

Background: Integrin α6β4 is overexpressed in pancreatic cancer and enhances invasion.
Results: Integrin α6β4 coordinately up-regulates AREG, EREG, and MMP1 through DNA demethylation and NFAT5 that in turn enhances HGF-mediated invasion.
Conclusion: Integrin α6β4 stimulates HGF-dependent invasion through autocrine EGFR signaling.
Significance: HGF-stimulated invasion is dependent on autocrine EGFR signaling, thus implicating why EGFR inhibitors are effective in a complex tumor microenvironment.
Keywords: pancreatic cancer, protein secretion, receptor regulation, receptor tyrosine kinase, signal transduction, receptor protein-tyrosine kinase, receptor protein-tyrosine kinases
Abstract
Integrin α6β4 is up-regulated in pancreatic adenocarcinomas where it contributes to carcinoma cell invasion by altering the transcriptome. In this study, we found that integrin α6β4 up-regulates several genes in the epidermal growth factor receptor (EGFR) pathway, including amphiregulin (AREG), epiregulin (EREG), and ectodomain cleavage protease MMP1, which is mediated by promoter demethylation and NFAT5. The correlation of these genes with integrin α6β4 was confirmed in The Cancer Genome Atlas Pancreatic Cancer Database. Based on previous observations that integrin α6β4 cooperates with c-Met in pancreatic cancers, we examined the impact of EGFR signaling on hepatocyte growth factor (HGF)-stimulated migration and invasion. We found that AREG and EREG were required for autocrine EGFR signaling, as knocking down either ligand inhibited HGF-mediated migration and invasion. We further determined that HGF induced secretion of AREG, which is dependent on integrin-growth factor signaling pathways, including MAPK, PI3K, and PKC. Moreover, matrix metalloproteinase activity and integrin α6β4 signaling were required for AREG secretion. Blocking EGFR signaling with EGFR-specific antibodies or an EGFR tyrosine kinase inhibitor hindered HGF-stimulated pancreatic carcinoma cell chemotaxis and invasive growth in three-dimensional culture. Finally, we found that EGFR was phosphorylated in response to HGF stimulation that is dependent on EGFR kinase activity; however, c-Met phosphorylation in response to HGF was unaffected by EGFR signaling. Taken together, these data illustrate that integrin α6β4 stimulates invasion by promoting autocrine EGFR signaling through transcriptional up-regulation of key EGFR family members and by facilitating HGF-stimulated EGFR ligand secretion. These signaling events, in turn, promote pancreatic carcinoma migration and invasion.
Introduction
Integrins are heterodimeric transmembrane receptors responsible for detecting and integrating signals from the extracellular environment that provide a link between the extracellular matrix and cytoskeleton. Of the 24 mammalian integrin receptors, integrin α6β4 is overexpressed in nearly all pancreatic carcinomas where it stimulates malignant progression by promoting tumor cell migration, invasion, and cell survival (1, 2). In normal epithelial cells, integrin α6β4 contributes to stable attachment of cells to the basement membrane through formation of hemidesmosomes (3). However, during wound healing and in advanced carcinomas, integrin α6β4 is phosphorylated, and hemidesmosomes are subsequently disassembled. Upon disassembly, integrin α6β4 is redistributed to the actin cytoskeleton where it cooperates with growth factor receptor signaling to enhance cell migration and invasion (4, 5). Integrin α6β4 facilitates cell motility by promoting lamellipodia and filopodia formation (6) as well as activating key pathways downstream of growth factor receptors such as PI3K, MAPK, Src family kinases, and Rho family small GTPases (7, 8).
We have found that signaling through the integrin α6β4 dramatically alters the transcriptome, leading to the up-regulation of key pro-invasion and metastatic genes such as autotaxin/ENPP2 (9), S100A4 (10), and TIAM1 (11). In this study, we find that in pancreatic carcinoma cells the integrin α6β4 stimulates the expression of AREG2 and EREG, which are ligands for EGFR.
EGFR and associated EGF-like ligands are dysregulated in many cancers, including pancreatic, head and neck, breast, colorectal, lung, prostate, kidney, ovarian, brain, and bladder (12). Signaling through the EGFR pathway mediates multiple processes involved in tumor progression, including angiogenesis, invasion, migration, proliferation, and evasion of apoptosis (13). Consequently, particular attention has been given to the role of the EGFR pathway in the development of malignant phenotypes, resulting in this pathway being targeted by a substantial array of chemotherapeutics.
There are seven ligands known to bind and signal through EGFR as follows: EGF; transforming growth factor-α; betacellulin; heparin-binding EGF-like growth factor; epigen; AREG; and EREG. Typically after ligand binding, activated EGFR complexes are endocytosed, which leads to recruitment of the ubiquitin ligase c-Cbl. Recruitment of c-Cbl promotes ubiquitination, lysosomal targeting, and degradation of EGFR (14). However, AREG and EREG are unique in their downstream signaling following ligand-receptor binding. Binding of AREG or EREG to EGFR results in a transient recruitment of c-Cbl to EGFR and a reduced level of ubiquitination. This property permits EGFR recycling back to the plasma membrane where it may continue signaling (15, 16). As a result, AREG and EREG have been strongly implicated in tumor progression.
EGFR ligands are integral membrane proteins that typically function in a paracrine and autocrine manner (17). For AREG, this occurs when ADAM-17/TACE (18) or MMP1 (19) cleaves the membrane precursor pro-AREG, releasing it into the extracellular environment. This release creates feedback loops in primary and metastatic sites to promote tumor progression. AREG may also enter the bloodstream and travel to distant organs, acting as an endocrine signal (20), and thus potentially creating a favorable microenvironment (21). This property allows tumors to maintain a high rate of proliferation with a reduced requirement for exogenously supplied growth factors (13). Notably, AREG has been demonstrated to stimulate proliferation of pancreatic ductal cells and associate with an increased frequency of lymph node involvement in pancreatic cancer patients (22). Finally, AREG can induce EGF-independent cell growth by acting as a self-sufficient growth signal in serum-free conditions (23, 24). Likewise, EREG expression is up-regulated in pancreatic cancer and contributes to cell growth by binding to EGFR through paracrine and autocrine loops (25). Similar to AREG, EREG is also cleaved at the cell membrane by Adam-17/TACE (18). Once released, EREG can stimulate the majority of the ErbB heterodimer receptor combinations (26). Although the affinity of EREG to EGFR is lower compared with other EGFR ligands, its signaling potency is higher, thus making EREG a more effective signaling ligand (26).
In this study, we sought to understand how changes in the transcriptome mediated by integrin α6β4 signaling affect pancreatic tumor cell invasion. Because the β4 integrin subunit dimerizes exclusively with the α6 integrin subunit, we can study cellular regulation from the integrin α6β4 by modulating integrin β4 expression (11). We find that integrin α6β4 stimulates the expression of AREG and EREG as well as the ectodomain cleavage enzyme MMP1. We further provide evidence that HGF stimulates the secretion of AREG, which is dependent on integrin signaling pathways. This autocrine secretion in turn promotes HGF-stimulated migration and three-dimensional invasive growth of pancreatic carcinoma cells.
Experimental Procedures
Cell Lines and Cell Culturing
For this study, we used AsPC1, Suit-2, and Panc-1 clones with either high or low expression of the integrin α6β4, as described previously (11, 27). AsPC1, Suit-2, and Panc-1/3D7 cells were used as high expressers of integrin α6β4. Panc-1/1G4 and Panc-1/2G6 cells were used as low expressers of integrin α6β4. Panc-1/β4Δcyt clones lack the cytoplasmic domain of integrin β4 and were used as a dominant negative for integrin α6β4. Suit-2 cells (obtained from Dr. Takeshi Iwamura, Miyazaki Medical College, Miyazaki, Japan) (28) and AsPC1 cells (obtained from American Type Culture Collection, ATCC) were maintained in RPMI 1640 medium. Clones generated from Panc-1 cells (from ATCC) were grown in Dulbecco's modified Eagle's medium (high glucose). All media were supplemented with 10% fetal bovine serum (Sigma), 1% penicillin, 1% streptomycin, and 1% l-glutamine (Gibco).
RNA Extraction and Real Time PCR
Cells were harvested using TRIzol reagent (Invitrogen) to extract total RNA according to the manufacturer's protocol. RNA purity was determined using the OD 260:280 ratio. cDNA was synthesized from 1 μg of RNA using the high capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA). Expression levels of AREG, EREG, and MMP1 were assessed by real time quantitative PCR (QPCR) using available probes, reagents, and a StepOnePlus real time PCR system from Applied Biosystems. Triplicate CT values were analyzed in Microsoft Excel using the comparative CT (ΔΔCT) method as described by the manufacturer (Applied Biosystems). The amount of target gene expressed (2−ΔΔCT) was determined by normalizing to the endogenous reference (18S or β-actin) and relative to one of the experimental samples. Data are representative of at least three separate determinations.
For experiments using 5-aza-2′-deoxycytidine (DAC), Panc-1/2G6 (β4 low), or Panc-1/β4ΔCyt, cells were treated with 5 μm DAC (Sigma; dissolved in DMSO) or DMSO control for 5 days in which fresh drug and medium were added every 24 h. After treatment, cells were harvested and mRNA levels of AREG and EREG measured by QPCR as described above.
Public Database Analysis
Correlations between gene expression (ITGB4, AREG, EREG, and MMP1) were analyzed by linear regression using the pancreatic adenocarcinoma dataset (n = 183) generated by the TCGA Research Network (cancergenome.nih.gov). The processed dataset was downloaded using University of California Santa Cruz Cancer Browser (29). Statistical analyses were performed with GraphPad software (La Jolla, CA).
