 24 h and then treated with (−)-maackiain (30 μm) or 17-AAG (5 μm) for 24 h before harvesting the cells. The extracts were immunoprecipitated (IP) with PKCδ antibody. Rabbit control IgG was used as a control. Cdc37 was detected by immunoblot analysis (IB). The arrow indicates Cdc37, and a band derived from IgG is indicated with an asterisk.
Effect of Quercetin on the Interaction of Hsp90 with PKCδ
We reported that quercetin suppresses PMA-induced up-regulation of H1R gene expression in HeLa cells (8). Quercetin inhibited the phosphorylation of Tyr311 residue on PKCδ. We investigated the effect of quercetin on the interaction between Hsp90 and PKCδ. Similar to 17-AAG, quercetin disrupted Hsp90-PKCδ interaction (Fig. 7), suggesting that quercetin is also an Hsp90 pathway inhibitor.
FIGURE 7.
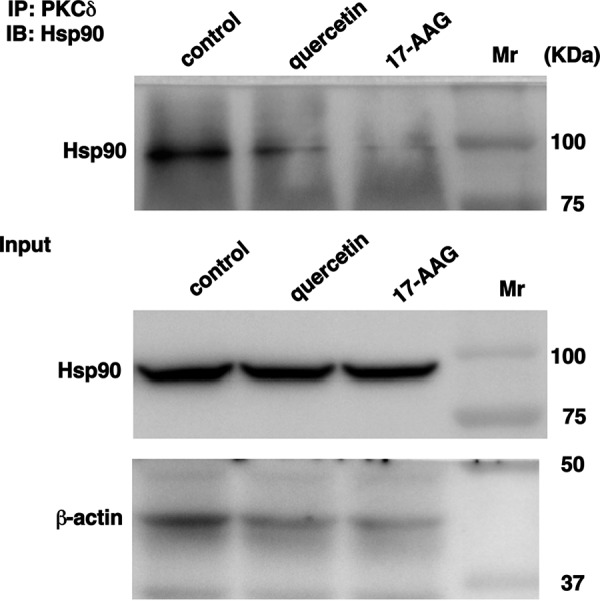
Effect of quercetin on the interaction of PKCδ with Hsp90. HeLa cells were serum-starved for 24 h and then treated with quercetin (50 μm) or 17-AAG (1 μm) for 1 h before harvesting the cells. The extracts were immunoprecipitated (IP) with PKCδ antibody. After centrifugation at 3,000 rpm for 2 min at 4 °C, Hsp90 was detected by immunoblot analysis (IB).
Effect of Celastrol on TDI-induced Nasal Symptoms and Up-regulation of H1R and IL-4 Gene Expression in TDI-sensitized Rats
Previously, we have demonstrated that pretreatment with racemic maackiain and quercetin suppresses TDI-induced nasal symptoms and up-regulation of H1R and IL-4 gene expression in TDI-sensitized rats (8, 14). In the present study, we investigated the effect of an additional Hsp90 inhibitor, celastrol, on TDI-induced nasal symptoms and up-regulation of H1R and IL-4 gene expression in the nasal mucosa of TDI-sensitized rats. Pretreatment with celastrol (1 mg/kg) for 1 week significantly reduced TDI-induced nasal symptoms and up-regulation of H1R and IL-4 gene expression (Fig. 8). These data suggest that Hsp90 inhibitors alleviate nasal symptoms in TDI-sensitized allergy model rats.
FIGURE 8.
