

Background: The IL-6 signaling complex consists of a hexameric structure essential for IL-6 cis- and trans-signaling.
Results: mAb 25F10 targets site IIb of IL-6R and disrupts hexamer assembly to selectively block trans-signaling.
Conclusion: Cis- and trans-signaling in mice utilize distinct mechanisms to mediate assembly of the IL-6R complex.
Significance: Therapeutic targeting of site IIb of IL-6R provides a unique mode of action for IL-6 inhibition.
Keywords: antibody, complex, interleukin 6 (IL-6), receptor, signaling
Abstract
The IL-6 signaling complex is described as a hexamer, formed by the association of two IL-6·IL-6 receptor (IL-6R)·gp130 trimers, with gp130 being the signal transducer inducing cis- and trans-mediated signaling via a membrane-bound or soluble form of the IL-6R, respectively. 25F10 is an anti-mouse IL-6R mAb that binds to both membrane-bound IL-6R and soluble IL-6R with the unique property of specifically inhibiting trans-mediated signaling events. In this study, epitope mapping revealed that 25F10 interacts at site IIb of IL-6R but allows the binding of IL-6 to the IL-6R and the recruitment of gp130, forming a trimer complex. Binding of 25F10 to IL-6R prevented the formation of the hexameric complex obligate for trans-mediated signaling, suggesting that the cis- and trans-modes of IL-6 signaling adopt different mechanisms for receptor complex assembly. To study this phenomenon also in the human system, we developed NI-1201, a mAb that targets, in the human IL-6R sequence, the epitope recognized by 25F10 for mice. Interestingly, NI-1201, however, did not selectively inhibit human IL-6 trans-signaling, although both mAbs produced beneficial outcomes in conditions of exacerbated IL-6 as compared with a site I-directed mAb. These findings shed light on the complexity of IL-6 signaling. First, triggering cis- versus trans-mediated IL-6 signaling occurs via distinctive mechanisms for receptor complex assembly in mice. Second, the formation of the receptor complex leading to cis- and trans-signaling biology in mice and humans is different, and this should be taken into account when developing strategies to inhibit IL-6 clinically.
Introduction
IL-6 is a pleiotropic cytokine mediating vital biological functions such as differentiation of B cells, induction of acute phase proteins, and regulation of hematopoiesis (1, 2). The established role of IL-6 in driving chronic inflammatory conditions (3), however, speaks to the flip side of activating this biology in patients. Thus, creating an optimal balance between controlling disease and maintaining homeostatic processes remains a goal. A key element to achieve this is to better understand the mechanisms by which IL-6 exerts its effects at a molecular level. IL-6 can signal using either a cis- or trans-mediated cascade; these cascades differ substantially in their cellular distribution (4). Indeed, cis-signaling is mediated by membrane-bound IL-6 receptor (mbIL-6R),3 which is expressed on only a limited number of cells, i.e. neutrophils, naive T cells, and hepatocytes. In contrast, for trans-signaling, the soluble form of the IL-6R (sIL-6R), which is generated by RNA alternative splicing or, more frequently, by proteolytic cleavage of mbIL-6R, is potentially able to stimulate all cells of the body (4). Upon IL-6 binding, mbIL-6R or sIL-6R recruits the ubiquitously expressed membrane protein gp130 that when dimerized activates JAK/STAT intracellular signaling pathways (5). Furthermore, although cis-mediated signaling appears to impact the vital, regulatory functions, trans-signaling is emerging as a driver of dysregulated inflammatory responses leading to disease (6).
The IL-6 signaling complex is thought to be a hexameric structure that assembles sequentially. As neither IL-6 nor IL-6R alone has an affinity for gp130, IL-6 binds first to an IL-6R, and the resulting dimer then binds to a gp130 molecule, forming a trimer. In turn, the trimer homodimerizes to form the hexameric signaling complex (7). The assembly of the hexameric complex is believed to be required for both cis- and trans-mediated signaling (8). Key interaction sites of the three proteins have been postulated (Fig. 1), highlighting points of contact and therefore interest for pharmaceutical medicine. Interaction site I is defined as the contact points between extracellular domains 2 (D2) and 3 (D3) of an IL-6R with IL-6 forming the IL-6·IL-6R dimer. Interaction site II involves the contact sites of the dimer with D2 and D3 of gp130 with sites IIa and IIb designating the IL-6/gp130 and IL-6R/gp130 interfaces, respectively. Finally, interaction site III refers to those of the two trimers with the IL-6·IL-6R dimer of the first trimer (i) making the contacts to bridge with D1 of the gp130 of the second trimer (ii). These contact points are designated as sites IIIa and IIIb for IL-6(i)/gp130(ii) and IL-6R(i)/gp130(ii) interfaces, respectively.
FIGURE 1.
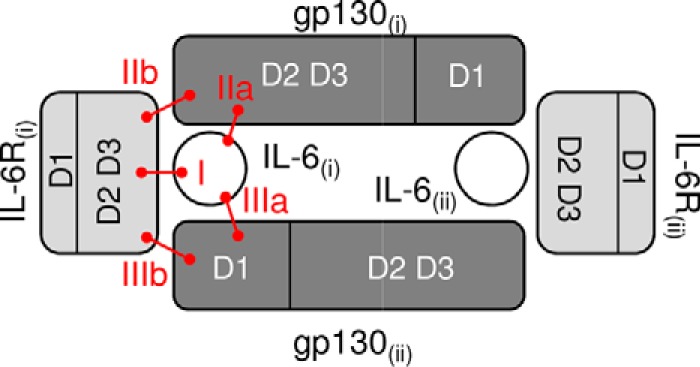
Schematic view of the interacting domains within the IL-6 hexameric signaling complex. IL-6 interacts with D2 and D3 of IL-6R (site I). Within this dimer, IL-6 and IL-6R are both involved in binding to D2 and D3 of gp130 through sites IIa and IIb, respectively. Additional interactions form the IL-6 signaling hexameric complex by assembling two dimers (i and ii) of IL-6·IL-6R·gp130 through D1 of gp130 (sites IIIa and IIIb). IL-6 is in white, IL-6R is in light gray, and gp130 is in dark gray.
Therapeutic monoclonal antibodies (mAbs) currently used to treat patients interfere at different sites of the IL-6 signaling complex. Tocilizumab (Actemra, Hoffmann-La Roche), an anti-human IL-6R mAb, for example, blocks the binding of IL-6 to IL-6R by targeting site I (9), whereas olokizumab (R-Pharm-UCB), an anti-human IL-6 mAb, blocks hexamer formation by targeting site IIIa (10). These clinically active molecules are believed to provide blockade of IL-6 signaling indiscriminately; i.e. both cis- and trans-mediated signaling are affected. Recently, however, the hypothesis that the biological consequences of inhibiting the two pathways are therapeutically divergent (for a review, see Ref. 11) has been supported using an engineered variant of soluble gp130, i.e. sgp130-hFc (12). Studies performed with sgp130-hFc have significantly advanced our appreciation of targeting IL-6 trans-signaling in disease.
Here, we further describe an antibody that targets mouse IL-6R (mIL-6R), 25F10, which inhibits trans- but not cis-signaling. Therefore, we set out to describe how 25F10 interferes with IL-6 biology. We demonstrate that 25F10 binds Glu-261 of mIL-6R, i.e. at site IIb, and based on the three-dimensional structure of the human IL-6 signaling complex should theoretically block the interaction with gp130. Interestingly, binding studies showed that 25F10 allows gp130 to interact with the IL-6·IL-6R complex. In addition, we demonstrate that the non-competitive nature of 25F10 inhibition is more beneficial than a competitive IL-6 mAb in shutting down inflammatory consequences driven by exacerbated IL-6 levels in mice. Collectively, these data suggest that in mice the hexameric complex assembly is required for IL-6 trans-signaling. Finally, we attempted to translate this unique mechanism of action to humans through the generation of a mAb targeting the same epitope on the human IL-6R (hIL-6R). Surprisingly, our results demonstrated that targeting a similar 25F10 epitope on the human protein led to inhibition of the IL-6·IL-6R complex binding to gp130, leading to an efficient inhibition in conditions of high IL-6 levels. Our data illustrate that assembly of the IL-6 signaling complex differs in mice as compared with humans. However, importantly, targeting the site IIb of IL-6R in either species provided a better outcome as compared with a site I-directed mAb in conditions of exacerbated IL-6 production.
