

Background: Phosphatidylinositides in the plasma membrane (PM) are pivotal for cellular functions.
Results: Superresolution imaging reveals homogeneous distribution of PI(4,5)P2, PI4P, and PI(3,4,5)P3 in the major area of the PM.
Conclusion: Phosphatidylinositides detected by PH domains are uniformly distributed in the major regions of the PM, with limited concentration gradients.
Significance: This result may imply a new working model of phosphatidylinositides at nanometer scale.
Keywords: β cell (B cell), inositol phospholipid, membrane lipid, metabolism, phosphatidylinositol, SNARE proteins, superresolution microscopy
Abstract
Both phosphatidylinositol 4-phosphate (PI4P) and phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2) are independent plasma membrane (PM) determinant lipids that are essential for multiple cellular functions. However, their nanoscale spatial organization in the PM remains elusive. Using single-molecule superresolution microscopy and new photoactivatable fluorescence probes on the basis of pleckstrin homology domains that specifically recognize phosphatidylinositides in insulin-secreting INS-1 cells, we report that the PI(4,5)P2 probes exhibited a remarkably uniform distribution in the major regions of the PM, with some sparse PI(4,5)P2-enriched membrane patches/domains of diverse sizes (383 ± 14 nm on average). Quantitative analysis revealed a modest concentration gradient that was much less steep than previously thought, and no densely packed PI(4,5)P2 nanodomains were observed. Live-cell superresolution imaging further demonstrated the dynamic structural changes of those domains in the flat PM and membrane protrusions. PI4P and phosphatidylinositol (3,4,5)-trisphosphate (PI(3,4,5)P3) showed similar spatial distributions as PI(4,5)P2. These data reveal the nanoscale landscape of key inositol phospholipids in the native PM and imply a framework for local cellular signaling and lipid-protein interactions at a nanometer scale.
Introduction
The plasma membrane of eukaryotic cells is composed of proteins and lipids in a complex topographic organization. Mounting evidence suggests that protein clusters or macroprotein complexes in a diversely confined area (1, 2) facilitate the efficiency, fidelity, and specificity of cellular signaling in time and space. A typical example is syntaxin1A, a key membrane protein that participates in membrane fusion and forms multiple dense clusters in the PM2 (3–5). Likewise, lipid rafts have been proposed as signaling platforms in the PM by forming nanoscale complexes that contain highly concentrated cholesterol, sphingolipid, and other proteins (6). However, whether a similar organization applies to phosphoinositides (PIs) in the PM is not clear.
Despite comprising only a small fraction of cellular phospholipids, PIs play essential roles in cell physiology. They consist of a family of seven polyphosphoinositides derived from reversible phosphorylation of the inositol ring at positions 3, 4, and/or 5 via distinct PI kinases and phosphatases (7, 8). Their different subcellular distribution and fast dynamic equilibrium facilitate their diverse functions. PI(4,5)P2 predominantly locates in the inner leaflet of the PM and participates in a variety of cellular processes (7, 9), including signaling transduction, actin dynamics, membrane trafficking, and ion channel modulation. It is mainly produced through phosphorylation of PI4P at the 5 position by PI4P 5 kinases, and it can also be converted into other PIs or second messengers such as inositol 1,4,5-triphosphate and diacylglycerol by phospholipase C (PLC) (10). PI4P synthesized by PI4KIIIα in the PM (11, 12) is critical for PM identity in addition to its role as the precursor for PI(4,5)P2 and other inositol phospholipids. Depletion of PI4P in the PM interrupts membrane protein functions and proper PM subcellular localization (12, 13). PI(3,4,5)P3 levels in the PM are much lower than PI(4,5)P2 levels, and they play important roles in actin dynamics, phagocytosis, and hormone secretion (14, 15). Therefore, it is crucial to understand how these phosphoinositides in the PM are organized spatially to serve their diverse functions.
PI(4,5)P2 directly interacts with proteins through multiple structured domains, such as the pleckstrin homology (PH) domain and other domains, including ENTH (epsin N-terminal homology)/ANTH (AP180 N-terminal homology), FERM (band four-point-one ezrin radixin moesin homology domains), FYVE (Fab1, YGLO23, Vps27, and EEA1), PX (phox), and TARF (TMV-associated RING finger) domains (16). It plays a critical role as a recruiter or cofactor for protein membrane targeting. The negative charges of PI(4,5)P2 (−3 to −5) also promote its interactions with proteins containing polybasic stretches of amino acids such as myristoylated alanine-rich C kinase substrate and GAP43 (17, 18). The discovery of the PH domain of PLCδ1 as a high-affinity PI(4,5)P2 biosensor (Kd = 2 μm) (19) allows the direct visualization of PI(4,5)P2 subcellular distribution and dynamics in live cells. PHPLCδ1 domain-based fluorescent proteins (FPs), together with PI(4,5)P2-specific antibodies, have revealed very diverse, yet sometimes controversial, organization patterns of PI(4,5)P2, ranging from uniform distribution (20–22) to large patches (23, 24) to small, dense puncta (24–28), depending on cell type and experimental conditions.
At a nanometer scale, the spatial organization of PI(4,5)P2 in the PM remains poorly understood, largely because of the lack of methods for precisely characterizing this at nanometer resolution as well as the rapid equilibrium among the seven PIs in live cells. Conventional light microscopy is limited by diffraction to a spatial resolution of over 200 nm. EM provides a much higher spatial resolution but usually requires harsh sample processing, which often distorts normal PI(4,5)P2 organization and introduces artifacts (29). FRET cannot be used to interrogate protein organization over distances of more than ∼10 nm. New techniques in superresolution optical imaging provide promising tools for this question. A recent study (30), using stimulated emission depletion (STED) microscopy (31), revealed abundant dense PI(4,5)P2 clusters (∼73 nm in diameter) in the PM sheets of PC-12 cells. This study reasoned that very high local concentrations of PI(4,5)P2 molecules in those nanodomains are required for syntaxin1A clustering through ionic protein-lipid interactions, although weak syntaxin1A self-association also produces syntaxin1A clustering (5). However, those abundant, dense PI(4,5)P2 nanodomains have not been observed using rapid freezing EM (29, 32), an approach that preserves the intact PM structure of live cells. In addition, the nanoscale organization of PI(4,5)P2 and PI4P in live cells is unknown so far, and it remains unclear whether the conclusion from fixed cells in those studies can equally apply to live cells.
Single-molecule localization microscopy accurately identifies the locations of individual molecules when they are photoactivated/converted at well separated locations and times. Therefore, it significantly improves the spatial resolution of light microscopy. This approach is promising to gain insights into the structure and function of macromembrane complexes in the PM. Single-molecule localization microscopy is given different names on the basis of the same principle, with slightly different imaging methods, such as photoactivated localization microscopy (PALM) (33), fluorescence photoactivation localization microscopy (34), stochastic optical reconstruction microscopy (35, 36), ground state depletion microscopy followed by individual molecule return (37), and direct stochastic optical reconstruction microscopy (dSTORM) (38). Of these methods, PALM uses photoactivatable FPs and has the advantages of high labeling specificity and density by genetically tagging photoactivatable FPs to any protein of interest (33, 39–44).
Insulin secretion from pancreatic β cells plays a critical role in plasma glucose homeostasis. Inositol lipid signaling contributes to diabetes development by regulating insulin secretion and its receptor signaling. Perturbations of different enzymes that regulate PI4P (45), PI(4,5)P2 (45, 46), and PI(3,4,5)P3 (47, 48) strongly affect insulin secretion. For example, inositol 5-phosphatase SH2 domain-containing inositol 5-phosphatase 2 (SHIP2) (49) and 3-phosphatase phosphatase and tensin homologue on chromosome 10 (PTEN) (50) tightly regulate insulin signal transduction and have been considered potential drug targets for diabetes treatment. Using insulin-secreting INS-1 cells as a model and combining single-molecule superresolution microscopy, we visualized the nanoscale organization of PI(4,5)P2 and two other key phosphoinositides in the PM. Surprisingly, PALM imaging uncovers a rather homogeneous PI(4,5)P2 distribution in the major areas of the PM in both fixed and live cells, and some areas with slightly higher PI(4,5)P2 concentration are sparsely distributed in the PM and cell periphery. These PI(4,5)P2-enriched membrane patches/domains have variable sizes and diverse shapes. PI4P and PI(3,4,5)P3 had similar uniform patterns in the PM as PI(4,5)P2 despite their different abundance. This study reveals a detailed nanoscale landscape of key signaling inositol lipids in the native plasma membrane, implying a new working model for local lipid signaling and lipid-protein interactions in the PM, a complex environment of live cells.
Materials and Methods
INS-1 Cell Culture, DNA Constructs, and Molecular Cloning
Insulin-secreting INS-1 832/13 cells (passages 50–65, from Dr. Christopher B. Newgard, Duke University) (51) were cultured at 37 °C in a 5% CO2 humidified incubator with RPMI 1640 culture medium (Cellgro) containing 11.1 mm glucose, 10 mm HEPES, 10 mm glutamine, 10 mm sodium pyruvate, 50 μm β-mercaptoethanol (all from Sigma), and 10% FBS (Atlanta Biological). The cells were seeded on round coverslips (#1.5, 18 mm, Warner Instruments) precoated with 30 μg/ml fibronectin (Millipore) for 24 h and then transfected with different plasmids using Lipofectamine 3000 (Life Technologies). All experiments were performed 48 h after transfection.
Multiple single-molecule localization probes that targeted to syntaxin1A and different PH domains were generated by PCR. PH domain plasmids of near-infrared fluorescent protein (iRFP)-PHPLCδ1, EGFP-PHOSH2, EGFP-PHOSBP, and mCherry-PHGRP1 were gifts from Dr. Pietro De Camilli (Yale). PAmCherry1 and mEos3.1 were PCR-amplified to replace the enhanced GFP (EGFP) in C1 and N1 cloning vectors (Clontech) with AgeI and BglII and AgeI and NotI, respectively. iRFP was PCR-amplified and inserted before PAmCherry1 with NheI and AgeI to make the iRFP-PAmCherry1-C1 vector. The PHPLCδ1, PHosh2, and PHGRP1 domains were then PCR-amplified and inserted after the iRFP-PAmCherry1 or mEos3.1 vector with BglII and BamHI to generate the new PALM probes iRFP-PAmCherry1-PHPLCδ1, iRFP-PAmCherry1-PHosh2, iRFP-PAmCherry1-PHGRP1, and mEos3.1-PHPLCδ1. peGFP(N1)-deltaCMV-rSyntaxin1a-meGFP (3) was from Dr. Wolfhard Almers (Addgene, plasmid 34631). rSyntaxin1a-meGFP was PCR-amplified and replaced EGFP in the N1 vector with flanking XhoI and NotI, and then PAmCherry1 was amplified and inserted before rSyntaxin1a with NheI and AgeI to create PAmCherry1-syntaxin1A-EGFP. deltaCMV-iRFP-PAmCherry1 and deltaCMV-mEos3.1 were generated by amplifying iRFP-PAmCherry1 and mEos3.1 flanked by NheI and NotI to replace rSyntaxin1a-meGFP in the deltaCMV-rsyntaxin1a-mEGFP plasmid.