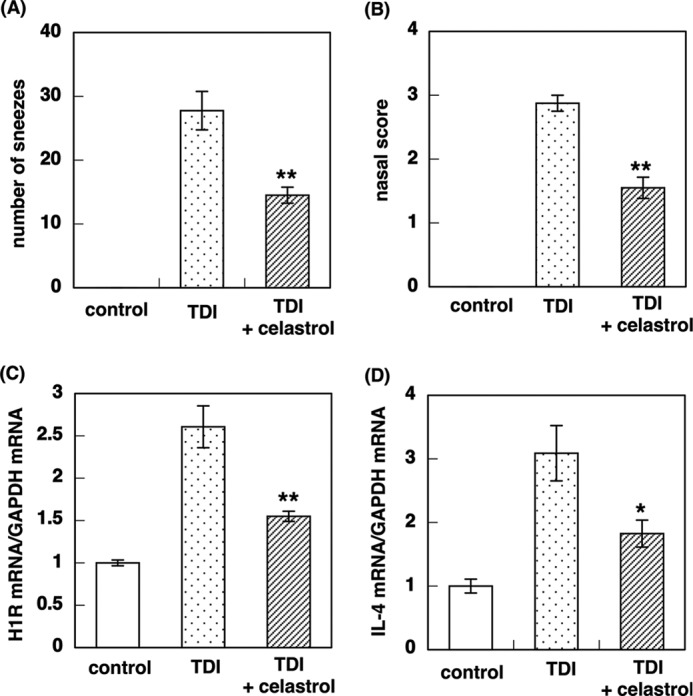
Effect of celastrol on TDI-induced nasal symptoms and up-regulation of H1R and IL-4 gene expression in TDI-sensitized rats. Rats were sensitized with 10 μl of 10% TDI in ethyl acetate for 2 weeks. After a 1-week interval, provocation was performed with 10 μl of 10% TDI. The control group was sensitized with only ethyl acetate. Celastrol (1 mg/kg) were administered once a day for 1 week. Celastrol was administered 1 h before TDI application when rats were sensitized with TDI. A, the number of sneezes was counted during the 10-min period just after TDI provocation. B, nasal scores (degrees of swelling and redness and watery rhinorrhea) were scored according to the criteria listed in Table 2 on a scale ranging from 0 to 3. Nasal mucosa samples were collected 4 h after TDI provocation, and total RNA was isolated. H1R (C) and IL-4 (D) mRNA levels were determined by real-time quantitative RT-PCR. Data are presented as the mean ± S.E. (error bars) (n = 4). **, p < 0.01; *, p < 0.05 versus TDI.
Discussion
In the present study, we showed that (−)-maackiain is a novel Hsp90 pathway inhibitor. (−)-Maackiain is a pterocarpan that is widely distributed in leguminous plants (24), and its anticancer and antimicrobial activity has been reported (25, 26). However, anti-allergic activity of (−)-maackiain had not yet been reported. We have shown that (−)-maackiain is the prominent component responsible for the anti-allergic activity of Kujin (14). Because (−)-maackiain did not show any antioxidant activity or inhibit PKCδ enzymatic activity, it is likely that its anti-allergic activity proceeded through an unknown mechanism. A fluorescence quenching study and immunoblot analysis revealed that Hsp90 is a target of (−)-maackiain, although we cannot exclude the possibility of the existence of other proteins bound to (−)-maackiain. A pull-down assay also demonstrated that (−)-maackiain binding to Hsp90 prevents Hsp90-PKCδ interaction. Studies using synthetic maackiain also suggest that there is another target for (−)-maackiain that enables it to suppress IL-4 expression. Identification of target protein for (−)-maackiain in IL-4 gene suppression is under investigation in our laboratory.
It has been reported that long-term inhibition of Hsp90 by 17-AAG down-regulates PKCδ expression (27). Hsp90 is one of the most abundant proteins in the cytoplasm, where it constitutes about 2% of total protein levels (28). However, it has been reported that Hsp90 translocates to the nucleus in response to stress and other stimuli (29, 30). Hsp90 does not have a nuclear localization signal sequence; therefore, it is thought that transport of Hsp90 to the nucleus depends on client proteins. Cytosolic Hsp90 can also be transported to other parts of the cell, including the extracellular matrix (31) and mitochondria (32). In our study, PMA stimulation induced translocation of the Hsp90-PKCδ complex to the Golgi. We do not yet know the molecular mechanism of this translocation. However, a recent report showed that p23, a type I transmembrane protein that belongs to the p24 endoplasmic reticulum/Golgi cargo family, is the anchoring protein for PKCδ, which binds to the C1b domain of PKCδ. This suggests the possibility that p23 plays an important role in driving the Golgi localization of the Hsp90-PKCδ complex (33). Co-chaperones are a critical component of the cytosolic Hsp90 folding pathway because their functions include targeting client proteins to Hsp90 and modulating Hsp90-ATPase activity or conformational changes (34). It was reported that Cdc37 is a universal kinase-specific co-chaperone in human cells, and Hsp90 and Cdc37 act in concert in chaperoning client kinases (23). However, our immunoprecipitation studies showed that Cdc37 did not bind to the Hsp90-PKCδ complex. Taipale et al. (23) reported similar data that demonstrated the binding of Cdc37-Hsp90 with PKCα, PKCβ, PKCγ, PKCϵ, PKCθ, PKCη, PKCζ, and PKCι but not with PKCδ.
