

Background: The activity of several Gq- and Gi-coupled receptors is modulated by the membrane potential.
Results: Voltage modulates catecholamine-mediated activation of Gs-coupled β1- and β2-adrenoceptors.
Conclusion: Voltage-dependence of β1-AR is due to alterations in the efficacy of catecholamines.
Significance: By modulating catecholamine efficacy on β1-ARs, voltage can modify receptor activity on a very fast time scale.
Keywords: adrenergic receptor, arrestin, fluorescence resonance energy transfer (FRET), G protein, G protein-coupled receptor (GPCR), kinetics, voltage-dependence
Abstract
G protein-coupled receptors (GPCRs) are membrane-located proteins and, therefore, are exposed to changes in membrane potential (VM) in excitable tissues. These changes have been shown to alter receptor activation of certain Gi-and Gq-coupled GPCRs. By means of a combination of whole-cell patch-clamp and Förster resonance energy transfer (FRET) in single cells, we demonstrate that the activation of the Gs-coupled β1-adrenoreceptor (β1-AR) by the catecholamines isoprenaline (Iso) and adrenaline (Adr) is regulated by VM. This voltage-dependence is also transmitted to G protein and arrestin 3 signaling. Voltage-dependence of β2-AR activation, however, was weak compared with β1-AR voltage-dependence. Drug efficacy is a major target of β1-AR voltage-dependence as depolarization attenuated receptor activation, even under saturating concentrations of agonists, with significantly faster kinetics than the deactivation upon agonist withdrawal. Also the efficacy of the endogenous full agonist adrenaline was reduced by depolarization. This is a unique finding since reports of natural full agonists at other voltage-dependent GPCRs only show alterations in affinity during depolarization. Based on a Boltzmann function fit to the relationship of VM and receptor-arrestin 3 interaction we determined the voltage-dependence with highest sensitivity in the physiological range of membrane potential. Our data suggest that under physiological conditions voltage regulates the activity of agonist-occupied β1-adrenoceptors on a very fast time scale.
Introduction
The classical way of activating G protein-coupled receptors (GPCRs)2 is initiated by the binding of extracellular ligands to the receptor. GPCR activity can also be modulated by the membrane potential (VM), which has first directly been shown for the regulation of the muscarinic M2 acetylcholine receptor (M2AChR) (1). Since then, several Gq- and Gi-coupled receptors of GPCR classes A and C were characterized with regard to their voltage-dependence (2–10). This voltage-dependence is an intrinsic property of the receptor molecule as shown by the measurement of “gating currents” in muscarinic receptors (11, 12) and depolarization-induced conformational changes of the α2A-adrenoreceptor (α2A-AR) measured with an intramolecular FRET-based biosensor (7). Measurements of GPCR-effector responses showed that also downstream signaling of, for example, certain glutamate receptors (2), the purinergic P2Y1 receptor (P2Y1R) (3, 13), dopaminergic D2 receptors (D2R) (14), and α2A-AR (7) was effected by VM. Depolarization induced changes in agonist affinity which either led to deactivation of M2AChR, metabotropic glutamate receptor 3 (mGluR3) and α2A-AR (1, 2, 7) or activation of M1AChR and mGluR1 (2, 11). The dissociation of agonists has been shown to be voltage-dependent with the association being unaffected (7, 15). Also increases in agonist potency have been reported for lysophosphatidic acid (LPA) receptors and P2Y1R (4, 13). In contrast, depolarization reduced the histamine potency at the histamine H4 receptor (H4R) (6). In addition to altered affinity or potency of agonists by VM there have been reports on agonist-specificity of voltage-dependence at the M2AChR (12) and D2SR (16). Despite increasing evidence for voltage-sensitive receptor function, the mechanism of voltage-dependence remains unclear.
β-Adrenoceptors (β-ARs) comprise another group of class A GPCRs of great clinical importance. Sympathetic stimulation of β-ARs with catecholamines is transduced into intracellular responses via coupling to Gs proteins to regulate, for example, heart rate and contractility (17). Of the three existing β-AR subtypes expressed in the (healthy) heart, β1-AR is the most abundant (18). With their seven-transmembrane spanning topology, β-ARs are constantly exposed to changes in VM in this excitable tissue. Therefore, it was the aim of this study to investigate voltage-sensitivity of the Gs-coupled β1-AR function and its transmission to downstream signaling by means of a combination of FRET and whole-cell voltage-clamp in single living HEK293 cells (Fig. 1A; 7, 19). With a FRET-based biosensor for β1-AR termed β1-AR sensor (20) we directly measured depolarization-mediated inactivating conformational changes occurring with very fast kinetics. FRET assays between β1-AR and either a G protein subunit (21) or arrestin 3 (Arr3) (22, 23) showed deactivation effects of depolarization on downstream signaling. The closely related β2-AR was also voltage-dependent but the extent of depolarization-induced deactivation was less pronounced than in β1-AR.
FIGURE 1.

Depolarization reduced β1-AR activation. A, schematic of FRET and electrophysiology measurements illustrated with a cell expressing a β1-AR sensor. Cells were excited at 430 nm (dark blue) and donor (F480, light blue) and acceptor (F535, yellow) emissions were recorded. Simultaneously, cells were superfused with buffer or agonist-containing buffer with a pressurized perfusion system (left), and the membrane potential was controlled in whole-cell voltage-clamp configuration (patch pipette and amplifier, right). B, representative measurement of the transiently transfected β1-AR sensor stimulated with 10 μm Adr. The FRET ratio (F535/F480) is shown before (above) and after (below) correction for photobleaching by subtraction of a mono-exponential curve (τ = 574 s) and normalization to the initial FRET value before stimulation. A black bar with agonist labeling above the ratio trace indicates the duration of application and type of agonist used. The time scale appears as a black bar in every graph. The bar below the traces shows the course of membrane potential (holding potential: −90 mV; test potential: +60 mV). C and D, mean ± S.E. of Iso-(C, 1 μm, n = 9) and Adr-(D, 10 μm, n = 8) stimulated β1-AR sensor in cells stably or transiently expressing the β1-AR sensor, respectively. Single traces were corrected for bleaching as indicated in B, normalized to the maximal agonist-induced amplitude and smoothed before averaging. The dashed lines indicate the baselines for the evaluation of FRET amplitudes which have been measured in individual experiments. E and F, to quantify the depolarization-induced reduction of FRET response the quotient of amplitudes during activation in depolarization and at holding potential was calculated and subtracted from 1 (100%) to obtain % deactivation. The summarized data at different concentrations of Iso (E, n = 8–10) or Adr (F, n = 5–8) are shown. **: p < 0.001, ***: p < 0.001; one way ANOVA with Bonferroni's Multiple Comparison Test was used as a statistical test.