Membrane Sheet Generation and Immunofluorescence Staining
PM membrane sheet preparation was adapted from previous studies (52, 53) with minor modifications. Clean coverslips were coated with 500 μg/ml poly-d-lysine for 1 h before experiments, and then they were placed on a prechilled (4 °C) metal plate for later use. INS-1 cells growing on coverslips were washed three times with ice-cold PBS buffer containing 1 mm EGTA and then excessive buffer was drained off with tissue paper at the edge of the coverslips. All subsequent steps were performed at 4 °C in a cold room, and all the buffers were prechilled unless specified otherwise. The coverslips with cells were placed face-down and put onto poly-d-lysine-coated sides of empty coverslips for 7 min. Then the flat PM sheets on the poly-d-lysine-coated coverslips were peeled off with tweezers, washed gently with ice-cold PBS immediately, and fixed with ice-cold 4% paraformaldehyde (PFA) + 0.2% glutaraldehyde for 15 min.
Phospholipid immunostaining was performed as described previously (22) with minor modifications. Coverslips with fixed PM sheets were washed three times (7 min each time) with ice-cold PBS containing 50 mm NH4Cl and quenched with 0.1% sodium borohydride in PBS for 7 min, followed by a wash with PBS (without 50 mm NH4Cl). Membrane sheets were then blocked for 45 min with PBS solution containing 5% (v/v) normal goat serum, 5% (v/v) BSA, and 50 mm NH4Cl and incubated with the primary PI(4,5)P2 antibody (catalog no. sc-53412, Santa Cruz Biotechnology, 1:300 dilution) in blocking solution for 1 h. After washing three times (10 min each time) with PBS containing 50 mm NH4Cl, samples were incubated with secondary F(ab′)2 goat anti-mouse Alexa Fluor 647 (catalog no. A-21237, Invitrogen, 1:300) in blocking buffer for 1 h and washed three times, followed by postfixation with 4% PFA + 0.2% glutaraldehyde for 15 min. Finally, the samples were washed three times (7 min each time) and stored in PBS at 4 °C before imaging. Tubulin immunostaining was performed according to a previous study (54).
To evaluate the optical properties of probes at single-molecule levels, different approaches were used. For deltaCMV-driven (4) iRFP-PAmCherry1 and mEos3.1 single-molecule experiments, transfected COS-7 cells grew for 48 h and were fixed with 4% PFA + 0.2% glutaraldehyde at room temperature. For Alexa Fluor 647 single-molecule tests, coverslips precoated with poly-d-lysine were incubated with diluted AF647 molecules in PBS for 15 min to allow the Alexa Fluor 647 molecules to settle down on the coverslips. The samples were washed three times with PBS afterward, incubated with dSTORM buffer, and imaged.
Spinning Disk Confocal and Total Internal Reflection Fluorescence (TIRF) Imaging
Fluorescence imaging was performed with a Nikon Ti-E Eclipse inverted microscope-based imaging system under the control of NIS-Elements AR software. The system was equipped with Ti-ND6-PFS Perfect Focus and multiple objectives (CFI Plan Fluor ×4; NA 0.13, WD 17.2 mm; CFI Plan APO λ ×20, NA 0.75, WD 1.00 mm; Plan APO VC ×60 oil, NA 1.4, WD 0.13 mm; and APO ×100 oil, NA 1.49, WD 0.12 mm), a spinning disk (CSU X-1, 10,000 rpm, Yokogawa), a Ti-TIRF motorized illuminator unit, and an Agilent MLC400 high-power monolithic laser combiner SP with 405-, 488-, 561-, and 642-nm lasers. The EMCCD (iXon X3 DU897, Andor) and Neo-sCMOS cameras (Andor, 6.5 × 6.5 μm pixel size) were installed at the left and right imaging ports of the microscope to acquire confocal and TIRF microscopy images, respectively.
For live-cell TIRF imaging of phosphoinositides, INS-1 cells were preincubated with extracellular buffer (ES) containing 135 mm NaCl, 5.6 mm KCl, 2.6 mm CaCl2, 1.2 mm MgCl2, 3 mm glucose, and 20 mm HEPES (pH adjusted to 7.3 with NaOH) for 30 min at 37 °C, and then the cells were imaged in a temperature-controlled chamber (35 °C) with continuous buffer perfusion. Cell stimulation was applied through the perfusion of different buffers as indicated. High-glucose buffer included 28 mm glucose in the ES, and 60 mm high K+ solution contained 80.6 mm NaCl, 60 mm KCl, 2.6 mm CaCl2, 1.2 mm MgCl2, 3 mm glucose, and 20 mm HEPES. Images were acquired at a narrow TIRF illumination angle with 500-ms exposure time at a 6-s interval with 2 × 2 binning. Fluorescence analysis and quantification for TIRF imaging were performed using NIS-Elements AR (Nikon). Briefly, regions of an ∼3–5-μm-wide square within individual cells were analyzed. Fluorescence intensity was background-subtracted by the intensity of an empty area adjacent to the cell and normalized to its baseline before stimulation. The peak changes in Fig. 6 were normalized to their baseline fluorescence intensity in each cell. All data are presented as mean ± S.E. Comparison was evaluated statistically by two-tailed Student's t test, and the significance level of difference are denoted by asterisks (*, p < 0.05; **, p < 0.01; ***, p < 0.005).
FIGURE 6.
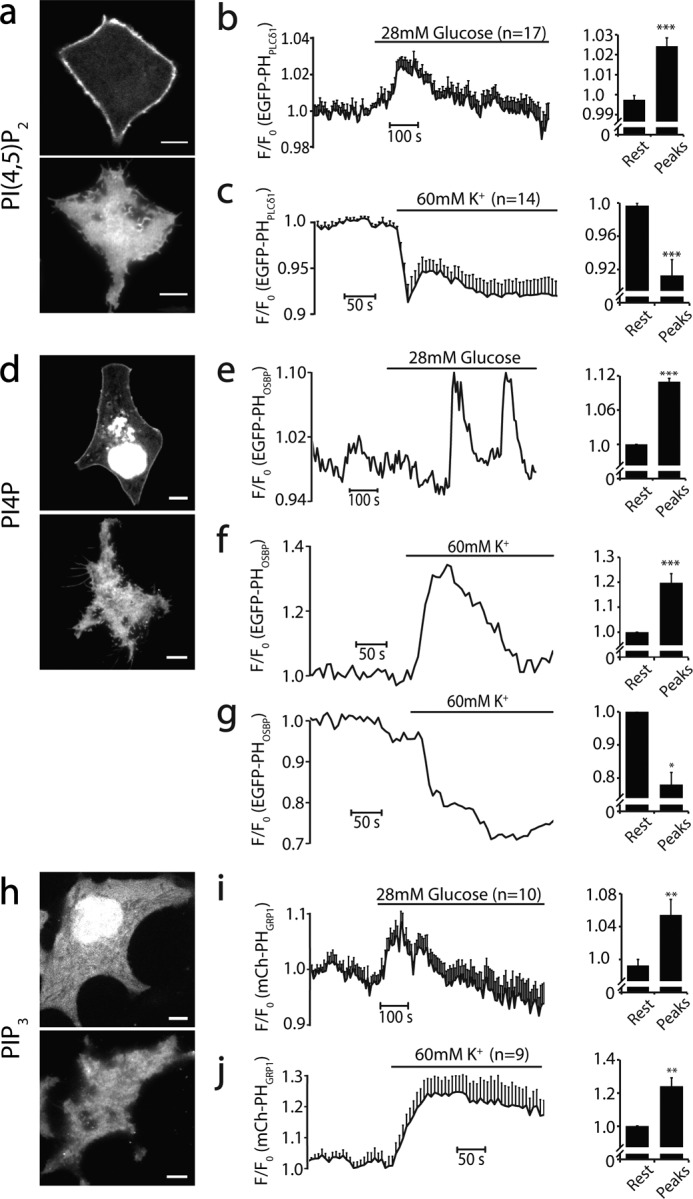
Dynamic changes of phosphoinositides in live cells in response to physiological stimulation at 35 °C. a, confocal (top panel) and TIRF (bottom panel) images of PI(4,5)P2 visualized with EGFP-PHPLCδ1 in INS-1 cells. Note the prominent PM localization of EGFP-PHPLCδ1. b, small but significant increase in EGFP-PHPLCδ1 fluorescence detected by TIRF after 28 mm glucose stimulation (n = 21 cells). c, the same as b but stimulated with 60 mm KCl (n = 14). d, confocal and TIRF images of PI4P visualized by EGFP-PHOSBP on an INS-1 cell. Note the prominent fluorescence in intracellular organelles of the INS-1 cell. e, PI4P oscillations induced by 28 mm glucose in a single INS-1 cell (4 of 24 cells). f and g, diverse PI4P concentration changes after 60 mm KCl stimulation in individual INS-1 cells. The majority of cells showed significant increases in PI4P levels on the PM (f, 24 of 31 cells), but some cells showed decreases (g, 6 of 31 cells), indicating complex PI4P metabolism among different INS-1 cells. h, confocal and TIRF images of mCherry-PHGRP1-labeled PI(3,4,5)P3 on an INS-1 cell PM. i, average responses of PI(3,4,5)P3 after 28 mm glucose (n = 12 cells) and 60 mm KCl stimulation (j, n = 10). For each condition, the baseline and the relative changes of upward/downward peaks were analyzed statistically and compared. *, p < 0.05; **, p < 0.01; ***, p < 0.005. Scale bars = 5 μm.
Single-molecule Point Localization Superresolution Imaging
Superresolution imaging was performed using Nikon-Ti Eclipse-based TIRF imaging system (×100 oil, 1.49 NA) as described above. Before imaging, diluted Tetraspeck beads (Invitrogen, 200-nm diameter, diluted in PBS containing an extra 50 mm MgCl2) were added to the imaging chamber and allowed to settle down for 10 min for offline drift correction, and then non-attached beads were washed away with PBS. Alexa Fluor 647 and PAmCherry1 were excited with 642- and 561-nm lasers, respectively, and the emission light was collected through the 700/75- and 600/50-nm filters, respectively, by a Neo-sCMOS camera at 2 × 2 binning (effective pixel size, 130 × 130 nm). The optimal density of individual fluorescence points was controlled by 405-nm laser intensity so that spatially isolated individual points could be identified unambiguously in each frame.
For dSTORM imaging, experiments were performed with imaging buffer containing 50 mm Tris-HCl, 10 mm NaCl, 10% glucose, 0.8 mg/ml glucose oxidase, 40 μg/ml catalase, and 10 mm cysteamine (Sigma) (pH 8.0). Image collection started when the majority of fluorescent molecules was isolated spatially after initial strong bleaching with the full power of 642-nm laser. In total, ∼30,000 images at 40 ms/frame were collected from individual cells.
For PALM imaging of cells expressing various iRFP-PAmCherry1 tagged to the PH domains or PAmCherry1-syntaxin1A-EGFP, cells were first identified by iRFP or EGFP to preserve the intact number of PAmCherry1 molecules. PALM images were acquired at 40 ms/frame for mEos3.1 and 50 ms/frame for PAmCherry1 until no more fluorescent probes could be activated. For imaging iRFP-PAmCherry1 probes without the PH domain in COS-7 cells, we imaged the cytosol space right above the PM by using slightly oblique rather than TIRF illumination. Because we used deltaCMV to drive the expression of iRFP-PAmCherry1, the expression levels of the probes were very low so that we could evaluate single molecular properties of the isolated individual probe in the cells.
