

Background: The EphA2 receptor tyrosine kinase can promote cell migration and cancer malignancy in the absence of ligand binding.
Results: We uncover a correlation between unliganded dimerization and tumorigenic signaling.
Conclusion: EphA2 pro-tumorigenic signaling is likely mediated by the EphA2 monomer.
Significance: A therapeutic strategy that aims at the stabilization of EphA2 dimers may be beneficial for the treatment of cancers linked to EphA2 overexpression.
Keywords: dimerization, membrane protein, receptor tyrosine kinase, signal transduction, thermodynamics
Abstract
The EphA2 receptor tyrosine kinase promotes cell migration and cancer malignancy through a ligand- and kinase-independent distinctive mechanism that has been linked to high Ser-897 phosphorylation and low tyrosine phosphorylation. Here, we demonstrate that EphA2 forms dimers in the plasma membrane of HEK293T cells in the absence of ephrin ligand binding, suggesting that the current seeding mechanism model of EphA2 activation is incomplete. We also characterize a dimerization-deficient EphA2 mutant that shows enhanced ability to promote cell migration, concomitant with increased Ser-897 phosphorylation and decreased tyrosine phosphorylation compared with EphA2 wild type. Our data reveal a correlation between unliganded dimerization and tumorigenic signaling and suggest that EphA2 pro-tumorigenic activity is mediated by the EphA2 monomer. Thus, a therapeutic strategy that aims at the stabilization of EphA2 dimers may be beneficial for the treatment of cancers linked to EphA2 overexpression.
Introduction
The Eph receptors comprise the largest family of receptor tyrosine kinases (RTKs)2 (1, 2). They are single pass transmembrane proteins, with an N-terminal extracellular ligand-binding region, a single transmembrane domain, and an intracellular region with tyrosine kinase activity. The extracellular region is composed of an N-terminal ligand-binding domain, a cysteine-rich domain, and two fibronectin repeats. The intracellular region encompasses a juxtamembrane domain, a kinase domain, a sterile α-motif domain, and a PDZ domain-binding motif at the C terminus.
The Eph receptors regulate cell-cell communication during development, and some of them are highly expressed in adult human epithelial cells. Although originally identified as regulators of axon guidance, the Eph receptors are now known to play profound roles in diverse physiological processes, such as angiogenesis (formation of new blood vessels from pre-existing ones), bone morphogenesis, inflammatory host responses, neurodegenerative diseases, and cancer (2, 3). The Eph receptors are unique among the RTKs, as their mechanism of activation is distinctly different and much more complex. These receptors form oligomers upon binding of their ligands (ephrins), which are anchored to opposing cells (4–8). In the oligomers, the Eph receptors cross-phosphorylate each other, particularly on two critical juxtamembrane domain tyrosine residues and on a tyrosine in the activation loop. The phosphorylated Eph receptors exert control over the strength of intercellular contacts and can mediate cell detachment (9–12). Cell contraction induced by Eph receptors that come in contact with ephrin ligands can lead to inhibition of cell migration/invasiveness as well as to inhibition of tumor growth (1, 2, 13, 14). Thus, ephrin-induced Eph receptor signaling is generally considered anti-tumorigenic.
Among the 14 Eph receptors, EphA2 is the one with the most profound links to cancer and is associated with poor prognosis (15–18). Intriguingly, EphA2 overexpression in cancer is often accompanied by the loss of its ligands (2, 19), suggesting that EphA2 behaves as an oncoprotein in the absence of ligand. In support of this view, recent work has demonstrated that EphA2 promotes cell migration and tumor malignancy in a ligand-independent manner via a process that is unique for EphA2 (20). This tumorigenic activity has been linked to high motility and stem-like properties of cells expressing high levels of Ser-897-phosphorylated EphA2 and low levels of tyrosine-phosphorylated EphA2 (20–23). The molecular basis for the EphA2 pro-tumorigenic activity, however, is largely unknown, and this lack of basic knowledge is a bottleneck in developing effective EphA2-targeted therapies.
The biophysical behavior of EphA2 has not been studied in the absence of ligand binding, under conditions that promote EphA2 oncogenic signaling. Some RTKs from other families, however, have been investigated and have been found to form unliganded dimers (24–26). We therefore asked whether EphA2 forms dimers in the absence of ligand binding and whether a link exists between EphA2 unliganded dimerization and EphA2 tumorigenic signaling. We measured EphA2 dimerization propensities in live cells using quantitative FRET (27–29). We sought to identify a mutant with compromised unliganded dimerization, and we gained new insight into EphA2 oncogenic signaling by comparing the biological effects of this mutant and wild-type EphA2.
Materials and Methods
EPHA2 Cloning and Mutagenesis
We obtained the plasmid encoding the monomeric Turquoise fluorescent protein (mTurq) from Prof. Paul S. Park (Case Western Reserve University, Cleveland, OH). The plasmid encoding the yellow fluorescent protein, eYFP, was a generous gift from Prof. M. Betenbaugh (The Johns Hopkins University). The A206K mutation was introduced in eYFP to render it monomeric. The gene encoding EphA2 was subcloned between the HindIII and KpnI restriction sites in the pcDNA 3.1(+) vector, which already contained a sequence encoding a 15-amino acid linker, followed by either mTurq or eYFP (30). Polymerase chain reaction (PCR) of the EPHA2 gene was performed using 5′-GGGCCCAAGCTTACCAGCAACATGGAGCTCCAGGCAGCC-3′ as a forward primer and 5′-CCCGGGGGTACCTTTGATGGGGATCCCCACAGTGTTCAC-3′ as a reverse primer. The ligation of the PCR products and the digested pcDNA 3.1(+) vector yielded the plasmid constructs pcDNA 3.1(+) EPHA2–15aa-mTurq and pcDNA3.1(+) EPHA2–15aa-eYFP.
Genes encoding three EphA2 mutants were generated using the QuikChange site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA), following the manufacturer's protocol. The first mutant contained a single L223R point mutation, and the gene was produced using 5′-GGCTCTGATGCACCTTCCCGGGCCACTGTGGCCGG-3′ as a forward and 5′-CCGGCCACAGTGGCCCGGGAAGGTGCATCAGAGCC-3′ as a reverse primer. The L223R/L254R mutant was generated from the L223R mutant using 5′-GCA GTG GAT GGC GAG TGG CGGGTGCCC ATT GGG CAG TGCC-3′ as a forward primer and 5′-GGCA CTG CCC AAT GGG CAC CCG CCA CTC GCC ATC CAC TGC-3′ as a reverse primer. The triple L223R/L254R/V255R mutant was obtained from the L223R/L254R mutant using 5′-GCAGTGGATGGCGAGTGGCGGCGGCCCATTGGGCAGTGCC-3′ as a forward primer and 5′-GGCACTGCCCAATGGGCCGCCGCCACTCGCCATCCACTGC-3′ as a reverse primer.