To our knowledge this is the first report directly addressing voltage-dependence of Gs protein-coupled receptors. Voltage-mediated alterations in agonist (or antagonist) efficacy, so far, have only been shown indirectly (12–14) with the exception of clonidine at the α2A-AR (7), and only for non-endogenous ligands. With FRET-based assays suited for the measurement of drug efficacy (7, 24) we identified efficacy as a major contributor to the voltage-dependent regulation of β1-AR activation. The contribution of decreased affinity during depolarization differed between Iso- and Adr-stimulated cells.
Experimental Procedures
Reagents
Isoprenaline, adrenaline, and noradrenaline were purchased from Sigma-Aldrich.
Plasmids
The plasmid containing the FRET-based human β1-AR sensor (Arg-389) as well as HEK293 cells stably expressing this sensor were kindly provided by Prof. Stefan Engelhardt, Technische Universität München, Germany. Cloning of the β1-AR sensor is described in Ref. 20.
Cell Culture and Transfection of HEK293 T Cells
Human embryonic kidney 293T (HEK293T) cells were cultured under standard conditions (25). Selection antibiotic G418 was added to the medium of HEK293 cells stably expressing the human β1-AR sensor (Arg-389). Transient transfection of HEK 293T cells was achieved with Effectene transfection reagent (Qiagen) following standard protocol in 6-cm culture dishes 2 days prior to experiments. We used the following cDNAs: Receptor activation: HEK 293 cell line stably expressing β1-AR sensor (kindly provided by Stefan Engelhardt) or cells transiently transfected with β1-AR sensor (0.5 μg) and pcDNA3 (0.5 μg). Receptor-arrestin interaction: human β1-YFP (Arg389) or human β2-YFP (0.4 μg), human GRK2 (0.5 μg), bovine CFP-arrestin 3 (CFP-Arr3) (0.6 μg). Receptor-G protein interaction: β1-YFP (Arg389) (0.4 μg), bovine Gαs-wt (2 μg), human Gβ1-wt (0.5 μg), bovine CFP-Gγ2 (0.2 μg) (21). Transfected cells were split onto poly-l-lysine-coated glass coverslips the day before measurements.
FRET Measurements and Electrophysiology
Fluorescence and electrophysiological measurements were performed simultaneously as described before (7) (for a schematic see Fig. 1A). In brief, an inverted Zeiss microscope (Axiovert 130) equipped with a dual-emission photometry system (TILL Photonics) was used to perform real-time live-cell FRET measurements. Cells were continuously superfused during measurements with either buffer (in mm: 137 NaCl, 5.4 KCl, 1 MgCl2, 10 HEPES, pH 7.3) or buffer containing agonist (isoprenaline (Iso), adrenaline (Adr), or noradrenaline (NA)) using a pressurized superfusion device (ALA Scientific Instruments). Donor (F480) and acceptor (F535) emissions were detected by photodiodes following short donor excitation at 430 nm with a Polychrome V light source (TILL Photonics), and data were processed using Patchmaster software (v2X52, HEKA). Sampling frequencies were 2.5 Hz (arrestin FRET assay) or 5 Hz (β1-AR sensor and receptor-G protein interaction). The ratio of acceptor over donor emission termed FRET ratio (F535/F480) was calculated. Pronounced photobleaching in 5 Hz frequency measurements of β1-AR sensor and of receptor-G protein interaction made corrections of FRET ratios of individual experiments necessary. Because of a poor signal to noise ratio, FRET ratios of experiments with the β1-AR sensor were smoothed (5-point smoothing, Savitzky-Golay) using Origin Pro 9.1 software (OriginLabs) before averaging. During FRET measurements cells were patched in whole-cell configuration, and the membrane potential (VM) was set to desired values with an EPC-10 amplifier (HEKA). Patch pipette resistances ranged from 4–8 MΩ, and pipettes were filled with internal buffer (in mm: 100 K+-aspartate, 40 KCl, 5 NaCl, 7 MgCl2, 20 HEPES, 10 EGTA, 0.025 GTP, 5 Na+-ATP).
Data Analysis and Statistics
Data were analyzed with Origin Pro 9.1 or GraphPad Prism 4 (GraphPad Software) and are displayed as individual experiments or mean ± S.E. of n individual measurements. Statistical analysis was performed with one way ANOVA with Bonferroni's posthoc test, paired Student's t test or F-test for fit comparison. Differences were considered statistically significant at p < 0.05.
Results
Membrane Potential Alters Activation of Gs-coupled β1-AR
To investigate the voltage-dependence of Gs-coupled β1-AR, FRET measurements were performed in HEK293 cells expressing an intramolecular FRET-based β1-AR sensor (20). This FRET sensor displays a high FRET signal in the inactive state. Stimulation with agonist induces a conformational change in GPCRs with the largest change being the outward movement and rotation of transmembrane helix 6 (TM 6) relative to the C terminus, which reflects receptor activation (26). This rearrangement leads to a decrease in FRET ratio due to an increase in distance between the two fluorophores YFP and Cerulean (Cer, CFP variant) inserted into the third intracellular loop and fused to the C terminus of β1-AR, respectively. To display the dependence of receptor activation on the membrane potential (VM), cells were simultaneously subjected to whole-cell voltage-clamp conditions (Fig. 1A). As FRET-based sensors are prone to pronounced photobleaching, measurements were corrected for this effect by subtraction of a mono-exponential function and were afterward normalized to the value prior to agonist stimulation or to the maximal amplitude evoked by agonist application. Upon stimulation with 10 μm Adr the FRET ratio (F535/F480) decreased indicating receptor activation. This decrease in FRET ratio was reversible by withdrawal of the agonists. Depolarization from a holding potential of −90 mV to +60 mV reduced the agonist-evoked FRET response (FRET amplitude), an effect which was also reversible upon repolarization (Fig. 1B). In the absence of agonists VM didn't significantly alter the FRET signal (n = 7, p = 0.22). This indicates that voltage-induced FRET changes in the presence of agonist were caused by conformational changes associated with alterations in receptor activity. To evoke similar responses, despite differences in agonist affinities (27), we used higher concentrations of Adr compared to Iso. Cells stimulated with either 1 μm Iso or 10 μm Adr were deactivated during depolarization to different degrees (Fig. 1, C and D). Note, the depolarization-evoked deactivation occurred with much faster kinetics than the deactivation by washout. Increasing concentrations of agonist showed a tendency to lessen depolarization-induced deactivation in Iso-stimulated cells (Fig. 1E). For Adr-stimulated receptors this depolarization-mediated effect was significantly attenuated with a maximal reduction of the depolarization effect of about 3-fold (Fig. 1F). This points toward a change in affinity playing a role in voltage-dependence of β1-AR, which is more pronounced for Adr than for Iso. Nevertheless, even saturating concentrations of either Iso or Adr could not completely block receptor deactivation by positive VM. About a 20% reduction of the FRET amplitude still remained when stimulated with 100 μm Iso or 500 μm Adr (Fig. 1, E and F). These initial experiments revealed a voltage-dependent regulation of receptor activation at the receptor level demonstrating that the Gs-coupled β1-AR is voltage-dependent.