For live-cell PALM, Tetraspeck beads were added directly to the imaging chamber for ∼10 min, and cells were imaged with continuous perfusion of extracellular buffer at 35 °C. PALM images were acquired at 10 ms/frame for 5 min, and every 1000 consecutive frames were pooled together as a single superresolution image (10 s/PALM frame). This high-speed acquisition may slightly overestimate the number and size of moving probes but should not affect the overall spatial distribution pattern of objects in each reconstructed frame (see Imaging Reconstruction below).
Point Localization and Superresolution Image Analysis
Image stacks were processed and reconstructed in MATLAB as described previously (42, 55). Individual fluorescent points were identified by a wavelet transform algorithm (56, 57) and localized by the local maximum with a mask of 5 × 5 pixels. All fluorescent points detected in a 7 × 7 pixel area were fitted with a two-dimensional Gaussian using the MLE_GPU algorithm (58) for precise molecular localization. Continuously detected points in the neighboring frames within a 65-nm distance (half-pixel width) were combined and fitted as a single molecule unless specified otherwise. For PALM imaging in fixed cells, fluorescent points detected within a 65-nm distance and a time window of 1.8 s for mEos3 (59) and 1.3 s for PAmCherry1 (60) were combined as the same single-molecule emission event to avoid overcounting because of photoblinking of the probes. In the case of PAmCherry1 (but not mESO3), such processes did not significantly affect PALM results, in good agreement with the negligible photoblinking of PAmCherry1 (60). For live-cell PALM, a small number of activated probes may dissociate from PI(4,5)P2 in the PM and freely diffuse around before bleaching, which can cause oversampling. To account for this potential effect, single-molecule signals in the neighboring frames within 130 nm, instead 65 nm, were combined into a single emission event. Minor position drift during acquisition was corrected with Tetraspeck beads for all experiments.
For dSTORM images, molecular clusters were analyzed with density-based spatial clustering of applications with noise analysis (61) to group and sort cluster members. Within each cluster, the standard deviation of the coordinates of members were calculated (as σ) and pooled together. For syntaxin1A clusters and PI(4,5)P2 membrane patch analysis, syntaxin1A or PI(4,5)P2 molecules with sparse distribution (noise-like) were removed with spatial filtering (62) at a density threshold of ∼4–5 molecules in a 75 × 75 nm area for syntaxin1A and 50 molecules in a 350 × 350 nm area for PI(4,5)P2. Then images were applied with density-based spatial clustering of applications with noise analysis, and σ in each cluster/patch was calculated. For syntaxin1A clusters and PI(4,5)P2-enriched patches, the size (in full width at half-maximum, FWHM) was calculated as 2.355σ, and the diameter of PI(4,5)P2 patches was calculated as 4σ. The molecule density of PI(4,5)P2 patches was analyzed after identifying each membrane patch and calculated by dividing the total molecule number in each patch by its area. Pair correlation analysis of syntaxin1A, PI(4,5)P2, iRFP-PAmCherry1, and simulated images was conducted by choosing eight 2.5 × 2.5 μm regions in the images and processed with a pair correlation code (63). In PI(4,5)P2 images, only areas that did not contain obvious PI(4,5)P2 patches were analyzed for pair correlation analysis. To analyze PI4P, PI(4,5)P2, and PI(3,4,5)P3 membrane density in the uniform areas, five to seven 2.5 × 2.5 μm regions in each cell and ∼10 cells in total for each probe (iRFP-PAmCherry1-PH labeled PI4P, PI(4,5)P2, and PI(3,4,5)P3) were analyzed to obtain their mean density.
Results
Assessment of the Single-molecule Localization Superresolution Approach
We estimated the spatial resolution of our single-molecule localization approach in our superresolution imaging system by using microtubules whose ultrastructure has been well characterized. Fig. 1, a—d, shows microtubule images under TIRF and dSTORM from the same COS-7 cell immunostained with Alexa Fluor 647 (AF647)-conjugated secondary antibody, demonstrating a significant improvement in the spatial resolution of the dSTORM image over its TIRF image. The pixelated complex microtubule structures in the TIRF image were clearly resolved in the dSTORM image (Fig. 1, c and d). Single-molecule imaging of AF647 attached to blank coverslips demonstrated a spatial resolution (FWHM) of ∼35 nm, with a localization precision (δ) of 14.7 ± 7.3 nm (n = 1649 molecules from 6 experiments) (Fig. 1e).
FIGURE 1.

Single-molecule superresolution imaging reveals fine structures of microtubules and syntaxin1A. a, TIRF image of microtubules in a large area of a COS-7 cell. b, superresolution image of the same area by dSTORM. c and d, enlarged views of the TIRF (c) and dSTORM images (d) of the boxed area in b. Note that the pixelation of microtubule structures under TIRF became clearly visible in dSTROM. e, histogram of the point localization precision of centroid positions. The localization uncertainty is determined as (σx + σy) / 2 for each isolated Alexa Fluor 647 molecule, where σx and σy are the standard deviations of the centroid position in x and y dimensions. The localization uncertainty (σ) is 14.73 ± 7.29 nm (n = 1649 molecules). The inset represents the centroid positions of a single Alexa Fluor 647 molecule during 16 photoblinkings, where the brighter spot showed two localizations in the same site. f and g, TIRF (f) and superresolution (g) images of PAmCherry1-labeled syntaxin1A in an INS-1 cell PM sheet. h, enlarged views of the boxed area in f. i, histograms of PAmCherry1-syntaxin1A-EGFP cluster size (top panel; median size, 68 nm in FWHM) and molecule number per cluster (bottom panel; 20.48 ± 6.68/cluster on average). Scale bars = 4 μm (b), 400 nm (d), 40 nm (e), 3 μm (g), and 500 nm (h).
Next we examined the PALM approach with syntaxin1A, a membrane protein with a typical cluster structure (3), in the plasma membrane sheets of INS-1 cells. The PM sheet preparation has the advantage of yielding structurally intact PM lawns with fewer cytoplasmic remnants (53, 64), and it avoids the minor contribution of a free cytosolic probe signal under whole-cell imaging as well as the detergent treatment, which can distort the native structure of the lipids in the PM (65). To perform syntaxin1A PALM imaging, we first generated PAmCherry1-syntaxin1A-EGFP as a dual-color PALM probe. PAmCherry1 is a photoactivatable monomeric red FP (40) with high photoactivation efficiency and little photoblinking (60, 66). EGFP was used to identify cells expressing PAmCherry1 before PALM imaging, and this is critical to preserve the original number of PAmCherry1 for quantitative PALM. To avoid repeatedly counting the same molecules resulted from photoblinking of a very small population (15%) of PAmCherry1 (60, 66), the molecules detected in the same location in neighboring frames within 1.3 s were combined and fitted as a single-molecule emission event (60) (see “Materials and Methods”). This approach effectively accounts for overcounting to obtain quantitative PALM images (60). In our case, this restriction only marginally decreased the total number of molecules detected in syntaxin clusters (data not shown).
PALM imaging showed a cluster organization of syntaxin1A in the INS-1 PM (Fig. 1, f–h); it was similar to the TIRF image but with a much higher spatial resolution. Syntaxin1A clusters had variable sizes, with a median FWHM of ∼68 nm (Fig. 1i), which is comparable with the size estimated in previous studies in PC-12 cells using STED (5), dSTORM (41, 67), and PALM (68). The molecular density in syntaxin1A clusters in the PM of INS-1 cells was 20.5 ± 6.7 per cluster (n = 95 from 7 cells), equivalent to 41.0 ± 13.4 per cluster after considering 50% photoactivation efficiency for PAmCherry1 (66). This number is comparable with the previous estimation of syntaxin1A number in each cluster (4, 5, 67), although this may be an underestimate because of the presence of unlabeled endogenous syntaxin1A.
Uniform Nanoscale Landscape of PI(4,5)P2 in the Major Areas of the PM from Fixed INS-1 Cells
After validating our superresolution imaging system, we generated and expressed iRFP-PAmCherry tagged PHPLCδ1 domain (19), the PI(4,5)P2 probe for PALM imaging in the INS-1 PM. iRFP (642-nm laser excitation) in this new probe was used to identify cells expressing the probe but avoid activating PAmCherry1 before PALM acquisition. TIRF imaging of PI(4,5)P2 revealed uniform PI(4,5)P2 probe fluorescence over the major region of the flat PM (Fig. 2a) with modest fluorescence fluctuations. A similar result was observed in fixed whole-cell samples (data not shown). These results are consistent with the results reported for COS-7 cells (22). More importantly, these results are comparable with the relatively uniform distribution of EGFP-PHPLCδ1 in live cells (see below), suggesting that PI(4,5)P2 organization in PM sheets is well preserved under our experimental conditions.
FIGURE 2.

Homogeneous spatial distribution of PI(4,5)P2 probes in the major region of the INS-1 cell PM at nanometer scale. a and b, TIRF and corresponding PALM image of the PM sheet from an INS-1 cell expressing iRFP-PAmCherry1-PHPLCδ1. b1 and b2, enlarged views of the boxed regions in b. Arrows indicate sparsely distributed PM microdomains enriched with PI(4,5)P2 probes in the flat PM (b1) and at the cell periphery (b2). c, cropped image corresponding to the boxed area in b1. d, histogram of PI(4,5)P2 microdomain size in INS-1 membrane sheets (mean diameter, 383.3 ± 14.3 nm). e and f, PALM images of iRFP-PAmCherry1 (without the PH domain) expressed in a COS-7 cell under PALM imaging. Each dot in the images represents one hypothetical iRFP-PAmCherry1 molecule, indicating its monomeric presence and negligible photoblinking. g and h, simulated images with randomly distributed points with the same density as in c. i, pair correlation analysis of PI(4,5)P2 from large areas that contained no PI(4,5)P2 microdomains for iRFP-PAmCherry1-PHPLCδ1, iRFP-PAmcherry1 single-molecule control, simulation data, and syntaxin1A data. Syntaxin1A data were used as a positive control. iRFP-PAmcherry1 and simulation data were used as negative controls. Scale bars = 3 μm (b), 500 nm (b1, b2, e, and g), and 200 nm (c, f, and h).
Accordingly, at the nanometer scale, PALM imaging uncovered a homogeneous appearance of PI(4,5)P2 fluorescent biosensors in the majority of the flat PM regions (Fig. 2, b1 and c). Quantitative analysis further revealed a uniform distribution of PI(4,5)P2 biosensors in these PM regions (Fig. 2i). This contrasts the typical cluster distribution of syntaxin1A but is similar to the simulated images or images obtained with a probe without the PH domain as negative controls (Fig. 2, e–h). Moreover, we observed some slightly PI(4,5)P2 enriched membrane patches with diverse shapes and sizes (383.3 ± 14.3 nm in diameter, n = 74 regions from 12 cells) (Fig. 2d). These microdomains were located randomly in the flat PM, cell periphery, and filopodium structures (arrows, Fig. 2, b1 and b2). PI(4,5)P2 distribution did not change with the expression levels of biosensors (data not shown). The homogenous organization of PI(4,5)P2 probes at nanometer scale in the major areas of the PM was in agreement with the data observed under quick freezing electron microscopy (29, 69), despite their lower labeling efficiency. No dense nanoclusters of PI(4,5)P2 probes were observed in INS-1 cells as in PC-12 cells (30).
