

Background: Acyl-CoA mutases catalyze radical-based carbon skeleton rearrangements.
Results: Crystal structures of isobutyryl-CoA mutase in complex with four different substrates reveal active site architecture and determinants of substrate specificity.
Conclusion: Identification of specificity-determining residues allows for prediction of new acyl-CoA mutase activities.
Significance: Improved understanding of acyl-CoA mutase substrate specificity is critical for biotechnological and engineering applications.
Keywords: adenosylcobalamin (AdoCbl), enzyme catalysis, metalloenzyme, substrate specificity, x-ray crystallography, acyl-CoA mutase
Abstract
Acyl-CoA mutases are a growing class of adenosylcobalamin-dependent radical enzymes that perform challenging carbon skeleton rearrangements in primary and secondary metabolism. Members of this class of enzymes must precisely control substrate positioning to prevent oxidative interception of radical intermediates during catalysis. Our understanding of substrate specificity and catalysis in acyl-CoA mutases, however, is incomplete. Here, we present crystal structures of IcmF, a natural fusion protein variant of isobutyryl-CoA mutase, in complex with the adenosylcobalamin cofactor and four different acyl-CoA substrates. These structures demonstrate how the active site is designed to accommodate the aliphatic acyl chains of each substrate. The structures suggest that a conformational change of the 5′-deoxyadenosyl group from C2′-endo to C3′-endo could contribute to initiation of catalysis. Furthermore, detailed bioinformatic analyses guided by our structural findings identify critical determinants of acyl-CoA mutase substrate specificity and predict new acyl-CoA mutase-catalyzed reactions. These results expand our understanding of the substrate specificity and the catalytic scope of acyl-CoA mutases and could benefit engineering efforts for biotechnological applications ranging from production of biofuels and commercial products to hydrocarbon remediation.
Introduction
Adenosylcobalamin (AdoCbl,3 coenzyme B12) is an organometallic enzyme cofactor for radical chemistry. Its reactivity is based on a unique covalent cobalt-carbon (Co–C) bond that is sufficiently weak to allow for reversible homolytic cleavage in enzyme active sites, generating a 5′-deoxyadenosyl radical in the presence of an appropriate substrate (1, 2). The radical can then abstract a substrate hydrogen atom and initiate difficult chemical transformations such as the carbon-skeleton rearrangements of acyl-CoA thioesters catalyzed by a major group of AdoCbl-dependent enzymes as part of primary and secondary metabolic pathways (Fig. 1) (3–5). These so-called acyl-CoA mutases allow for the degradation and biosynthesis of branched-chain compounds by altering the level of branching and therefore have biotechnological potential ranging from remediation of hydrocarbon pollution to synthesis of commercial products such as solvents and fragrances (4, 6, 7). To realize this potential and to engineer new activities, a better understanding of the factors governing substrate specificity in acyl-CoA mutases is required.
FIGURE 1.

Reversible interconversions catalyzed by characterized (a–e) and proposed (f and g) acyl-CoA mutases. See main text for details. Two variants of HCM, HCM1 and HCM2, use (S)-3-hydroxybutyryl-CoA and (R)-3-hydroxybutyryl-CoA, respectively. R group in g denotes alkyl groups. Stereochemistry of compounds in f and g is not unambiguously established (with the exception of 2-(1′-methylpentyl)succinyl-CoA and (2′-methylhexyl)malonyl-CoA described previously (20)).
The best characterized acyl-CoA mutase is methylmalonyl-CoA mutase (MCM), which is found in species ranging from bacteria to humans (3). MCM interconverts (R)-methylmalonyl-CoA and succinyl-CoA (Fig. 1a) in the degradation of odd-chain fatty acids, cholesterol, and branched amino acids or to supply methylmalonyl-CoA units for polyketide biosynthesis (8). Several related acyl-CoA mutases catalyze the interconversion of other branched-chain CoA thioesters (Fig. 1, b–e) as follows: isobutyryl-CoA mutase (ICM) (9) interconverts isobutyryl-CoA and n-butyryl-CoA (Fig. 1b) to provide isobutyryl extender units for polyketide biosynthesis (10); ethylmalonyl-CoA mutase (ECM) (11) interconverts (R)-ethylmalonyl-CoA and (S)-methylsuccinyl-CoA (Fig. 1d) in the ethylmalonyl-CoA pathway for acetate assimilation (12); and two variants of 2-hydroxyisobutyryl-CoA mutase (HCM1 and HCM2) interconvert 2-hydroxyisobutyryl-CoA and (S)-3-hydroxybutyryl-CoA (HCM1) (13) or (R)-3-hydroxybuyryl-CoA (HCM2) (14), for example in the catabolism of compounds bearing tert-butyl moieties (Fig. 1e). Furthermore, an ICM variant that is naturally fused to its G-protein metallochaperone MeaI, termed IcmF for ICM fused (15), was shown to catalyze the interconversion of pivalyl-CoA and isovaleryl-CoA in addition to its native ICM activity (16), and a pure pivalyl-CoA mutase (PCM) was recently characterized (Fig. 1c) (17). Finally, it was proposed that novel acyl-CoA mutases catalyze the interconversion of 2-(2′-aminophenyl)succinyl-CoA and (2′-aminobenzyl)malonyl-CoA and related compounds (Fig. 1f) in the anaerobic degradation of aromatic compounds such as indoleacetate (18, 19) as well as the interconversion of 2-(1′-methylalkyl)succinyl-CoA and (2′-methylalkyl)malonyl-CoA (Fig. 1g) in the anaerobic degradation of alkanes (20–23). Although these activities have not been biochemically verified, these studies suggest that the catalytic scope of acyl-CoA mutases might be much larger than originally anticipated.
Of these acyl-CoA mutases, only MCM (24–27), IcmF (28), and HCM1 (29) have been structurally characterized, and only MCM and HCM1 have been visualized with substrates bound. All three enzymes require two domains for catalytic activity as follows: a Rossmann-fold cobalamin (Cbl)-binding domain, which binds the AdoCbl cofactor in the “base-off/His-on” mode (24, 27, 30), and an (α/β)8 triose-phosphate isomerase (TIM) barrel, which binds the substrate. These two domains can be encoded on a single polypeptide (MCM, IcmF) or on separate polypeptides (HCM1). The Cbl-binding domain positions the AdoCbl into the TIM barrel, forming a buried active site cavity in which the free radical intermediates of catalysis are protected from oxidative quenching. Intriguingly, the TIM barrels of MCM and IcmF can undergo a dramatic conformational change, from a catalytically active closed state that resembles a typical TIM barrel to an unusual open state, in which the TIM barrel is split into two halves of four β-strands each with a cavity in the center of the barrel (25). For the substrate-free structure of homodimeric IcmF, one chain has AdoCbl positioned into a closed barrel, and the other has an open barrel with the Cbl cofactor displaced out of the active site (28). In contrast, for MCM, the open barrel is associated with the substrate-free form of the enzyme (25), whereas the substrate-bound structure is in the closed conformation with the substrate threaded through the barrel (24–26). It has been proposed that this conformational change may afford at least part of the substantial 1012 rate acceleration of Co–C bond homolysis that occurs upon substrate binding in MCM (25, 31, 32).
The large substrate-induced conformational rearrangement in MCM lies in stark contrast to the more subtle conformational changes that occur upon substrate binding in a different AdoCbl-dependent enzyme, glutamate mutase. Here, the ribose of the AdoCbl 5′-deoxyadenosyl group (5′-dAdo) undergoes pseudorotation from the C2′-endo conformation to the C3′-endo conformation when substrate binds, breaking the Co–C bond and repositioning the resulting 5′-deoxyadenosyl radical for hydrogen atom abstraction from substrate (33). No structural data exist as to whether the same change in ribose conformation is involved in reactivity of MCM or other acyl-CoA mutases. Given the limited number of mutases that have been structurally characterized, we still have much to learn about how these enzymes generate and control the highly reactive 5′-deoxyadenosyl radical upon substrate binding.
Furthermore, to engineer mutases for biotechnological applications, an understanding of the substrate binding determinants will also be required. Substrate-bound structures of MCM (25, 26) and HCM1 (29) as well as bioinformatic analyses (4, 6, 11, 13) have suggested that substrate specificity in acyl-CoA mutases is determined by the identity of a few key amino acids. Indeed, HCM1 carrying a single active site mutation has both considerable PCM and HCM2 activity and reduced HCM1 activity (29). All other attempts to rationally alter the substrate specificity of acyl-CoA mutases by mutagenesis, however, have failed (13, 34), indicating that our understanding of substrate specificity in acyl-CoA mutases is incomplete.
We recently reported crystal structures of IcmF from Cupriavidus metallidurans, which contains a G-protein domain in addition to the mutase domains, with AdoCbl in the ICM active site and GDP·Mg2+ in the G-protein active site (holo-IcmF·GDP) but without acyl-CoA substrates (28). Here, we report crystal structures of IcmF bound to AdoCbl, GDP·Mg2+, and all four known acyl-CoA substrates (pivalyl-CoA, isovaleryl-CoA, isobutyryl-CoA, and n-butyryl-CoA), revealing the mode of substrate binding and the determinants of substrate specificity in IcmF. Guided by the structural insight and bioinformatic analyses, we identify two classes of acyl-CoA mutases that likely catalyze novel AdoCbl-dependent reactions.
Experimental Procedures
Materials
Isobutyryl-CoA, n-butyryl-CoA, and isovaleryl-CoA were obtained from Sigma. Pivalyl-CoA was synthesized in a one-step procedure from pivalic anhydride (Sigma) and CoA (Sigma) (Fig. 2). Briefly, to a solution of 0.20 mmol of pivalic anhydride (40.6 μl) in 2 ml of anhydrous dimethylformamide were added 0.040 mmol of solid CoA hydrate (32 mg), 0.12 mmol of triethylamine (16.8 μl), and a catalytic amount of dimethylaminopyridine. The reaction was stirred for 30 min at 25 °C. Reaction progress was followed by thin layer chromatography in 1:1:1:1 1-butanol/acetic acid/ethyl acetate/water. The reaction was stopped by addition of 0.10 mmol of HCl followed by dilution with water, and the water/dimethylformamide mixture was removed by lyophilization. The solid product was dissolved in 400 μl of 95:5 water/acetonitrile and purified by HPLC on a 250 × 10-mm Targa C18 (5-μm pore size) reversed-phase column (Higgins Analytical). The final yield of pivalyl-CoA was 0.018 mmol (45%). ESI-MS (m/z): [M − 2H]2− calculated for C26H44N7O17P3S, 424.58; found, 424.58.