GPCR Downstream Signaling Is Attenuated at Positive VM
As a next step we tested whether those voltage-induced changes in receptor conformation translate into altered downstream signaling. Therefore, we transfected cells with YFP-labeled β1-AR (β1-YFP), CFP-Gγ2 subunit and unlabeled Gα and Gβ subunits to measure receptor-G protein interaction. Both Iso and Adr induced an increase in FRET reflecting the interaction of β1-AR and the heterotrimeric G protein (21), which was reversible upon washout of the ligands (Fig. 2, A and B). As seen for the β1-AR sensor in Fig. 1, the G protein pathway was sensitive to depolarization (+60 mV). Positive VM diminished the interaction between G protein subunits and receptor by 28.2 ± 5% (Iso) and 33.8 ± 5% (Adr), which was reversible upon repolarization of the cell (Fig. 2, A and B). This reduction in ligand potency during depolarization was accompanied by the aforementioned reduction in efficacy indicated by a faster offrate induced by depolarization than by ligand washout (Figs. 2, A and B and 5, C and D).
FIGURE 2.

VM alters G protein and arrestin 3 interaction with β1-AR. A and B, interaction of G protein and β1-AR was measured in cells transfected with β1-YFP, CFP-Gγ2 and unlabeled Gαs and Gβ subunits. Cells were stimulated with 1 μm Iso (A, n = 7) or 10 μm Adr (B, n = 7). The measurements were corrected for bleaching before averaging (mean ± S.E.). C and D, arrestin 3 interaction with β1-AR was analyzed in cells expressing β1-YFP, CFP-Arr3 and GRK2 (C: 100 nm Iso, n = 8 and D: 1 μm Adr, n = 6; mean ± S.E.). The membrane potential was depolarized from −90 mV to +45 mV. The red box marks the section of the details in E. Small schematic insets in A and C show the FRET assays corresponding to the figure data. E, an overlay of sections of averaged data (means ± S.E.) with saturating concentrations of Iso (n = 8), Adr (n = 8), and NA (n = 8) shows the depolarization-induced reduction of receptor-arrestin 3 interaction. Individual experiments were normalized to the maximal amplitude before averaging. F, FRET amplitudes to calculate %-deactivation by depolarization during activation with sub- and saturating concentrations of agonist were measured as follows: −90 mV amplitude: baseline to dashed line (C and D), +45 mV amplitude: baseline to curve. Iso (100 nm, n = 8; 10 μm, n = 8) and Adr (1 μm, n = 10; 100 μm, n = 8). The calculation and quantification was performed like in Fig. 1E and F. ***: p < 0.001, one way ANOVA with Bonferroni's Multiple Comparison Test was used as a statistical test.
FIGURE 5.

Agonist efficacy of β1-AR is regulated by voltage. Washout- and depolarization-induced deactivation of the β1-AR sensor (A and B) or of the disruption of G protein-receptor interaction (C and D) and arrestin 3-receptor interaction (E and F) were compared. Small schematic insets show the FRET assays corresponding to the figure data. A, in the average trace of Fig. 1C the dark gray (voltage) and light gray (washout) squares indicate where offrates were determined by fitting the average to a mono-exponential function (β1-AR sensor, n = 9, mean ± S.E.). C and E, data of Fig. 2, B and C were normalized to the maximal FRET amplitude in sections indicated in A and used for fitting and overlay of voltage- and washout-induced off rates (C: n = 7, E: n = 8). To increase clearness the error bars only point in one direction. The arrows indicate the time point when cells were depolarized (+60 mV (C)/+45 mV (E)) or when the agonist was withdrawn (−10 μm Adr (C), −100 nm Iso (E)). B, D, and F summarize the statistical analysis of koff-values of the sections indicated in A in Iso- or Adr-stimulated cells (B: n≥8, D: n = 7, F: n≥8). F-tests were used as statistical tests to compare fits and determine statistical significance: **: p < 0.01, ***: p < 0.001. #: The calculated koff-values were close to the detection limit of 200 ms (D) and 400 ms (F) (sampling frequencies of 5 Hz and 2.5 Hz, respectively).
In addition to G proteins, arrestins play an important role in GPCR-mediated signaling and desensitization (28). To confirm that voltage-dependence is also transmitted to the arrestin signaling pathway, cells transfected with β1-YFP, CFP-arrestin 3 (CFP-Arr3), and GPCR kinase 2 (GRK 2) were subjected to depolarization. GRKs phosphorylate the receptor in a ligand-dependent manner, which then leads to arrestin recruitment to and interaction with the phosphorylated receptor (29). Thus, an arrestin-receptor interaction FRET assay shows an increase in FRET amplitude upon stimulation with either Iso or Adr. To assure a stable whole-cell configuration and precise voltage control during long-term recordings, the depolarizing step was reduced from +60 mV to +45 mV. Arrestin 3-receptor interaction was disrupted during depolarization of the plasma membrane and restored after repolarization (Fig. 2, C and D). These data nicely show that both G protein and arrestin 3 signaling are negatively regulated by depolarization of the plasma membrane.
We also analyzed the effect of depolarization on receptor-arrestin 3 interaction in cells stimulated with saturating concentrations of agonists. Noradrenaline (NA) was included as a ligand because of its importance in regulating heart function through the sympathetic nervous system (18). Measurements with NA, Adr, or Iso were normalized to the maximal amplitude prior to depolarization and the data of corresponding sections to the one marked in D were overlaid (Fig. 2E). Depolarization induced the dissociation of arrestin 3 from β1-AR even when the receptor was saturated with NA, Iso, or Adr (Fig. 2E). The extent of inhibition was comparable for cells stimulated with NA and Adr, which is why further experiments were performed only with Adr. Compared with NA- and Adr-stimulated cells, the deactivation by depolarization was more pronounced during Iso-stimulation. To compare depolarization-induced deactivation during stimulation with non- and saturating concentrations of agonist with this arrestin-assay, the measurements in Fig. 2, C and D were repeated with 10 μm Iso and 100 μm Adr (data not shown). Agonist-induced amplitudes were measured at depolarization and set in relation to the amplitudes at −90 mV at the corresponding time point (on the dashed lines in Fig. 2, C and D). The comparison of non-saturating and saturating concentrations showed that the degree of depolarization-induced dissociation of arrestin 3 from β1-AR was similar using moderate concentrations of Adr or Iso (Fig. 2F). In saturation, about 50 and 25% of the depolarization effect remained during Iso and Adr stimulation, respectively (Fig. 2F). This remaining effect arises from a change in drug efficacy.