Migration and Invasion Assays
Transwell chemotactic migration assays were performed as described previously (11). Briefly, for EGFR inhibition, cells were left untreated or treated with EGFR tyrosine kinase inhibitor (TKI) (0.1 nm PD153035) or EGFR neutralizing antibody (4 μg/ml LA1) for 30 min. Transwell chambers were coated with 15 μg/ml laminin-1 for migration assays or with 10 μg of Matrigel for invasion assays (Biosciences). For all assays, the bottom chambers of Transwell (6.5-mm diameter, 8-μm pore size; Corning) were coated with 15 μg/ml laminin-1. Cells (5 × 104) were placed into the top Transwell chamber and allowed to migrate for 4 h or to invade for 6 h, in the presence of EGFR TKI or neutralizing antibody, toward serum-free medium containing 50 ng/ml HGF + 250 μg/ml BSA or BSA alone as a control. Cells that did not migrate or invade were removed from the top chamber using a cotton swab. Cells attached to the bottom of the chamber were fixed with 100% methanol, stained using 3% crystal violet in 2% ethanol, and counted visually using an inverted microscope. Results are reported as the mean number of cells migrated per mm2 from triplicate determinations. Data reported are representative of at least three separate experiments.
Three-dimensional Culture
For three-dimensional culture of pancreatic carcinoma cells, cells (5 × 103) in 200 μl of low serum media (5% FBS in high glucose DMEM) and 2% Matrigel were seeded onto solidified growth factor reduced Matrigel (BD Biosciences; 100 μl per well of 8-well chamber slide). EGFR TKI and EGFR neutralization antibody were added the next day. Cells were fed with media containing 5% FBS and 2% Matrigel every 2 days in the presence of EGFR TKI and EGFR antibody, as indicated. Three-dimensional cultures were maintained for 10–14 days before imaging. Phase contrast images of randomly chosen fields were taken with a Nikon Ti-E inverted microscope using Nikon Elements software. Images were cropped in Adobe Photoshop and assembled in Adobe Illustrator CS5.
Gene Knockdown by RNAi
For siRNA treatment, cells from 70% confluent cultures were trypsinized, rinsed three times, and resuspended in serum-free DMEM. Cells (3 × 106) were electroporated without or with 200 nm non-targeting siRNA (Dharmacon, Inc.) or 200 nm siRNA specific for NFAT5 (nuclear factor of activated T-cells 5) as described previously (9) and replated under normal culturing conditions. Cells were collected after 72 h and assessed for AREG and EREG expression by QPCR as described above.
For stable knockdown of AREG and EREG, lentivirus was produced by combining MISSION constructs for packaging (psPAX2), envelope (pDM2G) vectors, and shRNA for AREG or EREG or a non-targeting vector (pLKO.1) at a 4:2:1 ratio (all vectors obtained from Sigma). Polyethyleneimine (Polysciences) was combined with the DNA mixture at a 3:1 DNA to polyethyleneimine ratio and added to 70% confluent HEK 293LTV cells that had been passaged 24 h prior. Conditioned media were collected 24 and 48 h post-transfection and centrifuged at 5,000 × g for 10 min, and viral supernatant was collected. Panc-1/3D7 cells were passaged to 70% confluence in 10-cm dishes, and 4 ml of viral supernatant was added in combination with 8 μg/ml hexadimethrine bromide (Polybrene, Sigma). Cells were placed under puromycin selection (2 μg/ml) for 3 days, and gene expression was measured by QPCR 24 h following removal of puromycin. If knockdown efficiency was at least 90%, migration and invasion assays were performed the following day as described above.
AREG ELISA
Pancreatic carcinoma cells (1.5–2.0 × 105) were plated in 6-well plates in complete culture medium. The following day, cells were rinsed twice with PBS and then placed in serum-free medium containing 250 μg/ml BSA. For HGF stimulation experiments, cells were rinsed after 4 h in serum-free medium and then stimulated with HGF (10 or 50 ng/ml, as noted). After 24 h, conditioned medium was harvested, cleared by centrifugation, volume recorded, and AREG content analyzed by ELISA (human amphiregulin ELISA kit, catalog no. ELH-AR-001, RayBiotech, Inc.) using recombinant human AREG as a standard. Cells were counted by hemocytometer at the conclusion of each experiment to confirm equal cell numbers. Data from triplicate determinations are reported as picograms of AREG/ml/105 cells with standard deviation of the mean. For all experiments, data presented are representative of at least three separate determinations.
For inhibitor studies, cells were pretreated for 30 min with DMSO only, 50 μm PD98059 or 20 μm U0126 (MEK/MAPK), 15 μm H-89 (PKA), 2.5 μm GF109203X (PKC), 1 μm PP2 (Src), or 20 μm LY294002 (PI3K) after the initial serum starvation steps. Medium was then changed, and cells were stimulated with 50 ng/ml HGF in the presence or absence of inhibitor for 24 h before assessing the conditioned medium for AREG as described above. For DAC experiments, cells were treated with 5 μm DAC or DMSO as a control for 5 days, rinsed with PBS, and serum-starved for 4 h in serum-free medium plus 250 μg/ml BSA, and then stimulated with 50 ng/ml HGF in the presence of DAC or DMSO as a control. The next day, conditioned medium was analyzed for AREG expression levels as described above.
Immunoprecipitation and Immunoblot Analysis
Suit-2 cells were plated on laminin-1-coated plates for 3 h and then treated with EGFR TKI (PD153035) for 30 min prior to 5 or 30 min of stimulation with 50 ng/ml HGF, as noted. Cells were harvested in cold RIPA buffer containing 50 mm sodium fluoride, 10 mm sodium pyrophosphate, and 1 mm sodium orthovanadate plus protease inhibitors. EGFR or c-Met was immunoprecipitated from cell lysates using EGFR Erbitux (Lilly, cetuximab) or c-Met-specific (Santa Cruz Biotechnology, sc-161) antibodies, respectively, coupled to protein A/G-agarose beads (Pierce). Beads were washed four times, and then immunoprecipitates were separated by 6% SDS-PAGE, transferred, and immunoblotted for phosphotyrosine (PY20), EGFR (Santa Cruz Biotechnology, sc-03), or c-Met (Santa Cruz Biotechnology, sc-161), as indicated. Alternatively, cell lysates (80 μg) from control or HGF-treated cells were separated by 8% SDS-PAGE and immunoblotted with antibodies against p-EGFR for site p-Y845 (catalog no. 2231S), p-Y1045 (catalog no. 2237s), p-Y1068 (catalog no. 2234S), or p-Y1173 (catalog no. 4407S) (from Cell Signaling) using total EGFR (catalog no. sc-03, Santa Cruz Biotechnology) and/or actin (Sigma) as loading controls. Data are representative of three separate experiments.
Results
We (1) and others (2, 30, 31) have shown that integrin α6β4 is up-regulated in 92–100% of pancreatic adenocarcinomas. Furthermore, this integrin can promote pancreatic carcinoma invasiveness (11), in part through transcriptional regulation (10, 11). Transcriptional profiling of integrin α6β4-responsive genes (characterized in Ref. 11) revealed that integrin α6β4 is associated with the substantial up-regulation of genes within the EGFR pathway, including the EGFR ligands AREG and EREG and MMP1, a pro-tumorigenic protease that has been previously shown to cleave the pro-form of AREG and EREG, promoting autocrine release (19). These findings were validated by QPCR analysis. As shown in Fig. 1A, AREG and EREG are induced over 1,000-fold and MMP1 over 300-fold in cells with high integrin α6β4 expression compared with low expressing clones and the parental Panc-1 population. The contribution of integrin α6β4 was confirmed by stably reducing integrin α6β4 using integrin β4-targeting shRNA in the high integrin β4-expressing Panc-1 clone 3D7 (Fig. 1B) or by expressing a dominant-negative form of integrin β4, which lacks the cytoplasmic domain, into the Panc-1 cells (β4Δcyt; Fig. 1A). Furthermore, overexpression of a wild-type integrin β4 subunit into the low integrin β4-expressing clone (2G6) stimulated expression of AREG, EREG, and MMP1 (Fig. 1C). To confirm that these genes correlate with integrin α6β4 expression in human pancreatic tumors, we analyzed the TCGA pancreatic ductal adenocarcinoma database, which contains gene expression data as measured by RNAseq (see “Experimental Procedures”). We find that each of these genes positively and significantly correlates with expression of the integrin β4 subunit (ITGB4; Fig. 1D). These data demonstrate that integrin α6β4 controls expression of AREG, EREG, and MMP1 in pancreatic carcinoma cells.
FIGURE 1.

Integrin α6β4 controls the mRNA expression of AREG, EREG, and MMP1. A–C, RNA was isolated from various Panc-1 clones, including Panc-1 parental cells, Panc-1 clones with low (1G4, 2G6) or high (3D7) expression of integrin α6β4, or a clone expressing a dominant-negative β4 that lacks the cytoplasmic domain (β4Δcyt) (A); from Panc-3D7 cells expressing a non-targeting shRNA construct (3D7) or cells in which β4 integrin was stably knocked down with a specific shRNA (3D7-β4 shRNA) (B); and from two Panc-2G6 clones overexpressing wild-type β4 (wt β4, C). RNA was assessed by QPCR (A–C) for expression of AREG (top panels), EREG (middle panels), or MMP1 (bottom panels). Values were normalized to 18S and reported relative to Panc-2G6 using the 2−ΔΔCT method, as described under the “Experimental Procedures.” Experiments are depicted as the mean relative mRNA expression ± standard deviation of triplicate determinations. D, these findings were validated in an external dataset (TCGA), where by linear regression the gene expression levels of the integrin β4 subunit (ITGB4) were found to positively correlate with expression of AREG, EREG, and MMP1 in patient-derived pancreatic adenocarcinomas with p < 0.0001.