FIGURE 2.

Synthetic scheme for pivalyl-CoA synthesis. DMAP is dimethylaminopyridine.
Protein Expression, Purification, and Crystallization
N-terminally His-tagged IcmF from C. metallidurans was expressed and purified as described previously (15, 16, 28). Purified IcmF was supplemented with AdoCbl (Sigma), GDP (Sigma), and MgCl2 to generate holo-IcmF·GDP and crystallized at 25 °C using the hanging drop vapor diffusion technique. 1 μl of a protein solution (11.7 mg/ml IcmF in 100 mm NaCl, 50 mm HEPES, pH 7.5, 1 mm GDP, 3 mm MgCl2, 300 μm AdoCbl) was mixed with 1 μl of a precipitant solution (0.7–0.75 m potassium sodium tartrate, 0.2 m ammonium acetate, 0.1 m imidazole, pH 7.0–7.7, 3% (v/v) ethylene glycol) on a glass coverslip. The coverslip was sealed with grease over a reservoir containing 500 μl of the precipitant solution without ethylene glycol. Triangular crystals appeared within 3 weeks and grew to full size within 6 weeks. To generate crystals of holo-IcmF·GDP bound to n-butyryl-CoA, isobutyryl-CoA, isovaleryl-CoA, or pivalyl-CoA, pre-formed holo-IcmF·GDP crystals were transferred to 2 μl of a soak solution containing the precipitant, 2 mm GDP, 3 mm MgCl2, and 5 mm of the corresponding substrate in three steps of about 30 s each, with successive increases in the substrate concentration from 1.25 to 2.5 to 5 mm. After soaking, crystals were transferred in two steps of increasing glycerol concentration into a cryogenic solution containing the precipitant, 2 mm GDP, 3 mm MgCl2, 5 mm substrate, and 20% (v/v) glycerol, incubated in that solution for 15 s, and then flash-frozen in liquid nitrogen. All crystallization and soaking procedures were carried out in a dark room under red light to prevent cleavage of the AdoCbl Co–C bond before ligand binding.
Data Collection and Processing
All IcmF crystals belong to space group R32 (denoted as H32 by the PDB). All data were collected at the Advanced Photon Source (Argonne, IL) at beamline 24ID-C at a temperature of 100 K and a wavelength of 0.9795 Å (12,658 eV). Data for holo-IcmF·GDP bound to isobutyryl-CoA, n-butyryl-CoA, and isovaleryl-CoA were collected using a Quantum 315 detector in 0.5° (isobutyryl-CoA) or 1° oscillation steps (n-butyryl-CoA and isovaleryl-CoA). Data for holo-IcmF·GDP bound to pivalyl-CoA were collected using a Pilatus 6MF detector in wedges of 20° in 0.5° oscillation steps. The crystal was displaced along its major macroscopic axis after each wedge.
All data were integrated in XDS and scaled in XSCALE (35). The same reflections as in the previously determined holo-IcmF·GDP data set (28) were marked for the free set of reflections in all data sets, corresponding to 5% of total reflections. All data collection statistics are summarized in Table 1.
TABLE 1.
Crystallographic data collection and refinement statistics
| Holo-IcmF·GDP with isobutyryl-CoA | Holo-IcmF·GDP with n-butyryl-CoA | Holo-IcmF·GDP with isovaleryl-CoA | Holo-IcmF·GDP with pivalyl-CoA | |
|---|---|---|---|---|
| Data collection | ||||
| Space group | R32:h | R32:h | R32:h | R32:h |
| Cell dimensions | ||||
| a, b, c (Å) | 318.0, 318.0, 343.4 | 316.8, 316.8, 342.7 | 317.6, 317.6, 343.5 | 317.5, 317.5, 343.0 |
| α, β, γ (°) | 90, 90, 120 | 90, 90, 120 | 90, 90, 120 | 90, 90, 120 |
| Resolution (Å)a | 35.0-3.40 | 35-3.50 | 35-3.45 | 100-3.40 |
| (3.49-3.40) | (3.59-3.50) | (3.54-3.45) | (3.49-3.40) | |
| No. of reflectionsa | 87,533 (6578) | 81,568 (6052) | 86,230 (6411) | 90,203 (6660) |
| Rsym (%)a | 12.6 (67.1) | 14.8 (95.0) | 13.7 (80.2) | 13.7 (101.5) |
| Rmeas (%)a | 14.3 (76.2) | 16.6 (106.0) | 15.7 (92.0) | 14.5 (112.7) |
| CC1/2a | 99.3 (66.0) | 99.4 (67.5) | 99.2 (65.2) | 99.8 (69.4) |
| 〈I/σ(I)〉a | 10.6 (2.0) | 12.7 (2.0) | 10.0 (2.0) | 15.4 (2.4) |
| Completeness (%)a | 96.9 (98.9) | 98.4 (99.2) | 99.0 (99.9) | 99.4 (99.8) |
| Multiplicitya | 4.3 (4.3) | 5.1 (5.1) | 3.8 (3.8) | 10.5 (10.2) |
| Refinement | ||||
| Resolution (Å) | 35.0-3.40 | 35.0-3.50 | 35.0-3.45 | 100-3.40 |
| Rcryst/Rfree | 0.189/0.209 | 0.185/0.209 | 0.192/0.214 | 0.183/0.201 |
| No. of atoms | ||||
| Protein | 16,176 | 16,169 | 16,160 | 16,143 |
| AdoCbl | 200 | 200 | 200 | 200 |
| GDP | 56 | 56 | 56 | 56 |
| Magnesium | 4 | 4 | 4 | 4 |
| Substrate | 80 | 80 | 81 | 81 |
| Average B-factors (Å2) | ||||
| Protein | 89.9 | 98.7 | 89.5 | 108.7 |
| Cbl | 86.0 | 98.5 | 90.6 | 108.5 |
| 5′-dAdo | 102.2 | 114.3 | 113.0 | 174.6 |
| GDP | 78.6 | 87.6 | 78.0 | 98.4 |
| Magnesium | 58.1 | 67.2 | 56.9 | 81.7 |
| Substrate | 120.1 | 120.2 | 128.2 | 133.7 |
| r.m.s.d. | ||||
| Bond lengths (Å) | 0.003 | 0.003 | 0.003 | 0.003 |
| Bond angles (°) | 0.59 | 0.59 | 0.60 | 0.59 |
| Rotamer outliers | 8 (0.5%) | 9 (0.6%) | 7 (0.4%) | 8 (0.5%) |
| Ramachandran statistics | ||||
| Favored | 2031 (96.4%) | 2028 (96.2%) | 2023 (96.1%) | 2025 (96.0%) |
| Allowed | 72 (3.5%) | 76 (3.6%) | 78 (3.7%) | 83 (3.9%) |
| Disallowed | 3 (0.1%) | 4 (0.2%) | 5 (0.2%) | 1 (0.1%) |
| Average estimated coordinate error (Å) | 0.4 | 0.4 | 0.4 | 0.4 |
a Values in parentheses indicate highest resolution bin.
Structure Building and Refinement
All structures were determined to resolutions ranging from 3.40 to 3.50 Å resolution (Table 1) by molecular replacement. First, the structure of holo-IcmF·GDP bound to isobutyryl-CoA was determined by molecular replacement with the structure of substrate-free holo-IcmF·GDP (PDB code 4XC6) (28) using rigid body refinement in PHENIX (36). To minimize existing model bias, 10 cycles of simulated annealing refinement were carried out in PHENIX. There was clear electron density for isobutyryl-CoA in one of the two protomers in the asymmetric unit. After insertion of the substrate, the model was adjusted to account for any changes in the protein environment by iterative cycles of manual model building in COOT (37, 38) and refinement in PHENIX. The structures of holo-IcmF·GDP bound to n-butyryl-CoA, isovaleryl-CoA, and pivalyl-CoA were determined by molecular replacement with the structure of isobutyryl-CoA-bound holo-IcmF·GDP using rigid body refinement in PHENIX. For each structure, there was clear electron density for the corresponding substrate in one of the two protomers in the asymmetric unit. We only modeled the given substrate in our structures instead of a substrate/product mixture because the crystal/substrate incubation times were short relative to IcmF's turnover rate (15, 16). The models were adjusted to account for any changes in the protein environment by iterative cycles of manual model building in COOT and refinement in PHENIX. Side chains with limited electron density were truncated at the last atom with visible electron density. Initial stages of refinement included B-factor refinement for individual atoms. Final stages of refinement included TLS parameterization using one TLS group per chain (39). Strict noncrystallographic symmetry restraints were applied in early cycles of refinement. In advanced stages of refinement, noncrystallographic symmetry restraints were loosened for residues involved in crystal contacts as well as selected residues that were in substantially different environments due to conformational differences between the two chains of IcmF in the asymmetric unit.
Parameter files for Cbl were generously provided by Oliver Smart at Global Phasing (Cambridge, UK). Refinement restraints for 5′-dAdo, isobutyryl-CoA, n-butyryl-CoA, isovaleryl-CoA, and pivalyl-CoA were generated using the Grade Web Server (40). Refinement restraints for GDP were generated using the electronic Ligand Builder and Optimization Workbench (elBOW) (41) implemented in PHENIX.
Crystallographic refinement of the four structures of holo-IcmF·GDP bound to substrates yielded models that possess low free R-factors, excellent stereochemistry, and small root mean square deviations (r.m.s.d.) from ideal values for bond lengths and angles. The final models of holo-IcmF·GDP bound to isobutyryl-CoA, n-butyryl-CoA, isovaleryl-CoA, or pivalyl-CoA include residues 21–1093 (of 1093) for chain A and residues 22–1093 (of 1093) for chain B, lacking the hexahistidine tag and residues at the N terminus. The model of holo-IcmF·GDP bound to isobutyryl-CoA additionally lacks residues 285, 530–537, and 1013–1014 in chain A and residues 592, 904–906, and 1011–1018 in chain B. The model of holo-IcmF·GDP bound to n-butyryl-CoA lacks residues 285, 530–536, and 1014 in chain A and residues 592–593, 905–906, and 1011–1018 in chain B. The model of holo-IcmF·GDP bound to isovaleryl-CoA lacks residues 284–285, 530–536, and 1013–1014 in chain A and residues 592–593, 904–906, and 1011–1018 in chain B. The model of holo-IcmF·GDP bound to pivalyl-CoA lacks residues 530–536 and 1012–1014 in chain A and residues 592–593 and 1011–1018 in chain B. For all models, each chain contains bound cobalamin and GDP·Mg2+ and an additional Mg2+ in the GDP-binding site; chain A contains bound substrate and 5′-dAdo, and chain B contains the nucleotide portion of the substrate. All refinement statistics are summarized in Table 1. The models were validated using simulated annealing composite omit maps calculated in CNS (42, 43). Model geometry was analyzed using MolProbity (44) and ProCheck (45). Figures were generated using PyMOL (46). Crystallography software packages were compiled by SBGrid (47).