Voltage-dependence Occurs Within the Physiological Range of Membrane Potential
So far we could show voltage-dependence on the receptor level as well as the inhibition of downstream signaling at positive potentials. To determine whether the observed voltage-dependence occurs within the physiological range of VM we used the arrestin FRET assay to measure the relation of Arr3-β1-AR interaction and VM. β1-AR was activated with either a non-saturating concentration of Iso or Adr and VM was changed stepwise to different values. An example of a single measurement for an Adr-stimulated cell is given in Fig. 3A. Stepwise increase of VM gradually reduced the receptor-arrestin 3 interaction whereas hyperpolarization to −120 mV further enhanced the interaction. FRET amplitudes induced by test potentials were set relative to the corresponding amplitude at the holding potential of −90 mV (dashed lines in Fig. 3A), and results were plotted against VM (Fig. 3B). The data were then fit to a Boltzmann function which yielded similar half-maximal effects of interaction for Iso- and Adr-stimulated cells (V0.5 = −28 ± 22 mV (Iso) and V0.5 = −27 ± 8 mV (Adr)). Consequently, the voltage-sensitivity of β1-AR lies perfectly within the physiological range of membrane potential.
FIGURE 3.
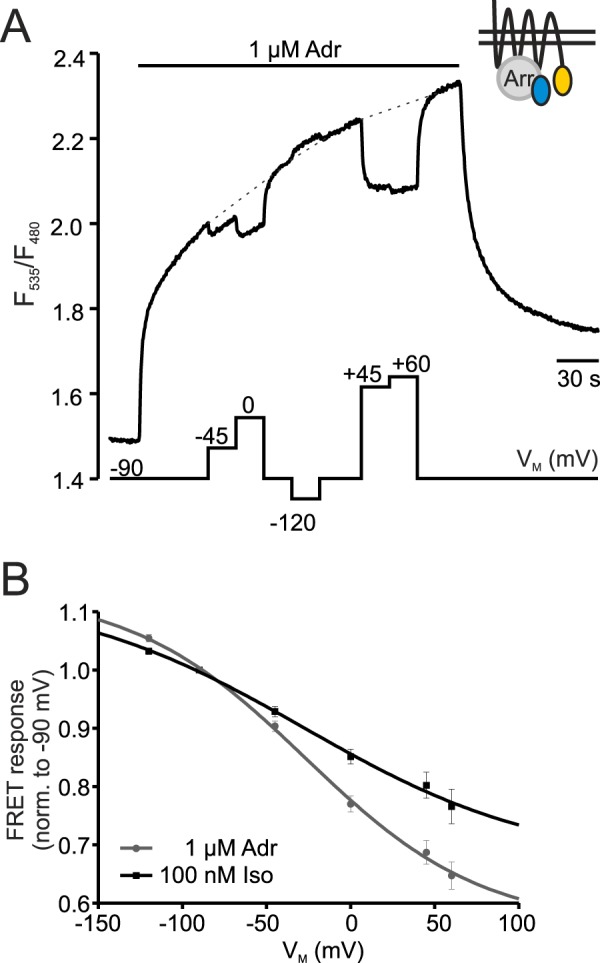
Voltage-dependence occurs within the physiological range of VM. A, representative single cell measurement shows the FRET ratio of a cell transfected with β1-YFP, CFP-Arr3 (see schematic inset), and GRK2 at various VM-steps. The amplitudes were measured during hyper- or depolarization steps relative to the amplitude at −90 mV (dashed lines) and plotted against VM (B). B, VM/FRET response ratio of Iso- and Adr-stimulated cells were fit to a Boltzmann function (Iso: black, n = 14, Adr: gray, n = 11; R2 of fits: 0.99).
Minor Alteration of β2-Adrenoreceptor Activation by VM
Next, we investigated whether the voltage-dependent effects seen so far were unique to the β1-AR or whether the activation of the closely related Gs-coupled β2-adrenoreceptor (β2-AR) is also regulated by VM. For this purpose, cells were transfected with an arrestin FRET assay exchanging β1-AR-YFP for β2-AR-YFP together with GRK2 and CFP-Arr3. Depolarization to +45 mV only had a minor effect on the interaction even in cells activated with submaximal concentrations of Iso (Fig. 4A). The average reduction of arrestin 3-β2-AR interaction at depolarization was 9.9 ± 1%. In saturation, this effect was abolished for cells stimulated with Adr (Fig. 4B, red trace) and almost absent when cells were stimulated with Iso (Fig. 4B, black trace). For better comparison of depolarization effects in cells stimulated with the two agonists, individual measurements were normalized to the maximal amplitude prior to the depolarizing step before averaging. Compared with β1-AR, membrane potential did not greatly alter the activation of β2-AR.
FIGURE 4.
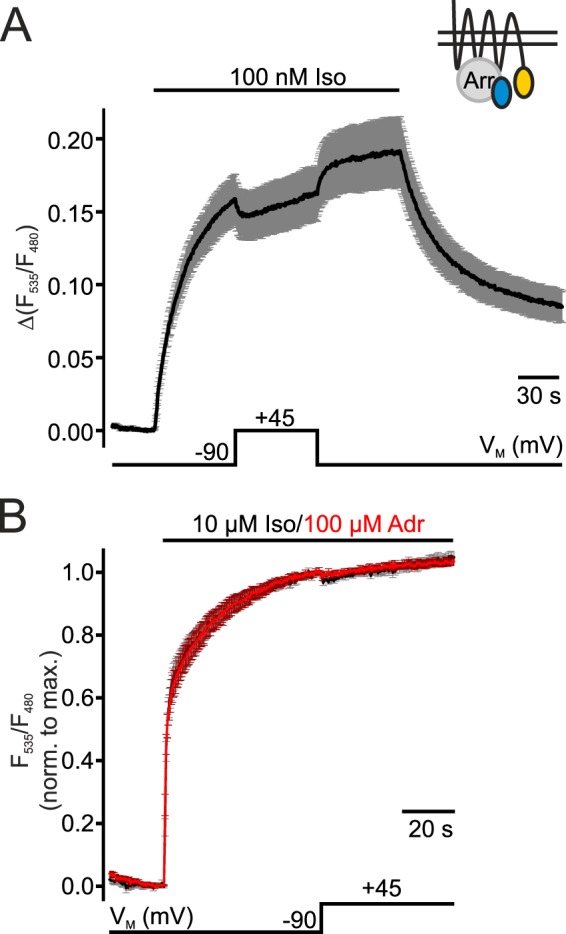
Voltage-sensitivity of the interaction of β2-AR and arrestin 3. Means ± S.E. of cells transfected with β2-YFP, CFP-Arr3 and GRK2 are shown. A small schematic inset shows the FRET assay used. A, cells were stimulated with 100 nm Iso. The single traces were normalized to the FRET value before stimulation and averaged (n = 9, mean ± S.E.). B, stimulation was induced with 10 μm Iso (black trace, n = 7) or 100 μm Adr (red trace, n = 8). Single measurements were normalized to the maximal amplitude prior to depolarization (+45 mV) before averaging (mean ± S.E.).