Cell Culture and Transfection
HEK293T cells were purchased from American Type Culture Collection (ATCC) (Manassas, VA). The cells were cultured in Dulbecco's modified Eagle's medium (DMEM), supplemented with 10% fetal bovine serum (FBS, Hyclone), 3.5 g/liter d-glucose (19.4 mm), and 1.5 g/liter sodium bicarbonate (17.9 mm) in 35-mm glass bottom dishes (MatTek Corp.). The cells were co-transfected with either pcDNA3.1(+) EPHA2–15aa-mTurq and pcDNA3.1(+) EPHA2–15aa-eYFP or their mutants using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Twelve hours after transfection, cells were serum-starved for 12 h. Prior to imaging, the starvation medium was replaced with hypo-osmotic medium to induce osmotic stress and swelling under reversible conditions as described (31).
Two-photon Microscopy of Cells under Reversible Osmotic Stress
Imaging was performed with a spectrally resolved two-photon microscope with line-scanning capabilities (32, 33). An ultrashort pulse laser (MaiTaiTM, Spectra-Physics, Santa Clara, CA), which generates femtosecond mode locked pulses at wavelengths between 690 and 1040 nm, was used as the excitation source for the fluorophores. Details of the microscope have been discussed previously (32, 33). Measurements were performed in cells under reversible osmotic stress. The swelling step was necessary because the membrane of cell is highly “wrinkled,” with cells possessing 2–3 times the membrane needed to sustain their shape (34, 35). When the membrane is stretched, the effective three-dimensional receptor concentrations, calibrated using purified fluorescent protein solutions of known concentrations, are easily converted into two-dimensional receptor concentrations in the plasma membrane (29).
To study unliganded dimerization, we imaged only free-standing membranes of swollen cells. HEK293T cells are known to express ephrins (4), and this protocol ensured that EphA2 receptors did not interact with ephrins on opposing cells. To ensure that no soluble ligand is present, the transfected cells were cultured under starvation conditions prior to the experiments. The cell culture medium was replaced just before imaging, thereby removing any ephrin-A that may have been cleaved from the cell surface (18, 36).
Measurements of Dimerization Propensities
EphA2 dimerization propensities were measured using quantitative FRET (29). The receptor concentrations were varied over a wide range, and the concentrations in the plasma membranes were measured, along with the FRET efficiencies (27, 28). The total receptor concentration and the dimeric receptor fraction were calculated for each membrane segment, and data from many cells were combined to yield dimerization curves. From the FRET data, we extracted two parameters that describe the dimerization process. The first parameter is the dimerization constant K. From the optimal dimerization constant K, we calculate the dissociation constant Kdiss = 1/K and we report it in Table 1, in units of receptors/μm2. We also calculate the dimer stability with respect to a standard state defined as K0 = 1 nm2/receptor as shown in Equation 1,
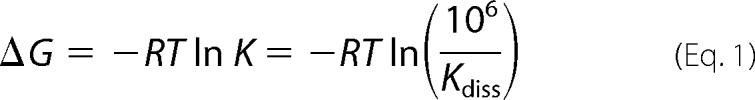 |
The second parameter is the structural parameter “Intrinsic FRET”, Ẽ, which depends on the dimer structure, in particular on the distance d between the fluorescent proteins in the dimer according to Refs. 27 and 28 and is shown in Equation 2,
 |
Here, Ro is the Förster radius of the FRET pair (29). For mTurq and eYFP, Ro is 54.5 Å.
TABLE 1.
Parameters describing the stability and structure of EphA2 unliganded dimers
Kdiss is the dissociation constant (receptors/μm2); ΔG is the dimerization free energy; Ẽ is the intrinsic FRET efficiency; and d is the calculated distance between the fluorescent proteins in the EphA2 dimers. Kdiss and Ẽ are determined from the fit of the dimerization model to the FRET data, and the uncertainties are the 95% confidence intervals from the fit. ΔG and d are calculated using Equations 1 and 2, respectively.
| Kdiss | ΔG | Ẽ | d | |
|---|---|---|---|---|
| receptors/μm2 | kcal/mol | Å | ||
| Wild-type EphA2 | 210 ± 50 | −5.0 ± 0.2 | 0.67 ± 0.03 | 48 ± 1 |
| L223R | 310 ± 60 | −4.8 ± 0.1 | 0.65 ± 0.03 | 49 ± 1 |
| L223R/L254R | 320 ± 100 | −5.1 ± 0.1 | 0.46 ± 0.03 | 56 ± 1 |
| L223R/L254R/V255R | 1200 ± 250 | −4.0 ± 0.2 | 0.72 ± 0.04 | 47 ± 1 |
Cell Migration Assay
HEK293T cells were cultured for 12 h following transfection. Cells were serum-starved overnight. A cell suspension with 1 × 106 cells/ml was prepared in serum-free medium containing 0.5% BSA. The CytoSelectTM cell haptotaxis assay kit (CellBiolabs) was used to assay the migratory properties of cells. This kit contains inserts with polycarbonate membrane with pores of 8 μm, coated with collagen I on the bottom side. The inserts were loaded with 300 μl of the cell suspension. 500 μl of medium containing 10% FBS was placed in the lower well of the migratory plate. Cells were incubated for 4 h at 37 °C. After that, the serum-free medium was aspirated from the inserts, and the inserts were cleaned using cotton swabs to remove non-migratory cells. The inserts were transferred to clean wells containing 300 μl of 1× Lysis Buffer/CyQuant® GR dye solution, supplied with the kit. After a 10-min incubation at room temperature, 200 μl of the lysate was transferred to a well in a 96-well plate. The fluorescence of the CyQuant® GR dye solution, which is directly proportional to the number of migratory cells, was measured in a plate reader.
To estimate the EphA2 concentrations in these migration experiments, we analyzed the EphA2 concentrations in the FRET experiments under the same transfection conditions (2 μg of DNA per well). Although the variation in concentration was large as expected, the mean was about ∼800 receptors/μm2.
Western Blots
For Western blots, HEK293T cells were cultured and transfected using Lipofectamine 2000. Twenty four hours post-transfection, cells were lysed in lysis buffer (25 mm Tris-Cl, 0.5% Triton X-100, 20 mm NaCl, 2 mm EDTA, and phosphate and protease inhibitors, Roche Applied Science). The lysed samples were collected and subjected to centrifugation at 15,000 × g for 15 min at 4 °C. The lysates were collected and stored at −20 °C. The amounts of total protein in the lysates were measured with the BCA protein assay kit (Bio-Rad). The lysates were loaded into 3–8% NuPAGEHNovexHTris acetate mini gels (Invitrogen). The proteins were transferred onto a nitrocellulose membrane and blocked using 5% nonfat milk in 1× TBST. Total EphA2 expression, Ser-897 phosphorylation, and Tyr-772 phosphorylation were probed using anti-EphA2 antibodies (Millipore), anti-phospho-Ser-897 antibodies (Cell Signaling), and anti-phospho-Tyr-772 antibodies (Cell Signaling), respectively. Anti-mouse HRP-conjugated antibodies (Promega) and anti-rabbit HRP-conjugated antibodies (Promega) were used as secondary antibodies to visualize EphA2 expression and phosphorylation bands, respectively. The membranes were incubated with Amersham Biosciences ECL PlusTM Western blotting detection reagent (GE Healthcare) for 2 min and were exposed for 1–60 s in Chemidoc molecular imager (Bio-Rad) to detect the antibody staining.