Phylogenetic and Bioinformatic Analyses
For calculation of a phylogenetic tree, sequences of different acyl-CoA mutase substrate-binding domains were retrieved from the genomic BLAST interface (February 5, 2015) at the National Center for Biotechnology Information (NCBI) or from the Integrated Microbial Genomes and Metagenomes database of the Joint Genome Institute of the United States Department of Energy (img.jgi.doe.gov). A representative set of 200 sequences was manually selected for alignment. MCM sequences were chosen to cover eukaryotes, archaea, and all major bacterial phyla that contain MCM and had >5 genome sequences reported (Proteobacteria (including α-, β-, γ-, and δ-proteobacteria), Actinobacteria, Bacteroidetes, Chlorobi, Chloroflexi, Cyanobacteria, Deferribacteres, Deinococci, Firmicutes, Planctomycetes, Spirochaete, Synergistes, and Verrucomicrobia). Additional sequences were chosen to cover characterized HCMs, ECMs, ICMs, IcmFs, and PCMs as well as putative uncharacterized mutases. A phylogenetic tree was then calculated using the following workflow implemented in the Phylogeny.fr web server (48). Sequences were aligned using MUSCLE (49), and the alignment was trimmed to homologous regions using Gblocks (50). A phylogenetic tree was constructed using the bootstrap method (100 bootstraps) and the LG substitution model (four substitution rate categories; γ-distribution parameter and proportion of invariable sites were estimated by the program) in PhyML (51) and visualized using TreeDyn (52). For validation, a complete phylogenetic tree was calculated using all sequences annotated as mutase substrate-binding domains (InterPro group IPR006098, accessed May 31, 2015, grouped together by >85% sequence identity) and using the same workflow. The two phylogenetic trees exhibited the same overall structure, confirming the observed grouping of sequences. The tree calculated from manually selected sequences was used for visualization.
Homology Modeling
Homology models of the uncharacterized mutase from Aromatoleum aromaticum (NCBI accession code WP_011236985.1) and of uncharacterized mutase 1 from Desulfatibacillum alkenivorans (NCBI accession code WP_012610856.1) were generated using the SWISS-MODEL Automated Comparative Protein Modeling Server (53) with the structure of substrate-bound MCM from Propionibacterium freudenreichii subsp. shermanii (PDB code 4REQ (26)) as template. The uncharacterized mutases from A. aromaticum and D. alkenivorans share 35 and 28% sequence identity with this template.
Results
Acyl-CoA Substrates Bind to the Catalytically Active Chain of IcmF
To visualize how IcmF binds its substrates, we sought to determine crystal structures of IcmF bound to its AdoCbl cofactor and the four different acyl-CoA molecules that all serve as substrates (15, 16). Whereas isobutyryl-CoA, n-butyryl-CoA, and isovaleryl-CoA are commercially available, the fourth substrate, pivalyl-CoA (2,2-dimethylpropionyl-CoA), was synthesized from pivalic anhydride via a one-step synthetic procedure (Fig. 2, see “Experimental Procedures”). Pre-formed holo-IcmF·GDP crystals were incubated with the different substrates, and structures were determined to resolutions ranging from 3.40 to 3.50 Å (Fig. 3 and Table 1). The resulting structures depict IcmF in complex with Cbl, the 5′-dAdo, and substrate in the mutase active site as well as GDP·Mg2+ in the G-protein active site (Fig. 3a). There is clear electron density for each of the four substrates as well as for the Cbl cofactor and the 5′-deoxyadenosyl group in chain A of these structures (Fig. 4, a–d), which is in the catalytically competent closed conformation. In chain B, which is in a catalytically inactive open conformation with the Cbl cofactor swung out of the active site and the TIM barrel substrate-binding domain split into two halves (28), electron density is only observed for the nucleotide portion of the substrate, whereas the remainder of the substrate is disordered (Fig. 5).
FIGURE 3.
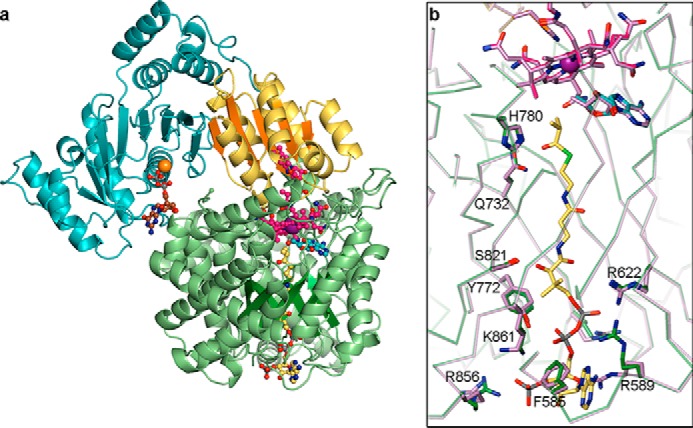
Overall structure of IcmF protomer and comparison of substrate-bound and substrate-free structures. a, overall structure of chain A of an IcmF dimer, shown in ribbon representation colored by domain. Cbl-binding domain is in yellow; TIM barrel substrate-binding domain is in green, and G-protein domain is in cyan. The linker region between the G-protein and substrate-binding domains is hidden for clarity. TIM barrel and Cbl-binding domain β-sheets are shown in darker colors for emphasis. Isobutyryl-CoA is threaded through the TIM barrel. Bound AdoCbl, GDP·Mg2+, and isobutyryl-CoA are shown in ball-and-stick representation with Cbl carbons in pink, 5′-dAdo carbons in cyan, cobalt in purple, GDP carbons in brown, Mg2+ in orange, and isobutyryl-CoA carbons in yellow. b, comparison of isobutyryl-CoA-bound (green) and substrate-free IcmF (lilac, PDB code 4XC6), revealing nearly identical structures. Selected residues contributing to substrate binding are shown in stick representation. AdoCbl and isobutyryl-CoA of the substrate-bound structure are shown as in a. AdoCbl of the substrate-free structure is shown in lilac.
FIGURE 4.

Overall mode of substrate binding in IcmF and comparison with MCM and HCM1. a, IcmF bound to Cbl (pink carbons; cobalt, purple), 5′-dAdo (cyan carbons), and isobutyryl-CoA (yellow carbons). 2Fo − Fc omit electron density (orange mesh) contoured at 1.0 σ is shown around the substrates and 5′-dAdo. IcmF is shown with the chain A TIM barrel substrate-binding domain in green and the Cbl-binding domain in yellow. The Cbl-coordinating His-39 as well as selected residues that bind substrate are shown as sticks, with hydrogen bonds, ionic interactions, and π-π interactions shown as black dashed lines. The 5′-dAdo C5′ and the locations of hydrogen abstraction on the substrates are shown as spheres, and red dashed lines connect the C5′ to the Cbl cobalt and the substrate carbons. b, IcmF bound to n-butyryl-CoA (orange carbons) displayed as in a. c, IcmF bound to pivalyl-CoA (maroon carbons) displayed as in a. d, IcmF bound to isovaleryl-CoA (light pink carbons) displayed as in a. e, comparison of isobutyryl-CoA-bound IcmF (green) and methylmalonyl-CoA-bound MCM (gray, PDB code 4REQ), revealing nearly identical structures and substrate binding modes. IcmF shown as in a. MCM-bound methylmalonyl-CoA, Cbl, and 5′-dAdo are shown with gray carbons. f, comparison of isobutyryl-CoA-bound IcmF (green) and hydroxyisobutyryl-CoA-bound HCM1 (lilac, PDB code 4R3U), revealing similar structures and substrate binding modes. IcmF shown as in a. HCM1-bound hydroxyisobutyryl-CoA, Cbl, and 5′-dAdo are shown with lilac carbons.
FIGURE 5.

Differences of substrate-binding domains of IcmF dimer. a, isobutyryl-CoA binding to IcmF chain A of the IcmF dimer, which is in a catalytically active conformation. Coloring as in Fig. 4. b, isobutyryl-CoA binding to IcmF chain B, which is in a catalytically inactive conformation. IcmF (green carbons), isobutyryl-CoA (yellow carbons), and 2Fo − Fc electron density (orange mesh), contoured at 1.0 σ, are shown as in Fig. 4a. Note that there is no electron density past the 5′-phosphate of the isobutyryl-CoA nucleotide moiety; therefore, additional atoms were not modeled. The nucleotide portion is bound by few specific interactions, as indicated by black dashed lines. Other interactions between IcmF and isobutyryl-CoA are disrupted because of the conformational change in IcmF chain B compared with IcmF chain A. c, different conformations of substrate-binding domains of IcmF chains A (dark green) and B (gray) isobutyryl-CoA-bound IcmF. Chains are superposed by TIM barrel β-strands. Cbl of chain A is shown as in Fig. 4a, Cbl of chain B is shown with carbons in black. Distances between corresponding Cα atoms are indicated in Å.
Comparison of the substrate-bound structures to that of substrate-free holo-IcmF·GDP (28) reveals that the structures match closely, with a Cα root mean square deviation (r.m.s.d.) of 0.3 Å for the entire IcmF dimer between the substrate-free and substrate-bound structures (Fig. 3b). Thus, both substrate-free and substrate-bound structures of IcmF have both open and closed conformations of the TIM barrel substrate-binding domain (Fig. 5c). In our structures, substrates appear to bind to the TIM barrel in both conformations, but only the TIM barrel in the closed conformation represents the catalytically active state with AdoCbl and substrate positioned for catalysis. In this chain (chain A), a few side chains, including those of Arg-589 and Arg-856, rearrange to engage in interactions with the substrates, but there are no large scale conformational changes upon substrate binding (Fig. 3b). To further evaluate the substrate binding mode, we focused on chain A in our structures.