Based on these correlations between the integrin β4 and members of the EGFR family, we sought to explain mechanistically how integrin α6β4 regulates their gene expression. It has previously been established that integrin β4 can signal downstream to transcription factors, including NFAT1 and NFAT5, to promote tumor cell invasion (10, 32). Considering that nuclear factor of activated T-cell consensus sites are found within the AREG and EREG sequences, we hypothesized that these genes could be targets for the nuclear factor of activated T-cells. To determine whether AREG and EREG are regulated by NFAT5 downstream of integrin α6β4 signaling, we used siRNA to target NFAT5. We find that both AREG and EREG expression are substantially decreased with NFAT5 knocked down in integrin β4 low cells (2G6; Fig. 2A) and even more robustly when integrin β4 is high (3D7; Fig. 2B). Knockdown of NFAT5 by siRNA was confirmed by QPCR (Fig. 2, C and D). These data indicate that the NFAT5 transcription factor is required for expression of AREG and EREG in response to signaling from the integrin α6β4.
FIGURE 2.
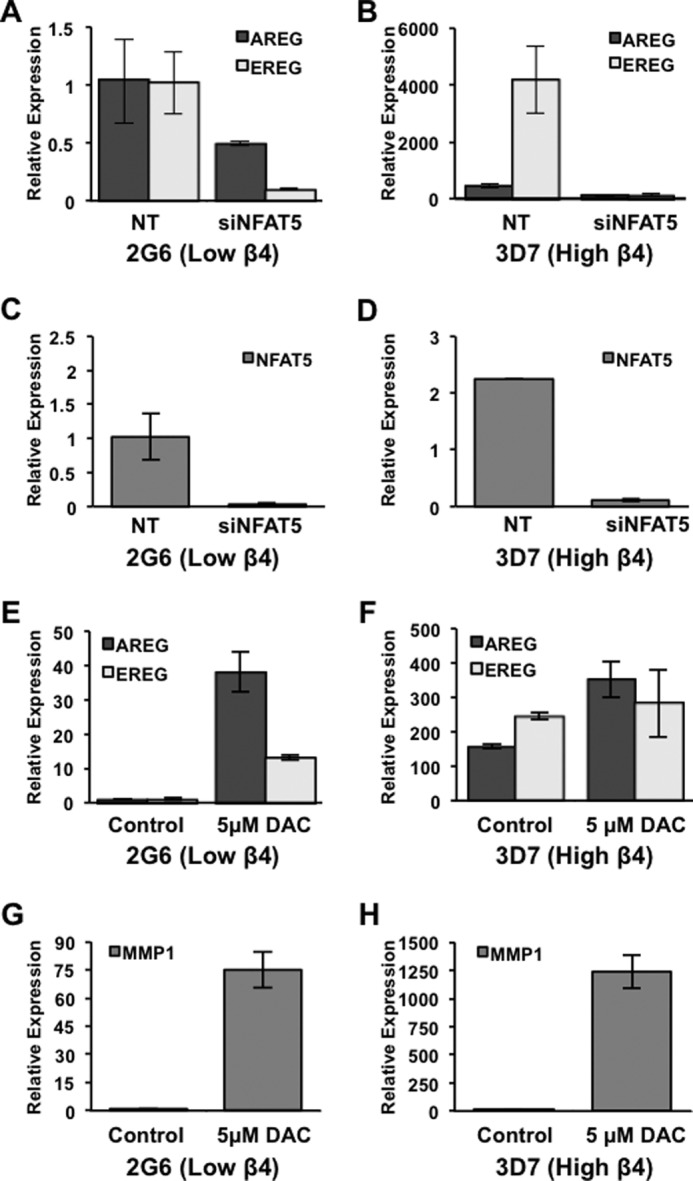
Integrin α6β4 regulates gene expression of AREG and EREG via NFAT5 and DNA demethylation. Panc-1 clones 2G6 and 3D7 were electroporated with siRNA specific for NFAT5 or non-targeting (NT) control. After 72 h in culture, RNA was harvested from treated cells and assessed for AREG and EREG expression (A and B) and for knockdown efficiency of NFAT5 (C and D) by QPCR. To examine DNA methylation, Panc-1 clones 2G6 and 3D7 were incubated for 5 days with or without 5 μm DAC in fresh medium daily, as noted. Cells were harvested, and gene expression levels of AREG, EREG, and MMP1 were measured by QPCR, as noted (E–H). Experiments depicted here are representative of at least three separate experiments. Data represent the mean ± S.D.
Another mechanism by which the integrin α6β4 may regulate gene expression of AREG and EREG is through DNA demethylation. We previously found that integrin α6β4 controls the DNA demethylation of select promoters in breast cancer, including metastasis-associated protein S100A4 (10). To determine whether AREG and EREG were similarly controlled by DNA demethylation, Panc-2G6 (β4 low) and Panc-β4Δcyt cells were treated with the DNA methyltransferase inhibitor, DAC, and assessed for expression by QPCR. We find that AREG, EREG, and MMP1 expression are dramatically up-regulated with DNA methyltransferase inhibition in Panc-2G6 cells (Fig. 2, E and G) and moderately up-regulated in Panc-3D7 cells (Fig. 2, F and H), indicating that gene expression is regulated by DNA methylation, in response to signaling from the integrin α6β4.
Integrin α6β4 promotes invasion by cooperating with growth factor receptors such as c-Met (4). Notably, c-Met and its associated ligand, HGF, are important players in pancreatic cancer (33). Therefore, we examined the individual contributions of AREG and EREG to HGF-mediated pancreatic cancer cell migration and invasion. Although it is well established that AREG and EREG contribute to cancer cell metastasis (25, 34), how autocrine EGFR secretion contributes to directed cell motility is less well defined. Therefore, we reduced AREG and EREG expression in cells with high integrin α6β4 (3D7) using target-specific shRNA, and we performed HGF-stimulated chemotaxis and chemoinvasion. Cells were assessed for AREG and EREG expression by QPCR (Fig. 3, E and F) the day prior to chemotaxis and chemoinvasion assays. Inclusion in chemotaxis and chemoinvasion required a minimum of 90% knockdown of AREG or EREG. As shown in Fig. 3, AREG or EREG knockdown results in decreased migration (Fig. 3, A and B) and invasion (Fig. 3, C and D) of Panc-3D7 cells. Notably, effective knockdown of one ligand resulted in decreased expression of the other ligand by about 50%. These data indicate that both AREG and EREG are required for HGF-mediated migration and invasion of pancreatic cancer cells.
FIGURE 3.
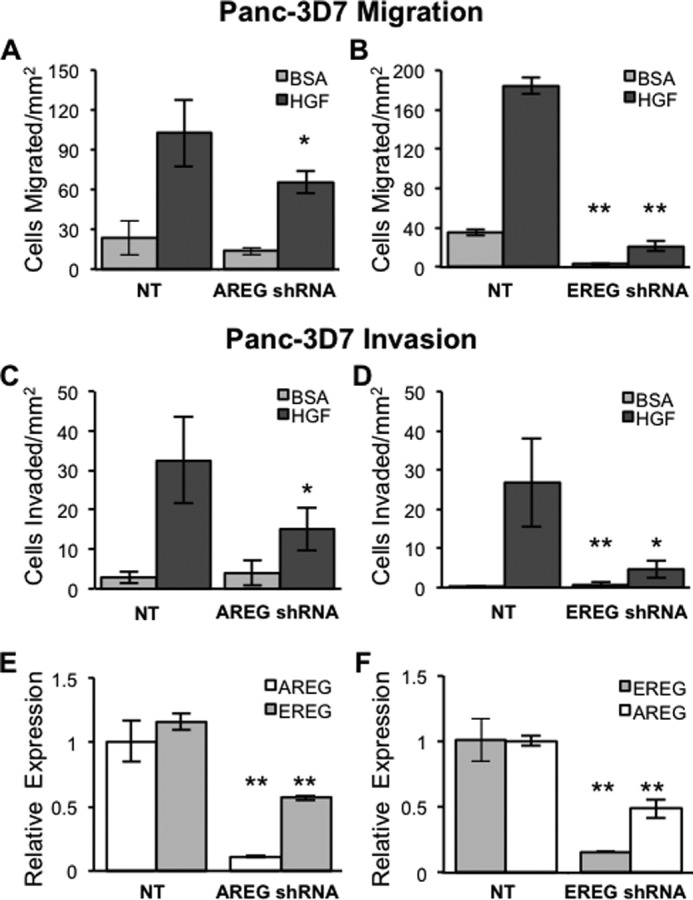
AREG and EREG are both required for chemotactic migration and invasion of pancreatic carcinoma cells. AREG or EREG expression levels were reduced using lentiviral delivery of specific shRNAs. Upon stable selection with puromycin, cells were assessed for AREG (E) and EREG (F) expression by QPCR. Populations that achieved a minimum of 90% reduction in expression of AREG or EREG specifically were assayed for migration or invasion the following day. A–D, cells (5 × 104) were placed into the top well of laminin-1-coated transwells for migration (A and C) or Matrigel-coated wells for invasion (B and D) with either BSA containing DMEM (DMEM/BSA) alone or DMEM/BSA with 50 ng/ml HGF in the bottom well, as described under “Experimental Procedures.” Cells were allowed to migrate for 4 h or invade for 6 h prior to harvest and quantification. Data depicted here are representative of at least three different experiments and represent the mean ± S.D. Statistical significance was calculated using a one-tailed t test in which *, p < 0.05, and **, p < 0.005. NT, non-targeting.
