

Background: FACT proteins SUPT16H and SSRP1 are identified as host factors that restrict HIV-1 replication.
Results: Biochemical and genetic evidences that SUPT16H and SSRP1 affect HIV-1/HTLV-1 transcription and latency are provided.
Conclusion: SUPT16H and SSRP1 suppress transcription of HIV-1/HTLV-1, and their presence may promote HIV-1 latency.
Significance: Identification of host factors necessary for HIV-1 latency is critical, which may benefit the development of novel HIV-1 latency-reversing agents.
Keywords: host-pathogen interaction, human immunodeficiency virus (HIV), RNA interference (RNAi), transcription repressor, viral transcription, CD4 T cell, FACT complex, latency, SSRP1, SUPT16H
Abstract
Our functional genomic RNAi screens have identified the protein components of the FACT (facilitates chromatin transcription) complex, SUPT16H and SSRP1, as top host factors that negatively regulate HIV-1 replication. FACT interacts specifically with histones H2A/H2B to affect assembly and disassembly of nucleosomes, as well as transcription elongation. We further investigated the suppressive role of FACT proteins in HIV-1 transcription. First, depletion of SUPT16H or SSRP1 protein enhances Tat-mediated HIV-1 LTR (long terminal repeat) promoter activity. Second, HIV-1 Tat interacts with SUPT16H but not SSRP1 protein. However, both SUPT16H and SSRP1 are recruited to LTR promoter. Third, the presence of SUPT16H interferes with the association of Cyclin T1 (CCNT1), a subunit of P-TEFb, with the Tat-LTR axis. Removing inhibitory mechanisms to permit HIV-1 transcription is an initial and key regulatory step to reverse post-integrated latent HIV-1 proviruses for purging of reservoir cells. We therefore evaluated the role of FACT proteins in HIV-1 latency and reactivation. Depletion of SUPT16H or SSRP1 protein affects both HIV-1 transcriptional initiation and elongation and spontaneously reverses latent HIV-1 in U1/HIV and J-LAT cells. Similar effects were observed with a primary CD4+ T cell model of HIV-1 latency. FACT proteins also interfere with HTLV-1 Tax-LTR-mediated transcription and viral latency, indicating that they may act as general transcriptional suppressors for retroviruses. We conclude that FACT proteins SUPT16H and SSRP1 play a key role in suppressing HIV-1 transcription and promoting viral latency, which may serve as promising gene targets for developing novel HIV-1 latency-reversing agents.
Introduction
The global rate of HIV-1 infection and the number of AIDS-related deaths have dramatically declined because of expanding access to highly active antiretroviral therapy (HAART).2 However, HIV-1 epidemic remains unsolved, and there is still no cure for HIV-1 infection, as well as a lack of valid HIV-1 vaccines (1). The HAART regimen remains the most effective treatment, but this only blocks active HIV-1 replication, leaving residual viraemia in most AIDS patients (2, 3). Viral loads readily rebound once the antiretroviral regimen is interrupted. Also, lifelong HAART is associated with significant adverse effects and increases the risk of multiple end organ diseases.
Ultrasensitive measurements confirm that residual viraemia is present with <50 copies/ml in the plasma of HAART-treated patients, initiating from a small set of cells harboring latent HIV-1 (4). Resting memory CD4+ T cells are the major, well defined latent reservoir, although other cells, including macrophages and hematopoietic stem cells, are permissible for low HIV-1 replication. HIV-1 infects activated CD4+ T cells and leads to their rapid death through cytopathic effects. Rarely (1/106), HIV-infected CD4+ T cells revert to a resting memory state, leading to silence of HIV-1 gene expression (5). In these resting cells, a comprehensive set of changes in the cellular environment prevents viral gene expression, and multiple cellular mechanisms facilitate HIV-1 latency, mostly through suppression of transcription. Cell surface receptors that inhibit T cell proliferation and differentiation, including PD-1, CTLA-4, and TIM-3, are turned on as negative feedback during T cell activation (6, 7). Key transcriptional initiating factors (NF-κB, NFAT) for HIV-1 gene expression are excluded from the nucleus. The P-TEFb (CDK9/Cyclin T) protein complex required for HIV-1 Tat-mediated RNA polymerase II activation is sequestered in the 7SK small nuclear ribonucleoprotein complex as an inactive form by HEXIM1 (8). The HIV-1 cDNAs tend to integrate within actively transcribed host genes in resting CD4+ T cells from HIV-1-infected individuals (9). In this scenario, transcriptional interference may occur that promotes HIV-1 latency (10, 11). Epigenetic regulation of the proviral 5′ long terminal repeat (LTR) region places another layer to control HIV-1 latency, which includes the events of chromatin modification and nucleosome reorganization by recruiting histone deacetylases (HDACs), histone methyltransferases, and DNA methyltransferases coordinately to silence the viral promoter (12, 13).
Thus, persistent HIV-1 latency is the major obstacle for HIV-1 elimination. Reversing HIV-1 latency for eradication requires the reactivation of integrated proviruses to induce a cytopathic effect to the reservoir cells, followed by reinforced HIV-specific immune responses to purge these cells. To reactivate latent HIV-1 proviruses, the cellular restrictive mechanisms suppressing HIV-1 transcription should be removed. A pharmacological approach seems effective for this purpose. Epigenetic suppression is reversed by inhibiting chromatin remodeling enzymes, such as HDACs, histone methyltransferases, and DNA methyltransferases. The HDAC inhibitors are currently one of the most promising HIV-1 latency-reversing agents, such as vorinostat (suberoylanilide hydroxamic acid (SAHA)) (14, 15). The NF-κB pathway is turned on by activating protein kinase C. Several drugs, such as TNF-α (16), prostratin (17), and bryostatin (18), are currently under investigation for treating HIV-1 latency through this mechanism. P-TEFb is activated by some small compounds to trigger its release from the inhibitory form in the 7SK small nuclear ribonucleoprotein complex, which allows more accessibility to HIV-1 Tat protein, such as hexamethylene bisacetamide (19) and disulfiram (20). However, most of these drugs are less potent when used individually in the in vitro resting CD4+ T cells or in vivo settings of clinical trials. Recently, it was uncovered that the size of the latent HIV-1 reservoir is much larger than previously estimated (21), which indicates the requirement of a combinatory regimen including a set of drugs targeting multiple steps of HIV-1 latency so that all integrated proviruses are reactivated.
Understanding the host restrictive machineries that facilitate HIV-1 latency will help identify new gene targets for HIV-1 anti-latency therapy. Using comprehensive RNAi functional genomic screens, our group recently identified a set of host factors that suppress HIV-1 transcription and promote its latency, including BRD4 (bromodomain-containing protein 4), which we have thoroughly studied previously (22, 23). Surprisingly, we also found that siRNAs targeting the two protein components of the FACT (facilitates chromatin transcription) complex, SUPT16H and SSRP1, significantly increase HIV-1 intracellular replication. Both components were ranked as top hits for HIV-1 restriction factors. The FACT protein complex is a well studied histone chaperone that removes the H2A-H2B dimer to facilitate polymerase II-driven transcription by destabilizing nucleosome structure and depositing core histones back afterward (24, 25). It seems that FACT proteins play an opposite role (negative versus positive) in regulating HIV-1 transcription as opposed to transcriptional regulation of host genes (25, 26). Their activity in HIV-1 transcription and latency needs further characterization. This will provide insight into the general functions of FACT in regulating transcription and the complex host mechanisms involving FACT proteins that modulate HIV-1 latency. By targeting these proteins, more effective novel latency-reversing agent regimens can be designed for HIV-1 anti-latency therapy.
Experimental Procedures
Cells and Plasmids
HEK293 and HEK293T cells were maintained in DMEM supplemented with 10% FBS. The monocytic THP89GFP cells were kindly provided by David Levy (New York University) (27), which were cultured in complete medium (RPMI 1640, 10% FBS, 1× glutamine, 1× MEM nonessential amino acid solution, 20 mmol/liter HEPES). U1/HIV, J-LAT A2, and MT-2 cells were obtained from the National Institutes of Health AIDS reagent repository and cultured in RPMI 1640 medium with 10% FBS. Primary CD4+ Helper T cells were purchased (Sanguine Biosciences, Lonza). pCDNA-Tat, pQCXIP-FLAG-Tat, HIV-LTR-luciferase, and pRL-TK-Renilla vectors were described previously (22). HTLV-1 LTR-luciferase, BC12-Tax, and pB-His6-Tax, were kindly provided by Chou-Zen Giam (Uniformed Services University of the Health Sciences) (28). pCDNA-V5-SSRP1 was constructed through Gateway® LR cloning technology using pDONR223-SSRP1 entry vector and pCDNA-DEST40 destination vector (Life Technologies). SUP16H, SSRP1, or nontargeting (NT) shRNAs were cloned into pAPM lentiviral vector (29) or pINDUCER10 vector (30), using XhoI and EcoRI sites.
Small Molecules
Prostratin (SC-203422) and SAHA (sc-220139) were purchased (Santa Cruz Biotechnology). Compounds were used at the following concentrations: Prostratin (1 μm) and SAHA (0.5 μm). Drug-treated cells were cultured in the presence of compounds for 24 h and subjected to flow cytometry assays on a FACSCalibur flow cytometer (Becton Dickinson), and results were acquired using BD CellQuest software and analyzed using FlowJo vx.0.7 program. Puromycin and doxycycline for cell treatment were purchased from Fisher Scientific. Propidium iodide for cell cycle assay was purchased from MP Biomedicals and used according to the manufacturer's manual.