Experimental Procedures
Reagents
hIL-6, mIL-6, smIL-6R, shgp130-hFc, and smgp130-hFc recombinant proteins were purchased from R&D Systems; the anti-hIL-6R tocilizumab was purchased from Hoffmann-La Roche. The rat monoclonal antibodies anti-mIL-6R mAb 25F10 and 2B10 (rat IgG1) and the recombinant human and mouse IL-6R complexes (IL-6Rc; IL-6 and sIL-6R bound by a peptide linker) were generated as described previously (13, 14). 1F7, a rat anti-mIL-6R mAb that blocks the interaction between IL-6·IL-6R and gp130, was generated by immunizing rats with mIL-6Rc. An isotype control mAb for rat IgG1 was produced in-house (clone mAb35). The generation of the fully human anti-hIL-6R mAb, NI-1201 (described in United States Patent 8,034,344 (31), was generated by immunization of HuMAbTM mice (licensed from Medarex, now part of Bristol-Myers Squibb Co.) using CHO cells expressing hIL-6R or hIL-6Rc at their surface and hIL-6Rc in Ribi adjuvant (Sigma). Splenocytes were fused with the Sp2/0 myeloma fusion partner as described previously (15). Subsequent screening of hybridomas was performed on mock transfected CHO cells or CHO cells expressing hIL-6, hIL-6R, or hIL-6Rc at their surface using the 8200 cellular detection system (Fluorometric Microvolume Assay Technology, Applied Biosystems). The Fab of NI-1201 was generated by enzymatic cleavage of NI-1201 using a Fab preparation kit (Pierce) following the manufacturer's instructions. Fab fragments were analyzed by electrophoresis on NuPAGE 4–12% Bis-Tris minigels (Invitrogen) in reducing and non-reducing conditions and stained with Coomassie Blue (Invitrogen).
IL-6R Sequence Alignment
Sequence alignment of the D3 domains of mIL-6R (UniProtKB number P22272, amino acids (aa) 212–319), hIL-6R (UniProtKB number P08887, aa 214–329), and rat IL-6R (rIL-6R) (UniProtKB number NP_058716.2, aa 212–319) was generated using Clone Manager (Sci-Ed).
Molecular Cloning of hIL-6R and hIL-6RT264E
The cDNA encoding mature hIL-6R (UniProtKB number P08887, full length) was amplified by PCR from peripheral blood mononuclear cell-derived cDNA and cloned in the pCR4-TOPO vector (Invitrogen). This construct was then subcloned into the corresponding vector pEAK8 (Edge Biosystems). Based on the alignment of human and mouse IL-6R sequences, Thr-264 of the hIL-6R corresponds to Glu-261 of the mIL-6R. The mutation T264E (UniProtKB number P08887) was introduced by site-directed mutagenesis using the QuikChange Lightning site-directed mutagenesis kit (Agilent Technologies). All constructs were verified by DNA sequencing (Fasteris).
Molecular Cloning and Cell Culture for hIL-6RcWT and hIL-6RcT264E Production
The cDNAs encoding shIL-6R (IL-6R) (UniProtKB number P08887, aa 1–333) and hIL-6 (UniProtKB number P05231, aa 29–212) were fused by a synthetic DNA linker coding for the amino acid sequence RGGGGSGGGGSVE and cloned in the pCR4-TOPO vector. Following a subsequent PCR step, a hexahistidine tag was introduced at the C terminus of the cytokine-coding sequence. These constructs were then subcloned into corresponding pEAK8 vectors for expression of the secreted soluble forms. All constructs were verified by DNA sequencing (Fasteris). PEAK cells (Edge Biosystems) were transfected with DMEM (Sigma-Aldrich) containing TransIT-LT1 transfection reagent (MirusBio) and DNA. On day 1 post-transfection, 0.5–2 μg/ml puromycin (Sigma-Aldrich) was added for cell selection. Cells were amplified and seeded in disposable CELLine bioreactors (Integra) for protein production in complete DMEM. Cell culture supernatants were harvested 7–10 days later. Proteins of interest were purified on a nickel-nitrilotriacetic acid affinity chromatography resin (Qiagen) and eluted using an ÄKTA Prime chromatography system (GE Healthcare). Purified proteins were analyzed by electrophoresis on NuPAGE 4–12% Bis-Tris minigels in reducing and non-reducing conditions and stained with Coomassie Blue (data not shown).
In Vitro Functional Assays for Murine IL-6 Cis- and Trans-signaling
Functional assays were performed as described previously (14) except that T1165 cell proliferation was evaluated using the CellTiter-Glo® luminescent reagent (Promega) following the manufacturer's instructions for the cis-signaling assay. A mixture of mIL-6 (0.5 μg/ml) and smIL-6R (1 μg/ml) was used for the trans-signaling assay.
Animals
IL-6-deficient (IL-6−/−) C57BL/6 mice were kindly provided by Prof. Simon A. Jones (Cardiff, UK). Experiments were performed with 8-week-old C57BL/6J female mice (Charles River Laboratories) and DBA/1J male mice (Janvier Labs). C57BL/6J female mice were injected intraperitoneally on days 0, 2, 4, and 7 with 50 μg of oligodeoxynucleotides (ODNs) containing a CpG motif, CpG-ODN 1826 (Invivogen). Antibodies were injected intravenously on day 7 at 10 mg/kg. DBA/1J mice received simultaneously a single intradermic injection in the right back of emulsified complete Freund's adjuvant (CFA; BD Biosciences) and an intravenous injection of mAbs at 10 mg/kg. Emulsion was performed as described previously (14). In both models, plasma was harvested 24 h after mAb injection. Experimental protocols involving mice were conducted according to licenses from the Swiss and United Kingdom veterinary offices for animal experimentation.
Measurement of Serum Amyloid A (SAA) Plasma Levels
SAA concentrations were determined using a mouse SAA1 ELISA kit (Life Diagnostics) following the manufacturer's instructions.
Pharmacokinetics of 25F10
25F10 was administered intravenously to wild-type (WT) C57BL/6 and IL-6−/− C57BL/6 mice at 10 mg/kg. At different time points following mAb injection, mice were sacrificed (three to five mice per group and per time point), and blood samples were collected in heparinized tubes (BD Biosciences). The concentration of unbound mAb in plasma was quantified by ELISA. Briefly, streptavidin-coated microplates (Roche Applied Science) were coated with biotinylated mIL-6R (produced in house; in vitro biotinylation on AviTagTM using the biotin ligase BirA (Avidity)) at 2 μg/ml in PBS for 1 h at 37 °C. After three washes, a standard curve of 25F10 and plasma samples were plated and incubated for 1 h at 37 °C. Finally, plates were washed three times, incubated with an HRP-labeled donkey anti-rat (heavy + light) antibody (Jackson ImmunoResearch Laboratories) for 1 h at 37 °C, and washed five times. The signal was revealed by adding tetramethylbenzidine (Sigma-Aldrich) for 30 min at room temperature and blocked by the addition of 1 n sulfuric acid. Absorbance was read at 450 nm using an Epoch microplate reader (BioTek), and data were analyzed with Gen5 software (BioTek). Values below the lower limit of detection are reported as lower limit of detection/2.
Binding Experiments Using Surface Plasmon Resonance
Binding experiments were carried out at 25 °C in duplicate using a BIAcore 2000 instrument (GE Healthcare). The surface of CM5 chips (GE Healthcare) was activated with a mixture of 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride and N-hydroxysuccinimide (200 and 50 mm, respectively) and coated with an anti-human Fc following the manufacturer's instructions (GE Healthcare, Human Antibody Capture kit). Then chip surfaces were quenched with 1 m ethanolamine. A reference surface was generated by activation and quenching without any protein coating. Smgp130-hFc or shgp130-hFc was captured by the anti-human Fc-coated chip. Injections were performed at 20 μl/min at the following concentrations in 0.01 m HEPES pH 7.4, 0.15 m NaCl, 3 mm EDTA, 0.005% v/v Surfactant P20 buffer (GE Healthcare): smgp130-hFc, 10 μg/ml; shgp130-hFc, 10 μg/ml; mIL-6Rc, 5 μg/ml; hIL-6Rc, 10 μg/ml; hIL-6Rc-T264E, 10 μg/ml; 25F10, 100 μg/ml in Fig. 4A and 5 μg/ml in Fig. 7G; NI-1201 Fab, 50 μg/ml. Following each binding assay, the chip surface was regenerated using 3 m magnesium chloride at 20 μl/min for 10 s followed by 1 min of stabilization in HBS-EP buffer. Using BIAevaluation v.4.1 software (GE Healthcare), data were double referenced with subtraction of the running buffer background signal and subtraction of the potential unspecific signal of injected proteins on the reference surface. Absence of nonspecific binding of the analytes was confirmed in each experiment.