The need for EGFR signaling during HGF-stimulated chemotaxis suggests that EGFR ligands may be released in response to HGF signaling in cooperation with the integrin α6β4. To test this hypothesis, we utilized a human AREG ELISA to assess AREG release from pancreatic cancer cells. For these experiments, we first analyzed AREG secretion under normal culturing conditions. As shown in Fig. 4A, we find that AREG is secreted by cells expressing high integrin α6β4 (AsPC1, Suit-2, and 3D7) but not in integrin β4-low cells (2G6). To determine whether HGF stimulates AREG release, cells were serum-starved for 4 h and then treated with HGF overnight. Conditioned medium was collected and tested for AREG levels by ELISA (Fig. 4B). These data show that HGF stimulated 2–4-fold greater AREG secretion versus controls in all three cell lines tested. Considering that DAC can induce AREG and EREG expression (Fig. 2C), we treated Panc-1 clones 2G6 and β4Δcyt with 5 μm DAC for 5 days to induce expression of AREG and test the impact of integrin α6β4 on the process of AREG secretion. After treatment, cells were assessed for HGF-stimulated AREG secretion by ELISA and compared with the Panc-3D7 cells. As shown in Fig. 4C, DAC induced AREG expression to a level approximately one-quarter that of untreated Panc-3D7 cells. With AREG secretion, however, the level of AREG in the medium from unstimulated and HGF-stimulated cells differed by 20–60-fold higher in Panc-3D7 cells compared with the Panc-β4Δcyt and Panc-2G6 cells. This observation implicates integrin α6β4 signaling in the secretion of AREG in addition to regulation by DNA demethylation. Taken together, these data indicate that the integrin α6β4 can drive both expression and subsequent release of AREG and that this release is amplified by cross-talk with the c-Met receptor.
FIGURE 4.
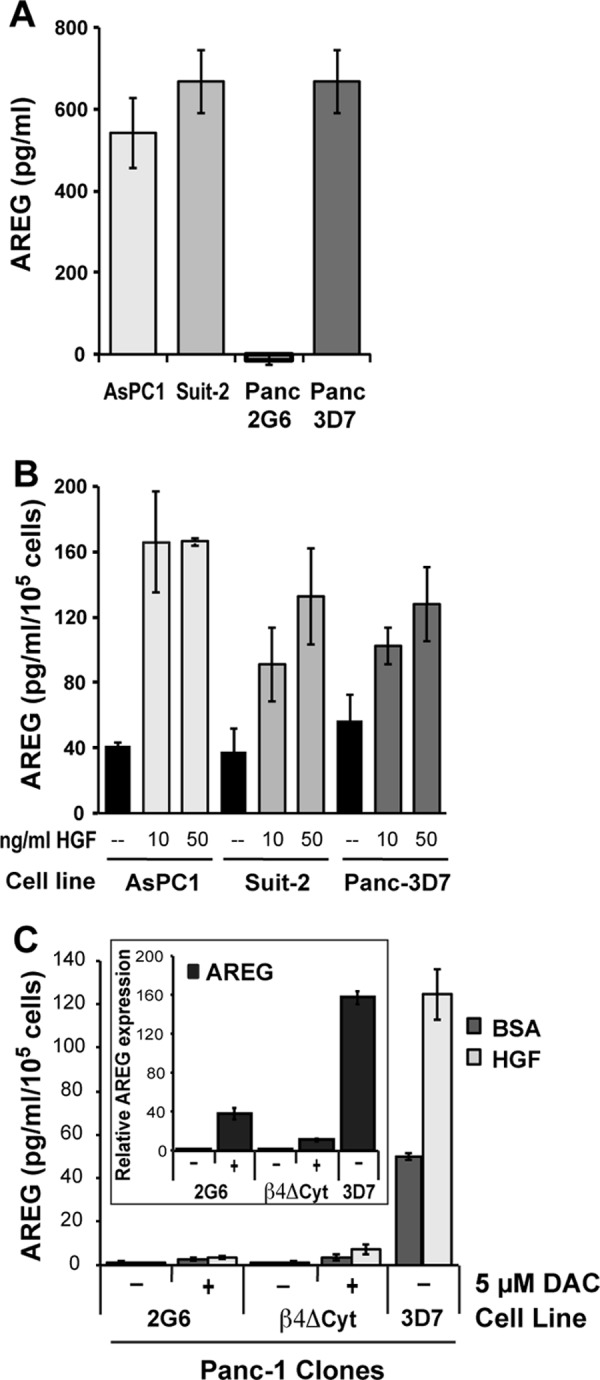
HGF stimulates secretion of AREG in cells with high integrin α6β4. A, AsPC1, Suit-2, Panc-2G6, and Panc-3D7 cells (1 × 105/well) were plated in 6-well dishes containing complete growth medium overnight. The next day, cells were washed with PBS, and culture medium was changed. After 24 h, the conditioned medium was assayed for AREG by ELISA (RayBiotech) and reported as picograms of AREG/ml. B, cells (1 × 105/well) were plated into 6-well dishes under normal culturing conditions. The following day, cells were serum-starved for 4 h before medium was changed, and HGF (0, 10, or 50 ng/ml, as noted) in serum-free medium was added. After 24 h, culture medium for each condition was assessed for AREG content by ELISA, and cells were counted to confirm equal cell numbers. C, cells were incubated for 5 days with or without 5 μm DAC in fresh medium daily. Cells were then replated at 1.5 × 105 cells into 35- mm dishes on the 4th day. The following day, cells were treated with either BSA or 50 ng/ml HGF as described in B and then harvested the following day for conditioned medium assayed by AREG ELISA. Data are reported as the mean picograms of AREG per ml of culture medium per 1 × 105 cells ± S.D. of triplicate determinations. Inset in C shows gene expression analysis of AREG to confirm that DAC induced gene expression in the same experiment.
To resolve which pathways mediate HGF-stimulated AREG release, specific inhibitors of growth factor signaling pathways enhanced by integrins, including MAPK, PKA, Src, and PI3K (7, 35–39), and the classical PKC pathway, were tested for their ability to block AREG secretion in response to HGF. As shown in Fig. 5, inhibitors of the MEK-ERK, PKC and PI3K pathways blocked HGF-mediated release of AREG in both Panc-3D7 and Suit-2 cells. Notably, MEK inhibitors also impeded basal secretion, thus suggesting that this pathway guides the basal secretion of AREG as well as HGF-stimulated secretion.
FIGURE 5.
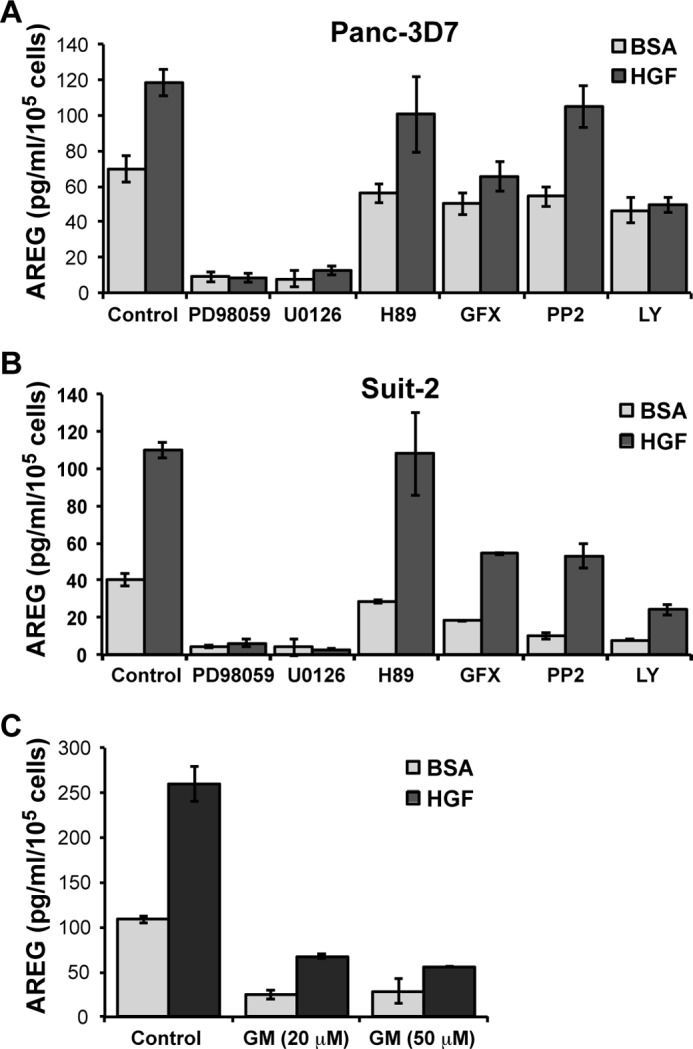
HGF-stimulated release of AREG from cells requires MAPK, PKC, and PI3K. Panc-3D7 (A) and Suit-2 (B) cells (2 × 105/well) were serum-starved for 4 h as described in Fig. 4B. Cells were then treated with inhibitors of MAPK/MEK (PD98059, U0126), PKA (H89), PKC (GF109203X (GFX)), Src (PP2), and PI3K (LY294002 (LY)) for 15 min as described under “Experimental Procedures.” Alternatively, Panc-3D7 cells (C) were treated with general metalloproteinase inhibitor GM6001 (GM) at the indicated concentration (20 or 50 μm). Cells were then stimulated with HGF in the presence of the indicated inhibitor for 24 h before conditioned medium was assessed for AREG content by ELISA, and cells were counted by a hemocytometer. Data are reported as picograms of AREG per ml of culture medium per 1 × 105 cells and are representative of at least three different experiments. Data are reported as the mean of triplicate determinations ± S.D.