Viruses
Lentviruses were produced by transfecting plasmids in HEK293T cells using TransIT®-293 transfection reagent (Mirus). VSV-G pseudotyped HIV-1 NL4–3 viruses were produced by co-transfecting pCG-VSV-G vector (22) with HIV-1 NL4–3-Luc (dEnv) plasmid (pNL4–3.Luc.R−.E−, National Institutes of Health AIDS reagent repository, no. 3418). pAPM or pINDUCER10 shRNA expression vectors were transfected in HEK293T cells with packaging vectors psPAX2 and pMD2.G (Addgene) (22). Cell supernatants containing lentiviruses were harvested and filtered though 0.45-μm filters (Millipore). Viruses were stored in aliquots at −80 °C for later use. To generate cell lines for stable expression of shRNAs, lentiviruses were transduced into cells (HEK293, J-LAT A2, U1/HIV, THP89GFP, or MT-2 cells). At 72 h post-transduction, puromycin (1 μg/ml) was added to the medium for stable selection.
Antibodies
The following antibodies were used in this study: mouse anti-SUPT16H (A-1), mouse anti-SSRP1 (D-7), rabbit anti-Cyclin T1 (CCNT1, H-245), rabbit anti-GAPDH (FL-335), goat anti-mouse IgG-HRP, and goat anti-rabbit IgG-HRP (Santa Cruz Biotechnology); mouse anti-V5 (Invitrogen); rabbit anti-FLAG (Rockland); mouse anti-FLAG (Sigma); and unlabeled rabbit and mouse IgGs (Southern Biotech). Monoclonal mouse anti-Tax antibody (4C5) was kindly provided by Chou-Zen Giam (Uniformed Services University of the Health Sciences).
Luciferase Reporter Assays
HEK293 cells that stably express SUPT16H or SSRP1 or NT shRNA were infected with VSV-G pseudo-typed HIV-1 NL4–3-Luc (dEnv) viruses. At 24 h postinfection, the luciferase unit was measured using the One-Glo® luciferase assay system (Promega) and normalized to total cell numbers. To measure the HIV-1 LTR activity, HEK293 cells that stably express SUPT16H or SSRP1 or NT shRNA were transfected with HIV-1 LTR-luciferase, pRL-TK-Renilla, and pCDNA-Tat vectors. At 48 h post-transfection, luciferase and Renilla units were measured using the Dual-Glo® luciferase assay system (Promega), and the relative luciferase unit was calculated. For HTLV-1 study, these cells were transfected with HTLV-1 LTR-luciferase, pRL-TK Renilla, and BC12-Tax vectors (28). At 48 h post-transfection, the relative luciferase unit was measured. All results were collected on a Luminoskan Ascent Microplate Luminometer (Thermo).
Co-immunoprecipitation
Co-immunoprecipitation assays followed the previously described protocol (31) with minor changes. Briefly, HEK293 cells in 10-cm tissue culture dishes were transfected with pQCXIP-FLAG-Tat. At 48 h post-transfection, cells were harvested and lysed in 1 ml of 1× radioimmune precipitation assay buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1% Nonidet P-40, 0.25% sodium deoxycholate, 1 mm EDTA, and protease inhibitor mixture; Pierce). Cell lysate was sonicated briefly, and the debris was spun down. The supernatant was transferred to a new tube and precleared by incubating with 50 μl of protein A/G beads (Pierce) for 2 h at 4 °C. The beads were removed, and the lysate was split equally for incubating with 2 μg of mouse anti-FLAG or IgG (mIgG) control antibody with rotation for overnight at 4 °C. 25 μl of protein A/G beads were added to each sample and incubated for another 2 h at 4 °C. The beads were washed three times with 1× radioimmune precipitation assay buffer and precipitated. Protein samples were eluted in 1× NuPAGE® LDS sample buffer (Life Technologies), and analyzed by SDS-PAGE and Western blots for endogenous SUPT16H or SSRP1. Similar assays were performed for Tax by transfecting pB-His6-Tax in HEK293 cells. To investigate the effect of SUPT16H on Tat-P-TEFb interaction, pQCXIP-FLAG-Tat was transfected in HEK293 cells stably expressing SUPT16H or NT shRNA. Cell lysate was immunoprecipitated using mouse anti-FLAG or mIgG antibody. Protein samples were analyzed by SDS-PAGE and Western blots for endogenous CCNT1. We also transfected pCDNA-V5-SSRP1 with or without pQCXIP-FLAG-Tat in HEK293 cells. Cell lysate was immunoprecipitated using mouse anti-V5 or mIgG antibody. Protein samples were analyzed by SDS-PAGE and Western blots for FLAG-Tat or endogenous SUPT16H.
Chromatin Immunoprecipitation
ChIP assays followed the previously described protocol (32) with minor changes. Briefly, HEK293 cells in 10-cm tissue culture dishes were infected with VSV-G pseudo-typed HIV-1 NL4–3-Luc (dEnv) viruses. At 48 h postinfection, the cells were cross-linked using 0.5% formaldehyde. After quenching the cross-linking reaction with 125 mm glycine, the cells were washed with cold 1× PBS and lysed in 1× CE buffer (10 mm HEPES-KOH, pH 7.9, 60 mm KCl, 1 mm EDTA, 0.5% Nonidet P-40, 1 mm DTT, and protease inhibitor mixture). Cell lysate was centrifuged at 700 × g for 10 min at 4 °C to pellet the nuclei. Nuclei pellet was resuspended in 1× SDS lysis buffer (1% SDS, 10 mm EDTA, 50 nm Tris-HCl, pH 8.1, and protease inhibitor mixture) to a final concentration of 3 × 107 cells/ml, and nuclear lysate was sonicated for 2 min using Fisher ScientificTM model 505 sonic dismembrator (1-s on and 1-s off cycles at 50% impulse) to fragment DNA to an average size of ∼ 500 bp. Cellular debris was spun down, and the supernatant was diluted 10-fold with 1× ChIP dilution buffer (0.01% SDS, 1% Triton X-100, 1.2 mm EDTA, 16.7 mm Tris-HCl, pH 8.1, 150 nm NaCl, and protease inhibitor mixture) and incubated with 5 μg of antibodies against SUPT16H, SSRP1, CCNT1, or control mIgG or rabbit IgG antibody, with rotation overnight at 4 °C. 50 μl of protein A/G beads were pre-equilibrated with 0.5 mg/ml BSA (Fisher Scientific) and 0.125 mg/ml calf thymus DNA (Trevigen) for 1 h at 4 °C and then added to each sample for incubation for another 2 h at 4 °C. Beads were collected, washed once each with low salt buffer (0.1% SDS, 1% Triton X-100, 2 mm EDTA, 20 mm Tris-HCl, pH 8.1, 150 mm NaCl), high salt buffer (0.1% SDS, 1% Triton X-100, 2 mm EDTA, 20 mm Tris-HCl, pH 8.1, 500 mm NaCl), and LiCl buffer (0.25 m LiCl, 1% Nonidet P-40, 1% sodium deoxycholace, 1 mm EDTA, 10 mm Tris-HCl, pH 8.1) and twice with 1× TE buffer (10 mm Tris-HCl, pH 8.1, 0.1 mm EDTA). Immunoprecipitated protein-DNA complexes were eluted twice with fresh elution buffer (1% SDS, 0.1 m NaHCO3) for 1 h and 15 min, respectively, at room temperature. Eluates and nuclear lysates (“input”) were heated at 65 °C for overnight to reverse cross-links in the presence of 0.2 m NaCl. Samples were then treated with 1 μl of 20 mg/ml proteinase K (Life Technologies), 10 μl of 2 m Tris-HCl (pH 6.5), and 10 μl of 0.5 m EDTA for 2 h at 50 °C. Released DNA was extracted by phenol/chloroform, precipitated by ethanol, and resuspended in 100 μl of water. 5 μl of each sample was used for semiquantitative PCR using primer sets for amplifying the LTR or nef region of HIV-1 genome. To investigate the effect of SUPT16H on P-TEFb LTR association, HEK293 cells stably expressing SUPT16H or NT shRNA were infected with VSV-G pseudo-typed HIV-1 NL4–3-Luc (dEnv) viruses and subjected to ChIP assays using rabbit CCNT1 or rabbit IgG antibody.
Real Time Quantitative PCR
To measure shRNA-mediated gene depletion or level of viral transcripts, total RNA was extracted from cells that stably express shRNAs using the RNeasy mini kit (Qiagen) and subsequently reverse transcribed using random hexamers (0.1 μm) and iScriptTM cDNA synthesis kit (Bio-Rad). The gene-specific primers in Table 1 (0.1 μm each) were mixed with reverse transcribed cDNA templates and iTaqTM Universal SYBR® Green supermix (Bio-Rad). The qPCR was performed on the CFX ConnectTM real time PCR detection system (Bio-Rad), in a 20-μl volume using the following program: 95 °C for 1 min and 40 cycles of 95 °C for 15 s and 60 °C for 30 s. GAPDH was used as an internal control.
TABLE 1.