The excellent single-molecule photophysical properties of PAmCherry1 (40, 66) and the PALM method make it possible to estimate PI(4,5)P2 probe density in the PM by direct molecular counting. PI(4,5)P2 probe surface density in the flat region of INS-1 cells was 1694.6 ± 319.6/μm2 (n = 64 areas from 12 cells) after considering 50% photoactivation efficiency of PAmCherry1 and 15% of PAmCherry1 blinking twice (66). PI(4,5)P2 probe density in the PI(4,5)P2 enriched membrane patches was 5468.2 ± 768.3/μm2 (n = 74 patches form 12 cells), which is only ∼3-fold higher than that in uniform regions. To our knowledge, this is the first estimation of molecular density of PI(4,5)P2 probes in the PM using direct molecule counting. According to the 1:1 stoichiometry and 2 μm Kd of PHPLCδ1 to PI(4,5)P2 (19, 70), our measurements set a low-end estimate of PI(4,5)P2 density in the PM. Given the stochastic nature of photoactivation, these data suggest a rather limited PI(4,5)P2 concentration gradient in the PM of native cells.
As a negative control of the PI(4,5)P2 PALM probes, we expressed iRFP-PAmCherry1 (without the PHPLCδ1 domain) at a very low level in COS-7 cells using a deltaCMV promoter (3). As expected, these probes were isolated diffusely in the cytosol and showed a typical monomeric presence with little photoblinking or molecular clustering (Fig. 2, e and f). These results are comparable with the simulated images (Fig. 2, g and h) in which molecules were placed randomly with the same density. Coexpression of the iRFP-PAmCherry1-PHPLCδ1 probe with oculocerebrorenal syndrome of Lowe (OCRL) 5-phosphotase domain dramatically decreased probe fluorescence in the PM both under TIRF and PALM, but did not change the PI(4,5)P2 distribution pattern (data not shown). We conclude that the new PI(4,5)P2 PALM probe developed here preserves the original photophysical and biochemical properties of PAmCherry1 (40, 60, 66) and the specific PI(4,5)P2 binding property of PHPLCδ1 in a cellular environment.
Superresolution Images of PI(4,5)P2 in the Fixed PM Is Sensitive to Photoblinking and Sample Fixation Conditions
It is noteworthy that photoblinking, a general photophysical property of most fluorescent probes, may introduce a false molecule clustering appearance during point localization superresolution imaging (59, 71) because of repeatedly counting the same set of blinking molecules. This phenomenon was confirmed in our initial efforts to image PI(4,5)P2 with a specific PI(4,5)P2 antibody and an AF647-conjugated secondary antibody that shows significant photoblinking (14 and 26 cycles, on average, under cysteamine and β-mercaptoethanol conditions, respectively) (72)). Superresolution imaging with AF647 in the PM of INS-1 cells generated an abundant dense cluster appearance (Fig. 3, a–c), an artifact caused by overcounting of single molecule blinking of AF647, as demonstrated by single molecular imaging on blank coverslips (Fig. 3, d and e). Compared with AF647, mEos3.1 is a bright, monomeric FP (42) and has much fewer photoblinking cycles (2.4 times on average) in ∼50% of mEos3.1 molecules (66). mEos3.1-tagged PI(4,5)P2 probes expressed in INS-1 cells produced superresolution images with much fewer dense clusters and a much lower point density in individual clusters (Fig. 3, f–h). Direct single-molecule imaging of mEos3.1 alone at low expression levels in COS-7 cells demonstrated a low level of clusters, suggesting the presence of minor photoblinking for mEos3 (Fig. 3, i and j). PAmCherry1 exhibited almost negligible levels of photoblinking and excellent monomeric properties in a cellular environment (66). When this probe was used for quantitative PALM imaging, as expected, the cluster appearance of the superresolution images almost disappeared (Fig. 2, e and f). These data suggest that single molecular superresolution imaging is prone to generating false fluorescence clusters because of photoblinking.
FIGURE 3.

PI(4,5)P2 spatial organization in the fixed PM is sensitive to probe blinking and sample fixation conditions. a, TIRF and a corresponding superresolution image of the INS-1 cell membrane sheet immunostained with a monoclonal PI(4,5)P2 antibody and a secondary antibody labeled with Alexa Fluor 647. The samples were fixed with 4% PFA + 0.2% glutaraldehyde at 4 °C. b, cropped image corresponding to the box in a. c, enlarged image of the boxed region in b. Note the cluster structure in b and c caused by photoblinking. d and e, superresolution images of Alexa Fluor 647 molecules on a blank coverslip as a control of Alexa Fluor 647 single-molecule photobehavior. Note the fluorescence cluster artifact caused by Alexa Fluor 647 photoblinking in e. f–h, similar to a–c but using INS-1 cells expressed with mEos3.1-PHPLCδ1. i, superresolution image of mEos3.1 expressed at isolated single-molecule levels in a COS-7 cell using the deltaCMV promoter. Note the cluster structure of the mEos3 molecule caused by moderate photoblinking. k, TIRF image of an INS-1 membrane sheet fixed at room temperature and immunostained by a monoclonal PI(4,5)P2 antibody and AF647-conjugated secondary antibody. Note the abundant dense puncta visible under TIRF that were rarely observed in live cells. l, dSTORM superresolution image of PI(4,5)P2 in k. m, enlarged TIRF and dSTORM images in the boxed area in l. n–p, similar to k–m, but, with fixation at 37 °C, PI(4,5)P2 dense cluster artifacts were observed similarly. q–s, similar to k–m but fixed at 4 °C with 0.2% glutaraldehyde in addition to 4% PFA. The display contrast in q was different compared with other TIRF images to avoid saturation. Note the relative even fluorescence image and the enhanced fluorescence intensity under TIRF in q–s. Scale bars = 3 μm (a, f, i, o, and r), 500 nm (b, d, g, i, m, p, and s), and 200 nm (c, e, h, and j).
Because the PI(4,5)P2 distribution patterns reported in the literature are quite variable, from uniform distribution (20–22) to large patches (23, 24) to dense puncta (24–27, 30), depending on cell type and experimental conditions, we wondered whether these discrepancies resulted from different experimental conditions. After comparing the conditions used commonly in immunocytochemistry, we found that both fixation temperature and fixative composition significantly affected PI(4,5)P2 distribution in INS-1 cells. The difference was already clearly visible in conventional TIRFM (Fig. 3, k and n). When the PM was fixed with 4% PFA at room temperature or at 37 °C, we observed abundant small dense clusters in the PM (Fig. 3, k, n, and q). dSTORM revealed abundant clusters with a size of 53.2 ± 2.0 nm (FWHM, n = 1308 puncta from 9 cells) at room temperature and 61.6 ± 3.4 nm (n = 1317 puncta from 8 cells) at 37 °C (Fig. 3, m and p). When samples were processed with 4% PFA + 0.2% glutaraldehyde at 4 °C, we observed a relatively uniform and enhanced TIRF fluorescent image of PI(4,5)P2 in the PM sheets (Fig. 3, a and q), a picture that mimicked those observed in live cells. Further dSTORM imaging revealed a high fluorescent cluster density because of repetitive photoblinking of AF647 molecules (Fig. 3e). Therefore, common sample processing conditions in immunocytochemistry can strongly disrupt PI(4,5)P2 patterns and compromise biological interpretation of the data, consistent with another study (22). We reasoned that experimental conditions that better preserve PI(4,5)P2 distribution in live cells should be used for inositol lipid imaging studies. In addition, other factors, such as detergent treatment (29, 73), antibody-induced cross-linking (29), and insufficient lipid fixing (73, 74), may also introduce artifacts and distort phosphoinositol lipid imaging at the nanometer scale.
Homogeneous Spatial Organization of PI(4,5)P2 in the Large Areas of the PM in Live Cells
To extend our conclusion from fixed samples to live cells, we performed PI(4,5)P2 superresolution imaging in live INS-1 cells. We first visualized syntaxin1A-mEos3.2 using live-cell PALM imaging to validate the approach (as positive controls). We chose mEos3 instead of PAmCherry1 as the PALM probe in live cells because of its better photon efficiency, brightness, and monomeric properties (42) despite a low level of photoblinking. Consistent with previous studies in PC-12 cells (3), syntaxin1A showed typical cluster structures in live-cell TIRFM under physiological conditions (35 °C, Fig. 4a). We next visualized syntaxin1A structure and dynamics under live-cell PALM (100-Hz acquisition) with 10-s time resolution. As in fixed cells, syntaxin1A showed a typical cluster organization under live-cell PALM (Fig. 4, b–d, and supplemental Movie 1). The size of syntaxin1A clusters was in the upper range of that in fixed cells, reflecting dynamic movement of syntaxin1A in live cells during the 10 s of image acquisition. Although most syntaxin1A clusters changed their locations and shapes rapidly, some clusters remained static, disappeared, or emerged during imaging (Fig. 4, c–f).
FIGURE 4.

Live-cell PALM imaging revealed homogeneous spatial distribution and fast dynamics of PI(4,5)P2 in INS-1 cells. a and b, TIRF and superresolution images of mEos3.2-labeled syntaxin1A in an INS-1 cell at 35 °C. c–e, enlarged images of the white box regions in b at 0 and 30 s. d, the intensity profile of the syntaxin1A cluster as indicated by the white line in c. e and f, the pseudocolor image of c (green) merged on e (red, f). Arrows indicate the generation, disappearance, and movement of syntaxin1A clusters during 30 s of imaging. g, TIRF image of PI(4,5)P2 in a live INS-1 cell expressed with mEos3.1-PHPLCδ1. The image was acquired rapidly using the green channel before PALM imaging (35 °C). h and i, sequential live-cell PALM images visualized at 10-s intervals. The insets show the intensity profiles of the local PI(4,5)P2 density along the same straight line 1 position indicated at different times. Note the completely different intensity profile changes in 10 s. j, live PALM image of the same cell 30 s after 60 mm K+ stimulation. k, time course of the average intensity of PI(4,5)P2 in the large area (box 2 in h, 3 × 3 μm) and small circles (circle 3, 4, and 5, 500-nm diameter) in h during 5 min of PALM imaging with an interval of 10 s. Note the rapid intensity fluctuations of local PI(4,5)P2 probe changes (circle 3, 4, and 5) compared with very small changes in the relatively broad area (box 2). l, enlarged PALM images of the box 2 region in h at the indicated times. Arrows indicate PI(4,5)P2-enriched membrane patches under physiological conditions. Scale bars = 3 μm (b and j) and 500 nm (f and l).