Data indicating that (−)-maackiain is an Hsp90 pathway inhibitor prompted us to investigate the anti-allergic effect of other Hsp90 inhibitors. We selected three Hsp90 inhibitors (i.e. 17-AAG, celastrol, and novobiocin). Hsp90 contains three highly conserved domains consisting of an N-terminal ATP-binding domain, a middle domain, and a C-terminal dimerization domain. 17-AAG is a geldanamycin derivative compound that competes with ATP for binding to Hsp90 (35). Novobiocin binds to the C-terminal dimerization domain and alters Hsp90-co-chaperon-client protein interaction (36), and celastrol also binds to C-terminal domain and inhibits interaction of Hsp90 with co-chaperones, such as Cdc37 or p23 (37, 38). Our data demonstrated that all three compounds suppress PMA-induced up-regulation of H1R gene expression in a dose-dependent manner and that 17-AAG inhibits PMA-induced translocation of PKCδ to the Golgi, phosphorylation of Tyr311 on PKCδ, and interaction of Hsp90 with PKCδ. Our animal experiments showed that maackiain and celastrol suppressed TDI-induced nasal symptoms and up-regulation of H1R and IL-4 gene expression in the nasal mucosa of TDI-sensitized rats (14) (this study). Thus, three compounds that interact with Hsp90 at different sites displayed effects associated with anti-allergic activity. These data suggest that Hsp90 is a promising target for anti-allergic drug development.
Because Hsp90 participates in stabilizing and activating many client proteins that are essential for constitutive cell signaling and adaptive responses to stress, many Hsp90 inhibitors that disrupt Hsp90-client protein interactions have been clinically evaluated as anticancer drugs (39). One of the drawbacks of Hsp90 inhibitors that bind to the N-terminal ATP binding domain of Hsp90 is that they tend to induce the heat shock response, which increases the expression of Hsp70 and Hsp90 (40), thereby limiting their clinical usefulness. Therefore, Hsp90 inhibitors that target the C-terminal domain of Hsp90 or the interaction of Hsp90 with co-chaperones have been developed. The competition of (−)-maackiain with ATP for Hsp90 binding suggests that (−)-maackiain binds to the neighborhood of the N-terminal ATP binding domain of Hsp90. However, (−)-maackiain does not induce Hsp90 expression. Animal studies also suggest that (−)-maackiain is less toxic and shows oral biological availability. These findings suggest the potential usefulness of (−)-maackiain for cancer treatment.
H1R gene expression is highly correlated with the severity of allergic symptoms, and compounds that suppress H1R gene expression alleviate allergic symptoms (4–8). TH2 cytokines are also suggested to play important roles in the pathogenesis of allergic inflammation (41). We have demonstrated the cross-talk between H1R signaling and TH2 cytokine signaling in patients with pollinosis (3, 42) and have shown that suppression of H1R signaling could inhibit TH2 cytokine signaling. Our data from animal studies showing that Hsp90 inhibitors suppressed both H1R and IL-4 gene up-regulation support the cross-talk between these two signaling, and consequently we consider suppression of H1R signaling to be crucial for the treatment of allergic diseases. We identified additional anti-allergic compounds, including epigallocatechin-3-O-gallate and quercetin, that suppress the up-regulation of H1R gene expression in HeLa cells and TDI-sensitized rats (5, 8). It has also been reported that epigallocatechin-3-O-gallate binds to the C-terminal domain of Hsp90 (43). Our data also show that quercetin is an Hsp90 pathway inhibitor. These findings suggest that the anti-allergic action of these compounds is also mediated by the inhibition of PKCδ activation through the disruption of Hsp90-PKCδ interaction.