Primers and shRNA oligonucleotides used for this study
| Primers | Sequences |
|---|---|
| LTR_for | 5′-CGA GAG CTG CAT CCG GAG TA-3′ |
| LTR_rev | 5′-TTG GCG TAC TCA CCA GTC GC-3′ |
| NEF_for | 5′-GGA ATG GAT GAC CCT GAG AG-3′ |
| NEF_rev | 5′-CCA CGT GAT GAA ATG CTA GG-3′ |
| SUPT16H_for | 5′-CGG GCA GCA TTA CTT ACA GA-3′ |
| SUPT16H_rev | 5′-TTC AGT CAA TCG CCT CTT TG-3′ |
| SSRP1_for | 5′-ATT CAA CCC AGG TGA AGA GG-3′ |
| SSRP1_rev | 5′-GTT TCC GCT TCT TCT CAT CC-3′ |
| GAPDH_for | 5′-GCC TCT TGT CTC TTA GAT TTG GTC-3′ |
| GAPDH_rev | 5′-TAG CAC TCA CCA TGT AGT TGA GGT-3′ |
| GAG_for | 5′-GAC GCT CTC GCA CCC ATC TC-3′ |
| GAG_rev | 5′-CTG AAG CGC GCA CGG CAA-3′ |
| GAG/POL (HTLV-1)_for | 5′-CCC TCC AGT TAC GAT TTC CA-3′ |
| GAG/POL (HTLV-1)_rev | 5′-GGC TTG GGT TTG GAT GAG TA-3′ |
| Initiation_for | 5′-GTT AGA CCA GAT CTG AGC CT-3′ |
| Initiation_rev | 5′-GTG GGT TCC CTA GTT AGC CA-3′ |
| Elongation_for | 5′-TGG GAG CTC TCT GGC TAA CT-3′ |
| Elongation_rev | 5′-TGC TAG AGA TTT TCC ACA CTG A-3′ |
| Non-targetin (NT) shRNA | 5′-CAC AAA CGC TCT CAT CGA CAA G-3′ |
| SUPT16H shRNA-1 | 5′-GGA AGA ACT TTG ATA TGG TAA T-3′ |
| SUPT16H shRNA-2 | 5′-GGG CTC TAA CCG TGG TTC CAG A-3′ |
| SSRP1 shRNA-1 | 5′-ACC GAG AGA AGA TCA AGT CAG A -3' |
| SSRP1 shRNA-2 | 5′-CGC GAT GAC TCA GGA GAA GAA A-3′ |
Generation of Primary CD4+ T Cell Model of HIV-1 Latency
To investigate the role of FACT protein in HIV-1 latency in primary cells, we used the primary cell model established by Dr. Vicente Planelles's group (33, 34) with slight modifications. Briefly, naïve CD4+ T cells (Sanguine Biosciences, Lonza) were stimulated with anti-human CD3 and anti-human CD28 antibodies (eBioscience) precoated on a Nunc-Immuno MaxiSorp plate (Thermo Scientific). Cells were incubated with complete medium supplemented with 10 ng/ml of TGF-β, 2 μg/ml of anti-human IL-12, and 1 μg/ml of anti-human IL-4 (R & D Systems) for 3 days. After activation, shRNA transduction was performed by spinoculation of cells with pINDUCER10-shNT/shSUPT16H/shSSRP1 lentiviruses at 1,741 × g for 2 h at 37 °C. The cells were then resuspended in complete medium supplemented with 100 IU/ml of rIL-2 and kept in culture for 4 days. The medium was changed every 2 days. On day 7, cells were infected with VSV-G pseudotyped HIV-1 NL4–3-Luc (dEnv) viruses and selected with puromycin (1 μg/ml) for stable transduction ofpINDUCER10 shRNAs. The cells were kept in culture for another 10 days and then treated with doxycycline (0.1 μg/ml) for 4 days to induce shRNA expression. On day 21, cells were analyzed by the luciferase reporter assays. An aliquot of activated CD4+ T cells were subjected to transduction, selection, and induction of pINDUCER10 shRNAs, and these cells were further used for RNA extraction to measure SUPT16H or SSRP1 depletion by reverse transcription and real-time qPCR. This study was approved by University of Rochester Medical Center Institutional Review Board for Protection of Human Subjects (no. RSRB00053667).
Results
RNAi Screens Identify FACT Proteins as Top HIV-1 Restriction Factors
We identified FACT proteins, SUPT16H and SSRP1, as top host restrictive modulators of HIV-1 replication, using multiple orthologous RNAi reagents coupled with integrated analytical tools (23). To calculate a gene-specific enrichment score based on the rank distribution of each individual RNAi reagent among all three screens performed by our group (Silencer Select, esiRNA, and SMARTpool), we used the RNAi gene enrichment ranking (RIGER) method that denotes the likelihood that the selected gene plays a role in the phenotype of interest (35, 36). We generated the average RIGER score (RIGER3) from three RIGER integrative approaches (the second best, weighted sum, and Kolmogorov-Smirnov) and ranked genes from the most likely host dependence factors to host restriction factors according to the RIGER3 score (Fig. 1A). Both FACT components, SUPT16H and SSRP1, were ranked as top HIV-1 restriction factors, among which were several known HIV-1 restriction factors including CCNK (37), BRD4 (22), and NELFCD (38).
FIGURE 1.

Depletion of FACT proteins increases HIV-1 transcription. A, RIGER method was applied to analyze screens performed using multiple orthologous RNAi reagents. Genes were ranked in order of their RIGER scores, from lowest to highest. RIGER analysis of these screens recognized several known host restriction factors (CCNK, BRD4, and NELFCD), as well as new ones, such as SUPT16H and SSRP1 FACT proteins. B and C, shRNAs (sh1, sh2) targeting SUPT16H or SSRP1 in lentiviral pAPM vector were transduced in HEK293 cells. HEK293 cells stably expressing shRNAs were lysed, separated by SDS-PAGE, and analyzed by Western blots using anti-SUPT16H (B) or anti-SSRP1 (C) antibody. GAPDH protein level was determined using an anti-GAPDH antibody to indicate equal loading of protein samples. The results were one representative from three independent experiments. D and E, HEK293 cells depleted of FACT proteins were infected with VSV-G pseudo-typed HIV-1 NL4–3-Luc (dEnv) viruses. At 24 h postinfection, the cells were subjected to measurement of luciferase activity that was normalized by cell numbers. F and G, vectors of HIV-1 LTR-luciferase, pTK-Renilla, and pCDNA-Tat were co-transfected in HEK293 cells stably expressing shSUPT16H (F) or shSSRP1 (G). At 48 h post-transfection, luciferase activity was measured and normalized to the Renilla signal. The relative light unit (RLU) of shSUPT16H (F) or shSSRP1 (G) expressing cells was normalized to shNT cells. The results throughout are the means of three independent experiments ± S.D. *, p < 0.05 using Student's t test.
Depletion of FACT Proteins Enhances HIV-1 LTR Promoter Activity
To confirm the effect of FACT proteins on HIV-1 replication, two sequence-unique shRNAs targeting SUPT16H or SSRP1 were synthesized and cloned in the pAPM lentiviral expression vector (29). SUPT16H or SSRP1 shRNAs were transduced in HEK293 cells, which were subjected to selection of shRNA stable expression. HEK293 cells expressing NT shRNA were created as a negative control. The NT shRNA expression in these cells did not affect the luciferase activity from VSV-G pseudo-typed HIV-1 NL4–3-Luc (dEnv) viruses as well as the HIV-1/HTLV-1 LTR-luciferase constructs (data not shown). The endogenous SUPT16H or SSRP1 protein expression was mostly silenced by their respective shRNA (Fig. 1, B and C). These cells were infected with VSV-G pseudo-typed HIV-1 NL4–3-Luc (dEnv) viruses and then subjected to luciferase assay at 24 h postinfection. Depletion of SUPT16H or SSRP1 clearly permitted better HIV-1 replication in HEK293 cells (Fig. 1, D and E). Furthermore, the HIV-1 LTR-luciferase assay showed that depletion of FACT proteins enhanced the HIV-1 Tat-mediated activation of the LTR promoter (Fig. 1, F and G). In this assay as well as following experiments, we used one of the two shRNAs targeting SUPT16H or SSRP1 (shRNA2 for SUPT16H and shRNA1 for SSRP1).
Tat Interacts with SUPT16H but Not SSRP1
We further confirmed the interaction of Tat with endogenous SUPT16H using an immunoprecipitation (IP) approach. The pQCXIP-FLAG-Tat vector was transiently transfected in HEK293 cells. At 48 h post-transfection, the cell lysate was prepared and subjected to IP experiments using a mouse anti-FLAG antibody or a control mIgG. Endogenous SUPT16H was readily co-precipitated with FLAG-Tat (Fig. 2A). However, SSRP1 seemed not to associate with FLAG-Tat in this IP assay (Fig. 2A). Co-transfection of pCDNA-V5-SSRP1 and pQCXIP-FLAG-Tat for IP experiments still failed to detect their protein interaction (Fig. 2B). These results indicate that Tat may associate with the FACT complex through SUPT16H. Furthermore, exogenous expression of FLAG-Tat did not interfere with the association of V5-SSRP1 with endogenous SUPT16H (Fig. 2F).
FIGURE 2.

FACT proteins associate with Tat-LTR. A, HEK293 cells were transiently transfected with a pQCXIP-FLAG-Tat vector. At 48 h post-transfection, cells were lysed for IP assays using an anti-FLAG or a mIgG antibody. Cell lysate and precipitated protein samples were separated by SDS-PAGE. Protein levels of SUPT16H, SSRP1, or FLAG-Tat were determined by Western blots using their respective antibody. B, IP assays were performed for HEK293 cells that were co-transfected with pCDNA-V5-SSRP1 and pQCXIP-FLAG-Tat vectors. At 48 h post-transfection, cells were lysed and incubated with an anti-V5 or mIgG antibody. Protein level of V5-SSRP1 or FLAG-Tat in cell lysate and precipitated samples was determined using their respective antibody. C and D, HEK293 cells were infected with VSV-G pseudo-typed HIV-1 NL4–3-Luc (dEnv) virus. At 48 h postinfection, cells were cross-linked using formaldehyde, and nuclei were isolated and lysed. Nuclear lysate was sonicated, precleared, and subjected to ChIP assays using anti-SUPT16H (C), anti-SSRP1 (D), or mIgG antibody. Precipitated DNA samples were released, extracted, and analyzed by semiquantitative PCR using primer sets amplifying the HIV-1 LTR promoter or nef region. E, the aforementioned cells were subjected to ChIP assays using anti-CCNT1 or rabbit IgG (rIgG) antibody. F, pCDNA-V5-SSRP1 vector, with pQCXIP-FLAG-Tat or the empty vector, was transfected in HEK293 cells. Co-immunoprecipitation assays were performed for these cells using an anti-V5 antibody. Precipitated protein samples were separated by SDS-PAGE and analyzed for SUPT16H by Western blot. The results throughout were from one representative from three independent experiments.