FIGURE 4.
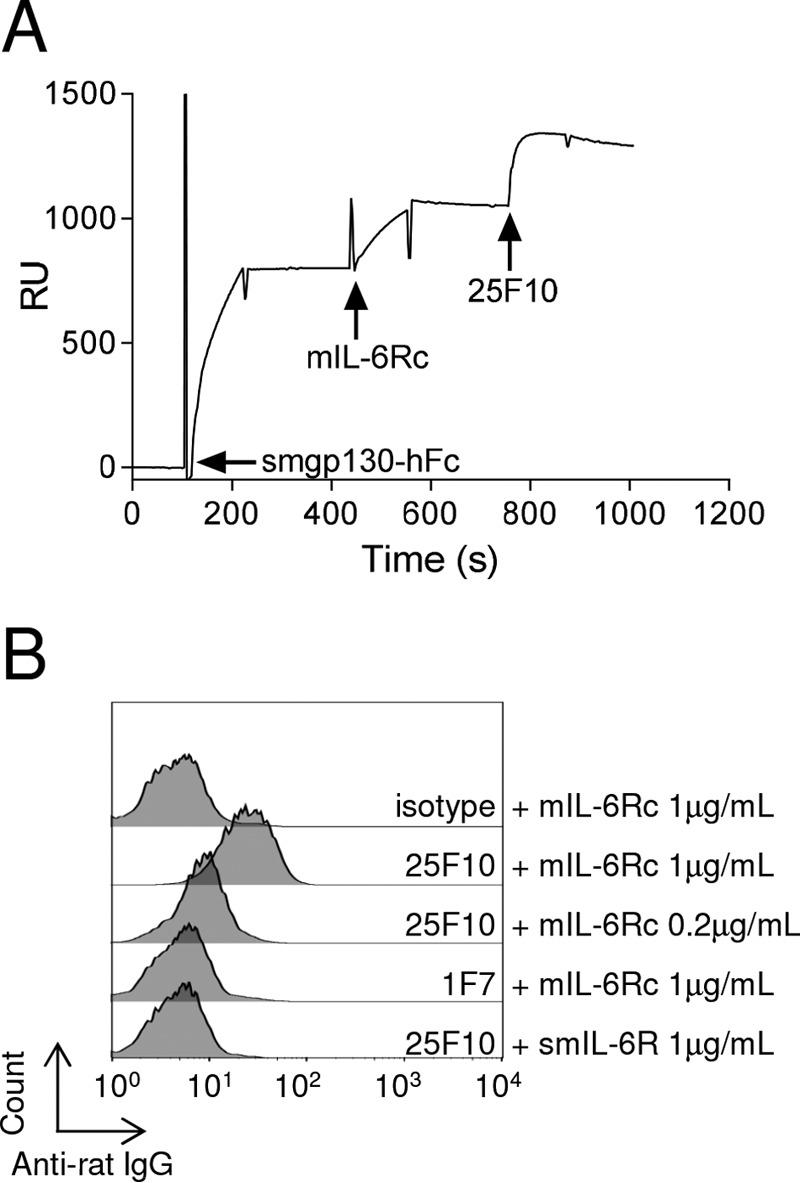
The trimer IL-6·IL-6R·gp130 assembles even in the presence of 25F10. A, an anti-human Fc was immobilized on a CM5 chip. Smgp130-hFc, mIL-6Rc, and 25F10 were sequentially injected at 10, 5, and 100 μg/ml, respectively. The sensorgram signal is shown as relative units (RU). Arrows indicate the start of an injection. Results are representative of at least two independent experiments. B, mIL-6Rc or smIL-6R (both at 1 μg/ml) was premixed with 5 μg/ml 25F10, isotype control, or 1F7, a rat anti-mIL-6R mAb that blocks the interaction between IL-6·IL-6R and gp130, prior to incubation for 15 min at 4 °C with NIH3T3 cells. mAbs bound to the cell surface were detected using an APC-coupled anti-rat IgG antibody, and cells were analyzed by flow cytometry.
FIGURE 7.

NI-1201 targets the same epitope on the human protein as 25F10 does for the mouse receptor but inhibits both IL-6 cis- and trans-signaling. A, PEAK cells were transiently transfected with wild-type or mutated hIL-6R as indicated in each panel. NI-1201 or isotype control was added to the cells, and mAbs bound to the surface were detected with anti-human IgG-APC and analyzed by flow cytometry (NI-1201, black line; isotype control, filled gray). Results are representative of at least two independent experiments. B and E, PEAK cells were transfected with pSIEM-STAT3-luciferase vector and incubated with serial dilutions of mAbs or isotype control as indicated and with 100 ng/ml hIL-6Rc WT or hIL-6Rc-T264E. Firefly luciferase activity was monitored 16 h later. C and F, PEAK cells were transfected with a vector containing hIL-6R or hIL-6R-T264E and 1 day later with pSIEM-STAT3-luciferase vector. They were then co-incubated with serial dilutions of mAbs or isotype control as indicated and with 10 ng/ml hIL-6. Data are expressed as the mean ± S.E. (error bars) and are representative of four independent experiments. D, anti-human Fc was immobilized on a CM5 chip. Shgp130-hFc, hIL-6Rc WT, and NI-1201 Fab were sequentially injected at 10, 10, and 50 μg/ml, respectively. Arrows indicate the start of the injection. Results are representative of at least two independent experiments. G, anti-human Fc was immobilized on a CM5 chip. Shgp130-hFc, mutated hIL-6Rc-T264E, and 25F10 were sequentially injected at 10, 10, and 5 μg/ml, respectively. Arrows indicate the start of an injection. Results are representative of at least two independent experiments. RLU, relative light units; RU, relative units.
NIH3T3 Staining for Flow Cytometry
NIH3T3 cells (ATCC) were maintained in DMEM (Sigma-Aldrich) supplemented with 10% heat inactivated FCS (Gibco) and 2 mm l-glutamine (Gibco). mIL-6Rc (1 and 0.2 μg/ml) or smIL-6R (1 μg/ml) and 25F10, 1F7, or isotype control (5 μg/ml) were incubated for 1 h at room temperature in PBS, 2% BSA and then cooled to 4 °C. In parallel, NIH3T3 cells were detached using an enzyme-free cell dissociation medium (Gibco) and washed in cold PBS, 2% BSA. NIH3T3 cells were incubated with the cold preincubated mixture for 15 min at 4 °C. Cells were washed in cold PBS, 2% BSA and then incubated with an allophycocyanin (APC)-labeled anti-rat Fc antibody (Jackson ImmunoResearch Laboratories) for 15 min at 4 °C. Cells were washed and resuspended in cold PBS, 2% BSA prior to acquisition on a FACSCalibur flow cytometer (BD Biosciences) and analyzed with FlowJo software (TreeStar).
Epitope Mapping of 25F10
The DNA sequences for extracellular hIL-6R (UniProtKB number P08887, aa 20–329); mouse-IL-6R-D1, -D2, or -D3 (UniProtKB number P22272, aa 20–117, 118–213, and 212–319, respectively); and rat-IL-6R-D3 (UniProtKB number NP_058716.2, aa 212–319) were synthesized by Eurofins. The DNA for hIL-6R was subcloned into pDisplay vector (Life Technologies) and digested to cleave the sequences of D1 (aa 20–121), D2 (aa 122–216), and D3 (aa 214–329). The DNA sequences for mouse-IL-6R-D1, -D2, or -D3 and rat-IL-6R-D3 were also digested and cloned into predigested pDisplay-hIL-6R for further expression at the cell surface. The pDisplay-hIL-6R-mD3 and pDisplay-hIL-6R-rD3 constructs were purified using a QIAprep Spin Miniprep kit (Qiagen). The mutation T264E was introduced among the pDisplay-hIL-6R using the QuikChange Lightning site-directed mutagenesis kit (Agilent Technologies). All constructs were verified by sequencing (Fasteris). PEAK cells were transiently transfected with 2 μg of each plasmid in 6-well plates using TransIT-LT1. Cell surface expression was tested 48 h post-transfection on a pool of transfectants with a phycoerythrin-labeled anti-hIL-6R antibody (BD Biosciences) binding the Ig-like domain of human IL-6R and an anti-c-myc (produced in house), which is expressed between the transmembrane domain and the protein of interest. Immunostaining was performed by incubating cells with 25F10 or an isotype control diluted to 5 μg/ml in PBS, 2% BSA for 30 min at 4 °C. Cells were washed in PBS, 2% BSA and then incubated with the secondary antibody, an APC-labeled anti-rat Fc antibody, for 30 min at 4 °C. Cells were washed and resuspended in PBS, 2% BSA prior to acquisition on a FACSCalibur flow cytometer and analyzed with FlowJo software.