Results
The EphA2 Receptor Forms Dimers in the Absence of Ligand Binding
To study EphA2 unliganded dimerization using FRET, we used full-length EphA2 tagged with fluorescent proteins (mTurquoise and YFP, a FRET pair) at its C terminus. The fluorescent proteins were attached through a (GGS)5 flexible linker, which has been previously shown not to affect dimerization (27). The (GGS)5 linker also lacks secondary structure, thus ensuring free rotation of the fluorescent proteins (39). HEK293T cells were co-transfected with plasmids encoding EphA2-YFP and EphA2-mTurq in 1:1 to 1:3 molar ratios. To achieve a broad range of EphA2 expression, the total amount of DNA was varied from 20 ng to 4 μg. The media were exchanged, and the cells were subjected to reversible osmotic stress following the protocol described previously (31), just before imaging. This treatment results in the disassembly of the caveolae and stretching of the plasma membrane (31), allowing determination of the two-dimensional EphA2 concentration in the membrane (29). This treatment is completely reversible (Fig. 1).
FIGURE 1.

Images of HEK293T cells expressing wild-type EphA2 and EphA2 mutants before, during, and after the application of reversible osmotic stress (31). The pre-swelling and post-swelling states are not distinguishable by fluorescence microscopy. This is consistent with previous work, which has demonstrated that the swelling is reversible and not lethal (31).
Spectral images of the swollen cells were captured in a two-photon microscope equipped with the OptiMiS detection system as described previously (32, 33); 277 individual cells expressing various levels of EphA2 were imaged in nine independent experiments. Homogeneous, diffraction-limited membrane regions with uniform fluorescence having lengths of ∼3 μm were chosen and analyzed (Fig. 2).
FIGURE 2.
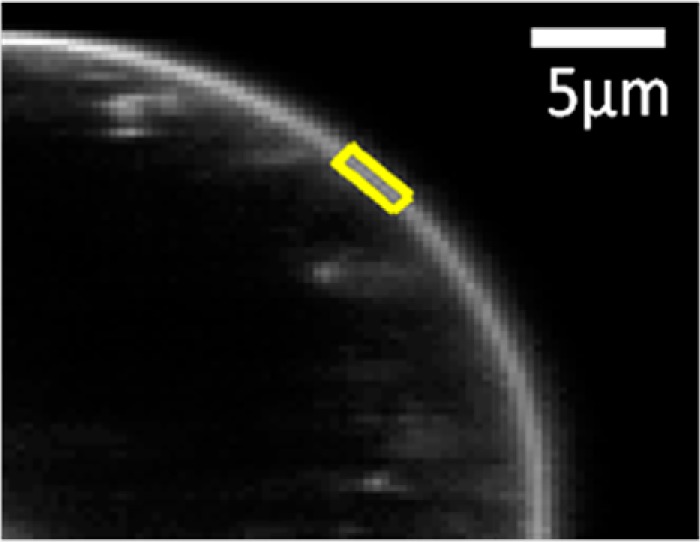
Selection of a membrane region in a cell under reversible osmotic stress. The cell exhibits large areas of stretched membrane with homogeneously distributed EphA2 fluorescence. A region of homogeneous diffraction-limited membrane fluorescence, 3–4 pixels in thickness (indicating stretched membrane) and ∼3 μm in length (yellow), is analyzed to produce one of the data points shown in Fig. 3, A and B.
The measured FRET efficiency, the donor concentration, and the acceptor concentration were calculated in each region (29). Fig. 3A shows FRET as a function of acceptor concentration, and Fig. 3B shows the donor versus the acceptor concentration. These data were used to determine the type of oligomer in the membrane that best describes the data. Different association models (dimer, trimer, tetramer, pentamer, and hexamer) were fitted to the data (40). The theoretical binding curves for a particular oligomer depend on two unknown parameters as follows: the monomer-oligomer association constant K and the intrinsic FRET, a structural parameter determined by the separation between the fluorescent proteins in the oligomer. The least squared error is calculated for each oligomer model, and the model that gives the lowest mean squared error (MSE) is considered the overall best model to represent the data. For wild-type EphA2, the best fit is achieved for the dimer model, which yields the lowest overall MSE (Fig. 4A).
FIGURE 3.
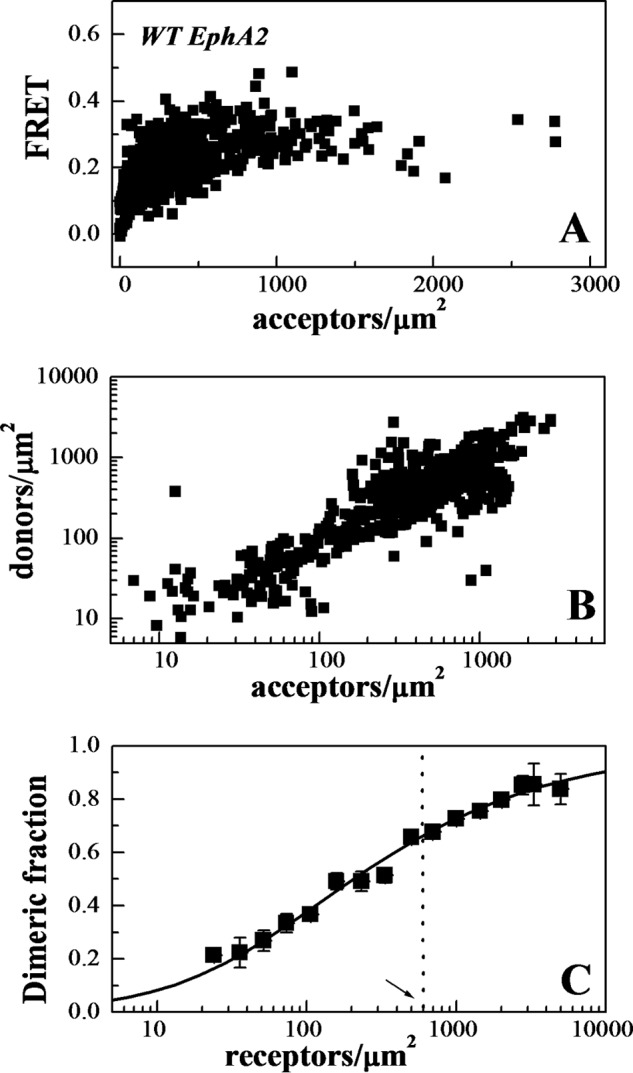
FRET measurements of EphA2 dimerization propensities. A, FRET as a function of EphA2-eYFP (acceptor) concentration for wild-type EphA2. Every data point represents a single membrane region. B, EphA2-mTurq (donor) concentration versus EphA2-eYFP (acceptor) concentration in each membrane region analyzed. C, dimeric fraction as a function of receptor concentrations. The dimeric fractions measured for individual membrane regions are binned, and the averages and the standard errors are shown. The solid line is the theoretical curve for the best fit dimerization model (29, 30). The dashed vertical line indicates 600 receptors/μm2, the reported EphA2 expression in A549 lung cancer cells (44).
FIGURE 4.