FACT Proteins Associate with the HIV-1 LTR Promoter
Because SUPT16H binds with Tat, we speculated that FACT proteins may be recruited to the HIV-1 LTR promoter. We used ChIP coupled with PCR assays to determine the occupancy of FACT proteins on the HIV-1 LTR promoter. HEK293 cells were infected with VSV-G pseudo-typed HIV-1 NL4–3-Luc (dEnv) viruses and then subjected to ChIP experiments using anti-SUPT16H, anti-SSRP1, or mIgG antibody. Co-precipitated DNA samples were extracted and analyzed using the primers that amplify the HIV-1 5′ LTR promoter. Both SUPT16H and SSRP1 undoubtedly associated with LTR promoter (Fig. 2, C and D). The results indicate that although SSRP1 has no direct contact with Tat, both SUPT16H and SSRP1 are recruited to HIV-1 LTR promoter. Our ChIP coupled with PCR experiments for CCNT1 illustrated that CCNT1 associated with LTR promoter as well (Fig. 2E).
SUPT16H Interferes with HIV-1 Transcriptional Elongation through P-TEFb
P-TEFb, comprised of cyclin-dependent kinase 9 (CDK9) and cyclin T1 (CCNT1), is a critical host factor that plays a key role in HIV-1 Tat-mediated transcription. CCNT1 has been shown to directly interact with Tat (39), which recruits P-TEFb to the LTR promoter to activate the promoter-proximal paused polymerase (40). We postulate that presence of SUPT16H may interfere with the association of P-TEFb with Tat-LTR and thus produce an inhibitory effect on HIV-1 transcription. We transiently transfected pQCXIP-FLAG-Tat in HEK293 cells that stably express shSUPT16H (sh2) and measured the interaction of FLAG-Tat and endogenous CCNT1 protein. Depletion of SUPT16H dramatically increased the interaction between FLAG-Tat and CCNT1 in HEK293 cells (Fig. 3A). Consistently, depletion of SUPT16H increased the occupancy of CCNT1 at the LTR promoter (Fig. 3B). We further determined whether FACT proteins affect transcription of latent HIV-1. The U1/HIV monocytic cell line is one of the most studied models of HIV postintegration latency. According to earlier studies, this cell line contains two integrated HIV-1 proviruses with Tat mutants (41). It is believed that these Tat mutants compromise viral transcription and lead to HIV-1 latency. shSUPT16H (sh2), shSSRP1 (sh1), or shNT was transduced in U1/HIV. Stable expression of shSUPT16H or shSSRP1 in these cells efficiently depleted FACT proteins (Fig. 4A). Depletion of SUPT16H or SSRP1 increased the level of HIV-1 gag viral transcript (Fig. 4B). To further discriminate whether FACT proteins affect HIV-1 transcriptional initiation or elongation, primer sets recognizing distinguished viral transcripts were used for SYBR® Green-based RT-qPCR (Table 1), following a previously reported protocol (22). Interestingly, both transcriptional initiation and elongation of HIV-1 were enhanced in U1/HIV cells depleted of FACT proteins (Fig. 4, C and D), indicating that FACT proteins interfere with HIV-1 transcription through a P-TEFb independent route, considering that P-TEFb mainly affects elongation (42). We also evaluated the effects of FACT proteins on HIV-1 latency in another monocytic cell line of HIV-1 latency, THP89GFP (27). Depletion of FACT proteins mildly increased HIV-1 reactivation (2–3-fold) with or without the presence of SAHA (Fig. 4, E and F).
FIGURE 3.
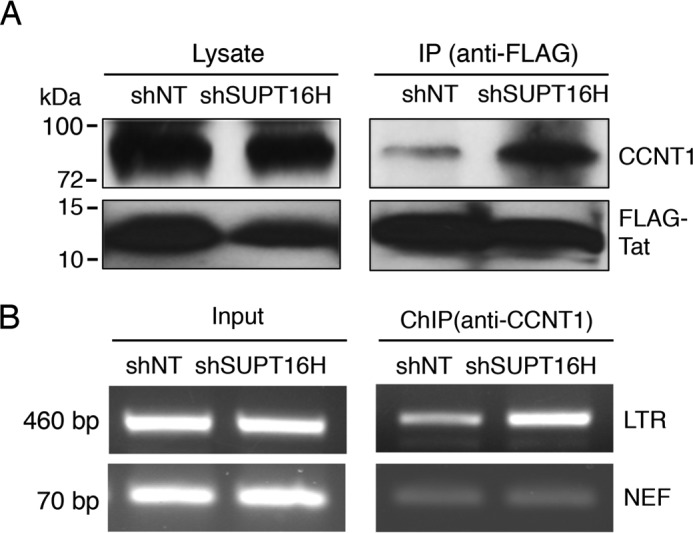
SUPT16H interferes with association of P-TEFb with Tat-LTR. A, HEK293 cells stably expressing shSUPT16H (sh2) or shNT were transiently transfected with a pQCXIP-FLAG-Tat vector and subjected to IP assays using an anti-FLAG antibody. Endogenous CCNT1 protein level in cell lysate and precipitated samples were analyzed by Western blots using an anti-CCNT1 antibody. B, ChIP assays were performed against CCNT1 in shSUPT16H (sh2) or shNT expressing HEK293 cells that were infected with VSV-G pseudo-typed HIV-1 NL4–3-Luc (dEnv) viruses. The results throughout were from one representative from three independent experiments.
FIGURE 4.

Depletion of FACT proteins enhances reversal of HIV-1 latency in monocytes. A, U1/HIV cells were transduced with pAPM-shSUPT16H (sh2), shSSRP1 (sh1), or shNT. Cell lysate was prepared, separated by SDS-PAGE, and analyzed by Western blots using anti-SUPT16H or anti-SSRP1 antibody. GAPDH protein level was determined using an anti-GAPDH antibody to indicate equal loading of protein samples. The results were from one representative from three independent experiments. B, cDNA samples from the aforementioned cells were subjected to qPCR assays to measure HIV-1 gag mRNA. The level of gag transcripts was normalized to shNT-expressing cells. C and D, cDNA samples from the aforementioned cells were subjected to qPCR assays to measure the HIV-1 initiation and elongation transcripts. The level of viral transcripts was normalized to shNT-expressing cells. E, THP89GFP cells were transduced with pAPM-shSUPT16H (sh2), shSSRP1 (sh1), or shNT. Cells were lysed, separated by SDS-PAGE, and analyzed by Western blots using anti-SUPT16H or anti-SSRP1 antibody. GAPDH protein level was determined using an anti-GAPDH antibody to indicate equal loading of protein samples. The results were one representative from three independent experiments. F and G, the aforementioned cells were treated with DMSO or SAHA (0.5 μm) for 24 h. Cells were analyzed by flow cytometry. GFP-expressing cells were sorted using a defined gate, and a percentage of GFP-positive cells was measured and normalized to shNT-expressing cells. The results throughout are the means of three independent experiments ± S.D. *, p < 0.05 using Student's t test.
Depletion of FACT Proteins Reverses Latent HIV-1 in CD4+ T Cells
Although monocytes are one source of residual HIV-1, resting CD4+ T cells count for the majority of HIV-1 latent reservoirs (43, 44). Thus, we then determined the role of FACT proteins in HIV-1 latency in CD4+ T cells. Using a J-LAT cell line (Jurkat cells latently infected with a HIV-1 virus encoding GFP), J-LAT A2, we confirmed that SUPT16H and SSRP1 were depleted because of the expression of their shRNAs (Fig. 5A) and that depletion of these proteins spontaneously reversed latent HIV-1 in J-LAT A2 cells (Fig. 5B). Similar to THP89GFP cells, silencing of FACT proteins further enhanced the reversing efficiency of SAHA (Fig. 5C) but not prostratin (data not shown). To rule out the possibility that this effect was due to the an indirect effect on cell cycle, we analyzed the cell cycle profiles of SUPT16H or SSRP1 depleted J-LAT A2 cells stained with propidium iodide by using flow cytometry. The results indicated that the loss of SUPT16H protein has no effect on cell cycle, whereas the loss of SSRP1 led to more cells entering the S phase (Fig. 5D). SSRP1 was previously shown to play a separate SUPT16H-independent role in regulation of gene transcription (45), which may explain its additional effect on cell cycle. However, the effect of SSRP1 depletion on the S phase did not correlate with the reversal of HIV latency, so this is not a probable cause. J-LAT cell lines are T-lymphoma cells that are not natural hosts for HIV-1. We further studied the FACT protein effects on HIV-1 latency using a more physiologically relevant in vitro system. The frequency of latent infection in CD4+ T cells is extremely low (<1 of 1–5 million) in AIDS patients. It is difficult to conduct genetic manipulation of FACT proteins in these cells to study their functions. We established HIV-1 latency in primary cells ex vivo by adapting a HIV-1 latency model using primary CD4+ T cells (33, 34). Naïve CD4+ T cells were activated and nonpolarized by treatment of anti-CD3/anti-CD28 antibodies, as well as TGF-β, anti-IL-4, and anti-IL-12 antibodies. To allow the timing of the FACT shRNA expression after establishment of HIV-1 latency, we cloned shSUPT16H (sh2) and shSSRP1 (sh1) into a tet-inducible shRNA lentiviral expression vector, pINDUCER10 (30). These shRNAs were stably transduced in activated CD4+ T cells. These cells were then spinoculated with VSV-G pseudo-typed HIV-1 NL4–3-Luc (dEnv). Latent infection was established through a long term culture of cells with recombinant rIL-2. SUPT16H or SSRP1 shRNA expression was induced by treating cells with doxycycline. Latency-reversing effects were determined by measuring luciferase activity (Fig. 6A). Our results using CD4+ T cells isolated from three donors showed that the mRNA levels of SUPT16H and SSRP1 were efficiently depleted by induced shRNA expression (Fig. 6B). Furthermore, depletion of FACT proteins indeed spontaneously facilitated reversing of latent HIV-1 in nonpolarized, memory CD4+ T cells (Fig. 6C).