In Vitro Functional Assays for Human IL-6 Signaling
On day 0, PEAK cells were transfected with pEAK8-hIL-6R WT, pEAK8-hIL-6R-T264E vector, or an irrelevant vector and on day 1 with pSIEM-STAT3-luciferase vector using Lipofectamine transfection reagent (Life Technologies). Transfected cells were seeded at 5 × 104 cells/well in 96-well white luminescence plates for adhesion. 5 h later they were incubated with serial dilutions of mAbs for 30 min at 37 °C. Then hIL-6 (10 ng/ml) was added on hIL-6R (WT or mutated)-transfected cells, whereas hIL-6Rc (100 ng/ml; WT or mutated) was added on cells transfected with the irrelevant vector. On day 3, culture medium was discarded, and cells were incubated with Steady Glo substrate (Promega) according to the manufacturer's instructions. Luminescence was read on a BMG Fluostar Optima (BMG Labtech).
When indicated, cis- and trans-signaling were studied using a proliferation assay of Ba/F3-hgp130-hIL-6R and Ba/F3-hgp130 cells described to be hIL-6- and hIL-6/shIL-6R-dependent, respectively (kindly provided by Prof. Jürgen Scheller, Düsseldorf, Germany). Cells were maintained in RPMI 1640 medium (Sigma-Aldrich) supplemented with 10% heat-inactivated FCS (Sigma-Aldrich), 2 mm l-glutamine, and 1 ng/ml hIL-6 or hIL-6Rc. On day 0, cells were washed with prewarmed RPMI 1640 medium to remove residual hIL-6 or hIL-6Rc and plated at 104 cells/well in a 96-well plate in RPMI 1640 medium supplemented with 0.5% FCS. Cells were incubated with mAbs at 10 nm and either increasing concentrations of hIL-6 (for the cis-signaling assay) or hIL-6 and shIL-6R at the indicated concentrations (for the trans-signaling assay) for 72 h at 37 °C with 5% CO2. Then proliferation was evaluated using the cell proliferation reagent WST-1 (Roche Applied Science) following the manufacturer's instructions. Absorbances at 450 and 650 nm were read using an Epoch microplate reader, and data were analyzed with Gen5 software by subtracting 450- and 650-nm absorbance values.
Results
IL-6 Trans-signaling-specific mAb, 25F10, Targets Site IIb of Mouse IL-6R
Two mAbs reported previously (14) and directed to the mouse IL-6R, 25F10 and 2B10, were evaluated for their ability to inhibit activities of IL-6. 25F10 selectively inhibits IL-6 trans-signaling (14, 16), whereas 2B10 blocks IL-6 binding to IL-6R and inhibits both cis- and trans-modes of IL-6 signaling (14) (Fig. 2A). 25F10, as well as 2B10, engages mbIL-6R (Fig. 2B), but the former is unable to inhibit cis-mediated proliferation of T1165 plasmacytoma cells (Fig. 2C). We therefore hypothesized that characterizing the site where 25F10 binds to the mIL-6R may lead to important insights into the extracellular molecular nature of cis- versus trans-signaling. Thus, the epitope of 25F10 was investigated. For this, chimeric human-mouse IL-6R constructs were created in PEAK cells by replacing extracellular domains of hIL-6R with D1, D2, or D3 of the mouse or rat protein (shown for D3 in Fig. 3A). As expected, 25F10 was unable to bind hIL-6R (hIL-6R WT; Fig. 3B). The replacement of the D1 or D2 sequences of hIL-6R with mouse D1 or D2, respectively, failed to restore 25F10 binding (hIL-6R-mD1 or hIL-6R-mD2; Fig. 3B), whereas the insertion of mouse D3 enabled 25F10 binding (hIL-6R-mD3; Fig. 3B). Despite 88% IL-6R sequence identity between mouse and rat IL-6R, 25F10 failed to bind the chimeric molecule containing the rat IL-6R-D3 sequence (hIL-6R-rD3; Fig. 3B). To determine the binding site of 25F10 on mIL-6R-D3, mutants were generated by inserting single substitutions of the mIL-6R-D3 within the D3 of hIL-6R. The strategy for choosing the point mutations was mainly based on targeting identical residues between rat and human but different from the mouse protein within a relevant region of D3, i.e. at the interface between IL-6R(i) and gp130(i). Among the numerous variants tested, 25F10 was only able to recognize the hIL-6R-T264E substitution (Fig. 3B). Based on the alignment of human and mouse IL-6R sequences, threonine in position 264 of the hIL-6R corresponds to glutamic acid in position 261 of the mIL-6R (Fig. 3A). Using this strategy, we observed that substituting other mouse residues juxtaposing T264E did not improve the binding of 25F10 to hIL-6R-D3 domain (data not shown), indicating that the Glu-261 in the mIL-6R is a critical amino acid within the epitope recognized by 25F10 (Fig. 3C). Taken together, our results show that the binding of 25F10 to mIL-6R requires the glutamic acid at position 261 in D3, which is a residue that has been described to be key in the interaction site IIb between IL-6R(i) and gp130(i) for the formation of the trimeric IL-6·IL-6R·gp130 complex (17).
FIGURE 2.

25F10 engages mbIL-6R but does not inhibit IL-6 cis-signaling, only trans-signaling. A, STAT3-luciferase-transfected PEAK cells were stimulated with mIL-6 (0.5 μg/ml) and smIL-6R (1 μg/ml), and after 18 h, firefly activity was measured in the presence or absence of varying concentrations of 25F10 or 2B10. mIL-6 and smIL-6R alone were used as negative controls. mIL-6 in combination with smIL-6R and an isotype control mAb at 0.67 × 10−8 m was used as a positive control. Error bars represent S.E. B, IL-6R+ T1165 cells were incubated for 30 min with 25F10, 2B10, or isotype control. Binding was assessed by flow cytometry. C, T1165 cells were stimulated with IL-6 (1 ng/ml), and after 48 h, cell proliferation was measured in the presence or absence of varying concentrations of 25F10 or 2B10. The results are presented as a percentage of proliferation normalized to the signal induced by IL-6 only. Error bars represent S.E. RLU, relative light units.
FIGURE 3.

25F10 binds the interaction site IIb of the IL-6 signaling complex. A, sequence alignment of D3 of mouse, rat, and human IL-6Rs. The residue responsible for the binding of 25F10 is indicated (▾). B, PEAK cells were transiently transfected with chimeric or mutated hIL-6R as indicated in each panel. 25F10 or isotype control was added to the cells, and mAbs bound to the surface were detected with an APC-coupled anti-rat IgG and analyzed by flow cytometry (25F10, filled dark gray; isotype control, filled light gray). Results are representative of at least two independent experiments. C, schematic view of the human IL-6 signaling hexameric complex generated with PyMOL software (Protein Data Bank code 1P9M) highlighting in green the amino acid responsible for the binding of 25F10 among its murine equivalent within the site IIb. The enlargement is a view of site IIb shown at higher magnification depicting Thr-264 among IL-6R and residues of gp130 involved in this interface (17).