Next we visualized PI(4,5)P2 spatial organization with PALM in live cells expressing mEos3.1-PHPLCδ1. In contrast to syntaxin1A clusters (Fig. 4a), TIRFM demonstrated a uniform fluorescence signal of mEos3.1-PHPLCδ1 in live cells (Fig. 4g). Because the PH domains are largely concentrated on the PM because of PI(4,5)P2 binding and TIRF illumination only activates <100 nm of space above the PM, the dominant signal was from the PI(4,5)P2 in the inner leaflet of the PM. This was supported further by similar results obtained in the PM sheets, which contained no cytosolic and unbound fluorescence probes (data not shown). PALM imaging showed homogeneous fluorescence with a marginal fluorescence difference in a large area of the PM, and we were unable to detect any prominent dense PI(4,5)P2 clusters (Fig. 4, g–j). Consistent with fixed cells, we observed slight PI(4,5)P2 enrichment in microdomains with diverse sizes (∼200–500 nm) in the flat PM regions and filopodium-like membrane protrusions. Continuous PALM imaging revealed dynamic changes of PI(4,5)P2 probes (Fig. 4, h and i, insets). Fig. 4k shows significant fluorescence changes over time in local (500-nm diameter circles) but not broad regions of the PM at rest, and these changes were faster (Fig. 4, h and i) than syntaxin1A. Upon 60 mm K+ stimulation, PI(4,5)P2 levels were reduced significantly globally (data not shown), consistent with TIRF imaging (Fig. 6). However, the overall spatial pattern of PI(4,5)P2 at nanoscale remained similar to that in fixed PM sheets (Fig. 4l).
The PI(4,5)P2 probe-enriched regions in live-cell PALM images may reflect active local lipid metabolism resulting from diverse signaling processes in live cells. The rapid fluoresce changes indicate a fast redistribution of PI(4,5)P2 probes because of local PI(4,5)P2 dynamics. For example, PI(4,5)P2 was more dynamic within thin membrane protrusions at the cell periphery (supplemental Movie 2). The tightly compartmentalized volume in these small structures limits the rapid diffusion of locally generated PI(4,5)P2 and promotes a higher local PI(4,5)P2 probe gradient. This is consistent with the high rate of actin turnover, which requires active PI(4,5)P2 interactions (75). The interpretation of live-cell PALM imaging requires extra caution because it is more complex than fixed cells due to multiple new factors, such as fast local PI(4,5)P2 dynamics, probe binding/unbinding, and diffusion in live cells. Our control experiments confirmed that the dominant signal in live-cell PALM is from the PI(4,5)P2-bound probes in the PM (data not shown). The consistent nanoscale pattern between live-cell PALM and the fixed PM sheet suggests a homogeneous distribution of PI(4,5)P2 with shallow PI(4,5)P2 gradients. If the highly concentrated PI(4,5)P2 nanoclusters were present in live cells, then one would expect to see brighter fluorescence spots with much steeper fluorescence gradients, as we observed for syntaxin1A. However, that did not occur in INS-1 cells.
PI4P and PI(3,4,5)P3 Nanoscale Organization in the Fixed Cell Membrane
PI4P, PI(4,5)P2, and PI(3,4,5)P3 are tightly interconnected inositol lipids in live cells (Fig. 5a). PI4P serves as one of the identity molecules of the PM (12, 13) in addition to its well known role as a metabolic precursor of PI(4,5)P2. PI(4,5)P2 can be further converted into PI(3,4,5)P3 to regulate actin dynamics (14), exocytosis (76), and AMPA receptor clustering (77).
FIGURE 5.

PI4P and PI(3,4,5)P3 show a relatively uniform distribution in the INS-1 PM. a, illustration of phosphoinositide metabolic pathways on the PM. b and c, TIRF and PALM images of PI4P on the PM sheet of an INS-1 cell expressed with iRFP-PAmCherry1-PHosh2. d1 and d2, enlarged images of the boxed regions in c. Note the relatively homogenous distribution of probes in the large area of the PM (d1). Arrows indicate a few PI4P-enriched patches (d2). e and f, TIRF and PALM image of PI(3,4,5)P3 labeled by the expressed iRFP-PAmCherry1-PHGRP1. g1 and g2, enlarged images of the boxed areas in f. Note that the surface abundance of the probes was significantly lower (f–g2). The enlarged views show sparsely distributed PI(3,4,5)P3 probes (g1) and, occasionally, PI(3,4,5)P3-enriched zones (arrows, g2) in the INS-1 PM. Scale bars = 3 μm (c and f) and 500 nm (d and g).
To examine the nanoscale distribution of these inositol lipids in the PM, we generated photo-activatible probes similarly by tagging iRFP-PAmCherry1 to the PHosh2 domain (12), which specifically binds PI4P, and to the PHGRP1 domain (78), which specifically binds to PI(3,4,5)P3. TIRF imaging of EGFP-PHosh2 showed a similar uniform spatial distribution in the PM (Fig. 5b) as EGFP-PHPLCδ1, but it had a lower intensity than EGFP-PHPLCδ1. This was further confirmed by EGFP-PHOSBP, another fluorescent probe that binds PI4P (12, 79). In addition, the prominent intracellular organelle distribution (in the nucleus and endoplasmic reticulum) was also observed in INS-1 cells using EGFP-PHOSBP, consistent with previous studies (12, 79). It has been shown that PHosh2 also binds PI(4,5)P2 in vitro (80), but a subsequent study has demonstrated a fairly specific binding of PI4P in the cells (11). This is also consistent with the different response of PI4P compared with PI(4,5)P2 in live cells (Fig. 6). Under PALM imaging, iRFP-PAmCherry1-PHOSH2 had an overall similar pattern as PI(4,5)P2 in the PM (Fig. 5c). PI4P was relatively homogeneous in the major PM area (Fig. 5, d1), accompanied by some PI4P-enriched membrane microdomains (Fig. 5, d2). Quantitative PALM imaging revealed an average density of 743.4 ± 114.89/μm2 (n = 54 areas from 13 cells) for PI4P probes in the PM of INS-1 cells.
In contrast, the fluorescence signal for PI(3,4,5)P3 detected by iRFP-PAmCherry1-PHGRP1 (78) was much weaker than that of both PI4P and PI(4,5)P2 in the PM under TIRF imaging. It exhibited a punctate appearance that looked like weak PI(3,4,5)P3 clusters because of the very low abundance and light diffraction of fluorescence probes (Fig. 5e). PALM imaging showed homogeneous, sparse molecules in the large regions of the PM (Fig. 5f). Most of these puncta, under TIRFM, contained only a few sparsely distributed PI(3,4,5)P3 probes slightly closer than their surrounding molecules (Fig. 5, g1). Accordingly, image simulation with randomly distributed molecules at the same density (data not shown) can generate a punctate appearance that is very similar to the data in PI(3,4,5)P3 images. This suggests a sparse but uniform nanoscale distribution of PI(3,4,5)P3 in the majority of PM regions. Quantitative PALM analysis in these regions gave a PI(3,4,5)P3 probe density of 406.2 ± 236.8/μm2 (n = 64 areas from 10 cells), which was much lower than that of PI(4,5)P2 and PI4P. In addition, we indeed observed a few fluorescent spots under TIRFM, and they turned out to be loose membrane microdomains relatively enriched in PI(3,4,5)P3 probes in PALM images (∼200–500 nm in diameter; Fig. 5, g2), presumably reflecting ongoing PI(3,4,5)P3-mediated signaling (14, 15). For both PI4P and PI(3,4,5)P3, the probe expression levels had little effect on their uniform nanoscale distribution patterns (data not shown), although the PI(3,4,5)P3 fluorescence signal appeared to be more heterogeneous at low expression levels under TIRF because of fluorescence light diffraction.
Physiological Dynamics of PI4P, PI(4,5)P2, and PI(3,4,5)P3 in Live Cells
We next visualized the molecular dynamics of PI4P, PI(4,5)P2, and PI(3,4,5)P3 in INS-1 cells in response to high concentrations of glucose, a physiological stimulation to trigger insulin secretion, and direct membrane depolarization with high-K+ solution. PI(4,5)P2 in live cells mainly localized on the PM, as evidenced by a “ring” shape of EGFP-PHPLCδ1 on the PM, with only faint and diffuse fluorescence in the cytosol under confocal microscopy (Fig. 6a). 28 mm glucose evoked a very small (only a few percent) but significant increase of EGFP-PHPLCδ1 intensity under TIRFM, and this response was opposite to the changes induced by 60 mm KCl depolarization (Fig. 6, b and c), in agreement with the previous study (81).
The PI4P probe EGFP-PHOSBP was distributed on the PM and some intracellular organelles (i.e. Golgi and endoplasmic reticulum) in INS-1 cells (Fig. 6d), consistent with previous studies (12, 79). We used very narrow TIRF illumination to avoid minor signal contamination from intracellular fluorescence. Glucose induced an oscillatory increase in PHOSBP-EGFP intensity (Fig. 6e), presumably mediated by PI4K activation. 60 mm KCl-evoked EGFP-PHOSBP changes varied greatly from cell to cell. The majority of cells (24 of 31 cells) exhibited fluorescence intensity increases, but some cells showed decreases (Fig. 6, f and g). This indicates the complex interactions and balancing of diverse signaling and metabolic pathways, and it is consistent with the independent cellular function of PI4P in addition to its role as a PI(4,5)P2 metabolic precursor (13, 81).
In contrast to PI4P and PI(4,5)P2, the PI(3,4,5)P3 probe mCherry-PHGRP1 (82) displayed abundant cytosolic fluorescence and a prominent nuclear signal, indicating an extremely low number of PI(3,4,5)P3 in the PM of live INS-1 cells. TIRF imaging of mCherry-PHGRP1 on the PM may be significantly compromised by cytosolic fluorescence signals, even with TIRF illumination. However, the relative structural changes should still represent the PM mCherry-PHGRP1 alteration because of the PM localization of signaling receptors that generate PI(3,4,5)P3. We also further narrowed the illumination angle to minimize the penetration depth of the evanescent wave during imaging. Under both 28 mm glucose and 60 mm KCl, TIRFM revealed a significant increase in PI(3,4,5)P3 in the PM (Fig. 6, h–j), suggesting PI3K activation through insulin autoreceptors (83) following insulin secretion induced by glucose or KCl. Importantly, we observed no PI(3,4,5)P3 steep gradients in either TIRF or PALM imaging in live INS-1 cells. This was also true under direct insulin (100 nm) stimulation to achieve stronger PI(3,4,5)P3 changes (data not shown).
We conclude that fluorescent probes on the basis of specific PH domains used in this study faithfully reflected the subcellular location and molecular behaviors of specific inositol lipids in vivo. Moreover, in live cells, PI4P, PI(4,5)P2 and PI(3,4,5)P3 are dynamically interconnected under physiological conditions and equilibrate rapidly through multiple metabolic pathways and/or signaling networks in response to diverse physiological stimulation.
Discussion
Phosphoinositol lipids are key signaling components in the PM, but their spatial organization at the nanometer scale remains poorly understood. We employed a novel single-molecule superresolution approach and dual-color biosensors to examine this issue. Surprisingly, PALM imaging revealed a relatively homogeneous spatial distribution pattern of PI(4,5)P2 probes in the majority of PMs in both fixed and live INS-1 cells, and sparse PI(4,5)P2-enriched membrane patches/domains were also observed. PI4P and PI(3,4,5)P3 displayed a very similar organization pattern as PI(4,5)P2 in INS-1 cells, but PI(3,4,5)P3 is much less abundant than the other two lipids. These data reveal the detailed nanoscale landscape of key phosphoinositol lipids in native cell PM, providing a framework for rethinking local cellular signaling and lipid-protein interactions at a nanometer scale.