Drug Efficacy Is the Main Target of Voltage-dependence
We briefly mentioned previously that we observed large differences in deactivation kinetics evoked by depolarization versus agonist withdrawal in measurements of the β1-AR sensor. Those differences could point toward a mechanism in which a change in efficacy underlies voltage-dependence of β1-AR activation. In earlier studies FRET-based intramolecular GPCR sensors have been described as very suitable tools to directly investigate intrinsic efficacy (24). Therefore, we analyzed voltage (i.e. depolarization)- and washout-mediated deactivation kinetics within the dark gray and light gray boxes (Fig. 5A) by fitting the corresponding sections of β1-AR sensor data presented in Fig. 1, C and D to a mono-exponential function. In cells expressing the β1-AR sensor poor signal-to-noise ratio only allowed mono-exponential fitting of the average trace in the marked sections. Comparison of off-rates determined in this manner showed an over 100-fold acceleration of voltage-induced over washout-induced deactivation of Iso-activated receptors ((k = 1.7 ± 0.5 s−1 versus k = 0.017 ± 0.003 s−1 or t1/2 = 0.4 s versus t1/2 = 40.9 s) (Fig. 5B). As mentioned before, the affinity of Adr towards the β1-AR is smaller than the one of Iso (27). This results in faster dissociation of adrenaline from the receptor during washout. Therefore, the difference between voltage- and washout-evoked dissociation rate was smaller in Adr-stimulated cells, but voltage still deactivated the receptor significantly faster (about 8-fold, k = 0.98 ± 0.2 s−1 versus k = 0.13 ± 0.006 s−1 or t1/2 = 0.7 s versus t1/2 = 5.5 s) than removal of Adr (Fig. 5B). The voltage-induced deactivation was comparable and fast in Iso- and Adr-stimulated cells. But does this efficacy phenomenon transmit to downstream signaling as well? Although measuring downstream consequences of receptor activation, the FRET assays detecting arrestin 3-receptor or G protein-receptor interaction are suitable to directly monitor drug efficacy. A prerequisite for a direct measurement of efficacy is that receptor number and coupling do not affect the measurement (24), which is fulfilled in these assays.
For kinetics analysis of receptor-G protein interaction or receptor-arrestin 3 interaction, FRET ratios of individual measurements (data of Fig. 2) were normalized to the maximal amplitude within the sections marked in Fig. 5A and averaged (Fig. 5, C and E). The arrows mark the time point at which cells were subjected to depolarization (+60 mV (C)/+45 mV (E)) or the agonist was withdrawn (−10 μm Adr (C), −100 nm Iso (E)). These overlays also nicely show a substantial acceleration in dissociation induced by depolarization compared to washout of agonists. For a quantitative analysis, averages were fit to a mono-exponential function and koff-values of voltage- and washout-induced disruption of receptor-transducer interaction were analyzed statistically (Fig. 5, D and F). The Iso-evoked interaction between receptor and G protein broke off 70-times faster upon depolarization (k = 2.2 ± 1.0 s−1 or t1/2 = 0.3 s) than when Iso was withdrawn from the cell (k = 0.033 ± 0.003 s−1 or t1/2 = 21.0 s). As seen above, the difference in kinetics in cells stimulated with Adr was not as pronounced but yet significant. An 11-fold increase in dissociation rate of G protein from the receptor was induced by depolarization to +60 mV (k = 2.3 ± 0.5 s−1 or t1/2 = 0.3 s) compared to washout (k = 0.21 ± 0.006 s−1 or t1/2 = 3.3 s) (Fig. 5D). Very similar results in terms of off kinetics were calculated by fitting the mentioned sections of data of arrestin 3-receptor interaction (Fig. 2, C and D) to mono-exponential decay functions (Fig. 5F). Also here significant differences were determined for voltage-induced (Iso: k = 0.32 ± 0.02 s−1 or t1/2 = 2.1 s; Adr: k = 1.37 ± 0.1 s−1 or t1/2 = 0.5 s) versus washout-induced (Iso: k = 0.031 ± 0.001 s−1 or t1/2 = 22.7 s; Adr: k = 0.099 ± 0.002 s−1 or t1/2 = 7.0 s) disruption of interaction in Iso- and Adr-stimulated cells. These values translate to 10- and 14-fold differences for Iso- or Adr-induced interaction of arrestin 3 and β1-AR, respectively. Note, that t1/2-values of the voltage-mediated disruption of receptor-transducer or -effector interaction occurred close to the detection limit of sampling frequency (G protein: t1/2 = 0.3 s, sampling frequency: 5 Hz; arrestin 3: t1/2 = 0.5 s (Adr stimulation), sampling frequency: 2.5 Hz). Measurement accuracy for the fast deactivation and disruption of interaction is limited and hence, the true kinetics could be even faster.
Next, we determined alterations in receptor affinity to Iso due to depolarization by analysis of the dissociation kinetics after withdrawal of agonist. Therefore, the decay of FRET between receptors and arrestins was measured twice in individual cells at −90 mV and at +45 mV in an alternating order. The data obtained at both voltages were normalized to the maximal amplitude and averaged (Fig. 6A). These data show an accelerated disruption of receptor-arrestin interaction at +45 mV compared to −90 mV. Fits of mono-exponential functions to the FRET decay yielded a significant but less than 2-fold increase in k-value at +45 mV (k = 0.044 ± 0.005 s−1 or t1/2 = 16.8 s) compared to −90 mV (k = 0.027 ± 0.003 s−1 or t1/2 = 28.4 s) (Fig. 6B), indicating a very moderate effect of voltage on the agonist affinity.
FIGURE 6.

Concentration-response curve yields significant reduction in efficacy by depolarization. The schematic inset shows the FRET-assay detecting interaction between β1-AR-YFP and CFP-Arr3 in the presence of unlabeled GRK2. A, cells were stimulated with 100 nm Iso and measured twice under the constant membrane potentials of −90 mV and +45 mV in an alternating order. Individual experiments were normalized to the maximal amplitudes and averaged (n = 9, mean ± S.E.). B, comparison and statistical analysis of koff-values at constant VM of the data in A. Statistical significance was determined by a paired samples t test, ***: p < 0.001. C, a representative measurement illustrates how data for a concentration-response-curve (D) were collected. Cells were first stimulated with a test concentration of Iso (here 1 μm). After washout of the test concentration the reference concentration of 10 μm Iso was applied. During both stimulations the membrane potential was switched between −90 mV and +45 mV. Amplitudes at +45 mV (A+45) and −90 mV (A-90) of the first stimulation were set in relation to the amplitude at −90 mV during application of 10 μm Iso (Aref). A two-point interpolation (dashed lines) was used to establish baselines for the calculation of the FRET amplitudes. This was necessary to account for the increase in the non-reversible fraction of the FRET signal over the time course of the experiment. D, concentration-response curve was drawn from summarized data collected as described in C at −90 mV and +45 mV at various test concentrations of Iso (n = 3–9 per data point). Paired Student's t-tests confirmed statistically significant differences between data points measured at +45 mV compared with −90 mV at all concentrations of Iso except for 10−10 m Iso.