Despite Targeting Site IIb with 25F10, the Trimeric IL-6·sIL-6R·gp130 Complex Can Assemble
Based on the results of epitope mapping, we next investigated whether 25F10 interfered with the assembly of the IL-6 signaling complex by binding to site IIb. Using surface plasmon resonance, we first confirmed that mIL-6Rc (a fusion protein of mIL-6 and smIL-6R) bound to immobilized smgp130-hFc (Fig. 4A). Surprisingly, 25F10 was able to bind the preformed complex of smgp130-hFc·mIL-6Rc with an apparent high affinity as illustrated by the slow rate of dissociation for 25F10 (Fig. 4A). These findings were also confirmed using a preincubated mixture of mIL-6 and smIL-6R added to immobilized smg130-hFc (data not shown). We next tested whether the ability of 25F10 to bind mIL-6Rc in the presence of mgp130 could be observed on cells. For this, murine fibroblast NIH3T3 cells were used as they display a high level of membrane gp130 and no detectable mbIL-6R (data not shown). NIH3T3 cells were incubated with the preformed complex of mIL-6Rc·25F10, and a fluorochrome-labeled anti-rat IgG was used to detect cell surface binding. No fluorescence signal was detected when mIL-6Rc was in the presence of a mouse IgG1 control or an anti-IL-6R mAb, namely 1F7 (which inhibits mIL-6Rc binding to mgp130) (Fig. 4B). When incubated with mIL-6Rc, 25F10 was detected at the cell surface, confirming the ability for this mAb to remain engaged to mIL-6R while in an IL-6·IL-6R·gp130 complex (Fig. 4B). Moreover, decreasing the mIL-6Rc concentration resulted in a reduced signal of 25F10 binding, suggesting a mIL-6Rc-dependent binding to the cell surface (Fig. 4B). Thus, the binding of 25F10 to mIL-6Rc does not prevent the interaction with membrane-bound gp130. Collectively, these results show that 25F10 binding to site IIb of mIL-6R does not impact the assembly of the IL-6·sIL-6R·gp130 trimeric complex.
25F10 Does Not Interfere with Assembly of the IL-6·mbIL-6R·gp130 Complex in Vivo
The above results demonstrate the selectivity of 25F10 for IL-6 trans-signaling while retaining a capacity to bind both soluble and membrane forms of IL-6R. Furthermore, we were surprised to see that the binding of this inhibitory mAb to an epitope described to be important for the IL-6R(i) and gp130(i) interaction (17) did not disrupt the assembly of the trimeric IL-6·sIL-6R·gp130 complex. To extend the above observations, we next tested whether 25F10 also allows assembly of the trimeric IL-6·membrane IL-6R·gp130 complex in a relevant physiological setting. mAbs targeting membrane-bound antigens are typically subject to target-driven elimination (18). Therefore, if 25F10 allows the trimeric IL-6·mbIL-6R·gp130 complex assembly, then its target-driven elimination should be mediated not only by the slow internalization rate of mbIL-6R (19) but also by the higher internalization rate of gp130 complexed to IL-6·IL-6R (20–22). To test this hypothesis, 25F10 was administered as a single intravenous injection to WT or IL-6−/− mice. In WT mice, 25F10 can theoretically interact with various complexes involving IL-6, mbIL-6R, sIL-6R, and gp130 at the cell surface (Fig. 5A, upper diagram), whereas in IL-6−/− mice, the mAb can bind only to mbIL-6R (Fig. 5A, lower diagram). Plasma concentrations of the unbound 25F10 were assessed at different time points postinjection. The concentration of 25F10 over time in IL-6−/− mice showed a lower clearance of the mAb than in WT mice (Fig. 5B). These data confirm the ability of 25F10 to bind the assembled IL-6·mbIL-6R·gp130 complex in vivo.
FIGURE 5.

In the presence of 25F10, a tetrameric complex, i.e. IL-6·IL-6R·25F10·gp130, is formed in vivo. A, schematic view of the different ways of target-driven elimination of 25F10 in WT versus IL-6−/− mice. In WT mice, 25F10 interacts with its targets mbIL-6R and sIL-6R and allows the formation of four distinct complexes at the cell membrane that drive the elimination of 25F10 from the circulation. In IL-6−/− mice, target-driven elimination of 25F10 is only able to be mediated by mbIL-6R as the other formats shown in the top panel cannot form in the absence of IL-6. B, 25F10 mAb was intravenously administered to WT or IL-6−/− mice at 10 mg/kg. Plasma samples were obtained at the indicated time points following injection, starting at 1 h postdosing, and the mAb concentration was analyzed by ELISA. Data are expressed as the mean ± S.E. (error bars) (n = 3–4 mice). mb, cell membrane; LLOD, lower limit of detection of the assay.
Targeting Site IIb of the Mouse IL-6R in Vivo Affords Enhanced Abrogation of IL-6 Responses
Several studies suggest that, when IL-6 concentrations exceed the levels of sIL-6R and sgp130, IL-6 signaling can occur systemically. Indeed, sIL-6R and sgp130 are thought to act as a buffer system to prevent systemic IL-6 signaling (6). Thus, we hypothesized that in such conditions of high IL-6, 25F10, i.e. the non-competitive IL-6 inhibitor targeting the IL-6R site IIb, would be more potent than 2B10, a competitive inhibitor of IL-6. Using an acute mouse model of inflammation characterized by a cytokine storm, we compared the efficacy of 25F10 versus 2B10 to control the IL-6-induced biomarker SAA (23). Naive mice receiving repeated injections of CpG-ODN on days 0, 2, 4, and 7 develop clinical features of hypercytokinemia (24) where IL-6 concentrations in plasma increase by 1000-fold by day 7.4 Using this protocol, mice received 10 mg/kg anti-IL-6R mAb 2B10 or 25F10 concomitantly with CpG-ODN administration on day 7, and SAA levels were assessed 24 h later. Although 2B10 was unable to reduce the CpG-ODN-induced SAA levels, 25F10 significantly abrogated the IL-6 response (Fig. 6A). To support the above data using a second model, 25F10 or 2B10 was injected concomitantly with a single administration of emulsified CFA. In this acute model, IL-6 concentrations in plasma are only moderate (i.e. 20 pg/ml 6 h post-CFA injection, reaching 60 pg/ml at 24 h; data not shown). In these conditions, 2B10 completely shut down the IL-6-induced SAA response, whereas 25F10 only reduced the signal 5-fold as compared with the isotype control group (Fig. 6B). These findings were further confirmed in mice where SAA levels were measured following a single dose of CpG (data not shown). Collectively, these in vivo studies highlight a potential therapeutic advantage in targeting site IIb as a mechanism to more effectively inhibit IL-6 trans-signaling-mediated responses in conditions associated with heightened IL-6 levels.
FIGURE 6.

In vivo IL-6 signaling blockade with 25F10 affords enhanced abrogation of IL-6 responses under inflammatory conditions. A, mice were injected with 50 μg of CpG-ODN on days 0, 2, 4, and 7. Mice were treated with mAbs intravenously at 10 mg/kg simultaneously with the last CpG injection (on day 7). Plasma was harvested 24 h after the last CpG injection, and SAA plasma concentrations were measured by ELISA. B, mice were treated with mAbs intravenously at 10 mg/kg simultaneously with the CFA intradermic injection. Plasma was harvested 24 h after the injections, and SAA plasma concentrations were measured by ELISA. Data are expressed as the mean. Statistical analyses were performed between the indicated group and the isotype control group values. ns, not significant; *, p < 0.05; **, p < 0.01 were obtained using the one-tailed non-parametric Mann-Whitney U test.
Targeting the Equivalent 25F10 Epitope on Human IL-6R Fails to Confer a Trans-signaling-only Blockade
The superior efficacy displayed by blocking IL-6 signaling with 25F10 in specific inflammatory conditions prompted us to investigate whether targeting the equivalent epitope in the human receptor would confer a similar mode of action. Thus, an anti-human equivalent of 25F10, NI-1201, recognizing the Thr-264 residue of hIL-6R corresponding to the equivalent epitope on mIL-6R, was generated. The binding specificity of NI-1201 for hIL-6R WT (Fig. 7A, left panel) conferred by the residue Thr-264 was confirmed by the abrogation of the binding to the variant form hIL-6R-T264E (Fig. 7A, right panel). To study the ability of NI-1201 to affect IL-6 signaling, cis-signaling was assessed using STAT3-luciferase activity in hIL-6R-transfected cells, and trans-signaling was induced by incubating STAT3-luciferase reporter cells with hIL-6Rc, a fusion protein of hIL-6 and shIL-6R. Unexpectedly, NI-1201 inhibited both IL-6 trans- and cis-signaling (Fig. 7, B and C, respectively). Next, we used surface plasmon resonance to investigate the potential for hIL-6Rc·shgp130-hFc complex to assemble in the presence of NI-1201. Here, the NI-1201-derived Fab fragment was used to avoid nonspecific binding of the mAb to the capture antibody on the chip. Unlike 25F10, we observed that the NI-1201 Fab failed to bind the preformed hIL-6Rc·shgp130-hFc trimeric complex (Fig. 7D). These data suggest that the knowledge of the trans-signaling specificity afforded by the 25F10 epitope on mIL-6R is not transferrable to the human form of the receptor.