Acyl-CoA Substrates Are Threaded through the TIM Barrel Substrate-binding Domain
The four substrate-bound IcmF structures are nearly identical, with Cα r.m.s.d. values smaller than 0.2 Å between all structures. The acyl-CoA substrates are bound in the same overall fashion: the nucleotide portion is positioned on the surface of the N-terminal face of the TIM barrel, the phosphopantetheine moiety is threaded through the center of the IcmF TIM barrel substrate-binding domain, and the acyl group is positioned adjacent to the Cbl cofactor and the 5′-deoxyadenosyl group in the active site cavity (Figs. 4, a–d and 5a). This mode of substrate binding is very similar to that observed in the related acyl-CoA mutases MCM (25, 26) and HCM1 (Fig. 4, e and f) (29), and many of the specific interactions are conserved. In IcmF, the thioester carbonyl is stabilized by hydrogen bonds from Gln-732 and His-780, securing the acyl group in the active site (Fig. 4, a–d). The interaction with His-780 in particular is likely important for catalysis, as mutation of the homologous His (His-244) to Gln or Ala in MCM reduced kcat by 102 to 103 and drastically increased the rate of oxidative inactivation (54). The phosphopantetheine arm and the nucleotide portion of the substrate are stabilized by additional interactions such as electrostatic interactions between the phosphate groups and the positively charged residues Arg-589, Arg-622, Arg-728, Arg-856, and Lys-861 (Figs. 4, a–d, and 5a). Furthermore, Tyr-772 hydrogen bonds with a phosphate oxygen, Phe-585 engages in π-π stacking interactions with the adenine base, and Ser-821 forms a hydrogen bond to the phosphopantetheine hydroxyl group (Figs. 4, a–d, and 5a). Together, the interactions to the phosphopantetheine and the nucleotide plug the access tunnel to the active site, thereby protecting the reactive catalytic intermediates.
IcmF Substrate-bound Structures Suggest Two Conformations of the 5′-Deoxyadenosyl Group
To gain insight into the effect of substrate binding on the AdoCbl cofactor, the electron density for AdoCbl in the substrate-bound IcmF structures was analyzed. The electron density is best fit by a mixture of species: cleaved AdoCbl with 5′-dAdo in the C3′-endo conformation and a Co–C distance of 3.2–3.5 Å and uncleaved AdoCbl with 5′-dAdo in the C2′-endo conformation and a Co–C distance of 2.2 Å (Fig. 6a). Although the resolutions of our structures are moderate, this assignment is supported by the previous observation of 5′-dAdo in both the C3′-endo and C2′-endo conformations in the substrate-bound structure of the related AdoCbl-dependent enzyme glutamate mutase (Fig. 6, b–d) (33). In the C3′-endo conformation, the 5′-dAdo C5′ atom is close to the hydrogen atom abstraction site on the substrate and could initiate catalysis (Fig. 6c, see below). Notably, the 5′-dAdo is stabilized by different interactions in the C2′-endo and C3′-endo conformations. The C2′-endo conformation is stabilized by hydrogen bonds from Tyr-779 to the ribose O2′ and from Glu-905 to the ribose O3′ (Fig. 6d). In the C3′-endo conformation, the interaction with Tyr-779 is disrupted; instead, now the ribose O3′ also forms a hydrogen bond to Glu-905, and the ribose O4′ forms a hydrogen bond to Gln-865 (Fig. 6c). Similar changes in the hydrogen bonding patterns of the two 5′-dAdo conformers were observed for glutamate mutase (Fig. 6, c and d) (33). Glu-905 is also conserved in glutamate mutase as Glu-330, and mutation of this Glu-330 to Asp, Gln, or Ala leads to a drastic reduction in activity, supporting its importance for catalysis (55).
FIGURE 6.

5′-dAdo conformational changes in IcmF and glutamate mutase. a, 2Fo − Fc omit electron density (orange mesh) contoured at 1.0 σ around Cbl and 5′-dAdo of n-butyryl-CoA bound IcmF. 5′-dAdo can be modeled in the C3′-endo conformation (cyan carbons) and in the C2′-endo conformation (light blue carbons). In the C2′-endo conformation, the C5′ is close to the Cbl cobalt, whereas in the C3′-endo conformation, the C5′ is pointed toward the substrate (orange carbons, dashed red line). Cbl is shown with carbons in pink and cobalt in purple. b, glutamate mutase active site (PDB code 1I9C) (33), revealing the presence of two 5′-dAdo conformers, C2′-endo (pink carbons) and C3′-endo (purple carbons), in the presence of glutamate (gray carbons). As in IcmF, the 5′-dAdo C5′ is close to the Cbl cobalt in the C2′-endo conformation (dashed red line) and pointed toward the location of hydrogen abstraction on the substrate in the C3′-endo conformation (dashed red line). Cbl is shown with carbons in light pink and Co in purple. c, comparison of the 5′-dAdo C3′-endo conformations in IcmF (cyan carbons) and glutamate mutase (purple carbons). Dashed red lines connect the 5′-dAdo C5′ and the corresponding substrate. Cbl is shown as in b. In both proteins, 5′-dAdo is stabilized by interactions (dashed black lines) to amino acid side chains (IcmF in green and glutamate mutase in pink). IcmF Gln-865 contributes to 5′-dAdo binding, but the corresponding Arg-66 in glutamate mutase does not. d, comparison of the 5′-dAdo C2′-endo conformations in IcmF (light blue carbons) and glutamate mutase (pink carbons). Protein side chains and Cbl colored as in c. Again, 5′-dAdo is stabilized by specific interactions (dashed black lines) to amino acid side chains. IcmF Tyr-779 contributes to 5′-dAdo binding, but the corresponding Pro-218 in glutamate mutase (hidden for clarity) does not. IcmF Asn-901 corresponds to glutamate mutase Lys-326 but does not contribute to 5′-dAdo binding.
IcmF Active Site Is Arranged for Hydrogen Atom Abstraction from All Four Substrates
To investigate the catalytic mechanism and substrate specificity of IcmF, the binding modes of the four different substrate acyl groups were compared. All four acyl-CoA substrates are bound in the active site in a similar orientation (Fig. 7, a and b), with a β-carbon of the acyl group positioned within 3.6 Å of the 5′-dAdo C5′ (in the C3′-endo conformation, see above), which is in agreement with other distances reported for hydrogen atom transfer (56). The binding site for the acyl groups is lined by Gln-732 and His-780 (see above) as well as Gln-742 and Tyr-779 on one side, Phe-598, Thr-679, and Gln-865 on the other side, and the Cbl corrin ring on the bottom (Fig. 7a). These residues create a tight binding pocket for the substrate acyl groups.
FIGURE 7.

IcmF active site and comparison with MCM and HCM1 in wall-eyed stereo view. a, overlay of four substrate-bound IcmF structures, revealing similar substrate orientation. Shown are isobutyryl-CoA (yellow carbons), n-butyryl-CoA (orange carbons), pivalyl-CoA (maroon carbons), and isovaleryl-CoA (light pink carbons). The locations of hydrogen abstraction are shown as spheres, located within 3.6 Å of the 5′-dAdo (C3′-endo conformation, cyan carbons) C5′ atom (cyan sphere), as indicated by the dashed red lines. Residues in the substrate-binding site are shown with dark green carbons. The third methyl group of pivalyl-CoA clashes with Phe-598 (yellow dashed line), leading to a small rotation of the side chain (maroon carbons) in this structure. Hydrogen bonds from Gln-732 and His-780 to the thioester carbonyl are indicated as dashed black lines. Cbl is shown with carbons in pink and cobalt in purple. b, overlay of substrate-bound IcmF and MCM (26) (PDB code 4REQ). Isobutyryl-CoA, n-butyryl-CoA, IcmF-bound 5′-dAdo, IcmF-bound Cbl, and IcmF residues are shown as in a. The locations of hydrogen atom abstraction in both methylmalonyl-CoA (gray carbons) and succinyl-CoA (purple carbons) overlay with those of IcmF substrates (spheres). MCM-bound 5′-dAdo, MCM-bound Cbl, and MCM substrate-binding residues are shown with gray carbons. Gln-197 and His-244 are conserved in MCM and IcmF, whereas IcmF Phe-598 is replaced by MCM Tyr-89 and IcmF Gln-742 is replaced by Arg-209, putatively accounting for the switch in substrate binding specificity. Hydrogen bonds and ionic interactions are shown as dashed black lines. c, active site of substrate-bound MCM, shown as in b, highlighting interactions to substrate carboxylate groups. Only interactions to methylmalonyl-CoA are shown for clarity. d, active site of substrate-bound HCM1 (PDB code 4R3U) (29), highlighting interactions to substrate hydroxyl groups. Hydroxyisobutyryl-CoA is shown in lilac and (S)-3-hydroxybutyryl-CoA in blue. Only interactions to hydroxyisobutyryl-CoA are shown for clarity. Locations of hydrogen atom abstraction are shown as spheres.
The acyl chains of pivalyl-CoA and isovaleryl-CoA are positioned similarly to those of isobutyryl-CoA and n-butyryl-CoA in the active site. For pivalyl-CoA, two of the methyl groups overlay closely with those of isobutyryl-CoA, whereas the third methyl group points toward Phe-598, causing a small rotation of the side chain to accommodate the methyl group (Fig. 7a). Isovaleryl-CoA is bound in a similar orientation as n-butyryl-CoA, without notable side chain rearrangements. Thus, the active site accommodates pivalyl-CoA and isovaleryl-CoA with only minor adjustments, explaining the observed substrate promiscuity of IcmF. Nevertheless, pivalyl-CoA would likely bind more readily if Phe-598 was replaced by a slightly smaller side chain such as Leu, and indeed, a mutase carrying this substitution was recently reported to have higher pivalyl-CoA mutase activity (17).