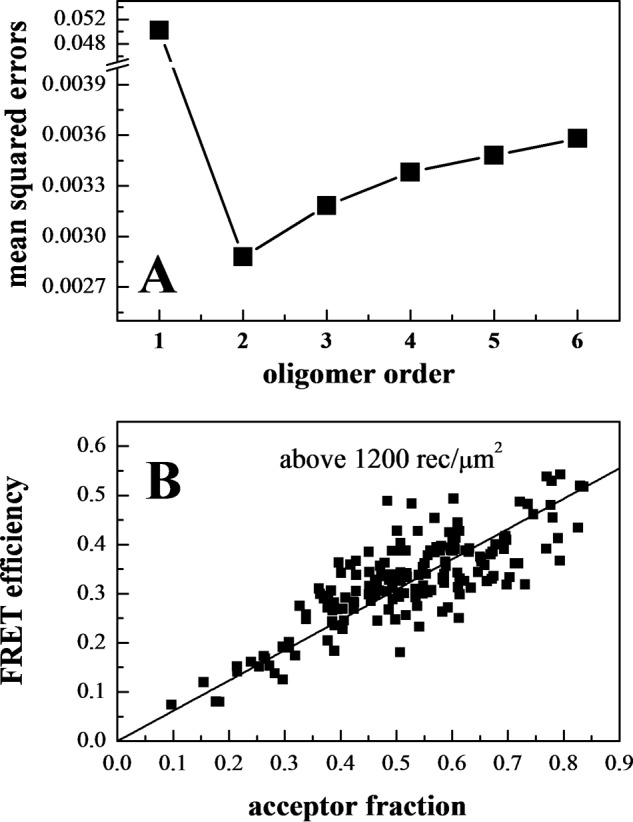
EphA2 forms dimers, not higher order oligomers. A, MSE versus oligomer order. The MSE is lowest for the dimer model. B, FRET as a function of acceptor fraction, for total EphA2 concentrations that exceed 1,200 receptors/μm2. Under these conditions, the EphA2 receptors are about 80% dimeric, and the FRET signal depends primarily on the acceptor fraction and not on the total concentration. The linear dependence is indicative of a dimer (41–43).
The dimerization curve in Fig. 3C is plotted for the best fit adjustable parameters in the monomer-dimer equilibrium model.
The two-dimensional dissociation constant, 210 receptors/μm2, corresponds to dimerization free energy of −5 kcal/mol (29) (see Equation 1). The intrinsic FRET, 0.67, corresponds to a separation of 48 Å between the fluorescent proteins in the wild-type EphA2 dimer, calculated under the assumption of free rotation of the fluorophores (justified here by the use of long flexible linkers for the attachment of the fluorescent proteins to the C terminus of EphA2 (27)).
In Fig. 3C, the data points follow closely the dimer prediction over the entire concentration range of the experiments, supporting the idea that EphA2 forms dimers in the absence of ephrin ligand binding. To further demonstrate the formation of dimers and not higher order oligomers, we plotted the FRET efficiency as a function of acceptor fraction, under conditions where dimerization does not strongly depend on concentration (Fig. 4B). In particular, we selected data points corresponding to membrane regions in which EphA2 concentration exceeded 1200 receptors/μm2, such that the dimeric fractions for all data points are ∼80%. Under these conditions, the dependence of FRET efficiency on the acceptor fraction is known to be linear for a dimer and non-linear for higher order oligomers (41–43). The data in Fig. 4B are well described by a linear function (p < 0.001), in support of the conclusion that EphA2 forms dimers in the absence of ligand binding.
The EphA2 dimerization curve in Fig. 3C allows us to directly evaluate the physiological relevance of EphA2 dimerization. EphA2 expression has been reported to be 600,000 receptors per cell in A549 lung cancer cells (44), which express moderately high EphA2 levels. Assuming a cell area of 1000 μm2, this corresponds to 600 receptors per μm2. At this concentration, EphA2 is 65% dimeric (arrow and dotted line in Fig. 3C). Therefore, the dimerization propensity of EphA2 is significant in a physiological context.
EphA2 Unliganded Dimers Are Stabilized by Leucine Zipper-like Interactions between the Cysteine-rich Domains
Having demonstrated that EphA2 forms dimers in the absence of ligand binding, we sought to identify receptor-receptor contacts that might be stabilizing these dimers. The solved crystal structure of the EphA2 extracellular domain, both in the absence of ligand and in complex with ephrin-A5, reveals that two distinct receptor-receptor interfaces contribute to EphA2 clustering (4, 5). One of these interfaces (called “clustering” interface (5) or interfaces B and D (4)) does not involve ligand. It is stabilized by interactions between the ligand-binding domains and between the cysteine-rich domains. The latter include a leucine zipper-like interaction involving Leu-223, Leu-254, and Val-255. To investigate whether these leucine zipper interactions may play a role in unliganded EphA2 dimerization, we mutated Leu-223, Leu-254, and Val-255 to Arg, and we studied whether the mutations affect EphA2 dimerization in the absence of ligand binding.
We generated three EphA2 mutants as follows: a single L223R mutant, a double L223R/L254R mutant, and a triple L223R/L254R/V255R mutant. We then studied the dimerization of the three mutant EphA2 receptors in HEK293T cells under reversible osmotic stress. We analyzed 501 cells in the case of the single L223R mutant, 380 cells in the case ofthe double L223R/L254R mutant, and 437 cells in the case of the triple L223R/L254R/V255R mutant. At least nine independent experiments were performed for each mutant. The apparent FRET efficiencies and the dimerization curves for the three mutants are shown in Fig. 5. The dimerization curves for the wild type and the mutants are compared in Fig. 6. The optimal parameters from the dimer model fitting are given in Table 1. The single L223R mutation has no effect on the measured dissociation constant and intrinsic FRET. The double L223R/L254R mutations have no effect on dimerization, but the intrinsic FRET for this mutant is lower (0.46 ± 0.03) as compared with the wild type (0.67 ± 0.03), corresponding to about an 8 Å increase in the average separation between the fluorescent proteins that are attached to the C termini. This suggests that the L223R/L254R mutations cause a structural change in the EphA2 dimer, which is propagated along the entire length of the receptor toward the C terminus.
FIGURE 5.

FRET data and dimerization curves for the three EphA2 mutants studied here.
FIGURE 6.