FIGURE 5.

Depletion of FACT proteins enhances reversal of HIV-1 latency in J-LAT cells. A, pAPM shRNA targeting SUPT16H or SSRP1 were transduced in J-LAT A2 cells. Cells with stable expression of shRNAs were lysed, separated by SDS-PAGE, and analyzed by Western blots using anti-SUPT16H or anti-SSRP1 antibody. The GAPDH protein level was determined using an anti-GAPDH antibody to indicate equal loading of protein samples. The results were one representative from three independent experiments. B and C, aforementioned cells were treated with DMSO, SAHA (0.5 μm), or prostratin (1 μm) for 24 h. The cells were then analyzed by flow cytometry. GFP-expressing cells were sorted using the defined gate, and the percentage of GFP-positive cells was measured and normalized to shNT-expressing cells. *, p < 0.05 using Student's t test. D, cell cycle analysis. J-LAT A2 cells stably expressing SUPT16H or SSRP1 shRNA were stained with propidium iodide and further analyzed by flow cytometry. Cells expressing shNT serve as a negative control. Flow cytometry plots were one representative from three independent experiments. Cell phase percentages were averaged from three independent experiments and represented in a 100% stacked bar graph. SSRP1 depletion led to changes of cell cycle (p < 0.05, Student's t test).
FIGURE 6.

Depletion of FACT proteins enhances reversal of HIV-1 latency in primary T cells. A, procedures are illustrated for generation of primary CD4+ T cell model of HIV-1 latency to study the latency-reversing effect of FACT proteins. It was adapted from Refs. 33 and 34. Colored bars indicate T cell differentiation and activation states. B, primary CD4+ T cells isolated from three donors were cultured ex vivo. Activated CD4+ T cells were transduced with pINDUCER10-shSUPT16H (sh2), shSSRP1 (sh1), or shNT. Cells stably expressing shRNAs were selected by treating cells with puromycin. shRNA expression was induced with doxycycline. Total RNAs were extracted from these cells and analyzed by reverse transcription and qPCR for measuring the transcripts of FACT proteins. Level of SUPT16H or SSRP1 transcript was normalized to shNT-expressing cells for individual donor. C, pINDUCER10-shSUPT16H or shSSRP1 stably transduced primary memory CD4+ T cells were infected with VSV-G pseudo-typed HIV-1 NL4–3-Luc (dEnv) viruses. Cells were kept in long term culture to permit HIV-1 latency. HIV-1 latently infected cells were treated with doxycycline to induce shRNA expression. Luciferase activity was measured for cells depleted of SUPT16H or SSRP1 and normalized to shNT-expressing cells. The results throughout are the means of three independent experiments ± S.D. * indicates p < 0.05 using Student's t test.
FACT Proteins Elicit Similar Effects on HTLV-1 Transcription
HTLV-1 is a human retrovirus that shares common features of transcriptional regulation with HIV-1, including the use of P-TEFb (46, 47). We expect that FACT proteins elicit similar effects on HTLV-1 transcription. Depletion of FACT proteins increased the luciferase expression driven by the HTLV-1 LTR promoter in HEK293 cells that were transfected with HTLV-1 LTR-Luciferase, pTK-Renilla, and BC12-Tax vectors (Fig. 7, A and B). The co-immunoprecipitation experiments in HEK293 cells transfected with pB-His6-Tax also confirmed that exogenously expressed Tax protein associated with endogenous SUPT16H (Fig. 7C). We further investigated the activity of FACT proteins on HTLV-1 transcription and latency in a human T cell leukemia cell line, MT-2, which is transformed with HTLV-1 (48). Stable expression of shSUPT16H or shSSRP1 in these cells efficiently depleted FACT proteins (Fig. 7D). Depletion of SUPT16H or SSRP1 increased the expression level of the HTLV-1 gag/pol gene assayed by qPCR (49), when the cells were treated with SAHA (Fig. 7E).
FIGURE 7.
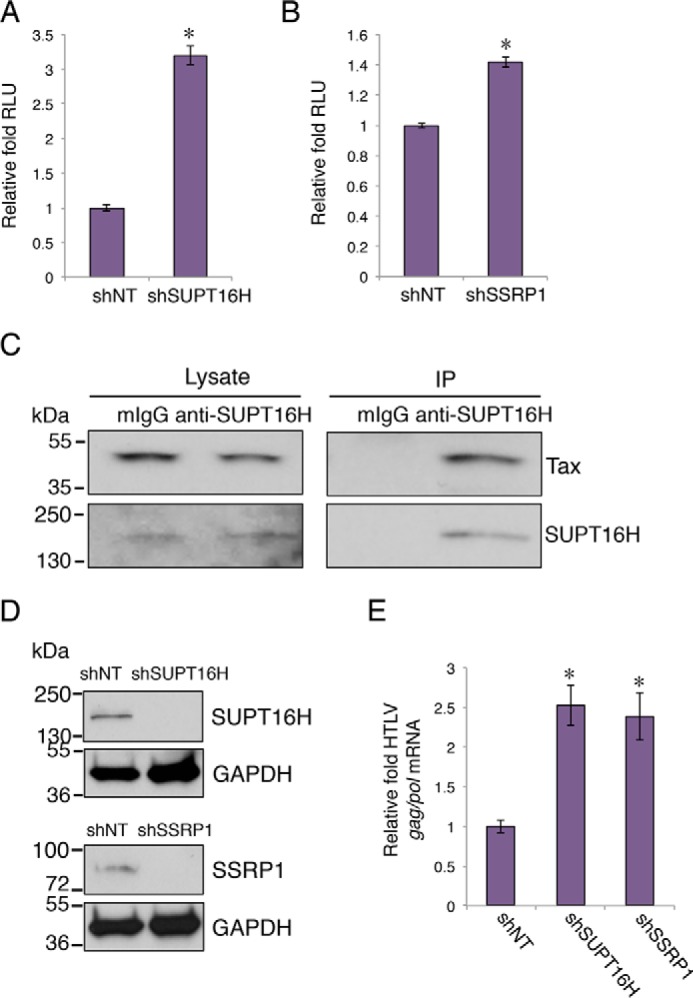
FACT proteins demonstrate similar effect on HTLV-1 transcription. A and B, the vectors HTLV-1 LTR-luciferase, pTK-Renilla, and BC12-Tax, were co-transfected in HEK293 cells stably expressing shRNAs of SUPT16H (A) or SSRP1 (B). At 48 h post-transfection, luciferase activity was measured and normalized to the Renilla signal. The relative light unit (RLU) of shSUPT16H or shSSRP1 expressing cells was normalized to shNT cells. C, HEK293 cells were transfected with pB-His6-Tax vector. At 48 h post-transfection, cells were lysed and subjected to IP assays using an anti-SUPT16H or mIgG antibody. Cell lysate and precipitated protein samples were separated by SDS-PAGE. Protein level of His6-Tax was determined by Western blots using a mouse anti-Tax antibody (4C5). The results were one representative from three independent experiments. The results were one representative from three independent experiments. D, HTLV-1 transformed MT-2 cells were stably transduced with pAPM-shSUPT16H (sh2), shSSRP1 (sh1), or shNT. Cells were lysed, separated by SDS-PAGE, and analyzed by Western blots using anti-SUPT16H or anti-SSRP1 antibody. The GAPDH protein level was determined using an anti-GAPDH antibody to indicate equal loading of protein samples. The results were one representative from three independent experiments. E, cDNA samples from the aforementioned cells treated with SAHA (0.5 μm) were subjected to qPCR assays to measure the HTLV-1 gag/pol mRNA. Level of viral transcripts was normalized to shNT cells. The results throughout are the means of three independent experiments ± S.D. *, p < 0.05 using Student's t test.
Discussion
HIV-1 only encodes 12 genes and undertakes comprehensive interplay with host cellular machineries to fulfill its life cycle, especially silencing the gene expression of integrated proviral HIV-1 resulting in latency. Consequentially, the fate of HIV-1 proviruses is mostly dependent upon the cellular environment and host genetic function. Host factors regulating HIV-1 transcription are the key to control the switch between latency and reactivation of HIV-1 proviruses. Earlier studies focusing on host genes that are required for HIV-1 transcription have lead to the identification of major regulators that promote either initiation or elongation of LTR transcription. The LTR promoter alone is able to initiate transcription very efficiently. Host transcriptional activators, such as NF-κB, NFAT, and C/EBPβ, recognize binding sites at the LTR promoter and further recruit chromatin-remodeling proteins (HATs, SWI-SNF complexes, and demethylases). These proteins modify nucleosomes at the LTR and lead to induction of transcription (50–52). The LTR promoter establishes only nonprocessive (basal) transcription in the absence of Tat and exclusively depends on Tat for transcriptional elongation. Tat recruits P-TEFb, a critical host protein complex required for HIV-1 transcription, to phosphorylate the paused RNA polymerase II at the LTR and activates it for efficient elongation. However, host cells also develop the mechanisms to silence HIV-1 transcription, which is necessary for establishing and maintaining latency. There are a few factors reported previously, such as CBF1 (53), NELFCD (38), and HDACs (54), but they play a relatively general role in transcriptional suppression with a lack of specificity to HIV-1. From our unbiased RNAi screens and further characterization of prioritized genes, we were able to identify a set of host factors that restrict HIV-1 replication through transcriptional suppression, including BRD4 and FACT proteins (SUPT16H and SSRP1) (Fig. 1A). These endeavors have allowed us to identify novel host regulators for HIV-1 transcription and further study their functions in HIV-1 latency and reactivation. Our goal is to improve the understanding of host-HIV interactions at latent phase, so that we can manage or manipulate them for elimination of residual HIV-1 in reservoir cells. Our work has also demonstrated that this RNAi-mediated functional genomic approach is equally useful for identification of host factors that restrict HIV-1 replication, because multiple earlier screens mainly focused on identifying host dependence factors for HIV-1 (55–57). The newly identified transcriptional suppressors, SUPT16H and SSRP1, seem to have similar function for other retroviruses (HTLV-1), regulating its transcription and latency, indicating that many host factors restricting HIV-1 may be evolutionarily conserved and play a common role in the replication of retroviruses in general (Fig. 8).