Finally, comparing the positioning of the isobutyryl-CoA and n-butyryl-CoA substrates allows us to probe the stereospecificity of the ICM reaction, which has been studied in stand-alone ICM (as opposed to the IcmF fusion protein) from Streptomyces cinnamonensis. IcmF-bound isobutyryl-CoA is positioned for hydrogen atom abstraction from the pro-S methyl group, which is located within 3.5 Å of the 5′-dAdo C5′, whereas the pro-R methyl group is farther away at a distance of 3.9 Å (Fig. 8, left box). For n-butyryl-CoA, modeling of hydrogen atoms with ideal geometry positions the pro-S hydrogen on C3 toward the 5′-dAdo C5′, an ideal position for hydrogen atom abstraction (Fig. 8, right box). Both of these observations match previous stereochemical investigations on ICM (57, 58). Our observed modes of substrate binding also provide an explanation for the observed partial breakdown of stereospecificity in ICM, for which a small amount of hydrogen atom abstraction occurs from the pro-R methyl group of isobutyryl-CoA (57). The isobutyryl group likely has rotational flexibility in the active site, with the preferred mode of binding as observed in our structure and an alternative mode of binding with the pro-S methyl group pointing toward Phe-598, as observed for pivalyl-CoA (Fig. 7a). The two possible modes of binding would lead to the observed breakdown of stereospecificity, as originally hypothesized (57).
FIGURE 8.

Stereochemical course of isobutyryl-CoA mutase reaction. The chemical mechanism shown at the bottom was established based on stereochemical studies (57). Following Co–C bond homolysis (step not shown), the 5′-dAdo radical abstracts a hydrogen atom (red) from the pro-S methyl group of isobutyryl-CoA (blue). The isobutyryl-CoA radical rearranges to the n-butyryl-CoA radical, which then re-abstracts the hydrogen atom from 5′-deoxyadenosine. The hydrogen atom ends up in the pro-S position. In the reverse reaction, the 5′-dAdo radical abstracts the pro-S hydrogen from n-butyryl-CoA. The structures of IcmF bound to isobutyryl-CoA (left) and n-butyryl-CoA (right) support the proposed stereochemistry. Isobutyryl-CoA (yellow carbons) positions its pro-S methyl group next to the 5′-dAdo group (cyan carbons), whereas n-butyryl-CoA positions its pro-S hydrogen (white sticks) toward the 5′-dAdo group. The red dashed line connects the 5′-dAdo C5′ to the closest hydrogen atom. Hydrogens are modeled based on ideal geometry. Cobalamin is shown with pink carbons and cobalt as a purple sphere.
Specific Active Site Substitutions Allow for Binding of Aliphatic CoA Thioesters
Comparing IcmF to MCM and HCM1 reveals that the active site architectures and the substrate positions of these acyl-CoA mutases are nearly identical (Fig. 7, b–d). The identity of a few amino acid side chains, however, is distinct to account for the different substrates. In particular, Phe-598 and Gln-742 in IcmF are replaced by a tyrosine (Tyr-89) and an arginine (Arg-207) in MCM (numbering as in MCM crystal structures from P. freudenreichii subsp. shermanii), which form specific contacts to the carboxylate groups of the MCM substrates (Fig. 7, b and c) (15, 25). In IcmF, the smaller Gln and Phe side chains increase the size and hydrophobicity of the active site, thereby allowing for accommodation of the hydrophobic substrates (Fig. 7, a and b). In HCM1, the glutamine is conserved (Gln-208), accommodating the substrate methyl group, but IcmF Phe-598 is replaced by an isoleucine (Ile-90). This replacement creates space for the substrate hydroxyl groups and an additional hydrogen-bonding aspartate in the active site (Asp-117, Fig. 7d). Superposition of IcmF and MCM also shows that the 5′-dAdo group is slightly shifted (Fig. 7b), but the significance of this shift, if any, is unclear.
Overall, the high structural similarity between MCM, HCM1, and IcmF as well as the sequence similarity (>20–30% identity) between substrate-binding domains of acyl-CoA mutases suggest that other acyl-CoA mutases have similar structures and active site architectures. Within this architecture, the substrate binding specificity of these acyl-CoA mutases is likely governed by the identity of a few residues in the substrate-binding domain, as suggested by a number of studies. For example, a Y89F/R207Q MCM double mutant was shown to bind isobutyryl-CoA and n-butyryl-CoA, whereas wild-type MCM does not appear to bind these thioesters (34). We note that in this case the mutant enzyme undergoes suicide inactivation during turnover, likely because second-sphere interactions are important to control the radical intermediates, but mutase binding specificity was clearly altered. Recently, it was also demonstrated that a single point mutation in HCM1, D117V, confers pivalyl-CoA mutase activity (29), further suggesting that these mutases can be tailored toward different substrates.
Bioinformatic Analyses Reveal New Classes of Acyl-CoA Mutases
To classify acyl-CoA mutases and predict their substrate specificities, we performed phylogenetic and bioinformatic analyses on the substrate-binding domains. The currently available sequences with homology to substrate-binding domains of characterized MCMs, ECMs, ICMs, IcmFs, or HCMs cluster into distinct groups in a phylogenetic tree according to the reaction catalyzed (Figs. 9 and 10 and supplemental material). Each cluster with characterized members contains a series of signature sequences indicating the substrate specificity (Figs. 9 and 11). Two major determinants are at the positions of MCM Tyr-89/IcmF Phe-598 (determinant 1) and MCM Arg-207/IcmF Gln-742 (determinant 2); MCMs and ECMs have Tyr and Arg at these positions, allowing them to bind charged carboxylate-bearing substrates (see above); ICMs and IcmFs have Phe and Gln; HCM1s have Ile or Val with an additional Asp (determinant 1) and Gln (determinant 2); and HCM2s have Ile and Ser. The most recently characterized PCM from Xanthobacter autotrophicus clusters with six other mutases. All of these mutases have Leu and Asn in the specificity positions, which provides space to bind the bulky pivalyl group (Figs. 9 and 11). MCMs and ECMs are further distinguished by two additional substitutions near the active site: a His and an Asn in MCMs are replaced by a Gly and a Pro in ECMs (Fig. 11), as described previously (11). Beyond these differences, catalytically important residues, such as the His and Gln contacting the substrate thioester carbonyl (Fig. 7) and the Glu contacting the 5′-dAdo ribose hydroxyl groups (Fig. 6d), are conserved in all sequences (Fig. 11), indicating that all sequences represent functional mutases.
FIGURE 9.

Simplified phylogenetic tree of acyl-CoA mutase substrate-binding domains. The sequences of substrate-binding domains cluster into distinct groups according to their substrate specificity. Larger clusters of characterized mutases are simplified as triangles. Clusters of thus far uncharacterized mutases are shown with organisms as indicated. The amino acid identities in the specificity determinant positions are indicated on the right (see main text and Fig. 11 for explanation of specificity determinant positions). IcmF from C. metallidurans is highlighted in blue, and two uncharacterized mutases and the most recently characterized PCM from X. autotrophicus are highlighted in red and discussed in the main text. The tree was rooted by midpoint rooting. Full tree is shown in Fig. 10 and in the supplemental material.
FIGURE 10.

Full phylogenetic tree of acyl-CoA mutase substrate-binding domains (left side, top half of tree; right side, bottom half of tree). Red numbers represent certainty of branch assignment. A larger version of this figure is provided as supplemental material.
FIGURE 11.

Sequence alignment of important regions of acyl-CoA mutases. Sequences were selected to represent most characterized acyl-CoA mutase classes, using sequences of structurally characterized (P. freudenreichii (24, 25), Homo sapiens MCM (27), and C. metallidurans IcmF (28)) or biochemically characterized mutases (P. freudenreichii MCM and H. sapiens MCM, C. metallidurans IcmF (15, 16), S. cinnamonensis ICM (57, 71), Aquincola tertiaricarbonis HCM1 (13), Rhodobacter sphaeroides ECM (11), and X. autotrophicus PCM (17)) when available. Two sequences from each of the two clusters of uncharacterized mutases were included, as well as two additional mutases encoded in the genome of A. aromaticum. Sequence determinant positions are highlighted by red boxes, positions proposed to distinguish MCMs and ECMs are highlighted by green boxes, and the unique AGGGGG stretch of uncharacterized mutases from A. aromaticum and Azoarcus toluclasticus is highlighted by a blue box. Other conserved catalytically important residues are labeled. Strict residue conservation is indicated by a yellow highlight, sequence similarity is indicated in red. Beginning of each sequence stretch is numbered on the left.
This bioinformatic analysis allows for a number of interesting observations. First, archaeal MCMs cluster with ICMs rather than with bacterial and eukaryotic MCMs (Fig. 9) as noted previously (5, 11, 59). Archaeal MCMs encode the substrate-binding and Cbl-binding domains on separate polypeptides, in notable contrast to most bacterial (see below) and all eukaryotic MCMs, which encode both domains on a single polypeptide. Most sequences annotated as archaeal MCMs have the characteristic features of MCMs, and MCM from the archaeon Pyrococcus horikoshii was recently shown to indeed have MCM activity (60). Notably, archaea appear to contain additional acyl-CoA mutases, including ICMs and several yet-uncharacterized mutases (Fig. 9), indicating that archaea use a variety of different AdoCbl-dependent reactions. The metabolic roles of these reactions remain to be determined.
Second, we observes two groups of MCMs, one containing MCMs from Firmicutes such as several Clostridium species and one containing MCMs from Thermotogae, that do not cluster with other bacterial MCMs (Figs. 9 and 10). Closer inspection reveals that these mutases contain the active site determinants of MCMs (Fig. 9) but are encoded on two separate polypeptides, in contrast to other known bacterial MCMs. Thus, both archaeal MCMs and a subgroup of bacterial MCMs resemble ICMs more closely than other MCMs, highlighting the complex evolutionary history of acyl-CoA mutases.
Third, the bioinformatic analysis reveals two phylogenetically distinct sequence clusters that do not contain characterized members (uncharacterized mutase clusters 1 and 2, see Fig. 9) and thus could represent new AdoCbl-dependent mutases. Analysis of the genomic context reveals that these mutase substrate-binding domains are encoded in larger operons that also encode a corresponding Cbl-binding domain, suggesting that they are active mutases. Uncharacterized mutase cluster 1 contains six mutases from different archaea and bacteria, including A. aromaticum (formerly Azoarcus strain EbN1). Uncharacterized mutase cluster 2 currently contains four sequences from different Deltaproteobacteria, including D. alkenivorans, a metabolically versatile bacterium (23, 61). The mutases in these clusters contain Tyr and Arg in the determinant positions, likely allowing them to bind carboxylate-bearing substrates (Fig. 11).