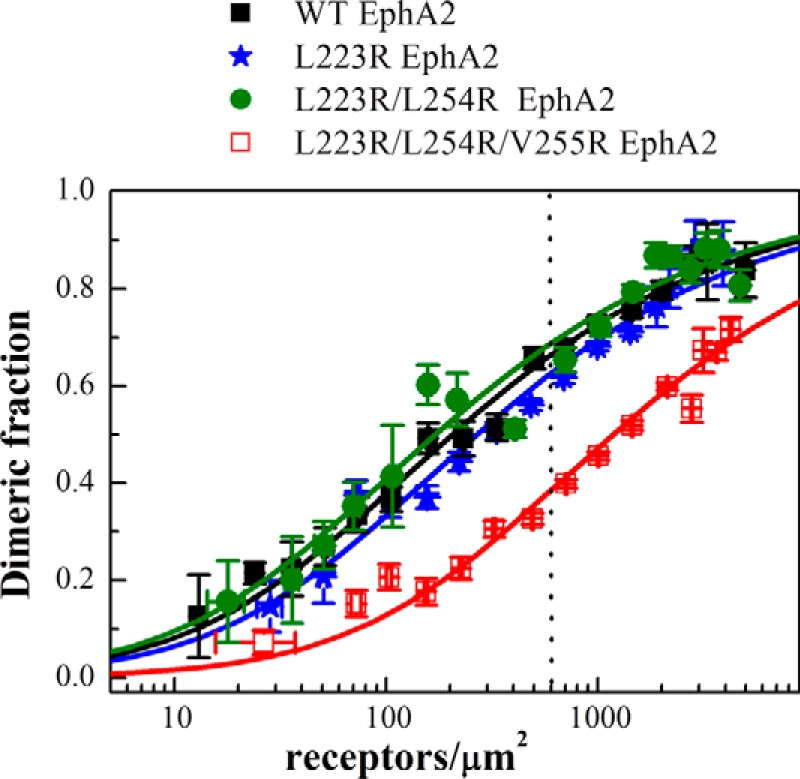
Dimerization curves for wild-type EphA2 and the three EphA2 mutants studied here. Although the effects of the single and the double mutations on dimerization are negligible, the triple mutation causes a significant decrease in dimerization in the absence of ligand binding. The dashed vertical line indicates 600 receptors/μm2, the reported EphA2 expression in A549 lung cancer cells (44).
Finally, the dimerization of the triple L223R/L254R/V255R mutant is significantly reduced as compared with the wild type, with the two-dimensional dissociation constant increased by an order of magnitude. However, the intrinsic FRET for the triple mutant is the same as the wild type (see Table 1). Thus, the triple mutation destabilized the EphA2 dimer, i.e. reduced the number of dimers, without inducing a measurable change in intrinsic FRET. For physiological expressions of 600 receptors/μm2, the fraction of dimeric receptors is reduced more than half, from 65 to 30% (see dashed line in Fig. 6). The substantial decrease in dimerization of the triple mutant suggests that the leucine zipper-like interactions between amino acids Leu-223, Leu-254, and Val-255 stabilize wild-type EphA2 unliganded dimers.
EphA2 Unliganded Dimerization Suppresses Cell Migration
EphA2 is known to promote cell migration and tumor progression in a ligand-independent manner via a process that is distinctive among RTKs. Expression of EphA2 can significantly enhance the serum-induced migration of HEK293T cells, independently of ligand binding and kinase activity (20). Here, we asked whether cell migration might be linked to unliganded EphA2 dimerization. We reasoned that the motility of wild-type EphA2 and the dimerization deficient L223R/L254R/V255R mutant will be different if such a link exists.
To answer this question, we compared the migratory ability of HEK293T cells, transfected with either wild-type EphA2 or the three EphA2 mutants. EphA2 concentrations in the plasma membrane in these experiments were estimated as ∼800 receptors/μm2, corresponding to 70% wild-type dimeric receptors. The migration assay was conducted using the Cell Biolabs CytoSelectTM cell haptotaxis assay kit (Cell Biolabs). Cells were placed in polycarbonate membrane inserts with 8-μm pores. The bottom sides of the inserts were coated with collagen I, and only the lower chamber contained 10% FBS. A migratory cell will pass through the pore of the inserts and move onto the collagen coating, in response to the collagen and FBS in the lower chamber. The measured fluorescence is directly proportional to the number of migratory cells. The relative number of migratory cells was measured in three independent experiments, and the averages and standard errors are shown in Fig. 7. Although the effects of the single and double mutations on cell migration were not statistically significant, the number of migratory cells expressing triple mutant EphA2 was significantly higher than the number of migratory cells expressing wild-type EphA2.
FIGURE 7.
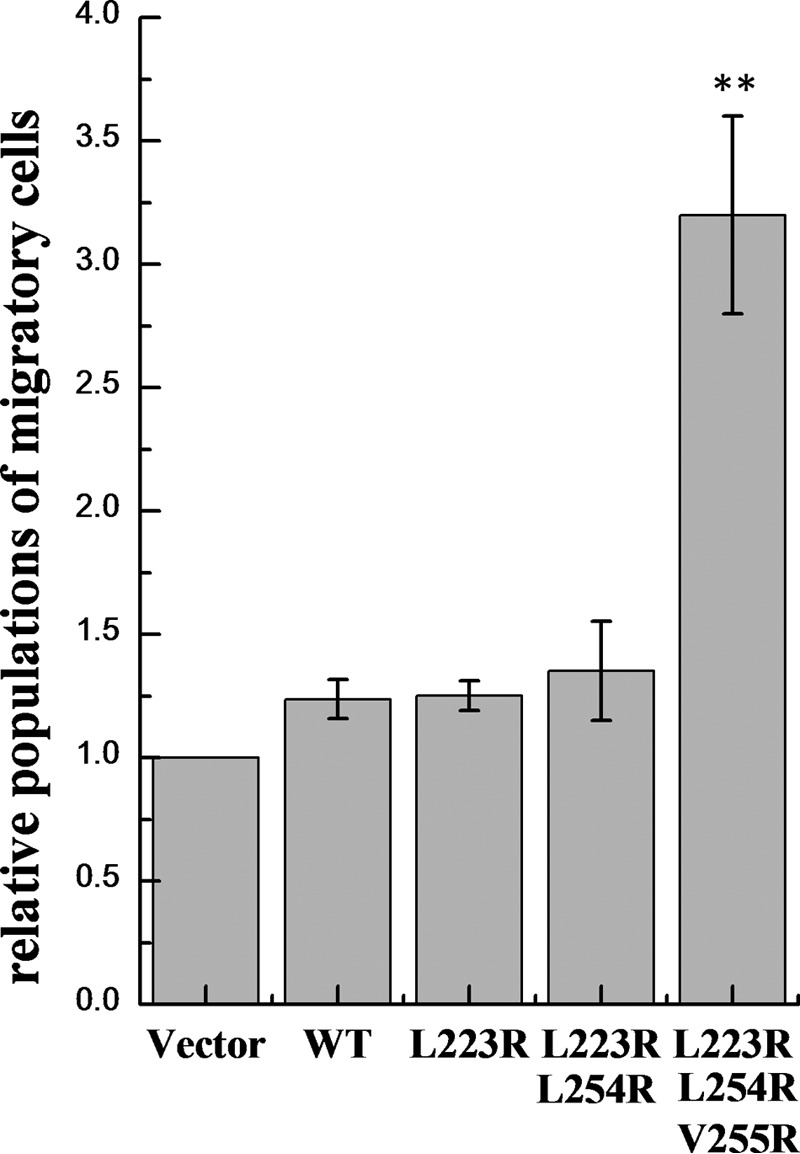
Results of the migration assay. Shown are means and standard errors from three independent experiments. The number of migratory HEK293T cells expressing the EphA2 triple mutant is significantly higher than the number of migratory cells expressing the wild type or the single and double mutants (p < 0.01 by analysis of variance). **, p < 0.01.