FIGURE 8.

A working model for FACT proteins exhibiting suppression of lentiviral transcription and facilitating viral latency through interaction with Tat/Tax, interfering with P-TEFb.
The FACT protein dimer (SUPT16H and SSRP1) was initially identified in mammalian cells as a critical chromatin-specific transcriptional elongation factor that facilitates nucleosome alteration and releases RNA polymerase II from a nucleosome-induced block to prompt productive transcription (24, 25). A yeast homologue is the CP complex, an abundant dimer of SPT16 (CDC68) and POB3 proteins, which is necessary for the transactivation of many genes (58). It is generally believed that the FACT complex facilitates transcriptional elongation based upon these studies. However, it is not completely understood how FACT can coordinate its assembly and disassembly activity to facilitate the sliding of RNA polymerase. Two major models describe the mechanism of FACT-mediated nucleosome reorganization: dimer eviction model and global accessibility/noneviction model (59). More intriguingly, studies in yeast have indicated that SPT16/CDC68 is important for the maintenance of chromatin-mediated repression in the absence of transactivation (58), but there is no such evidence in mammalian cells. Because of a lack of current studies focusing on this complex, we cannot rule out that FACT proteins may exert both promptive and suppressive effects on gene expression in human cells. Nevertheless, it is still surprising that the FACT complex, a general transcriptional facilitator, imposes an inhibitory effect on HIV-1 transcription, opposing earlier speculation (60). Our results have indicated that the presence of SUPT16H may interfere with interactions of P-TEFb with Tat-LTR (Fig. 3, A and B), which provides a novel and plausible explanation for FACT-mediated inhibition of HIV-1 transcription. Our tentative model indicates that LTR recruitment of FACT by Tat may generate a steric hindrance for P-TEFb association, resulting in reduced efficiency of RNA polymerase II (Fig. 8). Tax plays a very similar role as Tat, recruiting P-TEFb for stimulation of HTLV-1 transcription (46, 47, 61), which leads us to believe that FACT proteins may also interfere with P-TEFb required for HTLV-1 transcription through association with Tax (Fig. 7C). Further study of FACT proteins in human retroviral transcription, as well as more profound analysis of FACT-mediated transcriptional regulation at the genome-wide scale in retrovirus-infected cells, may help to unravel the complicated functions of FACT that could be modulated by viral components. In addition, FACT is also a multifunctional protein complex that regulates other cellular processes, such as DNA replication (62–64), DNA damage response (65, 66), and cell cycle progression (67, 68). Whether FACT-mediated nontranscriptional functions may affect HIV-1/HTLV-1 replication is unclear and requires further investigation.
The direct evidence that the FACT proteins affect HIV-1 transcription is their association with Tat and the LTR promoter. Our protein interaction studies have indicated that Tat associates with SUPT16H but not SSRP1, either endogenous or transiently expressed (Fig. 2, A and B). Interestingly, we found that the presence of Tat did not disturb the association between SUPT16H and SSRP1 (Fig. 2F). These studies suggest that SUPT16H directly binds to Tat. This binding may explain how FACT suppresses both HIV-1 transcriptional initiation and elongation (Fig. 4, C and D), because Tat increases initiation complex formation on LTR and stabilizes the complex during elongation (69, 70). Furthermore, our chromatin association studies have confirmed that both SUPT16H and SSRP1 are recruited to the LTR (Fig. 2, C and D). An intriguing explanation for SSRP1 association with LTR but not Tat could be that either SSRP1 may indirectly associate with Tat through SUPT16H but to a much lesser degree or that SSRP1 contains a high mobility group box so that its recruitment to LTR may be Tat-independent.
Because FACT proteins suppress HIV-1 transcription, we speculate that their presence may silence viral gene expression and promote HIV-1 latency. Earlier studies demonstrate that nucleosomes are precisely positioned at the HIV-1 LTR, and their organization is conserved (71–73). We expect that FACT proteins may impose a similar effect on the HIV-1 LTR chromatin across various cell models of HIV-1 latency. We used multiple cell models of HIV-1 latency to confirm that depletion of FACT proteins alleviates transcriptional suppression and reverses latent HIV-1 (Figs. 4–6). Most compellingly, we were able to validate FACT functions in HIV-1 latently infected primary CD4+ T cells (Fig. 6). In this model, activated CD4+ T cells were nonpolarized to mimic central memory CD4+ T cells to establish HIV-1 latency (33, 34). Depletion of FACT proteins in these cells spontaneously reversed HIV-1 latency, indicating that FACT proteins are main host factors for maintaining HIV-1 latency, and their removal may directly activate some of latent HIV-1 without need of other cell signaling activation. Another significant discovery is that depletion of FACT proteins synergized with the known latency-reversing agent, SAHA (Fig. 5C). Because SAHA blocks local histone deacetylation and allows loading of the transcriptional initiation complex onto the viral LTR to induce transcription, whereas depletion of FACT proteins enhances HIV-1 transcriptional elongation by increasing binding of P-TEFb to Tat-LTR, this may explain the enhanced reversing of latent HIV-1. Failure to produce any synergistic effects between FACT depletion and prostratin treatment may be due to prostratin quenching HIV-1 reversal, masking the inhibitory effect of FACT proteins. Future studies should also investigate the role of FACT proteins in establishment of HIV-1 latency in activated primary CD4+ T cells once they return to the resting stage. Although these results from primary cells are convincing, the more physiologically relevant HIV-1 latent reservoir cells to test ex vivo are CD8-depleted peripheral blood mononuclear cells isolated from HAART-treated AIDS patients with undetectable HIV-1 viral loads (22). However, it is difficult to perform genetic manipulation (shRNA-mediated knockdown or CRISPR/Cas9-mediated knockout) in these cells because the frequency of HIV-1 latently infected cells is extremely low (<1 of 1–5 million). The limited transduction efficiency of lentiviruses is unable to guarantee the successful delivery of shRNAs or sgRNAs (CRISPR/Cas9) into cells that actually harbor latent HIV-1 proviruses. Hence, we shall initiate the development and/or screening of small molecule compounds that are able to specifically disrupt the interaction of FACT proteins with Tat-LTR so that their suppressive effect on viral transcription is removed. These FACT inhibitors can be further tested in HIV-1 latent reservoirs of AIDS patients.
Author Contributions
J. Z. and S. E. conceived the project. J. Z. and H. H. designed the study and wrote the paper. H. H., N. S., D. P., S. S., and M. D. conducted the experiments. J. Z., H. H., N. S., and H. M. analyzed the results. K. G. and C.-Z. G. provided the reagents and advised the study. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Dr. David Levy at New York University for the gift of THP89GFP cells. We also acknowledge Drs. Stephen Dewhurst and Sanjay Maggirwar (University of Rochester Medical Center) for helpful discussions and preparation of the manuscript.
This work is supported by National Institutes of Health Grants R21AI116180 and R01GM117838 (to J. Z.) and in part by the University of Rochester Centers for AIDS Research Grant P30AI078498 from National Institutes of Health and the University of Rochester Center for Integrative Bioinformatics and Experimental Mathematics pilot grant. The authors declare that they have no conflicts of interest with the contents of this article.
- HAART
- highly active antiretroviral therapy
- HDAC
- histone deacetylase
- RIGER
- RNAi gene enrichment ranking
- LTR
- long terminal repeat
- SAHA
- suberoylanilide hydroxamic acid
- NT
- nontargeting
- qPCR
- quantitative PCR
- IP
- immunoprecipitation
- mIgG
- mouse IgG
- BRD4
- bromodomain-containing protein 4.