Nanoscale Organization of PI(4,5)P2 in the PM
The PM sheets used in this study bypass detergent treatment, which can cause clustering of PM molecules and non-preexisting domains (65). It also avoids the contamination of the cytosolic signal when the lipid abundance in the PM is low (i.e. PI(3,4,5)P3). These PM sheets preserve the intact fine structures of the PM and the associated proteins, such as cortical actin meshwork, vesicle tethers, and clathrin-coated pits (53, 64). A recent study has demonstrated abundant, dense PI(4,5)P2 clusters (∼73 nm in average) using STED imaging of PHPLCδ1-citrine and PI(4,5)P2 antibody in the PM sheets of PC12 cells (30). We wondered whether the dense clusters are general structures of PI(4,5)P2 in live cells, including the INS-1 cells in which we are interested. We first validated and characterized our superresolution imaging system using syntaxin1A, and we observed densely packed syntaxin1A clusters in the INS-1 PM, as reported in previous studies (4, 5). Unexpectedly, our data using PH domain PALM probes revealed an even nanoscale organization of PI(4,5)P2 in the major region of the INS-1 cell PM, in addition to some sparse and large PI(4,5)P2-enriched membrane patches or domains (383 ± 14 nm in diameter) with shallow concentration gradients. No dense PI(4,5)P2 nanoclusters or steep concentration gradients were present in the INS-1 cell PM.
This result is consistent with a recent freezing-fracture EM study using a GST-PHPLCδ1 probe in the open fibroblast membrane (69), where randomly distributed PI(4,5)P2 is observed in large areas of the PM. The diverse sizes of PI(4,5)P2-rich membrane patches/domains in INS-1 cells are comparable with the relatively higher-concentration PI(4,5)P2 regions (with a diameter of 60∼400 nm) in that study that contained caveolae and clathrin-coated pits, indicating functionally specialized membrane regions for PI(4,5)P2, such as clathrin-coated pits, exocytosis, membrane protrusions, or adhesion sites. Likewise, another rapid freezing EM study, using ultrathin sections of HEK293 cells expressing tandem PHPLCδ1 domains (GFP-PH-PH) and anti-GFP antibody, has also demonstrated an even distribution of PI(4,5)P2 in the PM (29).
Functional Implications of the Nanoscale Distribution of PI(4,5)P2 in Live Cells
As the major PM identity lipids (12, 13), both PI4P and PI(4,5)P2 concentrations should be relatively constant under physiological conditions. In supporting this idea, 28 mm glucose stimulation evoked only small (less than a few percent) concentration changes of PI(4,5)P2 in the PM (Fig. 6), although this glucose concentration is sufficiently high to trigger massive insulin secretion in vivo. With continuous membrane depolarization induced by 60 mm KCl, the changes were still very limited (<10% for PI(4,5)P2). Even after severe interruptions of major PI(4,5)P2 metabolic enzymes such as synaptojanin (84) and PIP kinase type 1γ (PIPK1γ) (85), PI(4,5)P2 changes in neurons were still less than 50% of control levels. The global stability of PI(4,5)P2 suggests that a sufficient number of free PI(4,5)P2, probably also PI4P, in the PM is readily available to rapidly initiate relevant signaling in live cells. It is likely that PI(4,5)P2 functions as a critical cofactor (or permission factor) with other proteins to regulate multiple cellular processes. The local availability of PI(4,5)P2 effector proteins, rather than PI(4,5)P2 itself, can offer both specificity and efficiency of signaling in space and time. This is consistent with the idea of coincidence detection for PI(4,5)P2 (16, 86). Therefore, densely packed nanodomains of PI(4,5)P2 appear to be unnecessary, although their transient presence and function in facilitating or maintaining the efficiency of some local signaling cannot be excluded. Furthermore, the PI(4,5)P2 diffusion coefficient in the PM inner leaflet was fast in live cells. It was estimated to be ∼1 μm2/s by fluorescence correlation spectroscopy using BODIPY-TMR-PI(4,5)P2 in J774A-1 macrophages (87) and ∼0.3–1 μm2/s by fluorescence recovery after photobleaching (FRAP) techniques (88, 89). This rapid diffusion poses another challenge to producing and maintaining the densely packed functional inositol lipid clusters in live cells. The facts of its high abundance and fast dynamics in the PM indicate a limited need for cells to maintain highly localized, steep PI(4,5)P2 gradients.
The shallow PI(4,5)P2 gradient regions observed under PALM may reflect certain functional zones where a relatively higher concentration of PI(4,5)P2 is preferred, such as clathrin-coated pits (69) and active actin-remodeling areas (supplemental Movie 2). Quantitative analysis demonstrated that the difference in PI(4,5)P2 probe density between the uniform regions and the PI(4,5)P2-enriched zones is only ∼3-fold, suggesting that shallow local concentration gradients may be sufficient to fine-tune diverse local signaling. Much less effort is required for cells to generate and remove such marginal concentration gradients in the PM than the dense nanodomains, and cells most likely use this more economical and flexible way to help effector proteins for local signaling. A few different mechanisms may be sufficient to locally enrich PI(4,5)P2 in live cells, including local recruitment of relevant kinases and phosphatases (12), restricted lateral diffusion by septin- (90), and actin-based (53) membrane “fence,” and local reversible sequestration by concentrated PI(4,5)P2 binding proteins.
Methodological Considerations for Studying Phosphoinositol Lipids in the PM
The single-molecule superresolution approach used in this study opens a new avenue to study phosphoinositol lipids and other macromolecular complexes in the PM. It has the following advantages. First, it provides nanoscale images (up to ∼35-nm resolution) using light microscopy and, therefore, avoids common caveats relevant to conventional EM, such as harsh sample processing and low labeling efficiency or specificity. Second, it can be applied directly to live cells (Fig. 4), and both fine structures and the dynamics of molecule complexes can be imaged under physiological conditions. Third, PALM imaging, combined with proper fluorescent probes and analysis, offers a new way to quantitatively determine molecule concentration/density at nanometer scale by direct molecule counting. Using PAmCherry1, whose single molecular photophysics have been well characterized in detail (40, 60, 66), we estimated that PI4P, PI(4,5)P2, and PI(3,4,5)P3 probe densities in the PM are ∼743, ∼1695, and ∼406/μm2, respectively. Live-cell TIRF imaging with different PH domains (Fig. 6) supports that these PH domain-based probes faithfully reflect the dynamic behaviors of inositol lipids in live cells. However, although they have been widely applied in inositol lipid studies in different cells, we cannot exclude the possibility that PH domains may not be able to efficiently access all phosphoinositides (i.e. those phosphoinositides bound by some sequestering proteins) in the PM because of space constraints.
Our experiments in INS-1 cells demonstrate that the fixation at low temperature (4 °C) with 4% PFA + 0.2% glutaraldehyde preserves PI(4,5)P2 spatial distribution in the PM sheet as in live cells, but the other conditions tested cause artificially dense PI(4,5)P2 clusters/puncta to different extents (Fig. 3). The variable experimental conditions may, at least in part, explain the diverse results in the literature regarding PI(4,5)P2 cellular distribution patterns.
It is noteworthy that the single-molecule superresolution approach may be compromised by various factors, such as label density, photo activation efficiency, and photoblinking of probes (44, 59, 60). Incomplete photoactivation causes an underestimate of molecule density, and overcounting leads to false molecule clusters (59, 71) (Fig. 3). Our data demonstrate that the extent of cluster appearance of individual molecules is correlated positively with the degree of photoblinking of fluorophores (AF647 ≫ mEos3.1 > PAmCherry1) (Fig. 3), which is problematic for molecule counting. To overcome this problem, we chose PAmCherry1 for quantitative PALM in fixed cells because of its almost negligible photoblinking and well defined single-molecule photophysical properties (40, 60, 66). Live-cell PALM is technically more challenging because of dynamic changes of live cells and a few other factors. Further development of the quantitative superresolution approach in live cell is obviously required.
In summary, our results unequivocally demonstrate the largely uniform nanoscale landscape of PI4P, PI(4,5)P2, and PI(3,4,5)P3 in the INS-1 cell PM, with limited spatial heterogeneity in some PM regions. The quantitative data revealed much shallower concentration gradients than thought previously for these PIs in the PM. This study suggests a different model of spatial organization of phosphoinositol lipids in the PM at the nanometer scale and implies an important framework to better understand the cellular signaling events of phosphoinositol lipids in a live cell membrane.
Author Contributions
C. J. and X. L. designed the experiments, analyzed the data, and wrote the manuscript. C. J. performed the experiments. Y. Z. and T. X. developed the software for superresolution imaging reconstruction. P. X. and T. X. contributed reagents and discussions.
Acknowledgments
We thank Dr. Christopher B. Newgard (Duke University) for sharing INS-1 cells; Pietro De Camilli (Yale) for DNA plasmids of PH domains; Mengyu Wang, Fan Fan, and Satyajit Mahapatra for technical support and discussions; and Meyer Jackson, Ed Chapman, and Tim Gomez for reading the manuscript and suggestions.
This work was supported by National Institutes of Health Grants R01DK093953, P30NS069271, and BRFSG-2014-07 (to X. L.) and American Heart Association Predoctoral Fellowship 14PRE20380168 (to C. J.). The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Movies 1 and 2.
- PM
- plasma membrane
- PI
- phosphoinositide
- PLC
- phospholipase C
- PH
- pleckstrin homology
- FP
- fluorescent protein
- PALM
- photoactivated localization microscopy
- dSTORM
- direct stochastic optical reconstruction microscopy
- PFA
- paraformaldehyde
- NA
- numerical aperture
- FWHM
- full width at half-maximum
- AF647
- Alexa Fluor 647
- PI4P
- phosphatidylinositol 4-phosphate
- PI(4,5)P2
- phosphatidylinositol 4,5-bisphosphate
- PI(3,4,5)P3
- phosphatidylinositol (3,4,5)-trisphosphate.