EphA2 Unliganded Dimerization Leads to Reduced Ser-897 and Enhanced Tyr-772 Phosphorylation
The migratory ability of cells expressing EphA2 in response to serum stimulation has been shown to be a consequence of EphA2 phosphorylation on Ser-897 (20). Furthermore, increased Ser-897 phosphorylation has been shown to correlate with decreased tyrosine phosphorylation and enhanced cell migration (20). Therefore, we hypothesized that compromised unliganded EphA2 dimerization will lead to increased Ser-897 phosphorylation and decreased tyrosine phosphorylation, leading to the observed changes in EphA2-mediated migration of HEK293T cells. To test this hypothesis, we examined the Ser-897 and Tyr-772 phosphorylation levels of wild-type and triple mutant EphA2 by Western blotting. This showed that Ser-897 phosphorylation is higher for the triple mutant than for the wild type, whereas phosphorylation of Tyr-772 in the activation loop is lower for the triple mutant than for the wild-type (Fig. 8). Thus, the decrease in unliganded dimerization due to the L223R/L254R/V255R mutations correlates with an increase in Ser-897 phosphorylation and a decrease in tyrosine phosphorylation, consistent with our hypothesis.
FIGURE 8.

Phosphorylation of wild-type EphA2 and the triple EphA2 mutant. Top, representative Western blot results. Bottom, quantification from three independent experiments. Shown are means and standard errors. The triple mutations led to increased EphA2 Ser-897 phosphorylation and reduced Tyr-772 phosphorylation. *, p < 0.05; **, p < 0.01.
Discussion
EphA2 Unliganded Dimer as an Intermediate in the Clustering Process
EphA2 receptors are known to cluster and thus mediate cell detachment and repulsion and ultimately control cell migration. The prevailing model of cluster formation is the so-called “seeding mechanism” model, based on crystal structures of isolated EphA2 extracellular domains, with and without bound ligands (4, 5). According to this model, 1) an Eph receptor binds to a ligand; 2) the ligand-bound receptors form a dimer via the so-called “dimerization” or “heterotetramerization” interface in the extracellular domains; and 3) these dimers assemble into larger clusters via the so-called “clustering interface” in the extracellular domains. Here, by probing the interactions of EphA2 in the plasma membrane, we demonstrate for the first time that EphA2 forms dimers in the absence of ligand binding. About 60% of the receptors are dimeric at expression levels that are known to be physiologically relevant. Thus, the assembly mechanism behind EphA2 clustering must involve the recruitment of pre-formed dimers, not just monomers, and the seeding mechanism model is thus incomplete.
The clustering interface in EphA2 crystal structures is stabilized, in part, by leucine zipper-like interactions in the cysteine-rich domain. The interface involves a large number of van der Waals contacts, including contacts between Leu-223, Leu-254, and Val-255. Previous work has characterized the significance of this receptor-receptor interface in the ligand-mediated EphA2 response, with the help of single and multiple residue mutations (4, 5). These studies have shown a larger effect due to the L223R/L254R/V255R mutations compared with the L223R single mutation and the L223R/L254R mutations (5), as well as a strong functional effect due to the L223R/L254R/V255R mutations (4). In particular, the L223R/L254R/V255R mutations significantly disrupted the ability of EphA2 to cluster and decreased its tyrosine phosphorylation in response to ephrin-A5 Fc.
Here, we introduced the same L223R/L254R/V255R mutations in EphA2, and we showed that the dimerization of EphA2 in the absence of ligand binding is significantly reduced due to these mutations. Thus, the same “clustering” interface mediates interactions in both liganded clusters and unliganded dimers. This finding suggests that EphA2 unliganded dimers are an intermediate in the process of EphA2 cluster formation.
Link between EphA2 Dimerization and EphA2 Oncogenic Signaling in the Absence of Ligand
We compared the biological activity of wild-type EphA2 and the L223R/L254R/V255R mutant, which exhibits compromised unliganded dimerization. We found that Ser-897 phosphorylation is higher in the triple mutant EphA2 than in the wild type, whereas Tyr-772 phosphorylation is lower. The migration of HEK293T cells expressing mutant EphA2 was significantly higher than the migration of cells expressing the wild-type receptor. This is consistent with the known correlation between EphA2 phosphorylation and cell migration (20). High Ser-897 phosphorylation and migratory ability, and low tyrosine phosphorylation, are considered signatures of EphA2 tumorigenic activity in the absence of ligand (20). Here, we demonstrate that these features correlate with a decrease in unliganded EphA2 dimerization.
A biochemical pathway that mediates EphA2 tumorigenic activity is triggered by the phosphorylation of Ser-897 by Akt (20). Therefore, one way to interpret our findings is to postulate that Akt can phosphorylate the EphA2 monomer but not the dimer. Thus, our data suggest that unliganded dimerization suppresses EphA2 tumorigenic activity.
Implications for Cancer
The ability to perform precise measurements of EphA2 dimerization allows us to calculate the density of EphA2 monomers on the cell surface as a function of total EphA2 levels. The results shown in Fig. 9A demonstrate that the increase in monomers at high EphA2 expression is substantially reduced due to dimerization, as compared with the case of no dimerization. Thus, EphA2 dimerization decreases the tumorigenic signaling activity of the receptor by partially depleting the monomer pool under high expression conditions.
FIGURE 9.
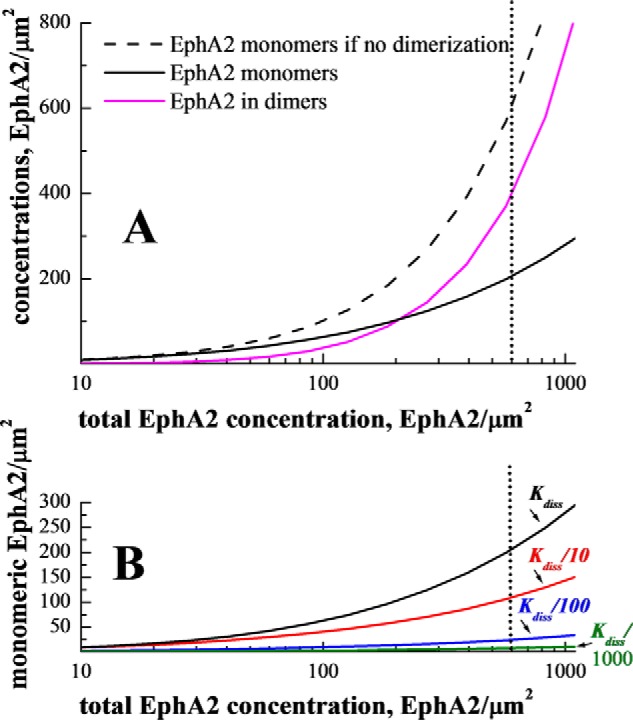
Predicted monomeric EphA2 populations as a function of EphA2 expression. A, monomeric and dimeric wild-type EphA2 concentrations (y axis) versus total EphA2 concentration (x axis), predicted from the measured dimerization curve in Fig. 3C. The solid magenta curve is the concentration of dimeric receptors, i.e. the product of total concentration and the measured dimeric fraction. The solid black curve is the monomer concentration, i.e. the product of total concentration and the measured monomeric fraction. The black dashed line corresponds to the case of no dimerization, when the monomer concentration is equal to the total concentration. For known physiological expression levels of 600 receptors per μm2 (44), the monomer concentration is decreased ∼3-fold due to the ability of EphA2 to form dimers in the absence of ligand binding. We propose that Akt phosphorylates only EphA2 monomers on Ser-897 to promote cell migration, and thus EphA2 unliganded dimerization provides protection against high migration and tumorigenic signaling. B, predicted EphA2 monomeric populations for wild-type EphA2 and for EphA2 dimers with higher stability. Such increased stability could be achieved through the use of engineered antibodies or peptides (see text). Black line, wild-type EphA2, Kd = 210 receptors/μm2; red line, Kd/10 = 21 receptors/μm2; blue line, Kd/100 = 2.1 receptors/μm2; green line, Kd/1000 = 0.21 receptors/μm2. Additional dimer stabilization will lead to dramatic reduction in EphA2 monomer populations.