The regulated release of EGFR ligands can be mediated by metalloproteinases such as MMP1 (19). The MMPs that release EGFR ligands can in turn be regulated by serine/threonine phosphorylation of their cytoplasmic domain by MAPK and other kinase pathways (40). Given our observation that MMP1 expression is up-regulated by integrin α6β4, we tested the role of MMP1 in HGF-mediated AREG secretion using the MMP chemical inhibitor GM6001. As shown in Fig. 5C, the MMP inhibitor strongly suppressed both basal and HGF-stimulated AREG secretion, thus suggesting a critical role for MMP1 in AREG secretion. Collectively, these data define an important role for integrin α6β4 in the coordinated regulation of autocrine EGFR ligand expression and regulated release.
We determined that AREG and EREG are required for pancreatic cancer cell migration and invasion and that HGF enhances integrin-mediated AREG secretion. Next, we sought to determine whether activation of EGFR signaling facilitates HGF-stimulated migration. For these experiments, we pretreated pancreatic carcinoma cells with either an EGFR protein-tyrosine kinase inhibitor (PD153035) or an EGFR function-blocking antibody (LA1) prior to assessing HGF-stimulated chemotaxis. As shown in Fig. 6, cells originating from a primary tumor with high integrin α6β4 expression (Panc-3D7 and Suit-2; Fig. 6, A and C) were sensitive to EGFR inhibition. Cells that had low integrin α6β4 (Panc-2G6, Fig. 6B) did not chemotax well toward HGF, and this limited chemotactic migration was not sensitive to EGFR signaling. However, inhibition of EGFR signaling in AsPC1 cells (Fig. 6D), which are derived from ascites (41), had no effect on cell migration toward HGF.
FIGURE 6.
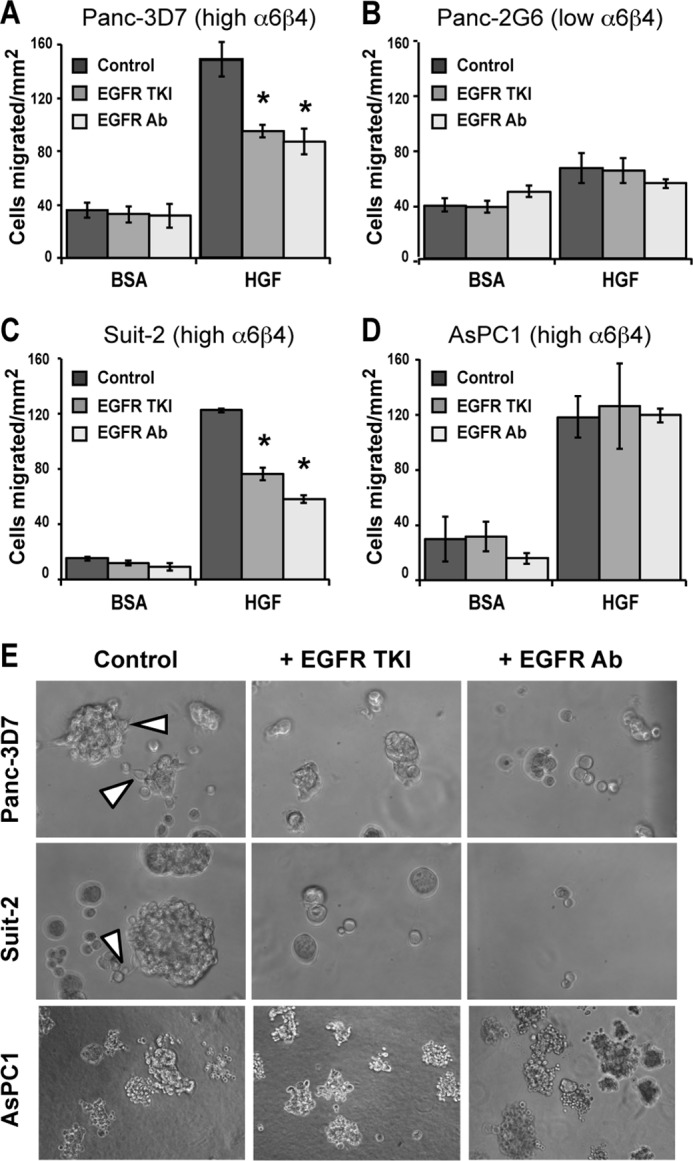
EGFR signaling is required for HGF-stimulated chemotaxis and invasive growth in pancreatic carcinoma cells overexpressing integrin α6β4. A–D, Panc-1 clones 3D7 (A) or 2G6 (B) or Suit-2 (C) or AsPC1 (D) cells were left untreated or treated with EGFR TKI (0.1 nm PD153035) or EGFR neutralizing antibody (4 μg/ml LA1) for 30 min. Cells (5 × 104) were placed into the top of laminin1-coated transwells and allowed to migrate for 4 h toward BSA or 50 ng/ml HGF in the presence of drug. Data depicted here are representative of at least three different experiments and represent the mean ± S.D. Asterisks represent a p value of <0.002 compared with controls (no drug). Other bars are not significantly different from their controls. E, Panc-3D7, Suit-2, or AsPC-1 cells (as noted) were grown in Matrigel under reduced serum conditions for 10 days as described under “Experimental Procedures.” EGFR TKI (0.1 nm PD153035) or EGFR antibody (4 μg/ml LA1) were added every 2 days at feeding. Note that both treatments limited growth of spheres and inhibited invasion from the spheres in Panc-3D7 and Suit-2 cells but not AsPC-1. Invasion of control cells is noted with arrowheads.
Considering that autocrine secretion of EGFR ligands creates a favorable microenvironment for proliferation and invasion, we tested the effect of EGFR inhibitors on three-dimensional invasive growth (42). Assessment of three-dimensional invasion combines the effects of invasion and proliferation in a more physiological three-dimensional environment. As shown in Fig. 6E, EGFR inhibitors blocked invasive growth and limited colonies to small acinar-like spherical masses in Panc-3D7 and Suit-2 cells, as indicated. Notably, these inhibitors did not affect AsPC1 cells in three-dimensional culture (Fig. 6E, bottom panels).
To determine how c-Met-EGFR cross-talk impacts receptor phosphorylation, we immunoprecipitated EGFR and c-Met from Suit-2 cells that were left unstimulated or stimulated with HGF in the presence or absence of an EGFR TKI. As shown in Fig. 7A, we found that HGF stimulation led to an increase in EGFR phosphorylation and that this phosphorylation was blocked by the EGFR TKI; however, c-Met phosphorylation was unaffected by EGFR signaling. We next examined the phosphorylation of individual tyrosines in EGFR in response to HGF. We found that treatment with HGF stimulated phosphorylation of EGFR at multiple sites. As shown in Fig. 7B, the phosphorylation site with the most dramatic increase in phosphorylation upon HGF treatment was tyrosine 845, which is phosphorylated by Src and leads to the activation of the kinase domain of EGFR (43). Phosphorylation of the Grb2- (Tyr-1068) and Shc (Tyr-1173)-docking sites were also enhanced by HGF stimulation, whereas the Cbl-binding site (Tyr-1045) was minimally impacted (Fig. 7B).
FIGURE 7.
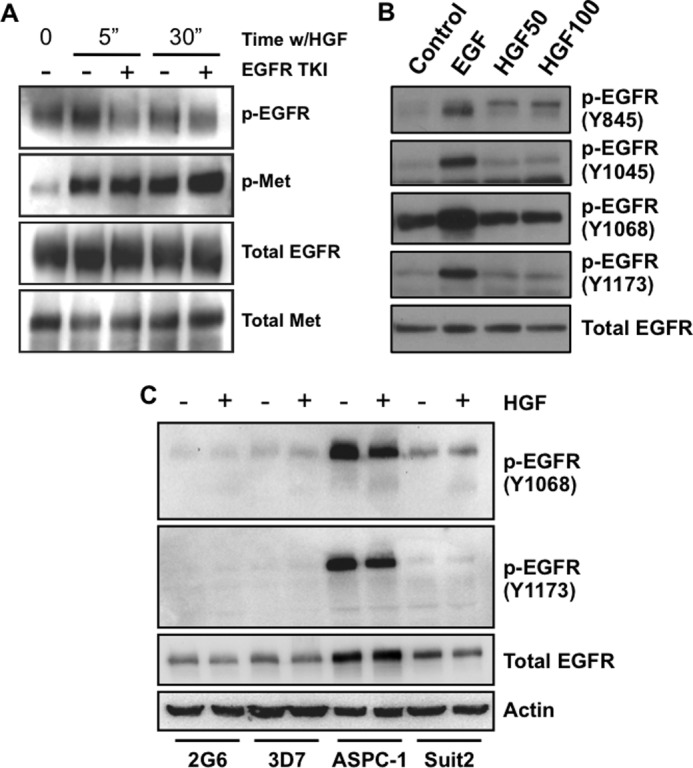
HGF stimulates EGFR phosphorylation, but EGFR signaling does not affect HGF-dependent phosphorylation of c-Met. A, Suit-2 cells were plated on laminin-1-coated plates for 3 h and then treated with EGFR TKI (PD153035) for 30 min prior to 5 or 30 min of stimulation with 50 ng/ml HGF where noted. EGFR or c-Met were immunoprecipitated from cell lysates and immunoblotted for phosphotyrosine (PY20), EGFR, or c-Met as indicated. B, Suit-2 cells were plated on laminin-1-coated dishes as in A and stimulated with 5 ng/ml EGF or 50 ng/ml HGF (HGF50) or 100 ng/ml HGF (HGF100) for 5 min as indicated before harvesting immunoblot analysis. C, Panc-2G6, Panc-3D7, AsPC-1, and Suit2 cells were seeded into 60-mm dishes, serum-starved overnight, and then stimulated with 50 ng/ml HGF for 5 min. Cell lysates were then immunoblotted with EGFR phosphotyrosine antibodies as noted (B and C). Total EGFR and actin were used as controls.