Furthermore, we investigated whether, by introducing the mouse Glu-261 residue into the hIL-6R-D3 region, 25F10 would have the ability to block trans-signaling. Thus, mutated versions of soluble hIL-6Rc (hIL-6Rc-T264E) and membrane-bound hIL-6R (hIL-6R-T264E) allowing 25F10 binding were generated. Importantly, the introduction of the T264E mutation had no impact on the human IL-6 cis- and trans-mediated signaling intensities (i.e. isotype control luminescence levels remain equivalent; Fig. 7, B and E and C and F). The ability of 25F10 to bind hIL-6R-T264E was demonstrated on transfected PEAK cells (data not shown). However, 25F10 was unable to inhibit human IL-6 trans- and cis-signaling (Fig. 7, E and F). Interestingly, surface plasmon resonance data showed that 25F10, as in the mouse system, was able to interact with the assembled hIL-6Rc-T264E·shgp130-hFc complex (Fig. 7G). Taken together, these data illustrate that the assembly of the IL-6 signaling components in mouse differs from that in human.
Targeting Site IIb of the Human IL-6 Signaling Complex Affords Beneficial Effects Compared with Targeting Site I
The above results demonstrate that the blockade of site IIb of hIL-6R using NI-1201 showed a higher potency on IL-6 trans-signaling (IC50 = 0.30 μg/ml) over IL-6 cis-signaling (IC50 = 7.39 μg/ml) (Fig. 7, B and C). Thus, we hypothesized that in conditions of exacerbated IL-6, similarly to 25F10, NI-1201 would exhibit beneficial effects compared with a competitive inhibitor form of an anti-hIL-6R mAb. To test this hypothesis, the cis- and trans-signaling-dependent proliferation of Ba/F3 cells stably transfected with hgp130-hIL-6R or hgp130, respectively, was evaluated. A dose response of hIL-6 was set up to evaluate the capability of NI-1201 versus tocilizumab, an anti-hIL-6R mAb that works as a competitive inhibitor, to inhibit cis-signaling. Then a dose response of NI-1201 versus tocilizumab was used with varying ratios of shIL-6R/hIL-6 (i.e. 1:50, 1:100, and 1:200) for the trans-signaling assay. Interestingly, the efficacies of NI-1201 and tocilizumab did not differentiate upon increasing concentrations of hIL-6 in the cis-signaling assay (Fig. 8A). However, when the relative amount of IL-6 increased in the trans-signaling assay, tocilizumab inhibited IL-6 signaling less efficiently than did NI-1201 (Fig. 8, B–D).
FIGURE 8.

NI-1201 abrogates more efficiently IL-6 trans-signaling activity in conditions of exacerbated IL-6 compared with tocilizumab. A, Ba/F3-hgp130-hIL-6R cells were stimulated with increasing concentrations of hIL-6 and 10 nm NI-1201 or tocilizumab. After 72 h, cell proliferation was measured. Data are expressed as the mean ± S.E. B–D, Ba/F3-hgp130 cells were stimulated with increasing hIL-6 concentrations and a fixed shIL-6R concentration (10 ng/ml) at the indicated molar ratios. After 72 h, cell proliferation was measured in the presence or absence of varying concentrations of NI-1201, tocilizumab, or isotype control. Data are expressed as the mean ± S.E. (error bars). TCZ, tocilizumab.
Discussion
Selectively targeting the proinflammatory properties of IL-6, emerging as mediated by IL-6 trans-signaling, is regarded as critical for successful intervention in disease (for a review, see Ref. 8). Here, through the use of anti-mouse IL-6R mAb 25F10, which exclusively blocks IL-6 trans-signaling, we shed new light on the assembly of the mouse IL-6R complex and how this information may be exploited for better targeted therapies.
Although the epitope of 25F10 resides in IL-6R site IIb, a site thought to be key for IL-6R(i) and gp130(i) interaction, we found that a stable complex comprising IL-6·IL-6R·25F10·gp130 was able to form at the cell membrane in the presence of the mAb. This unexpected finding may be explained by an observation made by Veverka et al. (17) proposing that the hIL-6·hIL-6R·hgp130 trimer is primarily driven by site III (i.e. D1 of gp130). Thus, the binding of 25F10 to IL-6R(i) would not impair the recruitment of gp130(ii) through site IIIb, allowing the formation of an IL-6·IL-6R·25F10·gp130 complex. However, we showed that, with the use of a fully human mAb, i.e. NI-1201, targeting site IIb of hIL-6R, we were able to prevent the binding of hgp130. Taken together, the 25F10 and NI-1201 mechanism of action studies suggest that the assembly of the IL-6·IL-6R·gp130 trimer is mainly driven by site II. More precisely, 25F10 binding allows gp130(i) to interact with IL-6R(i), whereas NI-1201 inhibits gp130(i) binding to IL-6R(i) probably through steric hindrance. This difference may be explained by alternative orientation with which mAbs are interacting with site IIb on IL-6R. The latter is supported by the fact that inserting the T264E mutation in hIL-6R is sufficient to allow the binding of hIL-6·hIL-6R-T264E·25F10 to hgp130. Interestingly, Thr-264 and Glu-261 are located within a loop of IL-6R (Fig. 3C), and as these regions display a relatively higher mobility compared with other secondary structures, this may lead to simultaneous binding of 25F10 and gp130(i) on IL-6R(i). Thus, the neutralizing activity of 25F10, allowing binding of IL-6(i) and gp130(i) to IL-6R(i), reflects its ability to impair the interaction between the two trimers, IL-6·IL-6R·gp130, i.e. on site III. Further studies dissecting the involvement of D1 and D2-D3 of gp130 in the IL-6·IL-6R·25F10·gp130 complex assembly are currently ongoing. Moreover, we cannot exclude that 25F10 induces a conformational change of mIL-6R to facilitate and allow the interaction with gp130 as has been suggested for other antigen-antibody interactions (10). Further studies aimed at solving the structure of IL-6·IL-6R·25F10 and IL-6·IL-6R·25F10·sgp130 would be needed to further our understanding related to this concept.
Although it was demonstrated in vitro that the IL-6·sIL-6R·25F10·gp130 complex is formed at the cell surface of mbIL-6R− NIH3T3 cells, it was important to extend this observation to a setting where mbIL-6R is expressed. The faster clearance of 25F10 in WT as compared with IL-6−/− mice supports the existence of an IL-6·mbIL-6R·25F10·gp130 complex at the cell membrane. To strengthen this hypothesis, we needed to exclude the fact that the lower levels of sIL-6R in IL-6−/− mice (25) may explain the slower elimination kinetics of 25F10. Thus, the non-competitive anti-mouse IL-6R mAb 1F7, which inhibits gp130 binding, was included as an important control and indeed demonstrated similar elimination profiles in WT and IL-6−/− mice (data not shown), suggesting that mbIL-6R indeed drives mAb clearance.
The IL-6 signaling complex has been postulated to be an assembly of IL-6·IL-6R·gp130 in a 2:2:2 stoichiometry, resulting in a hexamer thought to be identical for inducing cis- and trans-signaling (7). Our data showing that 25F10 uniquely blocks IL-6 trans-signaling through the binding to the IL-6·sIL-6R·gp130 complex suggests that the assembly of the hexamer is obligate for trans-signaling to occur in mice. IL-6 cis-signaling, however, is unaffected despite the formation of the IL-6·mbIL-6R·25F10·gp130 complex. As gp130 dimerization at the cell surface has been shown to be necessary for IL-6 signaling (26), the IL-6·mbIL-6R·25F10·gp130 complex must in some way interact with a second gp130 molecule for subsequent cis-signaling. Our data herein elicit questions on the required stoichiometry for IL-6 signaling to occur. The use of smgp130-hFc in binding experiments to address this question further adds bias as the hFc portion of the molecule promotes artificial gp130 dimerization. In an attempt to shed more light on this finding, therefore, we produced monomeric smgp130 (smgp130) and showed that 25F10 interacts with the mIL-6Rc·smgp130 complex. These data suggest that the IL-6·mbIL-6R·25F10·gp130 complex can assemble with one or two gp130 molecules. The 2:2:2 stoichiometry (IL-62·IL-6R2·gp1302) of the hIL-6 signaling complex has been debated extensively; it was initially thought to be a tetrameric conformation IL-61·IL-6R1·gp1302 (27–29) only to be refuted by crystallization experiments suggesting a hexameric complex (7). Taken together, our data propose a new model of IL-6 signaling in mice with the existence of a tetrameric complex IL-61·IL-6R1·gp1302 for cis-signaling and of a hexameric complex IL-62·IL-6R2·gp1302 for trans-signaling. Indeed, we suggest that 25F10 inhibits the hexamer assembly when bound to mb- and sIL-6R. Nevertheless, the IL-6·mbIL-6R·25F10·gp130 complex may be considered as an intermediary step to an IL-6·mbIL-6R·25F10·gp1302 complex (Fig. 9), whereas hexamer formation would be sufficient and necessary for trans-signaling. We propose, therefore, that cis-signaling, often associated to homeostatic functions (8), occurs through the formation of a tetrameric complex requiring less molecular energy than that of trans-signaling where the associated proinflammatory activity would require a hexameric complex assembly.