In addition to antihistamines, steroid nasal sprays have been frequently used recently to relive nasal symptoms. It is well known that the glucocorticoid receptor is a client protein of Hsp90, and it is maintained in its resting state by binding to Hsp90 in the absence of steroid. After steroid binds to the glucocorticoid receptor, the steroid-glucocorticoid receptor complex is released from Hsp90 and translocates into the nucleus, where the complex binds to a specific glucocorticoid-responsive element and regulates the transcription of steroid-susceptible proteins. It is known that steroid binds to AP-1 and down-regulates transcription of many proinflammatory cytokines and growth factors. Because of these mechanisms of action, it takes 1–3 days to exert the beneficial effects, although nasal topical steroids are effective for sneezing, watery rhinorrhea, and nasal mucosal swelling. However, some symptom has been shown to improve within 12 h (2). Our studies using TDI-sensitized rats demonstrated that dexamethasone suppressed TDI-induced H1R gene expression. We also showed that dexamethasone also suppressed histamine-induced up-regulation of H1R gene expression in HeLa cells. These findings and the present data suggest that this “acute” effect of steroids may be due to the suppression of H1R gene up-regulation.
In summary, we have shown that the underlying mechanism of the suppression of H1R gene up-regulation by (−)-maackiain is the inhibition of PKCδ activation through the disruption of Hsp90-PKCδ interaction. The data also suggest that Hsp90 is involved in H1R gene up-regulation, and the inhibition of Hsp90 could be a novel therapeutic strategy for allergic diseases. Hsp90 inhibitors may improve allergic symptoms or prevent development of allergic diseases, contributing to their clinical application for allergic diseases.
Author Contributions
Y. N., T. O., H. N., Y. S., Y. O., and H. N. performed the experimental work. H. M. designed the project and wrote the manuscript. Y. Y. and Y. K. analyzed the data and participated in the data interpretation. N. T. supervised the research and wrote the manuscript. H. F. conceived the project, supervised the research, and wrote the manuscript.
This work was supported in part by Grant-in-Aid 10103185 for Research on Allergic Disease and Immunology from the Ministry of Health Labor and Welfare of Japan (to H. F.), Grants-in-Aid for Scientific Research (B) 26305004 (to H. F.) and Scientific Research (C) 22580132 (to H. M.) and 15K07933 (to H. M.) from the Japan Society for the Promotion of Science, and by a grant from the Osaka Medical Research Foundation for Incurable Diseases. The authors declare that they have no conflicts of interest with the contents of this article.
- H1R
- histamine H1 receptor
- 17-AAG
- 17-(allylamino)-17-demethoxygeldanamycin
- Hsp90
- heat shock protein-90
- PMA
- phorbol 12-myristate 13-acetate
- TDI
- toluene-2,4-diisocyanate.
References
- 1.Nathan R. A., Meltzer E. O., Selner J. C., and Storms W. (1997) Prevalence of allergic rhinitis in the United States. J. Allergy Clin. Immunol. 99, S808–S814 [PubMed] [Google Scholar]
- 2.Okubo K., Kurono Y., Fujieda S., Ogino S., Uchio E., Odajima H., Takenaka H., and Japanese Society of Allergology. (2014) Japanese guideline for allergic rhinitis. Allergol. Int. 63, 357–375 [DOI] [PubMed] [Google Scholar]
- 3.Mizuguchi H., Hatano M., Matsushita C., Umehara H., Kuroda W., Kitamura Y., Takeda N., and Fukui H. (2008) Repeated pre-treatment with antihistamines suppresses transcriptional up-regulations of histamine H1 receptor and interleukin-4 genes in toluene-2,4-diisocyanate-sensitized rats. J. Pharmacol. Sci. 108, 480–486 [DOI] [PubMed] [Google Scholar]