Despite the protective effect of EphA2 dimerization, the monomer population is still significant in A549 lung cancer cells (44), i.e. about 200 monomers/μm2 or 200,000 monomers per cell (see dotted line in Fig. 9A). Because our work suggests that the EphA2 monomers are the pro-tumorigenic molecular species, a useful therapeutic strategy may be to reduce the monomer EphA2 population by stabilizing EphA2 unliganded dimers.
We can predict the degree of dimer stabilization that would be required to drastically reduce the EphA2 monomer population. In Fig. 9B, we show the predicted monomer populations when the dissociation constant is decreased by a factor of 10 (red), 100 (blue), and 1000 (green). We see that a decrease in the dissociation constant by a factor of 10, corresponding to EphA2 dimer stabilization by an additional 1.3 kcal/mol, will reduce the monomer population by half. A decrease in the dissociation constant by a factor of 100 (corresponding to EphA2 dimer stabilization by an additional 2.7 kcal/mol), will reduce the monomer population by an order of magnitude, from ∼200 to 20 monomers/μm2. Significant dimer stabilization, virtually eliminating the monomeric EphA2 population, may be possible through the use of bivalent antibodies, peptides, or ligands, binding to the extracellular domain of EphA2, provided that their geometry is suitably designed.
Author Contributions
D. R. S., E. B. P., and K. H. designed the experiments and wrote the paper. C. K. developed the FRET imaging and analysis methodologies and wrote the requisite software. D. R. S., N. G., and M. S. generated the plasmids. D. R. S., N. G., and M. S. performed the FRET experiments. D. R. S. and F. A. performed the functional assays.
Acknowledgment
We thank Michael Paul for technical help.
This work was supported by National Science Foundation Grant MCB 1157687, National Science Foundation Graduate Research Fellowship DGE-1232825 (to C. K.), and National Institutes of Health Grants GM068619 (to K. H.) and CA138390 (to E. B. P.). The authors declare that they have no conflicts of interest with the contents of this article.
- RTK
- receptor tyrosine kinase
- MSE
- mean squared error
- eYFP
- enhanced YFP.
References
- 1.Barquilla A., and Pasquale E. B. (2015) Eph receptors and ephrins: therapeutic opportunities. Annu. Rev. Pharmacol. Toxicol. 55, 465–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pasquale E. B. (2010) Eph receptors and ephrins in cancer: bidirectional signalling and beyond. Nat. Rev. Cancer 10, 165–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Funk S. D., Yurdagul A. Jr., Albert P., Traylor J. G. Jr., Jin L., Chen J., and Orr A. W. (2012) EphA2 activation promotes the endothelial cell inflammatory response a potential role in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 32, 686–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seiradake E., Harlos K., Sutton G., Aricescu A. R, and Jones E. Y. (2010) An extracellular steric seeding mechanism for Eph-ephrin signaling platform assembly. Nat. Struct. Mol. Biol. 17, 398–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Himanen J. P., Yermekbayeva L., Janes P. W., Walker J. R., Xu K., Atapattu L., Rajashankar K. R., Mensinga A., Lackmann M., Nikolov D. B., and Dhe-Paganon S. (2010) Architecture of eph receptor clusters. Proc. Natl. Acad. Sci. U.S.A. 107, 10860–10865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu K., Tzvetkova-Robev D., Xu Y., Goldgur Y., Chan Y. P., Himanen J. P., and Nikolov D. B. (2013) Insights into Eph receptor tyrosine kinase activation from crystal structures of the EphA4 ectodomain and its complex with ephrin-A5. Proc. Natl. Acad. Sci. U.S.A. 110, 14634–14639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wimmer-Kleikamp S. H., Nievergall E., Gegenbauer K., Adikari S., Mansour M., Yeadon T., Boyd A. W., Patani N. R., and Lackmann M. (2008) Elevated protein-tyrosine phosphatase activity provokes Eph/ephrin-facilitated adhesion of pre-B leukemia cells. Blood 112, 721–732 [DOI] [PubMed] [Google Scholar]
- 8.Janes P. W., Nievergall E., and Lackmann M. (2012) Concepts and consequences of Eph receptor clustering. Semin. Cell Dev. Biol. 23, 43–50 [DOI] [PubMed] [Google Scholar]
- 9.Fang W. B., Ireton R. C., Zhuang G., Takahashi T., Reynolds A., and Chen J. (2008) Overexpression of EPHA2 receptor destabilizes adherens junctions via a RhoA-dependent mechanism. J. Cell Sci. 121, 358–368 [DOI] [PubMed] [Google Scholar]
- 10.Pratt R. L., and Kinch M. S. (2002) Activation of the EphA2 tyrosine kinase stimulates the MAP/ERK kinase signaling cascade. Oncogene 21, 7690–7699 [DOI] [PubMed] [Google Scholar]
- 11.Wakayama Y., Miura K., Sabe H., and Mochizuki N. (2011) EphrinA1-EphA2 signal induces compaction and polarization of Madin-Darby Canine kidney cells by inactivating Ezrin through negative regulation of RhoA. J. Biol. Chem. 286, 44243–44253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miura K., Nam J. M., Kojima C., Mochizuki N., and Sabe H. (2009) EphA2 engages Git1 to suppress Arf6 activity modulating epithelial cell-cell contacts. Mol. Biol. Cell 20, 1949–1959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miao H., Burnett E., Kinch M., Simon E., and Wang B. (2000) Activation of EphA2 kinase suppresses integrin function and causes focal-adhesion-kinase dephosphorylation. Nat. Cell Biol. 2, 62–69 [DOI] [PubMed] [Google Scholar]
- 14.Nasreen N., Mohammed K. A., Lai Y., and Antony V. B. (2007) Receptor EphA2 activation with ephrinA1 suppresses growth of malignant mesothelioma (MM). Cancer Lett. 258, 215–222 [DOI] [PubMed] [Google Scholar]
- 15.Ireton R. C., and Chen J.. (2005) EphA2 receptor tyrosine kinase as a promising target for cancer therapeutics. Curr. Cancer Drug Targets 5, 149–157 [DOI] [PubMed] [Google Scholar]
- 16.Tandon M., Vemula S. V., and Mittal S. K. (2011) Emerging strategies for EphA2 receptor targeting for cancer therapeutics. Expert Opin. Ther. Targets 15, 31–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Biao-xue R., Xi-guang C., Shuan-ying Y., Wei L., and Zong-juan M. (2011) EphA2-dependent molecular targeting therapy for malignant tumors. Curr. Cancer Drug Targets 11, 1082–1097 [DOI] [PubMed] [Google Scholar]