References
- 1.Maartens G., Celum C., and Lewin S. R. (2014) HIV infection: epidemiology, pathogenesis, treatment, and prevention. Lancet 384, 258–271 [DOI] [PubMed] [Google Scholar]
- 2.Finzi D., Hermankova M., Pierson T., Carruth L. M., Buck C., Chaisson R. E., Quinn T. C., Chadwick K., Margolick J., Brookmeyer R., Gallant J., Markowitz M., Ho D. D., Richman D. D., and Siliciano R. F. (1997) Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 278, 1295–1300 [DOI] [PubMed] [Google Scholar]
- 3.Wong J. K., Hezareh M., Günthard H. F., Havlir D. V., Ignacio C. C., Spina C. A., and Richman D. D. (1997) Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science 278, 1291–1295 [DOI] [PubMed] [Google Scholar]
- 4.Shan L., and Siliciano R. F. (2013) From reactivation of latent HIV-1 to elimination of the latent reservoir: the presence of multiple barriers to viral eradication. BioEssays 35, 544–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eriksson S., Graf E. H., Dahl V., Strain M. C., Yukl S. A., Lysenko E. S., Bosch R. J., Lai J., Chioma S., Emad F., Abdel-Mohsen M., Hoh R., Hecht F., Hunt P., Somsouk M., Wong J., Johnston R., Siliciano R. F., Richman D. D., O'Doherty U., Palmer S., Deeks S. G., and Siliciano J. D. (2013) Comparative analysis of measures of viral reservoirs in HIV-1 eradication studies. PLoS Pathog. 9, e1003174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Katlama C., Deeks S. G., Autran B., Martinez-Picado J., van Lunzen J., Rouzioux C., Miller M., Vella S., Schmitz J. E., Ahlers J., Richman D. D., and Sekaly R. P. (2013) Barriers to a cure for HIV: new ways to target and eradicate HIV-1 reservoirs. Lancet 381, 2109–2117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Larsson M., Shankar E. M., Che K. F., Saeidi A., Ellegård R., Barathan M., Velu V., and Kamarulzaman A. (2013) Molecular signatures of T-cell inhibition in HIV-1 infection. Retrovirology 10, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ott M., Geyer M., and Zhou Q. (2011) The control of HIV transcription: keeping RNA polymerase II on track. Cell Host Microbe 10, 426–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Han Y., Lassen K., Monie D., Sedaghat A. R., Shimoji S., Liu X., Pierson T. C., Margolick J. B., Siliciano R. F., and Siliciano J. D. (2004) Resting CD4+ T cells from human immunodeficiency virus type 1 (HIV-1)-infected individuals carry integrated HIV-1 genomes within actively transcribed host genes. J. Virol. 78, 6122–6133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Han Y., Lin Y. B., An W., Xu J., Yang H. C., O'Connell K., Dordai D., Boeke J. D., Siliciano J. D., and Siliciano R. F. (2008) Orientation-dependent regulation of integrated HIV-1 expression by host gene transcriptional readthrough. Cell Host Microbe 4, 134–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Siliciano R. F., and Greene W. C. (2011) HIV latency. Cold Spring Harb. Perspect. Med. 1, a007096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coiras M., López-Huertas M. R., Pérez-Olmeda M., and Alcamí J. (2009) Understanding HIV-1 latency provides clues for the eradication of long-term reservoirs. Nat. Rev. Microbiol. 7, 798–812 [DOI] [PubMed] [Google Scholar]
- 13.Karn J. (2011) The molecular biology of HIV latency: breaking and restoring the Tat-dependent transcriptional circuit. Curr. Opin. HIV AIDS 6, 4–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Archin N. M., Liberty A. L., Kashuba A. D., Choudhary S. K., Kuruc J. D., Crooks A. M., Parker D. C., Anderson E. M., Kearney M. F., Strain M. C., Richman D. D., Hudgens M. G., Bosch R. J., Coffin J. M., Eron J. J., Hazuda D. J., and Margolis D. M. (2012) Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature 487, 482–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barton K. M., Burch B. D., Soriano-Sarabia N., and Margolis D. M. (2013) Prospects for treatment of latent HIV. Clin. Pharmacol. Ther. 93, 46–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duh E. J., Maury W. J., Folks T. M., Fauci A. S., and Rabson A. B. (1989) Tumor necrosis factor α activates human immunodeficiency virus type 1 through induction of nuclear factor binding to the NF-κB sites in the long terminal repeat. Proc. Natl. Acad. Sci. U.S.A. 86, 5974–5978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Williams S. A., Chen L. F., Kwon H., Fenard D., Bisgrove D., Verdin E., and Greene W. C. (2004) Prostratin antagonizes HIV latency by activating NF-κB. J. Biol. Chem. 279, 42008–42017 [DOI] [PubMed] [Google Scholar]
- 18.Sánchez-Duffhues G., Vo M. Q., Pérez M., Calzado M. A., Moreno S., Appendino G., and Muñoz E. (2011) Activation of latent HIV-1 expression by protein kinase C agonists: a novel therapeutic approach to eradicate HIV-1 reservoirs. Curr. Drug Targets 12, 348–356 [DOI] [PubMed] [Google Scholar]
- 19.Klichko V., Archin N., Kaur R., Lehrman G., and Margolis D. (2006) Hexamethylbisacetamide remodels the human immunodeficiency virus type 1 (HIV-1) promoter and induces Tat-independent HIV-1 expression but blunts cell activation. J. Virol. 80, 4570–4579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xing S., Bullen C. K., Shroff N. S., Shan L., Yang H. C., Manucci J. L., Bhat S., Zhang H., Margolick J. B., Quinn T. C., Margolis D. M., Siliciano J. D., and Siliciano R. F. (2011) Disulfiram reactivates latent HIV-1 in a Bcl-2-transduced primary CD4+ T cell model without inducing global T cell activation. J. Virol. 85, 6060–6064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ho Y. C., Shan L., Hosmane N. N., Wang J., Laskey S. B., Rosenbloom D. I., Lai J., Blankson J. N., Siliciano J. D., and Siliciano R. F. (2013) Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell 155, 540–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu J., Gaiha G. D., John S. P., Pertel T., Chin C. R., Gao G., Qu H., Walker B. D., Elledge S. J., and Brass A. L. (2012) Reactivation of latent HIV-1 by inhibition of BRD4. Cell Reports 2, 807–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu J., Davoli T., Perriera J. M., Chin C. R., Gaiha G. D., John S. P., Sigiollot F. D., Gao G., Xu Q., Qu H., Pertel T., Sims J. S., Smith J. A., Baker R. E., Maranda L., Ng A., Elledge S. J., and Brass A. L. (2014) Comprehensive identification of host modulators of HIV-1 replication using multiple orthologous RNAi reagents. Cell Reports 9, 752–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Orphanides G., Wu W. H., Lane W. S., Hampsey M., and Reinberg D. (1999) The chromatin-specific transcription elongation factor FACT comprises human SPT16 and SSRP1 proteins. Nature 400, 284–288 [DOI] [PubMed] [Google Scholar]
- 25.Belotserkovskaya R., Oh S., Bondarenko V. A., Orphanides G., Studitsky V. M., and Reinberg D. (2003) FACT facilitates transcription-dependent nucleosome alteration. Science 301, 1090–1093 [DOI] [PubMed] [Google Scholar]
- 26.Garcia H., Miecznikowski J. C., Safina A., Commane M., Ruusulehto A., Kilpinen S., Leach R. W., Attwood K., Li Y., Degan S., Omilian A. R., Guryanova O., Papantonopoulou O., Wang J., Buck M., Liu S., Morrison C., and Gurova K. V. (2013) Facilitates chromatin transcription complex is an “accelerator” of tumor transformation and potential marker and target of aggressive cancers. Cell Rep. 4, 159–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kutsch O., Benveniste E. N., Shaw G. M., and Levy D. N. (2002) Direct and quantitative single-cell analysis of human immunodeficiency virus type 1 reactivation from latency. J. Virol. 76, 8776–8786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhi H., Yang L., Kuo Y. L., Ho Y. K., Shih H. M., and Giam C. Z. (2011) NF-κB hyper-activation by HTLV-1 tax induces cellular senescence, but can be alleviated by the viral anti-sense protein HBZ. PLoS Pathog. 7, e1002025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pertel T., Hausmann S., Morger D., Züger S., Guerra J., Lascano J., Reinhard C., Santoni F. A., Uchil P. D., Chatel L., Bisiaux A., Albert M. L., Strambio-De-Castillia C., Mothes W., Pizzato M., Grütter M. G., and Luban J. (2011) TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature 472, 361–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meerbrey K. L., Hu G., Kessler J. D., Roarty K., Li M. Z., Fang J. E., Herschkowitz J. I., Burrows A. E., Ciccia A., Sun T., Schmitt E. M., Bernardi R. J., Fu X., Bland C. S., Cooper T. A., Schiff R., Rosen J. M., Westbrook T. F., and Elledge S. J. (2011) The pINDUCER lentiviral toolkit for inducible RNA interference in vitro and in vivo. Proc. Natl. Acad. Sci. U.S.A. 108, 3665–3670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Power D., Santoso N. G., Dieringer M., Yu J., Huang H., Simpson S., Seth I., Miao H., and Zhu J. (2015) IFI44 suppresses HIV-1 LTR promoter activity and facilitates its latency. Virology 481, 142–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jadlowsky J. K., Wong J. Y., Graham A. C., Dobrowolski C., Devor R. L., Adams M. D., Fujinaga K., and Karn J. (2014) Negative elongation factor is required for the maintenance of proviral latency but does not induce promoter-proximal pausing of RNA polymerase II on the HIV long terminal repeat. Mol. Cell. Biol. 34, 1911–1928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bosque A., and Planelles V. (2009) Induction of HIV-1 latency and reactivation in primary memory CD4+ T cells. Blood 113, 58–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bosque A., and Planelles V. (2011) Studies of HIV-1 latency in an ex vivo model that uses primary central memory T cells. Methods 53, 54–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luo B., Cheung H. W., Subramanian A., Sharifnia T., Okamoto M., Yang X., Hinkle G., Boehm J. S., Beroukhim R., Weir B. A., Mermel C., Barbie D. A., Awad T., Zhou X., Nguyen T., Piqani B., Li C., Golub T. R., Meyerson M., Hacohen N., Hahn W. C., Lander E. S., Sabatini D. M., and Root D. E. (2008) Highly parallel identification of essential genes in cancer cells. Proc. Natl. Acad. Sci. U.S.A. 105, 20380–20385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cheung H. W., Cowley G. S., Weir B. A., Boehm J. S., Rusin S., Scott J. A., East A., Ali L. D., Lizotte P. H., Wong T. C., Jiang G., Hsiao J., Mermel C. H., Getz G., Barretina J., Gopal S., Tamayo P., Gould J., Tsherniak A., Stransky N., Luo B., Ren Y., Drapkin R., Bhatia S. N., Mesirov J. P., Garraway L. A., Meyerson M., Lander E. S., Root D. E., and Hahn W. C. (2011) Systematic investigation of genetic vulnerabilities across cancer cell lines reveals lineage-specific dependencies in ovarian cancer. Proc. Natl. Acad. Sci. U.S.A. 108, 12372–12377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Khan S. Z., and Mitra D. (2011) Cyclin K inhibits HIV-1 gene expression and replication by interfering with cyclin-dependent kinase 9 (CDK9)-cyclin T1 interaction in Nef-dependent manner. J. Biol. Chem. 286, 22943–22954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang Z., Klatt A., Gilmour D. S., and Henderson A. J. (2007) Negative elongation factor NELF represses human immunodeficiency virus transcription by pausing the RNA polymerase II complex. J. Biol. Chem. 282, 16981–16988 [DOI] [PubMed] [Google Scholar]