Next, we compared the impact of HGF stimulation on EGFR phosphorylation in various cells to determine how the AsPC1 cells might differ from the other pancreatic carcinoma cell lines. We found that AsPC1 cells have markedly higher basal EGFR phosphorylation level (Fig. 7C) compared with other pancreatic cancer cell lines. Furthermore, HGF stimulation led to a slightly lower level of phosphorylation of Tyr-1068 and Tyr-1173 with HGF stimulation (Fig. 7C), which was a consistent finding among experiments. Collectively, these data indicate that HGF stimulates autocrine release of the EGFR ligands AREG and EREG and that this signaling in turn activates EGFR, which further propagates c-Met signaling. Furthermore, these data suggest that the high basal level of EGFR phosphorylation and insensitivity of this phosphorylation status to c-Met signaling may underlie the resistance of AsPC1 cells to EGFR inhibition.
Discussion
Integrin α6β4 is highly up-regulated in pancreatic cancer (1, 2) and potentiates an invasive phenotype in response to multiple growth factor receptors, including EGFR, c-Met, FGF, Ron, and lysophosphatidic acid receptors (4, 44–46). Our group (9) and others (32, 47, 48) have demonstrated that integrin α6β4 can activate important tumor-promoting transcription factors such as NFAT1, NFAT5, NFκB and AP-1. These factors, along with DNA demethylation of specific genes (10), alter the transcriptome to facilitate an invasive phenotype. Here, we expand on these observations and find that integrin α6β4 coordinates the increased expression of multiple components of the EGFR pathway, including a slight elevation in receptor expression (data not shown), de novo expression of the EGFR ligands AREG and EREG, and elevation of a key protease responsible for cleavage of these EGFR ligands into soluble growth factors. We find that both the transcription factor NFAT5 as well as DNA demethylation are required for expression and that integrin α6β4 is needed for the subsequent secretion of AREG and EREG. Using the TCGA dataset on pancreatic ductal adenocarcinoma, we find that AREG, EREG, and MMP1 correlate strongly with expression of the integrin β4 subunit, which is indicative of integrin α6β4 expression. This coordinated regulation of the EGFR ligands has been shown previously as a mechanism for pathway dependence in pancreatic cancer (49). Notably, we find that the combination of these genes permits pancreatic cancer cells to secrete EGFR ligands in response to HGF and, in turn, to promote the efficiency of HGF-mediated invasion and migration.
We further find that both AREG and EREG are required for HGF-mediated invasion and migration of pancreatic carcinoma cells. Notably, the contributions of AREG and EREG as pro-oncogenic factors in pancreatic cancer have previously been established (22, 50). Signaling from AREG, specifically autocrine secretion, has been implicated in chemoresistance, evasion of apoptosis, self-sufficiency in growth signals, proliferation, angiogenesis, invasion, and metastasis (20). In fact, AREG secretion levels in pancreatic cancer patients are elevated to the point that this ligand has been identified as a potential biomarker that is associated with a worse outcome (51). It may be that high levels of AREG are needed because of its low affinity for the receptor compared with other ligands. EREG, for example, is found at much lower concentrations but has a broader specificity for ErbB receptors and very high binding affinity, thus resulting in more potent signaling (26).
The involvement of integrin α6β4 in the regulation and secretion of growth factors that facilitate invasion appears to be an unexpectedly common event. Previous work from Mercurio and co-works (52) demonstrated that integrin α6β4 controls the Hif1α-mediated translation of VEGF, which then produces an autocrine effect to promote invasion and survival of breast carcinoma cells. Our laboratory has also previously shown that integrin α6β4 regulates the expression of the autocrine motility factor autotaxin (9) and S100A4/metastasin-1 (10), both of which can promote the migration and invasion of breast cancer cells. The ability of integrin α6β4 to promote the expression and secretion of factors such as autotaxin, S100A4, VEGF, AREG, and EREG suggests that autocrine secretion is a major function of integrin α6β4-mediated migration and invasion. Downstream of these secreted factors, integrin α6β4 is also well known for cooperating with various growth factor receptors, including EGFR, c-Met, and LPAR1 to amplify Rho family GTPase family members Rac (11, 53) and Rho (8, 54). These events in turn permit cell polarization and enhanced cell motility and invasion. Despite our understanding of these signaling pathways, how the regulated autocrine secretion helps drive migration and invasion is not well understood. According to our findings, cells can migrate in the absence of productive autocrine signaling (e.g. Panc-2G6), but this migration is not efficient. Furthermore, integrin-mediated signaling cooperates with HGF to stimulate AREG release (Fig. 5). Together, these observations suggest that integrin signaling and EGFR ligand secretion can amplify specific signals to promote pancreatic cancer cell migration and invasion.
These results led us to a model (Fig. 8) where signaling from the integrin α6β4 promotes enhanced transcription of MMP1, AREG, EREG, and EGFR, which are subsequently expressed at the cell surface. Activation of c-Met by HGF results in cooperative signaling with the integrin α6β4 leading to activation of MMP1. MMP1 cleaves AREG and EREG proproteins, releasing them into the extracellular space, allowing binding to and activation of EGFR. This amplified signaling network results in enhanced pancreatic cancer cell invasion and migration.
FIGURE 8.
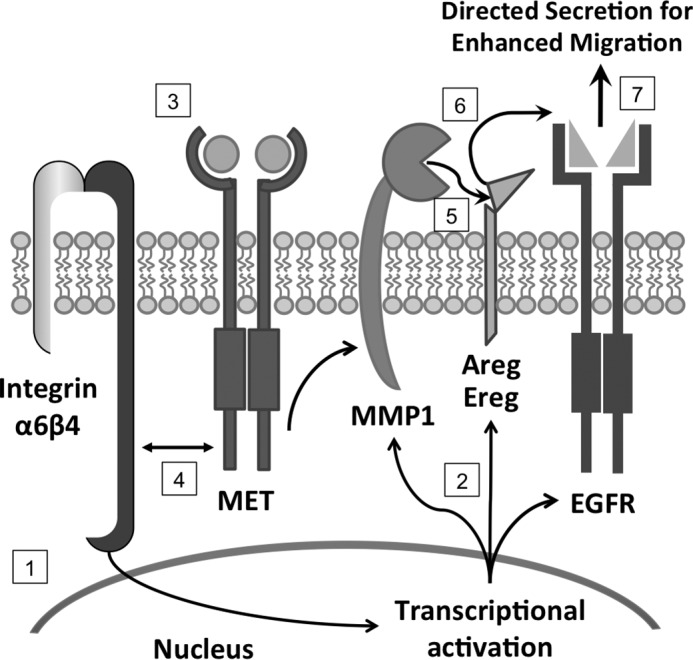
Cooperative signaling between EGFR, c-Met, and integrin α6β4 promotes pancreatic cancer cell migration and invasion. Integrin α6β4 signaling enhances transcription of EGFR ligands, AREG and EREG, as well as MMP1 and EGFR (1). Translated proteins are brought to the cell surface for activation (2). Upon HGF-mediated stimulation (3), c-Met and integrin signaling cooperate to activate MMP1 (4) through the MAPK, PKC, and PI3K pathways (data not shown). MMP1 cleaves AREG and EREG proproteins (5), which in turn are released and lead to autocrine activation of EGFR (6) and result in enhanced cell migration and invasion (7).
EGFR inhibitors have been quite effective in treating a variety of cancers (55). Despite this success, resistance is often a significant problem. The mechanisms governing resistance to EGFR-based treatments have been an active field of research. Perhaps the more pertinent question is why do EGFR inhibitors work at all given the prevalence and variety of growth factors present in the tumor stroma that could promote proliferation and invasion? The answer to this question may reside in the ability of EGFR to potentiate signaling from other growth factors. Our study demonstrates a surprising dependence of c-Met signaling on EGFR for more efficient guidance of tumor cell migration and invasion. Previous studies have highlighted the ability of lysophosphatidic acid signaling to cause transactivation of EGFR resulting in increased cancer cell invasion (56, 57). Accordingly, the effectiveness of EGFR inhibitors may be mediated, in part, by the dependence of other growth factor signaling on EGFR. c-Met signaling is well recognized as a mechanism of resistance for EGFR inhibitors (58). This is highlighted by the effective use of the c-Met inhibitor crizotinib in the clinic to treat EGFR inhibitor-resistant cancers such as non-small cell lung cancer (NSCLC) (59). Furthermore, a 2011 study by Siegfried and co-workers (62) found that EGFR signaling in NSCLC led to the delayed Src-dependent activation of the c-Met receptor in a manner that appears to be independent of HGF. They further discovered that inhibition of both growth factors was required for effective treatment of NSCLC growth in a xenograft model. Other mechanisms of resistance to EGFR inhibitors include the mutation, overexpression, or hyperactivation of EGFR (60, 61). Notably, we see in our studies that AsPC1 cells overexpress EGFR but also display dramatic hyperactivation of EGFR under unstimulated conditions, which may contribute to the inherent resistance of this cell line to EGFR inhibitors. Therefore, we propose that the ability for EGFR to cooperate with and potentiate signaling from other growth factors should be duly noted as a potent method for facilitating tumor invasion. Furthermore, by understanding these signaling events, other mechanisms of EGFR resistance may be uncovered.
In summary, our study demonstrates that integrin α6β4 signaling coordinately regulates the expression of the EGFR ligands AREG and EREG and the ectodomain cleavage protease MMP1. Furthermore, this integrin facilitates the regulated secretion of AREG in response to HGF stimulation, thus leading to cell polarization and enhanced migration and invasion. In total, we define a new coordinated autocrine signaling loop mediated by integrin α6β4 signaling as well as a novel dependence of HGF-c-Met signaling on the autocrine EGFR signaling pathway (Fig. 8). This observation may help to explain why EGFR inhibitors can be therapeutically effective in a complex tumor microenvironment composed of many different growth factors.