To further examine these uncharacterized mutases, we analyzed the sequences and generated homology models of these mutases. Mutases in uncharacterized cluster 2 have relatively low sequence similarity to MCM and IcmF, limiting the reliability of homology models. Current homology models reveal several structural changes in the active site, but do not provide conclusive evidence on the active site architecture. Notably, mutases in uncharacterized cluster 1 contain a stretch of six residues in the active site with the sequence AGGGGG (Fig. 11), replacing several residues, including an otherwise strictly conserved Gln that contacts the 5′-dAdo group (Gln-330 in MCM or Gln-865 in IcmF, Fig. 6d) by small Ala and Gly residues (Fig. 11). In addition, an otherwise conserved Phe in the active site (Phe-287 in MCM and Phe-823 in IcmF) is replaced by Asn. A homology model generated using the structure of substrate-bound MCM (see under “Experimental Procedures”) (25) reveals that the Gln to Ala substitution enlarges the active site cavity, which now appears ideally suited to bind substrates carrying larger substituents, and that the Phe to Asn substitution positions an additional hydrogen bonding partner in the active site (Fig. 12). Thus, these mutases may catalyze novel AdoCbl-dependent interconversions.
FIGURE 12.

Homology model of uncharacterized A. aromaticum mutase and comparison with MCM. a, structure of succinyl-CoA-bound MCM (PDB code 4REQ) (26), shown with protein, 5′-dAdo, and Cbl carbons in gray and succinyl-CoA carbons in violet. Cobalt is shown in purple. Succinyl-CoA is bound by specific hydrogen bonds and electrostatic interactions (black dashed lines). b, homology model of A. aromaticum mutase in the same orientation as MCM and with 5′-dAdo and Cbl shown as in a. Protein residues are shown with carbons in yellow. Substrate (pink carbons) is modeled into the active site by adding the aminophenyl group to MCM-bound succinyl-CoA without altering the positioning of succinyl-CoA. Interactions to the thioester carbonyl and the carboxylate are conserved (black dashed lines). Replacement of MCM Gln-330 by Ala-307 creates a cavity in the active site that could accommodate the aminophenyl group. Ala-307 is relatively close to the phenyl group (2.2 Å), but this region likely has significant flexibility because of the following stretch of Gly residues. In addition, replacement of MCM Phe-287 by Asn-263 in the uncharacterized mutase positions a potential hydrogen bonding partner for the amino group in the active site (dashed red line).
Discussion
Acyl-CoA mutases are a growing family of AdoCbl-dependent enzymes that perform challenging carbon skeleton rearrangements. Despite extensive studies, our understanding of catalysis and substrate specificity of acyl-CoA mutases remains incomplete, currently limiting their utility for biotechnological applications. Here, we report crystal structures of IcmF, an ICM variant, bound to four acyl-CoA substrates, revealing how this acyl-CoA mutase positions its substrates for catalysis. Together with bioinformatic analyses, these structures expand our understanding of catalysis and substrate specificity in acyl-CoA mutases and allow us to identify new classes of acyl-CoA mutases.
As observed previously in MCM (25, 26) and HCM1 (29), the IcmF acyl-CoA substrates are threaded through the 8-stranded β-barrel of the substrate-binding domain. Notably, TIM barrels typically feature a tightly packed hydrophobic core with the active site formed by loop regions at the periphery of the barrel and do not use the barrel core for substrate binding. The unique use of TIM barrels by acyl-CoA mutases and other AdoCbl-dependent enzymes is likely an adaptation to protect the radical-based intermediates that form during catalysis. The TIM barrels of acyl-CoA mutases are further distinguished by their ability to undergo a dramatic conformational change from a closed to an open conformation, splitting the barrel into two halves of four strands each (Fig. 5c). Initially observed in MCM (24, 25), we subsequently also captured IcmF in both these conformations (28), suggesting that TIM barrel flexibility is a general feature of acyl-CoA mutases. HCM1 has so far only been observed in the closed state (29), but only a single structure has been reported. In MCM, the barrel was captured in the open and closed conformations in the absence and presence of substrate, respectively, leading to the suggestion that the conformational change is substrate-induced (24, 25). In IcmF, however, we captured both open and closed conformations in the same structure in the absence of substrate (28), and here we again capture both conformations in the presence of substrate, with substrate binding to the already closed TIM barrel. The TIM barrel open conformation instead correlates with loss of the Cbl 5′-dAdo group and displacement of the Cbl out of the active site into a catalytically inactive conformation, possibly mediated by the cognate G-protein chaperone, which is absent in structures of MCM. Thus, we now have a series of snapshots depicting the TIM barrel in both open and closed conformations, indicating that these two conformations are in equilibrium, affected by the presence of substrates as well as by other factors such as presence of the G-protein chaperone and the cofactor state. Although TIM barrel opening could help product release and substrate binding and may play a role in triggering Co–C bond homolysis upon substrate binding (25), our IcmF structures indicate that the barrel does not absolutely need to open and close for every catalytic cycle. It appears that these barrel dynamics are inherent to acyl-CoA mutases, but further studies will be required to determine their role during catalysis and cofactor recycling.
In the active site, substrate binding and Co–C bond homolysis need to be tightly coupled to ensure a high catalytic rate while preventing generation of the 5′-deoxyadenosyl radical without substrate. Our structures suggest that in IcmF, the 5′-dAdo undergoes a conformational change from C2′-endo in intact AdoCbl to C3′-endo upon Co–C bond homolysis that propels the C5′ radical from its position above the Cbl cobalt toward the substrate for hydrogen atom abstraction. This pseudorotation of the 5′-dAdo ribose group appears ideally suited to bridge the 5.5–6.5 Å distance between the substrates and the Cbl in IcmF. Other AdoCbl-dependent mutases similarly position their substrates at the same distance from the Cbl, as determined from crystal structures (24, 29, 33) and by electron paramagnetic resonance spectroscopic studies of glutamate mutase (62) and MCM (63) under catalytic conditions. Pseudorotation of the 5′-dAdo ribose has also been suggested from structural and biochemical studies of glutamate mutase (33, 55) and from computational studies on MCM (64). Thus, given the combined structural, biochemical, and computational evidence, it appears that this mechanism of moving the active C5′ radical toward substrate is conserved in AdoCbl-dependent mutases such as acyl-CoA mutases. Notably, another group of AdoCbl-dependent enzymes, the eliminases, appear to employ a different conformational change; here, movement is proposed to occur by rotation about the 5′-dAdo N-glycosidic bond to bridge the larger distance of 11 Å between the substrate and the Cbl (65–67).
To accelerate Co–C bond homolysis, substrate binding likely modulates the interactions between the protein and the 5′-dAdo, for example by inducing large scale conformational changes such as the TIM barrel motions or by altering active site electrostatics or dynamics to destabilize the C2′-endo form or to stabilize the C3′-endo form (55, 68, 69). It is unclear how many molecular mechanisms AdoCbl enzymes use to afford the substantial 1012 enhancement in Co–C bond homolysis that accompanies substrate binding (1, 3, 70). The IcmF structures reported here suggest that the C2′-endo to C3′-endo transition that is promoted by substrate binding may be a more common mechanism for increasing Co–C homolysis rates than previously thought. These structures also cast doubt on the relevance of the TIM barrel motions to homolysis rates, if barrel opening and closing need not accompany every turnover. Although more studies are needed to understand the relationship between substrate binding and Co–C bond homolysis, it is clear that substrate positioning with respect to the AdoCbl is universally important. Both substrate radical generation by AdoCbl and AdoCbl regeneration following turnover require precise positioning of the substrate in the active site. Our structures of the acyl-CoA mutase IcmF reveal that all four substrates are positioned similarly, with a β-carbon pointed toward the 5′-dAdo for hydrogen atom abstraction. It appears that the active site has some flexibility, in particular at the position of Phe-598, allowing it to accommodate both sets of acyl-CoA substrates. Nevertheless, it appears that Phe-598 is a critical determinant for substrate specificity; smaller residues at this position allow for more facile binding of substrates with tertiary α-carbons, as observed in HCM and PCM.
With three different acyl-CoA mutases now known to have the same overall structure and mode of substrate binding, we can more reliably identify determinants of substrate specificity for other members of this class. Our bioinformatic analyses identify critical sequence determinants, similar to previous analyses (4, 11), for substrate specificity; the presence of a charged Arg and an additional Tyr is required for binding of substrates with carboxylate groups, whereas a panel of smaller groups allows for binding of different aliphatic substrates such as isobutyryl-CoA and hydroxyisobutyryl-CoA. These analyses allow us to look more closely at two groups of thus far uncharacterized mutases (Fig. 9) and to predict their activities.
The first cluster of uncharacterized mutases is found within larger operons that encode a putative hydantoinase, a thiolase, a CoA transferase, a tungsten-dependent (in archaea) or molybdenum-dependent oxidoreductase (in bacteria), and other enzymes. Although strains carrying these mutases are not well characterized, recent studies suggest that a strain related to A. aromaticum, Azoarcus evansii, employs this operon for anaerobic degradation of indoleacetate (18). The authors proposed that the degradation pathway involves the carbon skeleton rearrangement of 2-(2′-aminophenyl)succinyl-CoA to (2′-aminobenzyl)malonyl-CoA (Fig. 1f) by a novel acyl-CoA mutase and used similar bioinformatic analyses to identify the same cluster of uncharacterized mutases (18). Our understanding of substrate binding in acyl-CoA mutases now allows for a re-examination of this proposal. These mutases likely bind carboxylate-bearing substrates, as indicated by the presence of Tyr and Arg in the determinant positions (Fig. 11). Additional replacements around the active site, conserved within this cluster but not in other mutases, lead to a substantially enlarged active site cavity, which may be able to bind the aminophenyl group of the proposed substrate (Fig. 12, a and b). Similarly, these or related mutases could be involved in anaerobic degradation of ethylbenzene, which likely requires a carbon skeleton isomerization after it gets metabolized to (1-phenylethyl)succinyl-CoA (6, 19). Together, our bioinformatic and modeling studies suggest that these mutases represent a new class of acyl-CoA mutases and accept substrates with aromatic groups.