- 39.Wei P., Garber M. E., Fang S. M., Fischer W. H., and Jones K. A. (1998) A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell 92, 451–462 [DOI] [PubMed] [Google Scholar]
- 40.Ping Y. H., and Rana T. M. (2001) DSIF and NELF interact with RNA polymerase II elongation complex and HIV-1 Tat stimulates P-TEFb-mediated phosphorylation of RNA polymerase II and DSIF during transcription elongation. J. Biol. Chem. 276, 12951–12958 [DOI] [PubMed] [Google Scholar]
- 41.Emiliani S., Fischle W., Ott M., Van Lint C., Amella C. A., and Verdin E. (1998) Mutations in the tat gene are responsible for human immunodeficiency virus type 1 postintegration latency in the U1 cell line. J. Virol. 72, 1666–1670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fujinaga K., Cujec T. P., Peng J., Garriga J., Price D. H., Graña X., and Peterlin B. M. (1998) The ability of positive transcription elongation factor B to transactivate human immunodeficiency virus transcription depends on a functional kinase domain, cyclin T1, and Tat. J. Virol. 72, 7154–7159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Coleman C. M., and Wu L. (2009) HIV interactions with monocytes and dendritic cells: viral latency and reservoirs. Retrovirology 6, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chun T. W., Finzi D., Margolick J., Chadwick K., Schwartz D., and Siliciano R. F. (1995) In vivo fate of HIV-1-infected T cells: quantitative analysis of the transition to stable latency. Nat. Med. 1, 1284–1290 [DOI] [PubMed] [Google Scholar]
- 45.Li Y., Zeng S. X., Landais I., and Lu H. (2007) Human SSRP1 has Spt16-dependent and -independent roles in gene transcription. J. Biol. Chem. 282, 6936–6945 [DOI] [PubMed] [Google Scholar]
- 46.Cho W. K., Jang M. K., Huang K., Pise-Masison C. A., and Brady J. N. (2010) Human T-lymphotropic virus type 1 Tax protein complexes with P-TEFb and competes for Brd4 and 7SK snRNP/HEXIM1 binding. J. Virol. 84, 12801–12809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cho W. K., Zhou M., Jang M. K., Huang K., Jeong S. J., Ozato K., and Brady J. N. (2007) Modulation of the Brd4/P-TEFb interaction by the human T-lymphotropic virus type 1 tax protein. J. Virol. 81, 11179–11186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Harada S., Koyanagi Y., and Yamamoto N. (1985) Infection of HTLV-III/LAV in HTLV-I-carrying cells MT-2 and MT-4 and application in a plaque assay. Science 229, 563–566 [DOI] [PubMed] [Google Scholar]
- 49.Philip S., Zahoor M. A., Zhi H., Ho Y. K., and Giam C. Z. (2014) Regulation of human T-lymphotropic virus type I latency and reactivation by HBZ and Rex. PLoS Pathog. 10, e1004040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schiralli Lester G. M., and Henderson A. J. (2012) Mechanisms of HIV transcriptional regulation and their contribution to latency. Mol. Biol. Int. 2012, 614120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Van Duyne R., Guendel I., Narayanan A., Gregg E., Shafagati N., Tyagi M., Easley R., Klase Z., Nekhai S., Kehn-Hall K., and Kashanchi F. (2011) Varying modulation of HIV-1 LTR activity by Baf complexes. J. Mol. Biol. 411, 581–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rafati H., Parra M., Hakre S., Moshkin Y., Verdin E., and Mahmoudi T. (2011) Repressive LTR nucleosome positioning by the BAF complex is required for HIV latency. PLoS Biol. 9, e1001206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tyagi M., and Karn J. (2007) CBF-1 promotes transcriptional silencing during the establishment of HIV-1 latency. EMBO J. 26, 4985–4995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Keedy K. S., Archin N. M., Gates A. T., Espeseth A., Hazuda D. J., and Margolis D. M. (2009) A limited group of class I histone deacetylases acts to repress human immunodeficiency virus type 1 expression. J. Virol. 83, 4749–4756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brass A. L., Dykxhoorn D. M., Benita Y., Yan N., Engelman A., Xavier R. J., Lieberman J., and Elledge S. J. (2008) Identification of host proteins required for HIV infection through a functional genomic screen. Science 319, 921–926 [DOI] [PubMed] [Google Scholar]
- 56.König R., Zhou Y., Elleder D., Diamond T. L., Bonamy G. M., Irelan J. T., Chiang C. Y., Tu B. P., De Jesus P. D., Lilley C. E., Seidel S., Opaluch A. M., Caldwell J. S., Weitzman M. D., Kuhen K. L., Bandyopadhyay S., Ideker T., Orth A. P., Miraglia L. J., Bushman F. D., Young J. A., and Chanda S. K. (2008) Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell 135, 49–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhou H., Xu M., Huang Q., Gates A. T., Zhang X. D., Castle J. C., Stec E., Ferrer M., Strulovici B., Hazuda D. J., and Espeseth A. S. (2008) Genome-scale RNAi screen for host factors required for HIV replication. Cell Host Microbe 4, 495–504 [DOI] [PubMed] [Google Scholar]
- 58.Brewster N. K., Johnston G. C., and Singer R. A. (1998) Characterization of the CP complex, an abundant dimer of Cdc68 and Pob3 proteins that regulates yeast transcriptional activation and chromatin repression. J. Biol. Chem. 273, 21972–21979 [DOI] [PubMed] [Google Scholar]
- 59.Winkler D. D., and Luger K. (2011) The histone chaperone FACT: structural insights and mechanisms for nucleosome reorganization. J. Biol. Chem. 286, 18369–18374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Easley R., Van Duyne R., Coley W., Guendel I., Dadgar S., Kehn-Hall K., and Kashanchi F. (2010) Chromatin dynamics associated with HIV-1 Tat-activated transcription. Biochim. Biophys. Acta 1799, 275–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhou M., Lu H., Park H., Wilson-Chiru J., Linton R., and Brady J. N. (2006) Tax interacts with P-TEFb in a novel manner to stimulate human T-lymphotropic virus type 1 transcription. J. Virol. 80, 4781–4791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Abe T., Sugimura K., Hosono Y., Takami Y., Akita M., Yoshimura A., Tada S., Nakayama T., Murofushi H., Okumura K., Takeda S., Horikoshi M., Seki M., and Enomoto T. (2011) The histone chaperone facilitates chromatin transcription (FACT) protein maintains normal replication fork rates. J. Biol. Chem. 286, 30504–30512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Han J., Li Q., McCullough L., Kettelkamp C., Formosa T., and Zhang Z. (2010) Ubiquitylation of FACT by the cullin-E3 ligase Rtt101 connects FACT to DNA replication. Genes Dev. 24, 1485–1490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.VanDemark A. P., Blanksma M., Ferris E., Heroux A., Hill C. P., and Formosa T. (2006) The structure of the yFACT Pob3-M domain, its interaction with the DNA replication factor RPA, and a potential role in nucleosome deposition. Mol. Cell 22, 363–374 [DOI] [PubMed] [Google Scholar]
- 65.Heo K., Kim H., Choi S. H., Choi J., Kim K., Gu J., Lieber M. R., Yang A. S., and An W. (2008) FACT-mediated exchange of histone variant H2AX regulated by phosphorylation of H2AX and ADP-ribosylation of Spt16. Mol. Cell 30, 86–97 [DOI] [PubMed] [Google Scholar]
- 66.Dinant C., Ampatziadis-Michailidis G., Lans H., Tresini M., Lagarou A., Grosbart M., Theil A. F., van Cappellen W. A., Kimura H., Bartek J., Fousteri M., Houtsmuller A. B., Vermeulen W., and Marteijn J. A. (2013) Enhanced chromatin dynamics by FACT promotes transcriptional restart after UV-induced DNA damage. Mol. Cell 51, 469–479 [DOI] [PubMed] [Google Scholar]
- 67.Tan B. C., and Lee S. (2004) Nek9, a novel FACT-associated protein, modulates interphase progression. J. Biol. Chem. 279, 9321–9330 [DOI] [PubMed] [Google Scholar]
- 68.Morillo-Huesca M., Maya D., Muñoz-Centeno M. C., Singh R. K., Oreal V., Reddy G. U., Liang D., Géli V., Gunjan A., and Chávez S. (2010) FACT prevents the accumulation of free histones evicted from transcribed chromatin and a subsequent cell cycle delay in G1. PLoS Genet. 6, e1000964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Laspia M. F., Rice A. P., and Mathews M. B. (1989) HIV-1 Tat protein increases transcriptional initiation and stabilizes elongation. Cell 59, 283–292 [DOI] [PubMed] [Google Scholar]
- 70.Brady J., and Kashanchi F. (2005) Tat gets the “green” light on transcription initiation. Retrovirology 2, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Verdin E. (1991) DNase I-hypersensitive sites are associated with both long terminal repeats and with the intragenic enhancer of integrated human immunodeficiency virus type 1. J. Virol. 65, 6790–6799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Verdin E., Paras P. Jr., and Van Lint C. (1993) Chromatin disruption in the promoter of human immunodeficiency virus type 1 during transcriptional activation. EMBO J. 12, 3249–3259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Colin L., and Van Lint C. (2009) Molecular control of HIV-1 postintegration latency: implications for the development of new therapeutic strategies. Retrovirology 6, 111. [DOI] [PMC free article] [PubMed] [Google Scholar]