To determine the voltage-dependence of agonist efficacy and affinity, we measured concentration-response-curves for Iso at the two potentials of −90 mV and +45 mV. We stimulated cells with a test concentration of Iso first and applied the reference concentration of 10 μm Iso after washout of the test concentration to allow for normalization. During both stimulations, the membrane potential was switched between the holding potential and depolarization (Fig. 6C). FRET amplitudes arising from the stimulation with a given test concentration at −90 mV (A-90) and +45 mV (A+45) were measured relative to the reference concentration of 10 μm Iso at −90 mV (Aref). The analysis of FRET amplitudes was complicated by an increase in the non-recovering FRET signal, which is most likely attributed to an accumulation of receptor-arrestin complexes, presumably in clathrin-coated pits. We used a two-step interpolation of the baseline (dashed-lines) to at least partially account for the accumulation over time. The amplitudes at a test concentration were normalized to the reference amplitude (A-90/Aref or A+45/Aref) to enable comparison of amplitudes between cells. When 10 μm Iso was used as a test concentration the resulting FRET amplitude was normalized to the one measured subsequently. The normalized data were plotted against the Iso concentrations used and were fit to a sigmoidal function (Fig. 6D). High concentrations of Iso led to a fast accumulation of receptor-arrestin complexes which then reduced the fraction of available arrestins and receptors. This caused the second amplitude (Aref) to be comparatively smaller especially in measurements with high Iso concentrations (Fig. 6C). Hence, the data points collected at concentrations of 1 μm and higher exceed 100%. The apparent about 2-fold difference in EC50-values of the curves at −90 mV (EC50 = 2.5 × 10−8 m) and at +45 mV (EC50 =4.8 × 10−8 m) did not reach statistical significance (comparison of logEC50; p = 0.1084). In contrast, the maximal amplitude at depolarization was significantly lower than the one at the holding potential (Max+45 = 1.02, Max-90 = 1.17; p = 0.0066, F-test), indicating voltage-dependence of agonist efficacy.
The large and significant differences in off kinetics induced by depolarization or agonist withdrawal on the receptor level as well as in downstream signaling compared to only small changes in affinity (Figs. 1, E and F, 2F and 6B) corroborate the hypothesis of drug efficacy being a major contributor of voltage-dependent fine tuning of β1-AR activity.
Discussion
In this study we report for the first time that the activity of GPCRs signaling through Gs-proteins, here β1-AR and to a lesser extent β2-AR, is regulated by the membrane potential. We found this voltage-dependence to occur within the physiological range of membrane potential. Most importantly, we identified drug efficacy as a major target of voltage-dependence for both catecholamines isoprenaline and adrenaline. A reduction in agonist affinity only contributes to a minor extent. This is, to our knowledge, the first report where alterations in drug efficacy and not purely affinity of an endogenous full agonist underlie voltage-dependent receptor activation.
β-Adrenoceptors are members of the large superfamily of GPCRs and are important drug targets especially in the cardiovascular system. It is therefore important to fully understand the regulation of their activation. Here we demonstrate that the plasma membrane potential regulates the classical way of activation of β1-AR by the catecholamines isoprenaline and adrenaline. An active state crystal structure of β2-AR revealed rearrangements especially of the cytoplasmic ends of transmembrane domains (TM) 5, 6, and 7 compared to the inactive structure (26). The large outward movement of TM 6 transmits to intracellular loop 3 (IL3). Therefore, activation of receptors can be monitored directly with FRET-based intramolecular receptor fusion proteins with GFP variants in IL3 and at the C-terminal tail in intact living cells (24). With such a β1-AR receptor fusion protein named β1-AR sensor (20) we showed that depolarization inactivated the receptor on a very fast time scale (Figs. 1, C and D and 5, A and B). This direct influence of depolarization on receptor conformation, i.e. its deactivation, was shown for Iso- and Adr-stimulated cells. Theoretically, drastic changes of the membrane potential could influence, for example, the acceptor quantum yield resulting in unspecific alterations of the FRET signal especially in cells transfected with the β1-AR sensor. However, our findings that 1) no voltage-dependent changes of FRET were observed in the absence of agonists and 2) very similar results were observed with three different FRET assays, which show agonist-induced alterations in FRET in opposing directions, practically rule out that unspecific effects of voltage on the FRET signal affect our results. Despite stimulation with saturating concentrations of Iso or Adr, depolarization still deactivated the receptors by about 20%. The β1-AR sensor showed similar binding affinities for NA and Iso as well as similar downstream signaling like wildtype β1-AR (20). Our findings that β1-ARs are voltage-dependent can therefore be extrapolated to the native β1-AR.
Interaction measurements between β1-AR and the signaling partners Gs and arrestin 3 verified that voltage-dependence is passed on to downstream signaling (Fig. 2, A–D) as discussed for other voltage-sensitive GPCRs (reviewed in Refs. 9, 10 or e.g. 7, 12, 14). Both interactions were attenuated during depolarization and restored upon repolarization of the plasma membrane (Fig. 2). Although the effect was reduced under saturating conditions, the interaction between β1-AR and arrestin 3 was still hampered in cells either stimulated with the endogenous catecholamines NA and Adr or the synthetic catecholamine Iso (Fig. 2, E and F). This alteration of downstream signaling by membrane potential is an important requirement for voltage-dependence being of physiological relevance.
A good signal-to-noise ratio of the arrestin FRET assay enabled measurements of activation/VM relation curves which could be fit to a Boltzmann function (Fig. 3). Compared to voltage-gated ion channels the Boltzmann curve of β1-AR is shallow indicating a weaker voltage-dependence (30). As GPCRs do not possess a voltage-sensing domain like an S4-domain in ion channels a shallow curve was expected. Half-maximal effects of VM were reached within the physiological range of membrane potential at V50 = −28 mV (Iso) and V50 = −27 mV (Adr), which is in line with data published on the α2A-AR (7). The calculated z-score (charge movement across the membrane) for Adr-stimulated cells of z = 0.49 is similar to the z-scores reported for M2AChR (z = 0.85 and z = 0.58, (11, 12)) or α2A-AR (z = 0.5, (7)). The particularly shallow slope of the curve in Iso-stimulated cells resulted in a lower z-score of z = 0.36. The shallow curve arises from the overall smaller effect of depolarization on FRET amplitudes in Iso- compared to Adr-stimulated cells described for non-saturating conditions (Fig. 1).