- 4.Mizuguchi H., Kitamura Y., Kondo Y., Kuroda W., Yoshida H., Miyamoto Y., Hattori M., Fukui H., and Takeda N. (2010) Preseasonal prophylactic treatment with antihistamines suppresses nasal symptoms and expression of histamine H1 receptor mRNA in the nasal mucosa of patients with pollinosis. Methods Find. Exp. Clin. Pharmacol. 32, 745–748 [DOI] [PubMed] [Google Scholar]
- 5.Matsushita C., Mizuguchi H., Niino H., Sagesaka Y., Masuyama K., and Fukui H. (2008) Identification of epigallocatechin-3-O-gallate as an active constituent in tea extract that suppresses transcriptional up-regulations of the histamine H1 receptor and interleukin-4 genes. J. Trad. Med. 25, 133–142 [Google Scholar]
- 6.Dev S., Mizuguchi H., Das A. K., Maeyama K., Horinaga S., Kato S., Tamada M., Hattori M., Umehara H., and Fukui H. (2009) Kujin suppresses histamine signaling at the transcriptional level in toluene 2,4-diisocyanate-sensitized rats. J. Pharmacol. Sci. 109, 606–617 [DOI] [PubMed] [Google Scholar]
- 7.Nurul I. M., Mizuguchi H., Shahriar M., Venkatesh P., Maeyama K., Mukherjee P. K., Hattori M., Choudhuri M. S., Takeda N., and Fukui H. (2011) Albizia lebbeck suppresses histamine signaling by the inhibition of histamine H1 receptor and histidine decarboxylase gene transcriptions. Int. Immunopharmacol. 11, 1766–1772 [DOI] [PubMed] [Google Scholar]
- 8.Hattori M., Mizuguchi H., Baba Y., Ono S., Nakano T., Zhang Q., Sasaki Y., Kobayashi M., Kitamura Y., Takeda N., and Fukui H. (2013) Quercetin inhibits transcriptional up-regulation of histamine H1 receptor via suppressing protein kinase C-δ/extracellular signal-regulated kinase/poly(ADP-ribose) polymerase-1 signaling pathway in HeLa cells. Int. Immunopharmacol. 15, 232–239 [DOI] [PubMed] [Google Scholar]
- 9.Church M. K., and Casale T. B. (2012) in Allergy, 4th Ed. (Holgate S. T., Church M. K., Broide D. H., and Martinez F. D., eds) pp. 147–169, Elsevier, Amsterdam [Google Scholar]
- 10.Kitamura Y., Miyoshi A., Murata Y., Kalubi B., Fukui H., and Takeda N. (2004) Effect of glucocorticoid on upregulation of histamine H1 receptor mRNA in nasal mucosa of rats sensitized by exposure to toluene diisocyanate. Acta Otolaryngol. 124, 1053–1058 [DOI] [PubMed] [Google Scholar]
- 11.Das A. K., Yoshimura S., Mishima R., Fujimoto K., Mizuguchi H., Dev S., Wakayama Y., Kitamura Y., Horio S., Takeda N., and Fukui H. (2007) Stimulation of histamine H1 receptor up-regulates histamine receptor itself through activation of receptor gene transcription. J. Pharmacol. Sci. 103, 374–382 [DOI] [PubMed] [Google Scholar]
- 12.Mizuguchi H., Terao T., Kitai M., Ikeda M., Yoshimura Y., Das A. K., Kitamura Y., Takeda N., and Fukui H. (2011) Involvement of PKCδ/extracellular signal-regulated kinase/poly(ADP-ribose) polymerase-1 (PARP-1) signaling pathway in histamine-induced up-regulation of histamine H1 receptor gene expression in HeLa cells. J. Biol. Chem. 286, 30542–30551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mizuguchi H., Miyagi K., Terao T., Sakamoto N., Yamawaki Y., Adachi T., Ono S., Sasaki Y., Yoshimura Y., Kitamura Y., Takeda N., and Fukui H. (2012) PMA-induced dissociation of Ku86 from the promoter causes transcriptional up-regulation of histamine H1 receptor. Sci. Rep. 2, 916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mizuguchi H., Nariai Y., Kato S., Nakano T., Kanayama T., Kashiwada Y., Nemoto H., Kawazoe K., Takaishi Y., Kitamura Y., Takeda N., and Fukui H.. Maackiain is a novel anti-allergic compound that suppresses transcriptional up-regulation of the histamine H1 receptor and interleukin-4 genes. Pharmacol. Res. Perspect. 3, e00166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yoshimura Y., Shinkawa T., Taoka M., Kobayashi K., Isobe T., and Yamauchi T. (2002) Identification of protein substrates of Ca2+/calmodulin-dependent protein kinase II in the postsynaptic density by protein sequencing and mass spectrometry. Biochem. Biophys. Res. Commun. 290, 948–954 [DOI] [PubMed] [Google Scholar]