Similarly, the second cluster of uncharacterized mutases is encoded in operons responsible for anaerobic oxidation of long-chain alkanes. These operons contain an alkylsuccinate synthase of the glycyl radical enzyme family that is proposed to convert alkanes and fumarate to 2-(1′-methylalkyl)succinyl-CoA, which could then be isomerized by an acyl-CoA mutase to (2′-methylalkyl)malonyl-CoA (Fig. 1g) and further processed by β-oxidation (20–23). Notably, D. alkenivorans is indeed known to degrade long-chain alkanes under anaerobic conditions using such a pathway (22, 23). Thus, these uncharacterized mutases likely represent novel (2′-methylalkyl)malonyl-CoA mutases. Indeed, they contain Tyr and Arg in the determinant positions, which would allow them to accept carboxylate-containing substrates (Fig. 11). Unfortunately, our attempts to further model the active sites of these mutases failed due to the low sequence similarity to MCM and IcmF. Further biochemical characterization of these proteins will be required to establish their role in anaerobic alkane degradation.
Notably, A. aromaticum and D. alkenivorans as well as other strains bearing these novel mutases encode several additional acyl-CoA mutases in their genomes. A. aromaticum, for example, encodes MCM and IcmF (Figs. 9, 11), whereas D. alkenivorans encodes MCM as well as another uncharacterized mutase (Fig. 9). All of these strains are known to be metabolically flexible, and it is tempting to speculate that this ability in part stems from a diverse array of acyl-CoA mutases. These analyses highlight the complex evolutionary history of acyl-CoA mutases, which likely underwent specialization, frequent horizontal gene transfer, and different domain fusion events.
Altogether, our studies reveal important design principles of acyl-CoA mutases. The improved understanding of different acyl-CoA mutase classes and their metabolic versatility could help facilitate the rational and directed engineering of these acyl-CoA mutases for applications ranging from generation of branched-chain biofuels to hydrocarbon remediation.
Author Contributions
M. J. and C. L. D. designed the study and wrote the paper, with critical contributions from D. A. B., V. C., and R. B. V. C. purified IcmF. M. J. crystallized IcmF, determined the substrate-bound crystal structures, and performed bioinformatic analyses. D. A. B. contributed to bioinformatic analyses. All authors analyzed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgment
We thank Marcus Gibson for help with generation of the phylogenetic tree and Professor Barbara Imperiali for advice and help regarding the synthesis of pivalyl-CoA.
This work was supported, in whole or in part, by National Institutes of Health Grants GM069857 (to C. L. D.) and DK45776 (to R. B.), Training Grant T32 GM008313 (to D. A. B.). This work was also supported by an MIT Poitras pre-doctoral fellowship (to M. J.). This work is based in part on research conducted at the Advanced Photon Source on the Northeastern Collaborative Access Team beamlines, supported by the National Institutes of Health and Grant P41 GM103403 from NIGMS. This research used resources of the Advanced Photon Source, a United States Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. The authors declare that they have no conflicts of interest with the contents of this article.
This article was selected as a Paper of the Week.

This article contains supplemental Fig. S1.
The atomic coordinates and structure factors (codes 5CJT, 5CJU, 5CJV, and 5CJW) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- 5′-dAdo
- 5′-deoxyadenosyl group
- AdoCbl
- adenosylcobalamin
- Cbl
- cobalamin
- Co–C
- cobalt-carbon
- ECM
- ethylmalonyl-CoA mutase
- HCM
- hydroxyisobutyryl-CoA mutase
- ICM
- isobutyryl-CoA mutase
- MCM
- methylmalonyl-CoA mutase
- PCM
- pivalyl-CoA mutase
- r.m.s.d.
- root mean square deviation
- TIM
- triose-phosphate isomerase
- PDB
- Protein Data Bank.
References
- 1.Halpern J. (1985) Mechanisms of coenzyme B12-dependent rearrangements. Science 227, 869–875 [DOI] [PubMed] [Google Scholar]
- 2.Frey P. A. (2010) in Comprehensive Natural Products II Chemistry and Biology (Mander L., and Liu H. W., eds) pp. 501–546, Elsevier, Oxford, UK [Google Scholar]
- 3.Banerjee R. (2003) Radical carbon skeleton rearrangements: catalysis by coenzyme B12-dependent mutases. Chem. Rev. 103, 2083–2094 [DOI] [PubMed] [Google Scholar]
- 4.Cracan V., and Banerjee R. (2012) Novel B12-dependent Acyl-CoA mutases and their biotechnological potential. Biochemistry 51, 6039–6046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cracan V., and Banerjee R. (2013) Cobalt and corrinoid transport and biochemistry. Met. Ions Life Sci. 12, 333–374 [DOI] [PubMed] [Google Scholar]
- 6.Rohwerder T., and Müller H. (2007) in Vitamin B: New Research (Elliot C. M., ed) pp. 81–98, Nova Science Publishers, Hauppauge, New York [Google Scholar]
- 7.Rodriguez G. M., Tashiro Y., and Atsumi S. (2014) Expanding ester biosynthesis in Escherichia coli. Nat. Chem. Biol. 10, 259–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dayem L. C., Carney J. R., Santi D. V., Pfeifer B. A., Khosla C., and Kealey J. T. (2002) Metabolic engineering of a methylmalonyl-CoA mutase-epimerase pathway for complex polyketide biosynthesis in Escherichia coli. Biochemistry 41, 5193–5201 [DOI] [PubMed] [Google Scholar]
- 9.Brendelberger G., Retey J., Ashworth D. M., Reynolds K., Willenbrock F., and Robinson J. A. (1988) The enzymic interconversion of isobutyryl and n-butyrylcarba(dethia)-coenzyme A: a coenzyme B12-dependent carbon skeleton rearrangement. Angew. Chem. Int. Ed. Engl. 27, 1089–1090 [Google Scholar]
- 10.Vrijbloed J. W., Zerbe-Burkhardt K., Ratnatilleke A., Grubelnik-Leiser A., and Robinson J. A. (1999) Insertional inactivation of methylmalonyl coenzyme A (CoA) mutase and isobutyryl-CoA mutase genes in Streptomyces cinnamonensis: influence on polyketide antibiotic biosynthesis. J. Bacteriol. 181, 5600–5605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Erb T. J., Rétey J., Fuchs G., and Alber B. E. (2008) Ethylmalonyl-CoA mutase from Rhodobacter sphaeroides defines a new subclade of coenzyme B12-dependent Acyl-CoA mutases. J. Biol. Chem. 283, 32283–32293 [DOI] [PubMed] [Google Scholar]
- 12.Erb T. J., Berg I. A., Brecht V., Müller M., Fuchs G., and Alber B. E. (2007) Synthesis of C5-dicarboxylic acids from C2-units involving crotonyl-CoA carboxylase/reductase: the ethylmalonyl-CoA pathway. Proc. Natl. Acad. Sci. U.S.A. 104, 10631–10636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yaneva N., Schuster J., Schäfer F., Lede V., Przybylski D., Paproth T., Harms H., Müller R. H., and Rohwerder T. (2012) Bacterial acyl-CoA mutase specifically catalyzes coenzyme B12-dependent isomerization of 2-hydroxyisobutyryl-CoA and (S)-3-hydroxybutyryl-CoA. J. Biol. Chem. 287, 15502–15511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weichler M. T., Kurteva-Yaneva N., Przybylski D., Schuster J., Müller R. H., Harms H., and Rohwerder T. (2015) Thermophilic coenzyme B12-dependent acyl coenzyme A (CoA) mutase from Kyrpidia tusciae DSM 2912 preferentially catalyzes isomerization of (R)-3-hydroxybutyryl-CoA and 2-hydroxyisobutyryl-CoA. Appl. Environ. Microbiol. 81, 4564–4572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cracan V., Padovani D., and Banerjee R. (2010) IcmF is a fusion between the radical B12 enzyme isobutyryl-CoA mutase and its G-protein chaperone. J. Biol. Chem. 285, 655–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cracan V., and Banerjee R. (2012) Novel coenzyme B12-dependent interconversion of isovaleryl-CoA and pivalyl-CoA. J. Biol. Chem. 287, 3723–3732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kitanishi K., Cracan V., and Banerjee R. (2015) Engineered and native coenzyme B12-dependent isovaleryl-CoA/pivalyl-CoA mutase. J. Biol. Chem. 290, 20466–20476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ebenau-Jehle C., Thomas M., Scharf G., Kockelkorn D., Knapp B., Schühle K., Heider J., and Fuchs G. (2012) Anaerobic metabolism of indoleacetate. J. Bacteriol. 194, 2894–2903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kniemeyer O., Fischer T., Wilkes H., Glöckner F. O., and Widdel F. (2003) Anaerobic degradation of ethylbenzene by a new type of marine sulfate-reducing bacterium. Appl. Environ. Microbiol. 69, 760–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jarling R., Sadeghi M., Drozdowska M., Lahme S., Buckel W., Rabus R., Widdel F., Golding B. T., and Wilkes H. (2012) Stereochemical investigations reveal the mechanism of the bacterial activation of n-alkanes without oxygen. Angew. Chem. Int. Ed. Engl. 51, 1334–1338 [DOI] [PubMed] [Google Scholar]
- 21.Wilkes H., Rabus R., Fischer T., Armstroff A., Behrends A., and Widdel F. (2002) Anaerobic degradation of n-hexane in a denitrifying bacterium: further degradation of the initial intermediate (1-methylpentyl)succinate via C-skeleton rearrangement. Arch. Microbiol. 177, 235–243 [DOI] [PubMed] [Google Scholar]
- 22.Callaghan A. V., Gieg L. M., Kropp K. G., Suflita J. M., and Young L. Y. (2006) Comparison of mechanisms of alkane metabolism under sulfate-reducing conditions among two bacterial isolates and a bacterial consortium. Appl. Environ. Microbiol. 72, 4274–4282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Callaghan A. V., Morris B. E., Pereira I. A., McInerney M. J., Austin R. N., Groves J. T., Kukor J. J., Suflita J. M., Young L. Y., Zylstra G. J., and Wawrik B. (2012) The genome sequence of Desulfatibacillum alkenivorans AK-01: a blueprint for anaerobic alkane oxidation. Environ. Microbiol. 14, 101–113 [DOI] [PubMed] [Google Scholar]