- 18.Wykosky J., and Debinski W. (2008) The EphA2 receptor and EphrinA1 ligand in solid tumors: function and therapeutic targeting. Mol. Cancer Res. 6, 1795–1806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Macrae M., Neve R. M., Rodriguez-Viciana P., Haqq C., Yeh J., Chen C., Gray J. W., and McCormick F. (2005) A conditional feedback loop regulates Ras activity through EphA2. Cancer Cell 8, 111–118 [DOI] [PubMed] [Google Scholar]
- 20.Miao H., Li D. Q., Mukherjee A., Guo H., Petty A., Cutter J., Basilion J. P., Sedor J., Wu J., Danielpour D., Sloan A. E., Cohen M. L., and Wang B. (2009) EphA2 mediates ligand-dependent inhibition and ligand-independent promotion of cell migration and invasion via a reciprocal regulatory loop with Akt. Cancer Cell 16, 9–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miao H., Gale N. W., Guo H., Qian J., Petty A., Kaspar J., Murphy A. J., Valenzuela D. M., Yancopoulos G., Hambardzumyan D., Lathia J. D., Rich J. N., Lee J., and Wang B. (2015) EphA2 promotes infiltrative invasion of glioma stem cells in vivo through cross-talk with Akt and regulates stem cell properties. Oncogene 34, 558–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paraiso K. H., Das Thakur M., Fang B., Koomen J. M., Fedorenko I. V., John J. K., Tsao H., Flaherty K. T., Sondak V. K., Messina J. L., Pasquale E. B., Villagra A., Rao U. N., Kirkwood J. M., Meier F., et al. (2015) Ligand-independent EPHA2 signaling drives the adoption of a targeted therapy-mediated metastatic melanoma phenotype. Cancer Discov. 5, 264–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Binda E., Visioli A., Giani F., Lamorte G., Copetti M., Pitter K. L., Huse J. T., Cajola L., Zanetti N., DiMeco F., De Filippis L., Mangiola A., Maira G., Anile C., De Bonis P., et al. (2012) The EphA2 receptor drives self-renewal and tumorigenicity in stem-like tumor-propagating cells from human glioblastomas. Cancer Cell 22, 765–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Low-Nam S. T., Lidke K. A., Cutler P. J., Roovers R. C., van Bergen en Henegouwen P. M., Wilson B. S., and Lidke D. S. (2011) ErbB1 dimerization is promoted by domain co-confinement and stabilized by ligand binding. Nat. Struct. Mol. Biol. 18, 1244–1249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chung I., Akita R., Vandlen R., Toomre D., Schlessinger J., and Mellman I. (2010) Spatial control of EGF receptor activation by reversible dimerization on living cells. Nature 464, 783–787 [DOI] [PubMed] [Google Scholar]
- 26.Lin C. C., Melo F. A., Ghosh R., Suen K. M., Stagg L. J., Kirkpatrick J., Arold S. T., Ahmed Z., and Ladbury J. E. (2012) Inhibition of basal FGF receptor signaling by dimeric Grb2. Cell 149, 1514–1524 [DOI] [PubMed] [Google Scholar]
- 27.Sarabipour S., and Hristova K. (2015) FGFR3 unliganded dimer stabilization by the juxtamembrane domain. J. Mol. Biol. 427, 1705–1714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Del Piccolo N., Placone J., and Hristova K. (2015) Effect of thanatophoric dysplasia type I mutations on FGFR3 dimerization. Biophys. J. 108, 272–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen L., Novicky L., Merzlyakov M., Hristov T., and Hristova K. (2010) Measuring the energetics of membrane protein dimerization in mammalian membranes. J. Am. Chem. Soc. 132, 3628–3635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen L., Placone J., Novicky L., and Hristova K. (2010) The extracellular domain of fibroblast growth factor receptor 3 inhibits ligand-independent dimerization. Sci. Signal. 3, ra86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sinha B., Köster D., Ruez R., Gonnord P., Bastiani M., Abankwa D., Stan R. V., Butler-Browne G., Vedie B., Johannes L., Morone N., Parton R. G., Raposo G., Sens P., Lamaze C., and Nassoy P. (2011) Cells respond to mechanical stress by rapid disassembly of caveolae. Cell 144, 402–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Raicu V., Stoneman M. R., Fung R., Melnichuk M., Jansma D. B., Pisterzi L. F., Rath S., Fox M., Wells J. W., and Saldin D. K. (2009) Determination of supramolecular structure and spatial distribution of protein complexes in living cells. Nat. Photonics 3, 107–113 [Google Scholar]
- 33.Biener G., Stoneman M. R., Acbas G., Holz J. D., Orlova M., Komarova L., Kuchin S., and Raicu V. (2014) Development and experimental testing of an optical micro-spectroscopic technique incorporating true line-scan excitation. Int. J. Mol. Sci. 15, 261–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Adler J., Shevchuk A. I., Novak P., Korchev Y. E., and Parmryd I. (2010) Plasma membrane topography and interpretation of single-particle tracks. Nat. Methods 7, 170–171 [DOI] [PubMed] [Google Scholar]
- 35.Parmryd I., and Onfelt B. (2013) Consequences of membrane topography. FEBS J. 280, 2775–2784 [DOI] [PubMed] [Google Scholar]
- 36.Beauchamp A., Lively M. O., Mintz A., Gibo D., Wykosky J., and Debinski W. (2012) EphrinA1 is released in three forms from cancer cells by matrix metalloproteases. Mol. Cell. Biol. 32, 3253–3264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deleted in proof
- 38.Deleted in proof
- 39.Evers T. H., van Dongen E. M., Faesen A. C., Meijer E. W., and Merkx M. (2006) Quantitative understanding of the energy transfer between fluorescent proteins connected via flexible peptide linkers. Biochemistry 45, 13183–13192 [DOI] [PubMed] [Google Scholar]
- 40.Raicu V. (2007) Efficiency of resonance energy transfer in homo-oligomeric complexes of proteins. J. Biol. Physics 33, 109–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Adair B. D., and Engelman D. M. (1994) Glycophorin a helical transmembrane domains dimerize in phospholipid bilayers–a resonance energy transfer study. Biochemistry 33, 5539–5544 [DOI] [PubMed] [Google Scholar]
- 42.Li M., Reddy L. G., Bennett R., Silva N. D. Jr., Jones L. R., and Thomas D. D. (1999) A fluorescence energy transfer method for analyzing protein oligomeric structure: application to phospholamban. Biophys. J. 76, 2587–2599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schick S., Chen L., Li E., Lin J., Köper I., and Hristova K. (2010) Assembly of the M2 tetramer is strongly modulated by lipid chain length. Biophys. J. 99, 1810–1817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu Y., Lan X., Wu T., Lang J., Jin X., Sun X., Wen Q., and An R. (2014) Tc-99m-labeled SWL specific peptide for targeting EphA2 receptor. Nucl. Med. Biol. 41, 450–456 [DOI] [PubMed] [Google Scholar]