Author Contributions
B. L. C., M. C., T. K., K. A. D., S. M. W. H., and R. L. S. designed and performed the experiments. B. L. C., M. C., and K. L. O. prepared the figures and wrote the Experimental Procedures. B. L. C. and K. L. O. organized and wrote the manuscript with extensive input from M. C. All authors reviewed the results and approved the final version of the manuscript.
This work was supported by National Institutes of Health Grants T32 CA165990 (to B. L. C.), T32 CA160003 (to R. L. S.), IRG-85-001-25 (to M. C.), and R01 CA109136 (to K. L. O. and M. C.). The authors declare that they have no conflicts of interest with the contents of this article.
- AREG
- amphiregulin
- HGF
- hepatocyte growth factor
- EGFR
- epidermal growth factor receptor
- EREG
- epiregulin
- TCGA
- The Cancer Genome Atlas
- TKI
- tyrosine kinase inhibitor
- QPCR
- quantitative polymerase chain reaction
- DAC
- 5-aza-2′-deoxycytidine
- NSCLC
- non-small cell lung cancer
- MMP
- matrix metalloproteinase.
References
- 1.Cruz-Monserrate Z., Qiu S., Evers B. M., and O'Connor K. L. (2007) Upregulation and redistribution of integrin α6β4 expression occurs at an early stage in pancreatic adenocarcinoma progression. Mod. Pathol. 20, 656–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Logsdon C. D., Simeone D. M., Binkley C., Arumugam T., Greenson J. K., Giordano T. J., Misek D. E., Kuick R., and Hanash S. (2003) Molecular profiling of pancreatic adenocarcinoma and chronic pancreatitis identifies multiple genes differentially regulated in pancreatic cancer. Cancer Res. 63, 2649–2657 [PubMed] [Google Scholar]
- 3.Dowling J., Yu Q. C., and Fuchs E. (1996) β4 integrin is required for hemidesmosome formation, cell adhesion and cell survival. J. Cell Biol. 134, 559–572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Trusolino L., Bertotti A., and Comoglio P. M. (2001) A signaling adapter function for α6β4 integrin in the control of HGF-dependent invasive growth. Cell 107, 643–654 [DOI] [PubMed] [Google Scholar]
- 5.Gilcrease M. Z., Zhou X., Lu X., Woodward W. A., Hall B. E., and Morrissey P. J. (2009) α6β4 integrin crosslinking induces EGFR clustering and promotes EGF-mediated Rho activation in breast cancer. J. Exp. Clin. Cancer Res. 28, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rabinovitz I., Toker A., and Mercurio A. M. (1999) Protein kinase C-dependent mobilization of the α6β4 integrin from hemidesmosomes and its association with actin-rich cell protrusions drive the chemotactic migration of carcinoma cells. J. Cell Biol. 146, 1147–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lipscomb E. A., and Mercurio A. M. (2005) Mobilization and activation of a signaling competent α6β4 integrin underlies its contribution to carcinoma progression. Cancer Metastasis Rev. 24, 413–423 [DOI] [PubMed] [Google Scholar]
- 8.O'Connor K. L., Nguyen B. K., and Mercurio A. M. (2000) RhoA function in lamellae formation and migration is regulated by the α6β4 integrin and cAMP metabolism. J. Cell Biol. 148, 253–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen M., and O'Connor K. L. (2005) Integrin α6β4 promotes expression of autotaxin/ENPP2 autocrine motility factor in breast ductal carcinoma cells. Oncogene 24, 5125–5130 [DOI] [PubMed] [Google Scholar]
- 10.Chen M., Sinha M., Luxon B. A., Bresnick A. R., and O'Connor K. L. (2009) Integrin α6β4 controls the expression of genes associated with cell motility, invasion, and metastasis, including S100A4/metastasin. J. Biol. Chem. 284, 1484–1494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cruz-Monserrate Z., and O'Connor K. L. (2008) Integrin α6β4 promotes migration, invasion through Tiam1 upregulation and subsequent Rac activation. Neoplasia 10, 408–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Herbst R. S. (2004) Review of epidermal growth factor receptor biology. Int. J. Radiat. Oncol. Biol. Phys. 59, 21–26 [DOI] [PubMed] [Google Scholar]
- 13.Normanno N., De Luca A., Bianco C., Strizzi L., Mancino M., Maiello M. R., Carotenuto A., De Feo G., Caponigro F., and Salomon D. S. (2006) Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 366, 2–16 [DOI] [PubMed] [Google Scholar]
- 14.de Melker A. A., van der Horst G., Calafat J., Jansen H., and Borst J. (2001) c-Cbl ubiquitinates the EGF receptor at the plasma membrane and remains receptor associated throughout the endocytic route. J. Cell Sci. 114, 2167–2178 [DOI] [PubMed] [Google Scholar]
- 15.Stern K. A., Place T. L., and Lill N. L. (2008) EGF and amphiregulin differentially regulate Cbl recruitment to endosomes and EGF receptor fate. Biochem. J. 410, 585–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baldys A., Göoz M., Morinelli T. A., Lee M. H., Raymond J. R. Jr., Luttrell L. M., and Raymond J. R. Sr. (2009) Essential role of c-Cbl in amphiregulin-induced recycling and signaling of the endogenous epidermal growth factor receptor. Biochemistry 48, 1462–1473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Singh A. B., and Harris R. C. (2005) Autocrine, paracrine and juxtacrine signaling by EGFR ligands. Cell. Signal. 17, 1183–1193 [DOI] [PubMed] [Google Scholar]
- 18.Sahin U., Weskamp G., Kelly K., Zhou H. M., Higashiyama S., Peschon J., Hartmann D., Saftig P., and Blobel C. P. (2004) Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J. Cell Biol. 164, 769–779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu X., Wang Q., Hu G., Van Poznak C., Fleisher M., Reiss M., Massagué J., and Kang Y. (2009) ADAMTS1 and MMP1 proteolytically engage EGF-like ligands in an osteolytic signaling cascade for bone metastasis. Genes Dev. 23, 1882–1894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Busser B., Sancey L., Brambilla E., Coll J. L., and Hurbin A. (2011) The multiple roles of amphiregulin in human cancer. Biochim. Biophys. Acta 1816, 119–131 [DOI] [PubMed] [Google Scholar]
- 21.Eccles S. A. (2011) The epidermal growth factor receptor/Erb-B/HER family in normal and malignant breast biology. Int. J. Dev. Biol. 55, 685–696 [DOI] [PubMed] [Google Scholar]
- 22.Ebert M., Yokoyama M., Kobrin M. S., Friess H., Lopez M. E., Büchler M. W., Johnson G. R., and Korc M. (1994) Induction and expression of amphiregulin in human pancreatic cancer. Cancer Res. 54, 3959–3962 [PubMed] [Google Scholar]
- 23.Castillo J., Erroba E., Perugorría M. J., Santamaría M., Lee D. C., Prieto J., Avila M. A., and Berasain C. (2006) Amphiregulin contributes to the transformed phenotype of human hepatocellular carcinoma cells. Cancer Res. 66, 6129–6138 [DOI] [PubMed] [Google Scholar]
- 24.Johnson G. R., Saeki T., Gordon A. W., Shoyab M., Salomon D. S., and Stromberg K. (1992) Autocrine action of amphiregulin in a colon carcinoma cell line and immunocytochemical localization of amphiregulin in human colon. J. Cell Biol. 118, 741–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhu Z., Kleeff J., Friess H., Wang L., Zimmermann A., Yarden Y., Büchler M. W., and Korc M. (2000) Epiregulin is up-regulated in pancreatic cancer and stimulates pancreatic cancer cell growth. Biochem. Biophys. Res. Commun. 273, 1019–1024 [DOI] [PubMed] [Google Scholar]
- 26.Shelly M., Pinkas-Kramarski R., Guarino B. C., Waterman H., Wang L. M., Lyass L., Alimandi M., Kuo A., Bacus S. S., Pierce J. H., Andrews G. C., and Yarden Y. (1998) Epiregulin is a potent pan-ErbB ligand that preferentially activates heterodimeric receptor complexes. J. Biol. Chem. 273, 10496–10505 [DOI] [PubMed] [Google Scholar]
- 27.Lieber M., Mazzetta J., Nelson-Rees W., Kaplan M., and Todaro G. (1975) Establishment of a continuous tumor-cell line (panc-1) from a human carcinoma of the exocrine pancreas. Int. J. Cancer 15, 741–747 [DOI] [PubMed] [Google Scholar]
- 28.Iwamura T., Katsuki T., and Ide K. (1987) Establishment and characterization of a human pancreatic cancer cell line (SUIT-2) producing carcinoembryonic antigen and carbohydrate antigen 19-9. Jpn. J. Cancer Res. 78, 54–62 [PubMed] [Google Scholar]
- 29.Cline M. S., Craft B., Swatloski T., Goldman M., Ma S., Haussler D., and Zhu J. (2013) Exploring TCGA pan-cancer data at the UCSC cancer genomics browser. Sci. Rep. 3, 2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakamura T., Furukawa Y., Nakagawa H., Tsunoda T., Ohigashi H., Murata K., Ishikawa O., Ohgaki K., Kashimura N., Miyamoto M., Hirano S., Kondo S., Katoh H., Nakamura Y., and Katagiri T. (2004) Genome-wide cDNA microarray analysis of gene expression profiles in pancreatic cancers using populations of tumor cells and normal ductal epithelial cells selected for purity by laser microdissection. Oncogene 23, 2385–2400 [DOI] [PubMed] [Google Scholar]