- 16.Millson S. H., Truman A. W., Rácz A., Hu B., Panaretou B., Nuttall J., Mollapour M., Söti C., and Piper P. W. (2007) Expressed as the sole Hsp90 of yeast, the α and β isoforms of human Hsp90 differ with regard to their capacities for activation of certain client proteins, whereas only Hsp90β generates sensitivity to the Hsp90 inhibitor radicicol. FEBS J. 274, 4453–4463 [DOI] [PubMed] [Google Scholar]
- 17.Rowlands M. G., Newbatt Y. M., Prodromou C., Pearl L. H., Workman P., and Aherne W. (2004) High-throughput screening assay for inhibitors of heat-shock protein 90 ATPase activity. Anal. Biochem. 327, 176–183 [DOI] [PubMed] [Google Scholar]
- 18.Böhl M., Czupalla C., Tokalov S. V., Hoflack B., and Gutzeit H. O. (2005) Identification of actin as quercetin-binding protein: an approach to identify target molecules for specific ligands. Anal. Biochem. 346, 295–299 [DOI] [PubMed] [Google Scholar]
- 19.Kim J., Felts S., Llauger L., He H., Huezo H., Rosen N., and Chiosis G. (2004) Development of a fluorescence polarization assay for the molecular chaperon Hsp90. J. Biomol. Screen. 9, 375–381 [DOI] [PubMed] [Google Scholar]
- 20.Howes R., Barril X., Dymock B. W., Grant K., Northfield C. J., Robertson A. G. S., Surgenor A., Wayne J., Wright L., James K., Matthews T., Cheung K.-M., McDonald E., Workman P., and Drysdale M. J. (2006) A fluorescence polarization assay for inhibitors of Hsp90. Anal. Biochem. 350, 202–213 [DOI] [PubMed] [Google Scholar]
- 21.Grammatikakis N., Lin J. H., Grammatikakis A., Tsichlis P. N., and Cochran B. H. (1999) p50 (cdc37) acting in concert with Hsp90 is required for Raf-1 function. Mol. Cell. Biol. 19, 1661–1672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gray P. J. Jr., Prince T., Cheng J., Stevenson M. A., and Calderwood S. K. (2008) Targeting the oncogene and kinome chaperone CDC37. Nat. Rev. Cancer 8, 491–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Taipale M., Krykbaeva I., Koeva M., Kayatekin C., Westover K. D., Karras G. I., and Lindquist S. (2012) Quantitative analysis of HSP90-client interactions reveals principles of substrate recognition. Cell 150, 987–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shibata S., and Nishikawa Y. (1963) Constituents of Japanese and Chinese crude drugs. VII. Constituents of the roots of Sophora subprostrata and Sophora japonica. Chem. Pharm. Bull. 11, 167–177 [Google Scholar]
- 25.Aratanechemuge Y., Hibasami H., Katsuzaki H., Imai K., and Komiya T. (2004) Induction of apoptosis by maackiain and trifolirhizin (maackiain glycoside) isolated from sanzukon (Sophora subprostrate Chen et T. Chen) in human promyelotic leukemia HL-60 cells. Oncol. Rep. 12, 1183–1188 [PubMed] [Google Scholar]
- 26.Honda G., and Tabata M. (1982) Antidermatophytic substance from Sophora angustifolia. Planta. Med. 46, 122–123 [DOI] [PubMed] [Google Scholar]
- 27.Gould C. M., Kannan N., Taylor S. S., and Newton A. C. (2009) The chaperones Hsp90 and Cdc37 mediate the maturation and stabilization of protein kinase C through a conserved PXXP motif in the C-terminal tail. J. Biol. Chem. 284, 4921–4935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Borkovich K. A., Farrelly F. W., Finkelstein D. B., Taulien J., and Lindquist S. (1989) Hsp82 is an essential protein that is required in higher concentrations for growth of cells at higher temperatures. Mol. Cell. Biol. 9, 3919–3930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pratt W. B., Toft D. O. (1997) Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr. Rev. 18, 306–360 [DOI] [PubMed] [Google Scholar]