Although less abundant, also β2-ARs are expressed in the heart and the expression increases while β1-AR expression decreases in progression of heart failure (18). It was therefore interesting to investigate the β2-AR in respect to its voltage-sensitivity (Fig. 4). The interaction of β2-AR and arrestin 3 was only reduced by about 10% during depolarization when stimulated with a non-saturating concentration of Iso. In saturation with Adr or Iso a depolarization-mediated reduction of the interaction was hardly detectable. β2-AR activation appears to be less sensitive to regulation by the membrane potential.
We briefly mentioned before that the β1-AR deactivation as well as the disruption of β1-AR-effector interaction was sensitive to agonist concentration indicating a decline in agonist affinity. Alterations in binding affinity underlying voltage-dependence have been reported for the Gi-coupled receptors M2AChR, mGluR3, and α2A-AR (1, 2, 7). In saturation with Iso there was only a tendency toward a reduced depolarization effect on receptor deactivation. The concentration-response-curves of the receptor-arrestin interaction revealed a minor right-shift of about 2-fold due to depolarization, which did not reach significance (Fig. 6D). Similar in magnitude we observed a 2-fold increase in dissociation kinetics upon depolarization from −90 mV to +45 mV (Fig. 6B).
In case of stimulation with saturating Adr concentrations, the reduction was about 3-fold (Fig. 1F). In arrestin 3-β1-AR interaction assays, saturating concentrations led to a 2-fold (Iso) and 4-fold (Adr) decrease of the described depolarization effect (Fig. 2F). Therefore, the reduction of agonist affinity was only a minor contributor to β1-ARs voltage-dependence.
Changes in efficacy have been proposed as the underlying mechanism of voltage-dependence for certain antagonists of the P2Y1R (13), for some therapeutic agonists of the short splice variant of D2R (14) and for pilocarpine at the M2AChR (12). Only for clonidine at the α2A-AR (7) a change in efficacy has been directly measured with a FRET-based sensor, the other studies measured downstream effects of receptor activation and therefore only indirectly detected alterations in ligand efficacy. Intramolecular FRET-based receptor sensors are suitable tools to directly monitor intrinsic efficacy (7, 24). In addition, the FRET assays detecting arrestin 3-receptor or G protein-receptor interaction also allow direct measurement of drug efficacy since receptor number or coupling do not affect the measurement (24). During measurements with Iso, deactivation kinetics during depolarization were 100-, 70- and 10-fold faster than deactivation by washout on the receptor level or in the interactions between receptor and G protein or arrestin 3, respectively. This reduction in efficacy is also reflected in the concentration-response-curve where the maximal FRET amplitude is significantly reduced at +45 mV compared to −90 mV.
The differences were smaller during Adr stimulation as washout off-rates were faster due to a lower affinity of Adr toward the β1-AR compared to Iso (27). Yet, the acceleration of depolarization, versus washout-induced deactivation was about 8-fold, 11-fold, and 14-fold for receptor conformation and the interaction of β1-AR with G protein or arrestin 3, respectively. The observed differences in off kinetics could be even larger taking into account that fit time constants were close to the detection limit which influences measurement accuracy. With the change in affinity being of maximally 4-fold, an alteration in efficacy also mainly underlies the voltage-dependence in Adr-stimulated cells. Taken together, the very pronounced acceleration of deactivation kinetics during depolarization suggests a change in efficacy being a major contributor to voltage-sensitivity of β1-AR with some smaller contribution of decrease in affinity. Ligand specificity is apparent as the contribution of decreased affinity is more pronounced in Adr- than in Iso-stimulated cells. As the first endogenous full agonist, Adr shows decreases in efficacy in addition to a reduced affinity during depolarization. Under physiological conditions the membrane potential could therefore fine tune receptor function on a very fast time scale in the presence of agonist.
Our data expand the knowledge of voltage-dependent regulation of GPCRs to those coupling to Gs proteins with efficacy being an important contributor to this phenomenon. However, it remains unclear, on a molecular level, how changes in the membrane potential can be “sensed” by GPCRs. Therefore, it would be an invaluable achievement to create receptor mutants that lack voltage-dependence but exhibit otherwise wild-type-like properties.
Author Contributions
A. B. designed, performed, and analyzed experiments and wrote the manuscript. A. R. performed and designed experiments and helped with writing of the manuscript. M. B. designed the study and experiments and analyzed data and wrote the manuscript.
Acknowledgments
We thank Dr. Cornelius Krasel for his help with formatting the manuscript. The plasmid encoding for the β1-AR sensor and HEK293 cells stably expressing it were kindly provided by Prof. Stefan Engelhardt.
This work was supported by Deutsche Forschungsgemeinschaft as part of the SFB593 “Mechanisms of Cellular Compartmentalization and the Relevance for disease.” The authors declare that they have no conflicts of interest with the contents of this article.
- GPCR
- G protein-coupled receptor
- α2A-/β1-/β2-AR
- α2A-/β1-/β2-adrenoceptor
- M1/M2AChR
- muscarinic M1/M2 acetylcholine receptor
- P2Y1R
- purinergic P2Y1 receptors
- LPA
- lysophosphatidic acid
- D2R
- dopaminergic D2 receptor
- mGluR1/3
- metabotropic glutamate receptor 1/3
- H4R
- histamine H4 receptor
- VM
- membrane potential
- FRET
- Förster resonance energy transfer
- Iso
- isoprenaline
- Adr
- adrenaline
- NA
- noradrenaline
- Arr3
- arrestin 3
- GRK2
- G protein-coupled receptor kinase 2
- YFP
- yellow fluorescent protein
- CFP
- cyan fluorescent protein
- Cer
- Cerulean, variant of CFP
- TM
- transmembrane domain.