- 31.Gleason B., Adley B., Rao M. S., and Diaz L. K. (2005) Immunohistochemical detection of the β4 integrin subunit in pancreatic adenocarcinoma. J. Histochem. Cytochem. 53, 799–801 [DOI] [PubMed] [Google Scholar]
- 32.Jauliac S., López-Rodriguez C., Shaw L. M., Brown L. F., Rao A., and Toker A. (2002) The role of NFAT transcription factors in integrin-mediated carcinoma invasion. Nat. Cell Biol. 4, 540–544 [DOI] [PubMed] [Google Scholar]
- 33.Di Renzo M. F., Poulsom R., Olivero M., Comoglio P. M., and Lemoine N. R. (1995) Expression of the Met/hepatocyte growth factor receptor in human pancreatic cancer. Cancer Res. 55, 1129–1138 [PubMed] [Google Scholar]
- 34.So W. K., Fan Q., Lau M. T., Qiu X., Cheng J. C., and Leung P. C. (2014) Amphiregulin induces human ovarian cancer cell invasion by down-regulating E-cadherin expression. FEBS Lett. 588, 3998–4007 [DOI] [PubMed] [Google Scholar]
- 35.Plopper G. E., Huff J. L., Rust W. L., Schwartz M. A., and Quaranta V. (2000) Antibody-induced activation of β1 integrins stimulates cAMP-dependent migration of breast cells on laminin-5. Mol. Cell. Biol. Res. Commun. 4, 129–135 [DOI] [PubMed] [Google Scholar]
- 36.O'Connor K. L., and Mercurio A. M. (2001) Protein kinase A regulates Rac and is required for the growth factor-stimulated migration of carcinoma cells. J. Biol. Chem. 276, 47895–47900 [DOI] [PubMed] [Google Scholar]
- 37.Miranti C. K., and Brugge J. S. (2002) Sensing the environment: a historical perspective on integrin signal transduction. Nat. Cell Biol. 4, E83–E90 [DOI] [PubMed] [Google Scholar]
- 38.Guo W., and Giancotti F. G. (2004) Integrin signalling during tumour progression. Nat. Rev. Mol. Cell Biol. 5, 816–826 [DOI] [PubMed] [Google Scholar]
- 39.Rathinam R., and Alahari S. K. (2010) Important role of integrins in the cancer biology. Cancer Metastasis Rev. 29, 223–237 [DOI] [PubMed] [Google Scholar]
- 40.Park S., Jung H. H., Park Y. H., Ahn J. S., and Im Y. H. (2011) ERK/MAPK pathways play critical roles in EGFR ligands-induced MMP1 expression. Biochem. Biophys. Res. Commun. 407, 680–686 [DOI] [PubMed] [Google Scholar]
- 41.Chen W. H., Horoszewicz J. S., Leong S. S., Shimano T., Penetrante R., Sanders W. H., Berjian R., Douglass H. O., Martin E. W., and Chu T. M. (1982) Human pancreatic adenocarcinoma: in vitro and in vivo morphology of a new tumor line established from ascites. In Vitro 18, 24–34 [DOI] [PubMed] [Google Scholar]
- 42.Berquin I. M., Dziubinski M. L., Nolan G. P., and Ethier S. P. (2001) A functional screen for genes inducing epidermal growth factor autonomy of human mammary epithelial cells confirms the role of amphiregulin. Oncogene 20, 4019–4028 [DOI] [PubMed] [Google Scholar]
- 43.Sato K., Sato A., Aoto M., and Fukami Y. (1995) c-Src phosphorylates epidermal growth factor receptor on tyrosine 845. Biochem. Biophys. Res. Commun. 215, 1078–1087 [DOI] [PubMed] [Google Scholar]
- 44.O'Connor K. L., Shaw L. M., and Mercurio A. M. (1998) Release of cAMP gating by the α6β4 integrin stimulates lamellae formation and the chemotactic migration of invasive carcinoma cells. J. Cell Biol. 143, 1749–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mainiero F., Pepe A., Yeon M., Ren Y., and Giancotti F. G. (1996) The intracellular functions of α6β4 integrin are regulated by EGF. J. Cell Biol. 134, 241–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Santoro M. M., Gaudino G., and Marchisio P. C. (2003) The MSP receptor regulates α6β4 and α3β1 integrins via 14-3-3 proteins in keratinocyte migration. Dev. Cell 5, 257–271 [DOI] [PubMed] [Google Scholar]
- 47.Weaver V. M., Lelièvre S., Lakins J. N., Chrenek M. A., Jones J. C., Giancotti F., Werb Z., and Bissell M. J. (2002) β4 integrin-dependent formation of polarized three-dimensional architecture confers resistance to apoptosis in normal and malignant mammary epithelium. Cancer Cell 2, 205–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sun C., Liu X., Qi L., Xu J., Zhao J., Zhang Y., Zhang S., and Miao J. (2010) Modulation of vascular endothelial cell senescence by integrin β4. J. Cell. Physiol. 225, 673–681 [DOI] [PubMed] [Google Scholar]
- 49.Jimeno A., Tan A. C., Coffa J., Rajeshkumar N. V., Kulesza P., Rubio-Viqueira B., Wheelhouse J., Diosdado B., Messersmith W. A., Iacobuzio-Donahue C., Maitra A., Varella-Garcia M., Hirsch F. R., Meijer G. A., and Hidalgo M. (2008) Coordinated epidermal growth factor receptor pathway gene overexpression predicts epidermal growth factor receptor inhibitor sensitivity in pancreatic cancer. Cancer Res. 68, 2841–2849 [DOI] [PubMed] [Google Scholar]
- 50.Yokoyama M., Ebert M., Funatomi H., Friess H., Buchler M., Johnson G., and Korc M. (1995) Amphiregulin is a potent mitogen in human pancreatic-cancer cells–correlation with patient survival. Int. J. Oncol. 6, 625–631 [DOI] [PubMed] [Google Scholar]
- 51.Tun M. T., Pai R. K., Kwok S., Dong A., Gupta A., Visser B. C., Norton J. A., Poultsides G. A., Banerjee S., Van Dam J., Chen A. M., Friedland S., Scott B. A., Verma R., Lowe A. W., and Park W. G. (2012) Diagnostic accuracy of cyst fluid amphiregulin in pancreatic cysts. BMC Gastroenterol. 12, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lipscomb E. A., Simpson K. J., Lyle S. R., Ring J. E., Dugan A. S., and Mercurio A. M. (2005) The α6β4 integrin maintains the survival of human breast carcinoma cells in vivo. Cancer Res. 65, 10970–10976 [DOI] [PubMed] [Google Scholar]
- 53.Zahir N., Lakins J. N., Russell A., Ming W., Chatterjee C., Rozenberg G. I., Marinkovich M. P., and Weaver V. M. (2003) Autocrine laminin-5 ligates α6β4 integrin and activates RAC and NFκB to mediate anchorage-independent survival of mammary tumors. J. Cell Biol. 163, 1397–1407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.O'Connor K. L., Chen M., and Towers L. N. (2012) Integrin α6β4 cooperates with LPA signaling to stimulate Rac through AKAP-Lbc-mediated RhoA activation. Am. J. Physiol. Cell Physiol. 302, C605–C614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ciardiello F., and Tortora G. (2008) EGFR antagonists in cancer treatment. N. Engl. J. Med. 358, 1160–1174 [DOI] [PubMed] [Google Scholar]
- 56.Zhao Y., He D., Saatian B., Watkins T., Spannhake E. W., Pyne N. J., and Natarajan V. (2006) Regulation of lysophosphatidic acid-induced epidermal growth factor receptor transactivation and interleukin-8 secretion in human bronchial epithelial cells by protein kinase Cdelta, Lyn kinase, and matrix metalloproteinases. J. Biol. Chem. 281, 19501–19511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gschwind A., Prenzel N., and Ullrich A. (2002) Lysophosphatidic acid-induced squamous cell carcinoma cell proliferation and motility involves epidermal growth factor receptor signal transactivation. Cancer Res. 62, 6329–6336 [PubMed] [Google Scholar]
- 58.Engelman J. A., Zejnullahu K., Mitsudomi T., Song Y., Hyland C., Park J. O., Lindeman N., Gale C. M., Zhao X., Christensen J., Kosaka T., Holmes A. J., Rogers A. M., Cappuzzo F., Mok T., et al. (2007) MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 316, 1039–1043 [DOI] [PubMed] [Google Scholar]
- 59.Nanjo S., Yamada T., Nishihara H., Takeuchi S., Sano T., Nakagawa T., Ishikawa D., Zhao L., Ebi H., Yasumoto K., Matsumoto K., and Yano S. (2013) Ability of the Met kinase inhibitor crizotinib and new generation EGFR inhibitors to overcome resistance to EGFR inhibitors. PLoS ONE 8, e84700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sequist L. V., Waltman B. A., Dias-Santagata D., Digumarthy S., Turke A. B., Fidias P., Bergethon K., Shaw A. T., Gettinger S., Cosper A. K., Akhavanfard S., Heist R. S., Temel J., Christensen J. G., Wain J. C., et al. (2011) Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci. Transl. Med. 3, 75ra26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu W., Wu X., Zhang W., Montenegro R. C., Fackenthal D. L., Spitz J. A., Huff L. M., Innocenti F., Das S., Cook E. H. Jr., Cox N. J., Bates S. E., and Ratain M. J. (2007) Relationship of EGFR mutations, expression, amplification, and polymorphisms to epidermal growth factor receptor inhibitors in the NCI60 cell lines. Clin. Cancer Res. 13, 6788–6795 [DOI] [PubMed] [Google Scholar]
- 62.Stabile L. P., Rothstein M. E., Keohavong P., Lenzner D., Land S. R., Gaither-Davis A. L., Kim K. J., Kaminski N., and Siegfried J. M. (2010) Targeting of both the c-Met and EGFR pathways. Results in additive inhibition of lung tumorigenesis in transgenic mice. Cancers (Basel) 1;2(4):2153–2170 [DOI] [PMC free article] [PubMed] [Google Scholar]