- 24.Mancia F., Keep N. H., Nakagawa A., Leadlay P. F., McSweeney S., Rasmussen B., Bösecke P., Diat O., and Evans P. R. (1996) How coenzyme B12 radicals are generated: the crystal structure of methylmalonyl-coenzyme A mutase at 2 Å resolution. Structure 4, 339–350 [DOI] [PubMed] [Google Scholar]
- 25.Mancia F., and Evans P. R. (1998) Conformational changes on substrate binding to methylmalonyl-CoA mutase and new insights into the free radical mechanism. Structure 6, 711–720 [DOI] [PubMed] [Google Scholar]
- 26.Mancia F., Smith G. A., and Evans P. R. (1999) Crystal structure of substrate complexes of methylmalonyl-CoA mutase. Biochemistry 38, 7999–8005 [DOI] [PubMed] [Google Scholar]
- 27.Froese D. S., Kochan G., Muniz J. R., Wu X., Gileadi C., Ugochukwu E., Krysztofinska E., Gravel R. A., Oppermann U., and Yue W. W. (2010) Structures of the human GTPase MMAA and vitamin B12-dependent methylmalonyl-CoA mutase and insight into their complex formation. J. Biol. Chem. 285, 38204–38213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jost M., Cracan V., Hubbard P. A., Banerjee R., and Drennan C. L. (2015) Visualization of a radical B12 enzyme with its G-protein chaperone. Proc. Natl. Acad. Sci. U.S.A. 112, 2419–2424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kurteva-Yaneva N., Zahn M., Weichler M. T., Starke R., Harms H., Müller R. H., Sträter N., and Rohwerder T. (2015) Structural basis of the stereospecificity of bacterial B12-dependent 2-hydroxyisobutyryl-CoA mutase. J. Biol. Chem. 290, 9727–9737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Drennan C. L., Huang S., Drummond J. T., Matthews R. G., and Ludwig M. L. (1994) How a protein binds B12: a 3.0 Å x-ray structure of B12-binding domains of methionine synthase. Science 266, 1669–1674 [DOI] [PubMed] [Google Scholar]
- 31.Dowling D. P., Croft A. K., and Drennan C. L. (2012) Radical use of Rossmann and TIM barrel architectures for controlling coenzyme B12 chemistry. Annu. Rev. Biophys. 41, 403–427 [DOI] [PubMed] [Google Scholar]
- 32.Vlasie M. D., and Banerjee R. (2003) Tyrosine 89 accelerates Co-carbon bond homolysis in methylmalonyl-CoA mutase. J. Am. Chem. Soc. 125, 5431–5435 [DOI] [PubMed] [Google Scholar]
- 33.Gruber K., Reitzer R., and Kratky C. (2001) Radical shuttling in a protein: ribose pseudorotation controls alkyl-radical transfer in the coenzyme B12-dependent enzyme glutamate mutase. Angew. Chem. Int. Ed. Engl. 40, 3377–3380 [DOI] [PubMed] [Google Scholar]
- 34.Vlasie M. D., and Banerjee R. (2004) When a spectator turns killer: suicidal electron transfer from cobalamin in methylmalonyl-CoA mutase. Biochemistry 43, 8410–8417 [DOI] [PubMed] [Google Scholar]
- 35.Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 38.Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Painter J., and Merritt E. A. (2006) Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr. D Biol. Crystallogr. 62, 439–450 [DOI] [PubMed] [Google Scholar]
- 40.Smart O. S., Womack T. O., Sharff A., Flensburg C., Keller P., Paciorek W., Vonrhein C., and Bricogne G. (2011) Grade. Global Phasing Ltd., Cambridge, UK [Google Scholar]
- 41.Moriarty N. W., Grosse-Kunstleve R. W., and Adams P. D. (2009) Electronic ligand builder and optimization workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr. D Biol. Crystallogr. 65, 1074–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brunger A. T. (2007) Crystallography and NMR System. Version 1.2. Nat. Protoc. 2, 2728–2733 [DOI] [PubMed] [Google Scholar]
- 43.Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., and Warren G. L. (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 44.Chen V. B., Arendall W. B. 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Laskowski R. A., MacArthur M. W., Moss D. S., and Thornton J. M. (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Cryst. 26, 283–291 [Google Scholar]
- 46.Schrödinger L. L. (2014) The PyMOL Molecular Graphics System, Version 1.7.4, Schrödinger, LLC, New York [Google Scholar]
- 47.Morin A., Eisenbraun B., Key J., Sanschagrin P. C., Timony M. A., Ottaviano M., and Sliz P. (2013) Collaboration gets the most out of software. Elife 2, e01456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dereeper A., Guignon V., Blanc G., Audic S., Buffet S., Chevenet F., Dufayard J. F., Guindon S., Lefort V., Lescot M., Claverie J. M., and Gascuel O. (2008) Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 36, W465–W469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Edgar R. C. (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Castresana J. (2000) Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552 [DOI] [PubMed] [Google Scholar]
- 51.Guindon S., Dufayard J. F., Lefort V., Anisimova M., Hordijk W., and Gascuel O. (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321 [DOI] [PubMed] [Google Scholar]
- 52.Chevenet F., Brun C., Bañuls A. L., Jacq B., and Christen R. (2006) TreeDyn: toward dynamic graphics and annotations for analyses of trees. BMC Bioinformatics 7, 439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Biasini M., Bienert S., Waterhouse A., Arnold K., Studer G., Schmidt T., Kiefer F., Cassarino T. G., Bertoni M., Bordoli L., and Schwede T. (2014) SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 42, W252–W258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Thomä N. H., Evans P. R., and Leadlay P. F. (2000) Protection of radical intermediates at the active site of adenosylcobalamin-dependent methylmalonyl-CoA mutase. Biochemistry 39, 9213–9221 [DOI] [PubMed] [Google Scholar]
- 55.Román-Meléndez G. D., von Glehn P., Harvey J. N., Mulholland A. J., and Marsh E. N. (2014) Role of active site residues in promoting cobalt-carbon bond homolysis in adenosylcobalamin-dependent mutases revealed through experiment and computation. Biochemistry 53, 169–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vey J. L., and Drennan C. L. (2011) Structural insights into radical generation by the radical SAM superfamily. Chem. Rev. 111, 2487–2506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Moore B. S., Eisenberg R., Weber C., Bridges A., Nanz D., and Robinson J. A. (1995) On the stereospecificity of the coenzyme B12-dependent isobutyryl-CoA mutase reaction. J. Am. Chem. Soc. 117, 11285–11291 [Google Scholar]
- 58.Reynolds K. A., O'Hagan D., Gani D., and Robinson J. A. (1988) Butyrate metabolism in streptomycetes. Characterization of an intramolecular interchange rearrangement linking isobutyrate and butyrate in Streptomyces cinnamonensis. J. Chem. Soc. Perkin Trans. 1, 3195–3207 [Google Scholar]
- 59.Berg I. A., Kockelkorn D., Buckel W., and Fuchs G. (2007) A 3-hydroxypropionate/4-hydroxybutyrate autotrophic carbon dioxide assimilation pathway in archaea. Science 318, 1782–1786 [DOI] [PubMed] [Google Scholar]
- 60.Yabuta Y., Kamei Y., Bito T., Arima J., Yoneda K., Sakuraba H., Ohshima T., Nakano Y., and Watanabe F. (2015) Functional and structural characteristics of methylmalonyl-CoA mutase from Pyrococcus horikoshii. Biosci. Biotechnol. Biochem. 79, 710–717 [DOI] [PubMed] [Google Scholar]
- 61.So C. M., and Young L. Y. (1999) Isolation and characterization of a sulfate-reducing bacterium that anaerobically degrades alkanes. Appl. Environ. Microbiol. 65, 2969–2976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bothe H., Darley D. J., Albracht S. P., Gerfen G. J., Golding B. T., and Buckel W. (1998) Identification of the 4-glutamyl radical as an intermediate in the carbon skeleton rearrangement catalyzed by coenzyme B12-dependent glutamate mutase from Clostridium cochlearium. Biochemistry 37, 4105–4113 [DOI] [PubMed] [Google Scholar]
- 63.Mansoorabadi S. O., Padmakumar R., Fazliddinova N., Vlasie M., Banerjee R., and Reed G. H. (2005) Characterization of a succinyl-CoA radical-cob(II)alamin spin triplet intermediate in the reaction catalyzed by adenosylcobalamin-dependent methylmalonyl-CoA mutase. Biochemistry 44, 3153–3158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bucher D., Sandala G. M., Durbeej B., Radom L., and Smith D. M. (2012) The elusive 5′-deoxyadenosyl radical in coenzyme-B12-mediated reactions. J. Am. Chem. Soc. 134, 1591–1599 [DOI] [PubMed] [Google Scholar]
- 65.Masuda J., Shibata N., Morimoto Y., Toraya T., and Yasuoka N. (2000) How a protein generates a catalytic radical from coenzyme B12: x-ray structure of a diol-dehydratase-adeninylpentylcobalamin complex. Structure 8, 775–788 [DOI] [PubMed] [Google Scholar]
- 66.LoBrutto R., Bandarian V., Magnusson O. T., Chen X., Schramm V. L., and Reed G. H. (2001) 5′-Deoxyadenosine contacts the substrate radical intermediate in the active site of ethanolamine ammonia-lyase: 2H and 13C electron nuclear double resonance studies. Biochemistry 40, 9–14 [DOI] [PubMed] [Google Scholar]
- 67.Toraya T. (2014) Cobalamin-dependent dehydratases and a deaminase: radical catalysis and reactivating chaperones. Arch. Biochem. Biophys. 544, 40–57 [DOI] [PubMed] [Google Scholar]
- 68.Sharma P. K., Chu Z. T., Olsson M. H., and Warshel A. (2007) A new paradigm for electrostatic catalysis of radical reactions in vitamin B12 enzymes. Proc. Natl. Acad. Sci. U.S.A. 104, 9661–9666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jensen K. P., and Ryde U. (2005) How the Co–C bond is cleaved in coenzyme B12 enzymes: a theoretical study. J. Am. Chem. Soc. 127, 9117–9128 [DOI] [PubMed] [Google Scholar]
- 70.Brown K. L. (2005) Chemistry and enzymology of vitamin B12. Chem. Rev. 105, 2075–2149 [DOI] [PubMed] [Google Scholar]
- 71.Ratnatilleke A., Vrijbloed J. W., and Robinson J. A. (1999) Cloning and sequencing of the coenzyme B12-binding domain of isobutyryl-CoA mutase from Streptomyces cinnamonensis, reconstitution of mutase activity, and characterization of the recombinant enzyme produced in Escherichia coli. J. Biol. Chem. 274, 31679–31685 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.