FIGURE 9.
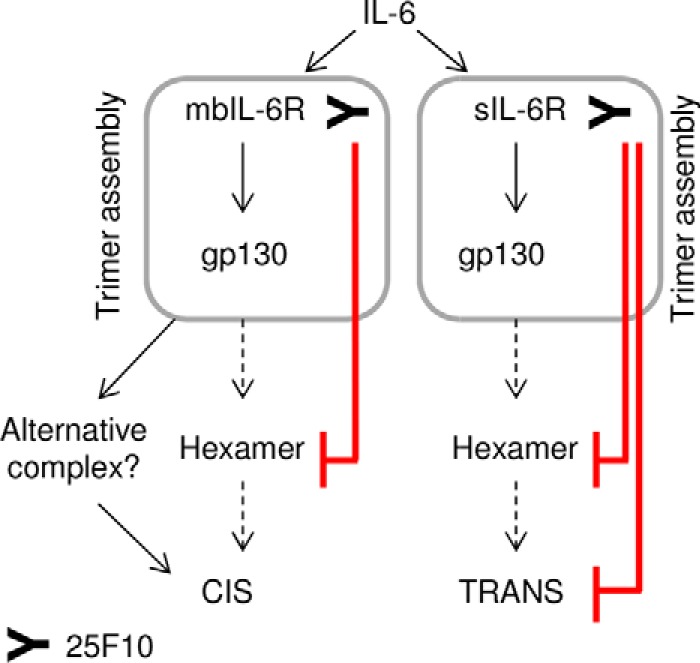
Model to illustrate the potential mechanism of action of 25F10. 25F10 inhibits the hexamer assembly when bound to mb- and sIL-6R (red lines). Nevertheless, the IL-6·mbIL-6R·25F10·gp130 complex may be considered as an intermediary step to induce cis-signaling, whereas hexamer formation would be sufficient and necessary for trans-signaling.
We showed how targeting site IIb of IL-6R would be favorable to inhibit an exacerbated IL-6-induced response, here measured by SAA induction typically associated with a cis-mediated event (30). The injection of anti-IL-6R mAbs to mice when IL-6 serum levels were maximal showed that 25F10 significantly reduced the IL-6-driven SAA response. This was in contrast to the poor blockade seen with 2B10, a competitive inhibitor of IL-6. This emphasizes how, by differentially interfering with the IL-6 signaling complex assembly, IL-6R-targeting mAbs are able to control responses in vivo driven by excessive IL-6 production. Surprised to see the contribution of trans-signaling in the aforementioned, we confirmed in two other inflammatory models with modest IL-6 production directly following a single insult (CFA or CpG) that the SAA response is primarily driven by cis-signaling. We also noted that 25F10, similarly to 2B10, induces a degree of mbIL-6R internalization and may indirectly desensitize cells to IL-6 (data not shown). This rate-limiting mechanism fails to account for the significant reduction of the SAA in 25F10-treated mice given repeat injections of CpG but may explain the moderate SAA inhibition induced by 25F10 in the CFA-induced acute phase response model. Collectively, these data confirm that SAA induction is primarily an IL-6 cis-signaling-driven event and suggest a trans-signaling contribution in situations where IL-6 levels exceed the buffering capacity of sIL-6R and sgp130.
The above data suggested that a mAb targeting site IIb of hIL-6R may afford similar consequences to hIL-6 signaling by translating the properties of 25F10. Introducing the 25F10 mouse epitope to hIL-6R (T264E) allows the binding of 25F10 and the trimer assembly (i.e. hIL-6·hIL-6R-T264E·25F10·hgp130). However, 25F10 was unable to inhibit human IL-6 trans-signaling. The 3-fold lower affinity of 25F10 for hIL-6Rc-T264E (KD = 39 nm) as compared with mIL-6Rc (KD = 13 nm; data not shown) cannot explain the loss of 25F10 efficacy. Nonetheless, using NI-1201, an antibody directed to site IIb of hIL-6R, the interaction of hIL-6Rc with gp130 was inhibited, leading to the abrogation of cis- and trans-signaling pathways. Overall, these data suggest that the complex assembly obligate for cis- and trans-signaling in mice differs from that in human. However, we cannot exclude that a specific inhibition of human IL-6 trans-signaling is possible through another IL-6R targeting approach distinct from blockade of site IIb. Nonetheless, the targeting of site IIb on human IL-6R with NI-1201 induced a more potent inhibition of IL-6 trans-signaling as compared with the site I-directed mAb tocilizumab in conditions of IL-6 high levels. This novel mechanism of action disrupting the IL-6 signaling complex assembly would be advantageous, therefore, in conditions where inflammatory milieus present with exacerbated local IL-6 levels often 100–1000-fold above sIL-6R concentrations (6).
Collectively, these data demonstrate that targeting different sites of the IL-6 signaling complex assembly affords distinct neutralizing consequences. Furthermore, we highlight a potential difference in the stoichiometry of the IL-6 signaling complex between mouse and human leading to cis- and trans-signaling that will be critical to consider when designing the next generation of IL-6 inhibitors.
Author Contributions
M. L., V. B., and W. F. conceived the study and wrote the paper. M. L., F. G., and G. W. J. performed the experiments (Figs. 2–7, Fig. 8, and IL-6−/− mouse experiment in Fig. 5, respectively). F. R. and G. M. constructed vectors for expression of mutant proteins. F. R., P. M., G. M., S. H., and R. L. generated antibodies and recombinant proteins. J. S. and S. A. J. reviewed the manuscript and contributed to experimental design. M. K.-V, Z. J., V. B., and W. F. edited the manuscript. All authors approved the final version of the manuscript.
Acknowledgments
We thank Serge Wolfersperger and Cyril Baudon for animal husbandry.
M. L., F. R., F. G., P. M., G. M., S. H., M. K.-V., Z. J., V. B., and W. F. are employees of Novimmune. S. A. J. is a consultant for Novimmune. F. G., M. K.-V., and W. F. are listed as inventors on United States Patent 8,034,344, “Anti-IL-6/IL-6R antibodies and methods of use thereof.”
V. Buatois, L. Chatel, L. Cons, S. Lory, F. Richard, C. Bracaglia, F. De Benedetti, C. de Min, M. H. Kosco-Vilbois, and W. G. Ferlin, manuscript submitted.
- mbIL-6R
- membrane-bound IL-6 receptor
- s
- soluble
- m
- mouse
- h
- human
- r
- rat
- sgp130-hFc
- soluble glycoprotein 130, human fragment crystallizable
- APC
- allophycocyanin
- CFA
- complete Freund's adjuvant
- Fab
- fragment antigen-binding
- D
- extracellular domain
- IL-6Rc
- IL-6R complex
- Bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- aa
- amino acids
- SAA
- serum amyloid A.