References
- 1.Engelman D. M. (2005) Membranes are more mosaic than fluid. Nature 438, 578–580 [DOI] [PubMed] [Google Scholar]
- 2.Spira F., Mueller N. S., Beck G., von Olshausen P., Beig J., and Wedlich-Söldner R. (2012) Patchwork organization of the yeast plasma membrane into numerous coexisting domains. Nat. Cell Biol. 14, 640–648 [DOI] [PubMed] [Google Scholar]
- 3.Barg S., Knowles M. K., Chen X., Midorikawa M., and Almers W. (2010) Syntaxin clusters assemble reversibly at sites of secretory granules in live cells. Proc. Natl. Acad. Sci. U.S.A. 107, 20804–20809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Knowles M. K., Barg S., Wan L., Midorikawa M., Chen X., and Almers W. (2010) Single secretory granules of live cells recruit syntaxin-1 and synaptosomal associated protein 25 (SNAP-25) in large copy numbers. Proc. Natl. Acad. Sci. U.S.A. 107, 20810–20815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sieber J. J., Willig K. I., Kutzner C., Gerding-Reimers C., Harke B., Donnert G., Rammner B., Eggeling C., Hell S. W., Grubmüller H., and Lang T. (2007) Anatomy and dynamics of a supramolecular membrane protein cluster. Science 317, 1072–1076 [DOI] [PubMed] [Google Scholar]
- 6.Lingwood D., and Simons K. (2010) Lipid rafts as a membrane-organizing principle. Science 327, 46–50 [DOI] [PubMed] [Google Scholar]
- 7.Di Paolo G., and De Camilli P. (2006) Phosphoinositides in cell regulation and membrane dynamics. Nature 443, 651–657 [DOI] [PubMed] [Google Scholar]
- 8.Vicinanza M., D'Angelo G., Di Campli A., and De Matteis M. A. (2008) Function and dysfunction of the PI system in membrane trafficking. EMBO J. 27, 2457–2470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Balla T. (2013) Phosphoinositides: tiny lipids with giant impact on cell regulation. Physiol. Rev. 93, 1019–1137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Berridge M. J., and Irvine R. F. (1984) Inositol trisphosphate, a novel second messenger in cellular signal transduction. Nature 312, 315–321 [DOI] [PubMed] [Google Scholar]
- 11.Balla A., Kim Y. J., Varnai P., Szentpetery Z., Knight Z., Shokat K. M., and Balla T. (2008) Maintenance of hormone-sensitive phosphoinositide pools in the plasma membrane requires phosphatidylinositol 4-kinase IIIα. Mol. Biol. Cell 19, 711–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nakatsu F., Baskin J. M., Chung J., Tanner L. B., Shui G., Lee S. Y., Pirruccello M., Hao M., Ingolia N. T., Wenk M. R., and De Camilli P. (2012) PtdIns4P synthesis by PI4KIIIα at the plasma membrane and its impact on plasma membrane identity. J. Cell Biol. 199, 1003–1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hammond G. R., Fischer M. J., Anderson K. E., Holdich J., Koteci A., Balla T., and Irvine R. F. (2012) PI4P and PI (4, 5) P2 are essential but independent lipid determinants of membrane identity. Science 337, 727–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Czech M. P. (2000) PIP2 and PIP3: complex roles at the cell surface. Cell 100, 603–606 [DOI] [PubMed] [Google Scholar]
- 15.Czech M. P. (2003) Dynamics of phosphoinositides in membrane retrieval and insertion. Annu. Rev. Physiol. 65, 791–815 [DOI] [PubMed] [Google Scholar]
- 16.Hammond G. R., and Balla T. (2015) Polyphosphoinositide binding domains: key to inositol lipid biology. Biochim. Biophys. Acta 1851, 746–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murray D., Arbuzova A., Hangyás-Mihályné G., Gambhir A., Ben-Tal N., Honig B., and McLaughlin S. (1999) Electrostatic properties of membranes containing acidic lipids and adsorbed basic peptides: theory and experiment. Biophys. J. 77, 3176–3188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McLaughlin S., and Murray D. (2005) Plasma membrane phosphoinositide organization by protein electrostatics. Nature 438, 605–611 [DOI] [PubMed] [Google Scholar]
- 19.Garcia P., Gupta R., Shah S., Morris A. J., Rudge S. A., Scarlata S., Petrova V., McLaughlin S., and Rebecchi M. J. (1995) The pleckstrin homology domain of phospholipase C-δ 1 binds with high affinity to phosphatidylinositol 4,5-bisphosphate in bilayer membranes. Biochemistry 34, 16228–16234 [DOI] [PubMed] [Google Scholar]
- 20.Milosevic I., Sørensen J. B., Lang T., Krauss M., Nagy G., Haucke V., Jahn R., and Neher E. (2005) Plasmalemmal phosphatidylinositol-4,5-bisphosphate level regulates the releasable vesicle pool size in chromaffin cells. J. Neurosci. 25, 2557–2565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Rheenen J., and Jalink K. (2002) Agonist-induced PIP(2) hydrolysis inhibits cortical actin dynamics: regulation at a global but not at a micrometer scale. Mol. Biol. Cell 13, 3257–3267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hammond G. R., Schiavo G., and Irvine R. F. (2009) Immunocytochemical techniques reveal multiple, distinct cellular pools of PtdIns4P and PtdIns (4, 5) P2. Biochem. J. 422, 23–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang S., Lifshitz L., Patki-Kamath V., Tuft R., Fogarty K., and Czech M. P. (2004) Phosphatidylinositol-4,5-bisphosphate-rich plasma membrane patches organize active zones of endocytosis and ruffling in cultured adipocytes. Mol. Cell. Biol. 24, 9102–9123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.James D. J., Khodthong C., Kowalchyk J. A., and Martin T. F. (2008) Phosphatidylinositol 4,5-bisphosphate regulates SNARE-dependent membrane fusion. J. Cell Biol. 182, 355–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Laux T., Fukami K., Thelen M., Golub T., Frey D., and Caroni P. (2000) GAP43, MARCKS, and CAP23 modulate PI(4,5)P(2) at plasmalemmal rafts, and regulate cell cortex actin dynamics through a common mechanism. J. Cell Biol. 149, 1455–1472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aoyagi K., Sugaya T., Umeda M., Yamamoto S., Terakawa S., and Takahashi M. (2005) The activation of exocytotic sites by the formation of phosphatidylinositol 4,5-bisphosphate microdomains at syntaxin clusters. J. Biol. Chem. 280, 17346–17352 [DOI] [PubMed] [Google Scholar]
- 27.Kabachinski G., Yamaga M., Kielar-Grevstad D. M., Bruinsma S., and Martin T. F. (2014) CAPS and Munc13 utilize distinct PIP2-linked mechanisms to promote vesicle exocytosis. Mol. Biol. Cell 25, 508–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang J., and Richards D. A. (2012) Segregation of PIP2 and PIP3 into distinct nanoscale regions within the plasma membrane. Biol. Open 1, 857–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van Rheenen J., Achame E. M., Janssen H., Calafat J., and Jalink K. (2005) PIP2 signaling in lipid domains: a critical re-evaluation. EMBO J. 24, 1664–1673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van den Bogaart G., Meyenberg K., Risselada H. J., Amin H., Willig K. I., Hubrich B. E., Dier M., Hell S. W., Grubmüller H., Diederichsen U., and Jahn R. (2011) Membrane protein sequestering by ionic protein-lipid interactions. Nature 479, 552–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hell S. W., and Wichmann J. (1994) Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy. Opt. Lett. 19, 780–782 [DOI] [PubMed] [Google Scholar]
- 32.Sato K., Ernstrom G. G., Watanabe S., Weimer R. M., Chen C. H., Sato M., Siddiqui A., Jorgensen E. M., and Grant B. D. (2009) Differential requirements for clathrin in receptor-mediated endocytosis and maintenance of synaptic vesicle pools. Proc. Natl. Acad. Sci. U.S.A. 106, 1139–1144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Betzig E., Patterson G. H., Sougrat R., Lindwasser O. W., Olenych S., Bonifacino J. S., Davidson M. W., Lippincott-Schwartz J., and Hess H. F. (2006) Imaging intracellular fluorescent proteins at nanometer resolution. Science 313, 1642–1645 [DOI] [PubMed] [Google Scholar]
- 34.Hess S. T., Girirajan T. P., and Mason M. D. (2006) Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys. J. 91, 4258–4272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rust M. J., Bates M., and Zhuang X. (2006) Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 3, 793–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu K., Zhong G., and Zhuang X. (2013) Actin, spectrin, and associated proteins form a periodic cytoskeletal structure in axons. Science 339, 452–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fölling J., Bossi M., Bock H., Medda R., Wurm C. A., Hein B., Jakobs S., Eggeling C., and Hell S. W. (2008) Fluorescence nanoscopy by ground-state depletion and single-molecule return. Nat. Methods 5, 943–945 [DOI] [PubMed] [Google Scholar]
- 38.Wombacher R., Heidbreder M., van de Linde S., Sheetz M. P., Heilemann M., Cornish V. W., and Sauer M. (2010) Live-cell super-resolution imaging with trimethoprim conjugates. Nat. Methods 7, 717–719 [DOI] [PubMed] [Google Scholar]
- 39.Szymborska A., de Marco A., Daigle N., Cordes V. C., Briggs J. A., and Ellenberg J. (2013) Nuclear pore scaffold structure analyzed by super-resolution microscopy and particle averaging. Science 341, 655–658 [DOI] [PubMed] [Google Scholar]
- 40.Subach F. V., Patterson G. H., Manley S., Gillette J. M., Lippincott-Schwartz J., and Verkhusha V. V. (2009) Photoactivatable mCherry for high-resolution two-color fluorescence microscopy. Nat. Methods 6, 153–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pertsinidis A., Mukherjee K., Sharma M., Pang Z. P., Park S. R., Zhang Y., Brunger A. T., Südhof T. C., and Chu S. (2013) Ultrahigh-resolution imaging reveals formation of neuronal SNARE/Munc18 complexes in situ. Proc. Natl. Acad. Sci. U.S.A. 110, E2812–2820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang M., Chang H., Zhang Y., Yu J., Wu L., Ji W., Chen J., Liu B., Lu J., Liu Y., Zhang J., Xu P., and Xu T. (2012) Rational design of true monomeric and bright photoactivatable fluorescent proteins. Nat. Methods 9, 727–729 [DOI] [PubMed] [Google Scholar]
- 43.Huang F., Hartwich T. M., Rivera-Molina F. E., Lin Y., Duim W. C., Long J. J., Uchil P. D., Myers J. R., Baird M. A., Mothes W., Davidson M. W., Toomre D., and Bewersdorf J. (2013) Video-rate nanoscopy using sCMOS camera-specific single-molecule localization algorithms. Nat. Methods 10, 653–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sengupta P., Van Engelenburg S., and Lippincott-Schwartz J. (2012) Visualizing cell structure and function with point-localization superresolution imaging. Dev. Cell 23, 1092–1102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Olsen H. L., Hoy M., Zhang W., Bertorello A. M., Bokvist K., Capito K., Efanov A. M., Meister B., Thams P., Yang S. N., Rorsman P., Berggren P. O., and Gromada J. (2003) Phosphatidylinositol 4-kinase serves as a metabolic sensor and regulates priming of secretory granules in pancreatic β cells. Proc. Natl. Acad. Sci. U.S.A. 100, 5187–5192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang P., Yeku O., Zong H., Tsang P., Su W., Yu X., Teng S., Osisami M., Kanaho Y., Pessin J. E., and Frohman M. A. (2011) Phosphatidylinositol-4-phosphate-5-kinase α deficiency alters dynamics of glucose-stimulated insulin release to improve glucohomeostasis and decrease obesity in mice. Diabetes 60, 454–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kolic J., Spigelman A. F., Smith A. M., Manning Fox J. E., and MacDonald P. E. (2014) Insulin secretion induced by glucose-dependent insulinotropic polypeptide requires phosphatidylinositol 3-kinase γ in rodent and human β-cells. J. Biol. Chem. 289, 32109–32120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cheng K. K., Lam K. S., Wu D., Wang Y., Sweeney G., Hoo R. L., Zhang J., and Xu A. (2012) APPL1 potentiates insulin secretion in pancreatic β cells by enhancing protein kinase Akt-dependent expression of SNARE proteins in mice. Proc. Natl. Acad. Sci. U.S.A. 109, 8919–8924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sleeman M. W., Wortley K. E., Lai K. M., Gowen L. C., Kintner J., Kline W. O., Garcia K., Stitt T. N., Yancopoulos G. D., Wiegand S. J., and Glass D. J. (2005) Absence of the lipid phosphatase SHIP2 confers resistance to dietary obesity. Nat. Med. 11, 199–205 [DOI] [PubMed] [Google Scholar]