- 30.Langer T., Rosmus S., and Fasold H. (2003) Intracellular localization of the 90 kDa heat shock protein (HSP90α) determined by expression of a EGFP-HSP90α-fusion protein in unstressed and heat stressed 3T3 cells. Cell Biol. Int. 27, 47–52 [DOI] [PubMed] [Google Scholar]
- 31.Tsutsumi S., and Neckers L. (2007) Extracellular heat shock protein 90: a role for a molecular chaperone in cell motility and cancer metastasis. Cancer Sci. 98, 1536–1539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kang B. H., Plescia J., Dohi T., Rosa J., Doxsey S. J., and Altieri D. C. (2007) Regulation of tumor cell mitochondrial homeostasis by an organelle-specific Hsp90 chaperone network. Cell 131, 257–270 [DOI] [PubMed] [Google Scholar]
- 33.Wang H., Xiao L., and Kazanietz M. G. (2011) p23/Tmp21 associates with protein kinase C δ (PKCδ) and modulates its apoptotic function. J. Biol. Chem. 286, 15821–15831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li J., Soroka J., and Buchner J. (2012) The Hsp90 chaperone machinery: conformational dynamics and regulation by co-chaperones. Biochim. Biophys. Acta 1823, 624–635 [DOI] [PubMed] [Google Scholar]
- 35.Guo W., Reigan P., Siegel D., Zirrolli J., Gustafson D., and Ross D. (2005) Formation of 17-allylamino-demethoxygeldanamycin (17-AAG) hydroquinone by NAD(P)H:quinone oxidoreductase 1: role of 17-AAG hydroquinone in heat shock protein 90 inhibition. Cancer Res. 65, 10006–10015 [DOI] [PubMed] [Google Scholar]
- 36.Yun B. G., Huang W., Leach N., Hartson S. D., and Matts R. L. (2004) Novobiocin induces a distinct conformation of Hsp90 and alters Hsp90-cochaperon-client interaction. Biochemistry 43, 8217–8229 [DOI] [PubMed] [Google Scholar]
- 37.Zhang T., Li Y., Yu Y., Zou P., Jiang Y., and Sun D. (2009) Characterization of celastrol to inhibit Hsp90 and Cdc37 interaction. J. Biol. Chem. 284, 35381–35389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chadli A., Felts S. J., Wang Q., Sullivan W. P., Botuyan M. V., Fauq A., Ramirez-Alvarado M., and Mer G. (2010) Celastrol inhibits Hsp90 chaperoning of steroid receptors by inducing fibrillization of the co-chaperone p23. J. Biol. Chem. 285, 4224–4231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Trepel J., Mollapour M., Giaccone G., and Neckers L. (2010) Targeting the dynamic HSP90 complex in cancer. Nat. Rev. Cancer 10, 537–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morimoto R. I., and Santoro M. G. (1998) Stress–inducible responses and heat shock proteins: new pharmacologic targets for cytoprotection. Nat. Biotechnol. 16, 833–838 [DOI] [PubMed] [Google Scholar]
- 41.Kitamura Y., Mizuguchi H., Ogishi H., Kuroda W., Hattori M., Fukui H., Takeda N. (2012) Preseasonal prophylactic treatment with antihistamines suppresses IL-5 but not IL-33 mRNA expression in the nasal mucosa of patients with seasonal allergic rhinitis caused by Japanese cedar pollen. Acta Oto-Laryngologica 132, 434–438 [DOI] [PubMed] [Google Scholar]
- 42.Shahriar M., Mizuguchi H., Maeyama K., Kitamura Y., Orimoto N., Horio S., Umehara H., Hattori M., Takeda N., Fukui H. (2009) Suplatast tosilate inhibits histamine signaling by direct and indirect down-regulation of histamine H1 receptor gene expression through suppression of histidine decarboxylase and IL-4 gene transcriptions. J. Immunol. 183, 2133–2141 [DOI] [PubMed] [Google Scholar]
- 43.Donnelly A., and Blagg B. S. (2008) Novobiocin and additional inhibitors of the Hsp90 C-terminal nucleotide-binding pocket. Curr. Med. Chem. 15, 2702–2717 [DOI] [PMC free article] [PubMed] [Google Scholar]