References
- 1.Ben-Chaim Y., Tour O., Dascal N., Parnas I., and Parnas H. (2003) The M2 muscarinic G-protein-coupled receptor is voltage-sensitive. J. Biol. Chem. 278, 22482–22491 [DOI] [PubMed] [Google Scholar]
- 2.Ohana L., Barchad O., Parnas I., and Parnas H. (2006) The metabotropic glutamate G-protein-coupled receptors mGluR3 and mGluR1a are voltage-sensitive. J. Biol. Chem. 281, 24204–24215 [DOI] [PubMed] [Google Scholar]
- 3.Martinez-Pinna J., Gurung I. S., Vial C., Leon C., Gachet C., Evans R. J., and Mahaut-Smith M. P. (2005) Direct voltage control of signaling via P2Y1 and other Gαq-coupled receptors. J. Biol. Chem. 280, 1490–1498 [DOI] [PubMed] [Google Scholar]
- 4.Martinez-Pinna J., Gurung I. S., Mahaut-Smith M. P., and Morales A. (2010) Direct voltage control of endogenous lysophosphatidic acid G-protein-coupled receptors in Xenopus oocytes. J. Physiol. 588, 1683–1693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sahlholm K., Nilsson J., Marcellino D., Fuxe K., and Arhem P. (2008) Voltage-dependence of the human dopamine D2 receptor. Synapse 62, 476–480 [DOI] [PubMed] [Google Scholar]
- 6.Sahlholm K., Nilsson J., Marcellino D., Fuxe K., and Arhem P. (2012) Voltage sensitivities and deactivation kinetics of histamine H3 and H4 receptors. Biochim. Biophys. Acta 1818, 3081–3089 [DOI] [PubMed] [Google Scholar]
- 7.Rinne A., Birk A., and Bünemann M. (2013) Voltage regulates adrenergic receptor function. Proc. Natl. Acad. Sci. 110, 1536–1541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moreno-Galindo E. G., Sánchez-Chapula J. A., Sachse F. B., Rodriguez-Paredes J. A., Tristani-Firouzi M., and Navarro-Polanco R. a (2011) Relaxation gating of the acetylcholine-activated inward rectifier K+ current (IKACh) is mediated by intrinsic voltage-sensitivity of the muscarinic receptor. J. Physiol. 7, 1755–1767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mahaut-Smith M. P., Martinez-Pinna J., and Gurung I. S. (2008) A role for membrane potential in regulating GPCRs? Trends Pharmacol. Sci. 29, 421–429 [DOI] [PubMed] [Google Scholar]
- 10.Parnas I., and Parnas H. (2010) Control of neurotransmitter release: From Ca2+ to voltage dependent G-protein coupled receptors. Pflügers Arch. Eur. J. Physiol. 460, 975–990 [DOI] [PubMed] [Google Scholar]
- 11.Ben-Chaim Y., Chanda B., Dascal N., Bezanilla F., Parnas I., and Parnas H. (2006) Movement of “gating charge” is coupled to ligand binding in a G-protein-coupled receptor. Nature 444, 1–13 [DOI] [PubMed] [Google Scholar]
- 12.Navarro-Polanco R. a, Moreno-Galindo E. G., Ferrer-Villada T., Arias M., Rigby J. R., Sánchez-Chapula J. A., and Tristani-Firouzi M. (2011) Conformational changes in the M2 muscarinic receptor induced by membrane voltage and agonist binding. J. Physiol. 7, 1741–1753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gurung I. S., Martinez-Pinna J., and Mahaut-Smith M. P. (2008) Novel consequences of voltage-dependence to G-protein-coupled P2Y1 receptors. Br. J. Pharmacol. 154, 882–889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sahlholm K., Barchad-Avitzur O., Marcellino D., Gómez-Soler M., Fuxe K., Ciruela F., and Arhem P. (2011) Agonist-specific voltage sensitivity at the dopamine D2S receptor - molecular determinants and relevance to therapeutic ligands. Neuropharmacology 61, 937–949 [DOI] [PubMed] [Google Scholar]
- 15.Ben Chaim Y., Bochnik S., Parnas I., and Parnas H. (2013) Voltage Affects the Dissociation Rate Constant of the m2 Muscarinic Receptor. PLoS ONE 8, e74354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sahlholm K., Marcellino D., Nilsson J., Fuxe K., and Arhem P. (2008) Voltage-sensitivity at the human dopamine D2S receptor is agonist-specific. Biochem. Biophys. Res. Commun. 377, 1216–1221 [DOI] [PubMed] [Google Scholar]
- 17.Kirstein S. L., and Insel P. A. (2004) Autonomic nervous system pharmacogenomics: a progress report. Pharmacol. Rev. 56, 31–52 [DOI] [PubMed] [Google Scholar]
- 18.Lymperopoulos A., Rengo G., and Koch W. J. (2013) Adrenergic Nervous System in Heart Failure: Pathophysiology and Therapy. Circ. Res. 113, 739–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tateyama M., and Kubo Y. (2013) Binding of Gq protein stabilizes the activated state of the muscarinic receptor type 1. Neuropharmacology 65, 173–181 [DOI] [PubMed] [Google Scholar]
- 20.Rochais F., Vilardaga J.-P., Nikolaev V. O., Bünemann M., Lohse M. J., and Engelhardt S. (2007) Real-time optical recording of β1 -adrenergic receptor activation reveals supersensitivity of the Arg389 variant to carvedilol. J. Clin. Invest. 117, 229–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hein P., Rochais F., Hoffmann C., Dorsch S., Nikolaev V. O., Engelhardt S., Berlot C. H., Lohse M. J., and Bünemann M. (2006) Gs activation is time-limiting in initiating receptor-mediated signaling. J. Biol. Chem. 281, 33345–33351 [DOI] [PubMed] [Google Scholar]
- 22.Krasel C., Bünemann M., Lorenz K., and Lohse M. J. (2005) Beta-arrestin binding to the beta2-adrenergic receptor requires both receptor phosphorylation and receptor activation. J. Biol. Chem. 280, 9528–9535 [DOI] [PubMed] [Google Scholar]
- 23.Ahles A., Rodewald F., Rochais F., Bünemann M., and Engelhardt S. (2015) Interhelical interaction and receptor phosphorylation regulate activation kinetics of different human β1-adrenoceptor variants. J. Biol. Chem. 290, 1760–1769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lohse M. J., Vilardaga J.-P., and Bünemann M. (2003) Direct optical recording of intrinsic efficacy at a G protein-coupled receptor. Life Sci. 74, 397–404 [DOI] [PubMed] [Google Scholar]
- 25.Vilardaga J.-P., Bünemann M., Krasel C., Castro M., and Lohse M. J. (2003) Measurement of the millisecond activation switch of G protein-coupled receptors in living cells. Nat. Biotechnol. 21, 807–812 [DOI] [PubMed] [Google Scholar]
- 26.Rasmussen S. G. F., Choi H.-J., Fung J. J., Pardon E., Casarosa P., Chae P. S., Devree B. T., Rosenbaum D. M., Thian F. S., Kobilka T. S., Schnapp A., Konetzki I., Sunahara R. K., Gellman S. H., Pautsch A., Steyaert J., Weis W. I., and Kobilka B. K. (2011) Structure of a nanobody-stabilized active state of the β(2) adrenoceptor. Nature 469, 175–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bond R. A., Bylund D. B., Eikenburg D. C., Hieble J. P., Hills R., Minneman K. P., and Parra S. (2015) Adrenoceptors: β1-adrenoceptor. IUPHAR/BPS Guide to Pharmacology [online] www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=28 [Google Scholar]
- 28.Shenoy S. K., and Lefkowitz R. J. (2005) Seven-Transmembrane Receptor Signaling Through beta-Arrestin. Sci. STKE Signal Transduct. Knowl. Environ. 308, cm10. [DOI] [PubMed] [Google Scholar]
- 29.Lohse M. J. (1993) Molecular mechanisms of membrane receptor desensitization. Biochim. Biophys. Acta 1179, 171–188 [DOI] [PubMed] [Google Scholar]
- 30.Bezanilla F. (2000) The voltage sensor in voltage-dependent ion channels. Physiol. Rev. 80, 555–592 [DOI] [PubMed] [Google Scholar]