References
- 1.Mihara M., Hashizume M., Yoshida H., Suzuki M., and Shiina M. (2012) IL-6/IL-6 receptor system and its role in physiological and pathological conditions. Clin. Sci. 122, 143–159 [DOI] [PubMed] [Google Scholar]
- 2.Peters M., Müller A. M., and Rose-John S. (1998) Interleukin-6 and soluble interleukin-6 receptor: direct stimulation of gp130 and hematopoiesis. Blood 92, 3495–3504 [PubMed] [Google Scholar]
- 3.Scheller J., Chalaris A., Schmidt-Arras D., and Rose-John S. (2011) The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim. Biophys. Acta 1813, 878–888 [DOI] [PubMed] [Google Scholar]
- 4.Scheller J., Garbers C., and Rose-John S. (2014) Interleukin-6: from basic biology to selective blockade of pro-inflammatory activities. Semin. Immunol. 26, 2–12 [DOI] [PubMed] [Google Scholar]
- 5.Heinrich P. C., Behrmann I., Haan S., Hermanns H. M., Müller-Newen G., and Schaper F. (2003) Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 374, 1–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rose-John S. (2012) IL-6 trans-signaling via the soluble IL-6 receptor: importance for the pro-inflammatory activities of IL-6. Int. J. Biol. Sci. 8, 1237–1247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boulanger M. J., Chow D. C., Brevnova E. E., and Garcia K. C. (2003) Hexameric structure and assembly of the interleukin-6/IL-6α-receptor/gp130 complex. Science 300, 2101–2104 [DOI] [PubMed] [Google Scholar]
- 8.Hunter C. A., and Jones S. A. (2015) IL-6 as a keystone cytokine in health and disease. Nat. Immunol. 16, 448–457 [DOI] [PubMed] [Google Scholar]
- 9.Mihara M., Kasutani K., Okazaki M., Nakamura A., Kawai S., Sugimoto M., Matsumoto Y., and Ohsugi Y. (2005) Tocilizumab inhibits signal transduction mediated by both mIL-6R and sIL-6R, but not by the receptors of other members of IL-6 cytokine family. Int. Immunopharmacol. 5, 1731–1740 [DOI] [PubMed] [Google Scholar]
- 10.Shaw S., Bourne T., Meier C., Carrington B., Gelinas R., Henry A., Popplewell A., Adams R., Baker T., Rapecki S., Marshall D., Moore A., Neale H., and Lawson A. (2014) Discovery and characterization of olokizumab: a humanized antibody targeting interleukin-6 and neutralizing gp130-signaling. MAbs 6, 774–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garbers C., Aparicio-Siegmund S., and Rose-John S. (2015) The IL-6/gp130/STAT3 signaling axis: recent advances towards specific inhibition. Curr. Opin. Immunol. 34, 75–82 [DOI] [PubMed] [Google Scholar]
- 12.Jostock T., Müllberg J., Ozbek S., Atreya R., Blinn G., Voltz N., Fischer M., Neurath M. F., and Rose-John S. (2001) Soluble gp130 is the natural inhibitor of soluble interleukin-6 receptor transsignaling responses. Eur. J. Biochem. 268, 160–167 [DOI] [PubMed] [Google Scholar]
- 13.Fischer M., Goldschmitt J., Peschel C., Brakenhoff J. P., Kallen K. J., Wollmer A., Grötzinger J., and Rose-John S. (1997) I. A bioactive designer cytokine for human hematopoietic progenitor cell expansion. Nat. Biotechnol. 15, 142–145 [DOI] [PubMed] [Google Scholar]
- 14.Lissilaa R., Buatois V., Magistrelli G., Williams A. S., Jones G. W., Herren S., Shang L., Malinge P., Guilhot F., Chatel L., Hatterer E., Jones S. A., Kosco-Vilbois M. H., and Ferlin W. G. (2010) Although IL-6 trans-signaling is sufficient to drive local immune responses, classical IL-6 signaling is obligate for the induction of T cell-mediated autoimmunity. J. Immunol. 185, 5512–5521 [DOI] [PubMed] [Google Scholar]
- 15.Buell G., Chessell I. P., Michel A. D., Collo G., Salazzo M., Herren S., Gretener D., Grahames C., Kaur R., Kosco-Vilbois M. H., and Humphrey P. P. (1998) Blockade of human P2X7 receptor function with a monoclonal antibody. Blood 92, 3521–3528 [PubMed] [Google Scholar]
- 16.Garbers C., Thaiss W., Jones G. W., Waetzig G. H., Lorenzen I., Guilhot F., Lissilaa R., Ferlin W. G., Grötzinger J., Jones S. A., Rose-John S., and Scheller J. (2011) Inhibition of classic signaling is a novel function of soluble glycoprotein 130 (sgp130), which is controlled by the ratio of interleukin 6 and soluble interleukin 6 receptor. J. Biol. Chem. 286, 42959–42970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Veverka V., Baker T., Redpath N. T., Carrington B., Muskett F. W., Taylor R. J., Lawson A. D., Henry A. J., and Carr M. D. (2012) Conservation of functional sites on interleukin-6 and implications for evolution of signaling complex assembly and therapeutic intervention. J. Biol. Chem. 287, 40043–40050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tabrizi M. A., Tseng C. M., and Roskos L. K. (2006) Elimination mechanisms of therapeutic monoclonal antibodies. Drug Discov. Today 11, 81–88 [DOI] [PubMed] [Google Scholar]
- 19.Dittrich E., Rose-John S., Gerhartz C., Müllberg J., Stoyan T., Yasukawa K., Heinrich P. C., and Graeve L. (1994) Identification of a region within the cytoplasmic domain of the interleukin-6 (IL-6) signal transducer gp130 important for ligand-induced endocytosis of the IL-6 receptor. J. Biol. Chem. 269, 19014–19020 [PubMed] [Google Scholar]
- 20.Dittrich E., Haft C. R., Muys L., Heinrich P. C., and Graeve L. (1996) A di-leucine motif and an upstream serine in the interleukin-6 (IL-6) signal transducer gp130 mediate ligand-induced endocytosis and down-regulation of the IL-6 receptor. J. Biol. Chem. 271, 5487–5494 [DOI] [PubMed] [Google Scholar]
- 21.Graeve L., Korolenko T. A., Hemmann U., Weiergräber O., Dittrich E., and Heinrich P. C. (1996) A complex of the soluble interleukin-6 receptor and interleukin-6 is internalized via the signal transducer gp130. FEBS Lett. 399, 131–134 [DOI] [PubMed] [Google Scholar]
- 22.Zohlnhöfer D., Graeve L., Rose-John S., Schooltink H., Dittrich E., and Heinrich P. C. (1992) The hepatic interleukin-6 receptor. Down-regulation of the interleukin-6 binding subunit (gp80) by its ligand. FEBS Lett. 306, 219–222 [DOI] [PubMed] [Google Scholar]
- 23.Gauldie J., Richards C., Harnish D., Lansdorp P., and Baumann H. (1987) Interferon β2/B-cell stimulatory factor type 2 shares identity with monocyte-derived hepatocyte-stimulating factor and regulates the major acute phase protein response in liver cells. Proc. Natl. Acad. Sci. U.S.A. 84, 7251–7255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Behrens E. M., Canna S. W., Slade K., Rao S., Kreiger P. A., Paessler M., Kambayashi T., and Koretzky G. A. (2011) Repeated TLR9 stimulation results in macrophage activation syndrome-like disease in mice. J. Clin. Investig. 121, 2264–2277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coles B., Fielding C. A., Rose-John S., Scheller J., Jones S. A., and O'Donnell V. B. (2007) Classic interleukin-6 receptor signaling and interleukin-6 trans-signaling differentially control angiotensin II-dependent hypertension, cardiac signal transducer and activator of transcription-3 activation, and vascular hypertrophy in vivo. Am. J. Pathol. 171, 315–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stuhlmann-Laeisz C., Lang S., Chalaris A., Krzysztof P., Enge S., Eichler J., Klingmüller U., Samuel M., Ernst M., Rose-John S., and Scheller J. (2006) Forced dimerization of gp130 leads to constitutive STAT3 activation, cytokine-independent growth, and blockade of differentiation of embryonic stem cells. Mol. Biol. Cell 17, 2986–2995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yawata H., Yasukawa K., Natsuka S., Murakami M., Yamasaki K., Hibi M., Taga T., and Kishimoto T. (1993) Structure-function analysis of human IL-6 receptor: dissociation of amino acid residues required for IL-6-binding and for IL-6 signal transduction through gp130. EMBO J. 12, 1705–1712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grötzinger J., Kurapkat G., Wollmer A., Kalai M., and Rose-John S. (1997) The family of the IL-6-type cytokines: specificity and promiscuity of the receptor complexes. Proteins 27, 96–109 [PubMed] [Google Scholar]
- 29.Pflanz S., Kurth I., Grötzinger J., Heinrich P. C., and Müller-Newen G. (2000) Two different epitopes of the signal transducer gp130 sequentially cooperate on IL-6-induced receptor activation. J. Immunol. 165, 7042–7049 [DOI] [PubMed] [Google Scholar]
- 30.Hoge J., Yan I., Jänner N., Schumacher V., Chalaris A., Steinmetz O. M., Engel D. R., Scheller J., Rose-John S., and Mittrücker H. W. (2013) IL-6 controls the innate immune response against Listeria monocytogenes via classical IL-6 signaling. J. Immunol. 190, 703–711 [DOI] [PubMed] [Google Scholar]
- 31.Ferlin W., Kosco-Vilbois M., Elson G., Leger O., and Guilhot F. (October 11, 2011) United States Patent 8,034,344