- 50.Butler M., McKay R. A., Popoff I. J., Gaarde W. A., Witchell D., Murray S. F., Dean N. M., Bhanot S., and Monia B. P. (2002) Specific inhibition of PTEN expression reverses hyperglycemia in diabetic mice. Diabetes 51, 1028–1034 [DOI] [PubMed] [Google Scholar]
- 51.Hohmeier H. E., Mulder H., Chen G., Henkel-Rieger R., Prentki M., and Newgard C. B. (2000) Isolation of INS-1-derived cell lines with robust ATP-sensitive K+ channel-dependent and -independent glucose-stimulated insulin secretion. Diabetes 49, 424–430 [DOI] [PubMed] [Google Scholar]
- 52.Sanan D. A., and Anderson R. (1991) Simultaneous visualization of LDL receptor distribution and clathrin lattices on membranes torn from the upper surface of cultured cells. J. Histochem. Cytochem. 39, 1017–1024 [DOI] [PubMed] [Google Scholar]
- 53.Morone N., Fujiwara T., Murase K., Kasai R. S., Ike H., Yuasa S., Usukura J., and Kusumi A. (2006) Three-dimensional reconstruction of the membrane skeleton at the plasma membrane interface by electron tomography. J. Cell Biol. 174, 851–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huang B., Jones S. A., Brandenburg B., and Zhuang X. (2008) Whole-cell 3D STORM reveals interactions between cellular structures with nanometer-scale resolution. Nat. Methods 5, 1047–1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chang H., Zhang M., Ji W., Chen J., Zhang Y., Liu B., Lu J., Zhang J., Xu P., and Xu T. (2012) A unique series of reversibly switchable fluorescent proteins with beneficial properties for various applications. Proc. Natl. Acad. Sci. U.S.A. 109, 4455–4460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Olivo-Marin J.-C. (2002) Extraction of spots in biological images using multiscale products. Pattern Recognit. 35, 1989–1996 [Google Scholar]
- 57.Zhang Y.-D., Chen L.-Y., and Xu T. (2010) Application of improved wavelet transform in biological particle detection. Prog. Biochem. Biophys. 37, 1144–1150 [Google Scholar]
- 58.Smith C. S., Joseph N., Rieger B., and Lidke K. A. (2010) Fast, single-molecule localization that achieves theoretically minimum uncertainty. Nat. Methods 7, 373–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Annibale P., Vanni S., Scarselli M., Rothlisberger U., and Radenovic A. (2011) Quantitative photo activated localization microscopy: unraveling the effects of photoblinking. PloS ONE 6, e22678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nan X., Collisson E. A., Lewis S., Huang J., Tamgüney T. M., Liphardt J. T., McCormick F., Gray J. W., and Chu S. (2013) Single-molecule superresolution imaging allows quantitative analysis of RAF multimer formation and signaling. Proc. Natl. Acad. Sci. U.S.A. 110, 18519–18524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ester M., Kriegel H.-P., Sander J. r., and Xu X. (1996) A density-based algorithm for discovering clusters in large spatial databases with noise. in Kdd [Google Scholar]
- 62.Jones S. A., Shim S.-H., He J., and Zhuang X. (2011) Fast, three-dimensional super-resolution imaging of live cells. Nat. Methods 8, 499–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sengupta P., Jovanovic-Talisman T., Skoko D., Renz M., Veatch S. L., and Lippincott-Schwartz J. (2011) Probing protein heterogeneity in the plasma membrane using PALM and pair correlation analysis. Nat. Methods 8, 969–975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Peters K. R., Carley W. W., and Palade G. E. (1985) Endothelial plasmalemmal vesicles have a characteristic striped bipolar surface structure. J. Cell Biol. 101, 2233–2238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Heerklotz H. (2002) Triton promotes domain formation in lipid raft mixtures. Biophys. J 83, 2693–2701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Durisic N., Laparra-Cuervo L., Sandoval-Álvarez A., Borbely J. S., and Lakadamyali M. (2014) Single-molecule evaluation of fluorescent protein photoactivation efficiency using an in vivo nanotemplate. Nat. Methods 11, 156–162 [DOI] [PubMed] [Google Scholar]
- 67.Bar-On D., Wolter S., van de Linde S., Heilemann M., Nudelman G., Nachliel E., Gutman M., Sauer M., and Ashery U. (2012) Super-resolution imaging reveals the internal architecture of nano-sized syntaxin clusters. J. Biol. Chem. 287, 27158–27167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rickman C., Medine C. N., Dun A. R., Moulton D. J., Mandula O., Halemani N. D., Rizzoli S. O., Chamberlain L. H., and Duncan R. R. (2010) t-SNARE protein conformations patterned by the lipid microenvironment. J. Biol. Chem. 285, 13535–13541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fujita A., Cheng J., Tauchi-Sato K., Takenawa T., and Fujimoto T. (2009) A distinct pool of phosphatidylinositol 4,5-bisphosphate in caveolae revealed by a nanoscale labeling technique. Proc. Natl. Acad. Sci. U.S.A. 106, 9256–9261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lemmon M. A., Ferguson K. M., O'Brien R., Sigler P. B., and Schlessinger J. (1995) Specific and high-affinity binding of inositol phosphates to an isolated pleckstrin homology domain. Proc. Natl. Acad. Sci. U.S.A. 92, 10472–10476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Annibale P., Vanni S., Scarselli M., Rothlisberger U., and Radenovic A. (2011) Identification of clustering artifacts in photoactivated localization microscopy. Nat. Methods 8, 527–528 [DOI] [PubMed] [Google Scholar]
- 72.Dempsey G. T., Vaughan J. C., Chen K. H., Bates M., and Zhuang X. (2011) Evaluation of fluorophores for optimal performance in localization-based super-resolution imaging. Nat. Methods 8, 1027–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Osborne S. L., Thomas C. L., Gschmeissner S., and Schiavo G. (2001) Nuclear PtdIns(4,5)P2 assembles in a mitotically regulated particle involved in pre-mRNA splicing. J. Cell Sci. 114, 2501–2511 [DOI] [PubMed] [Google Scholar]
- 74.Tanaka K. A., Suzuki K. G., Shirai Y. M., Shibutani S. T., Miyahara M. S., Tsuboi H., Yahara M., Yoshimura A., Mayor S., Fujiwara T. K., and Kusumi A. (2010) Membrane molecules mobile even after chemical fixation. Nat. Methods 7, 865–866 [DOI] [PubMed] [Google Scholar]
- 75.Saarikangas J., Zhao H., and Lappalainen P. (2010) Regulation of the actin cytoskeleton-plasma membrane interplay by phosphoinositides. Physiol. Rev. 90, 259–289 [DOI] [PubMed] [Google Scholar]
- 76.Khuong T. M., Habets R. L., Kuenen S., Witkowska A., Kasprowicz J., Swerts J., Jahn R., van den Bogaart G., and Verstreken P. (2013) Synaptic PI (3, 4, 5) P 3 is required for Syntaxin1A clustering and neurotransmitter release. Neuron 77, 1097–1108 [DOI] [PubMed] [Google Scholar]
- 77.Arendt K. L., Royo M., Fernández-Monreal M., Knafo S., Petrok C. N., Martens J. R., and Esteban J. A. (2010) PIP3 controls synaptic function by maintaining AMPA receptor clustering at the postsynaptic membrane. Nat. Neurosci. 13, 36–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Klarlund J. K., Guilherme A., Holik J. J., Virbasius J. V., Chawla A., and Czech M. P. (1997) Signaling by phosphoinositide-3,4,5-trisphosphate through proteins containing pleckstrin and Sec7 homology domains. Science 275, 1927–1930 [DOI] [PubMed] [Google Scholar]
- 79.Hammond G. R., Machner M. P., and Balla T. (2014) A novel probe for phosphatidylinositol 4-phosphate reveals multiple pools beyond the Golgi. J. Cell Biol. 205, 113–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Roy A., and Levine T. P. (2004) Multiple pools of phosphatidylinositol 4-phosphate detected using the pleckstrin homology domain of Osh2p. J. Biol. Chem. 279, 44683–44689 [DOI] [PubMed] [Google Scholar]
- 81.Wuttke A., Sågetorp J., and Tengholm A. (2010) Distinct plasma-membrane PtdIns(4)P and PtdIns(4,5)P2 dynamics in secretagogue-stimulated β-cells. J. Cell Sci. 123, 1492–1502 [DOI] [PubMed] [Google Scholar]
- 82.Várnai P., Bondeva T., Tamás P., Tóth B., Buday L., Hunyady L., and Balla T. (2005) Selective cellular effects of overexpressed pleckstrin-homology domains that recognize PtdIns(3,4,5)P3 suggest their interaction with protein binding partners. J. Cell Sci. 118, 4879–4888 [DOI] [PubMed] [Google Scholar]
- 83.Hagren O. I., and Tengholm A. (2006) Glucose and insulin synergistically activate phosphatidylinositol 3-kinase to trigger oscillations of phosphatidylinositol 3,4,5-trisphosphate in β-cells. J. Biol. Chem. 281, 39121–39127 [DOI] [PubMed] [Google Scholar]
- 84.Cremona O., Di Paolo G., Wenk M. R., Lüthi A., Kim W. T., Takei K., Daniell L., Nemoto Y., Shears S. B., Flavell R. A., McCormick D. A., and De Camilli P. (1999) Essential role of phosphoinositide metabolism in synaptic vesicle recycling. Cell 99, 179–188 [DOI] [PubMed] [Google Scholar]
- 85.Di Paolo G., Moskowitz H. S., Gipson K., Wenk M. R., Voronov S., Obayashi M., Flavell R., Fitzsimonds R. M., Ryan T. A., and De Camilli P. (2004) Impaired PtdIns(4,5)P2 synthesis in nerve terminals produces defects in synaptic vesicle trafficking. Nature 431, 415–422 [DOI] [PubMed] [Google Scholar]
- 86.Carlton J. G., and Cullen P. J. (2005) Coincidence detection in phosphoinositide signaling. Trends Cell Biol. 15, 540–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Golebiewska U., Kay J. G., Masters T., Grinstein S., Im W., Pastor R. W., Scarlata S., and McLaughlin S. (2011) Evidence for a fence that impedes the diffusion of phosphatidylinositol 4, 5-bisphosphate out of the forming phagosomes of macrophages. Mol. Biol. Cell 22, 3498–3507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yaradanakul A., and Hilgemann D. W. (2007) Unrestricted diffusion of exogenous and endogenous PIP 2 in baby hamster kidney and Chinese hamster ovary cell plasmalemma. J. Membr. Biol. 221, 175–175 [DOI] [PubMed] [Google Scholar]
- 89.Hammond G. R., Sim Y., Lagnado L., and Irvine R. F. (2009) Reversible binding and rapid diffusion of proteins in complex with inositol lipids serves to coordinate free movement with spatial information. J. Cell Biol. 184, 297–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Garrenton L. S., Stefan C. J., McMurray M. A., Emr S. D., and Thorner J. (2010) Pheromone-induced anisotropy in yeast plasma membrane phosphatidylinositol-4,5-bisphosphate distribution is required for MAPK signaling. Proc. Natl. Acad. Sci. U.S.A. 107, 11805–11810 [DOI] [PMC free article] [PubMed] [Google Scholar]