

Background: Akt translocation to the plasma membrane (PM) is modulated by calmodulin (CaM).
Results: CaM forms a tight complex with the PHD of Akt, which represents a novel class of CaM-binding domains.
Conclusion: The binding mode of CaM to Akt(PHD) suggests a synergistic role in membrane binding and subsequent activation.
Significance: Characterization of Akt-CaM is critical for understanding Akt activation.
Keywords: breast cancer; calmodulin (CaM); isothermal titration calorimetry (ITC); nuclear magnetic resonance (NMR); plasma membrane; Akt; phosphatidylinositol (3,4,5)-trisphosphate; pleckstrin homology domain
Abstract
The translocation of Akt, a serine/threonine kinase, to the plasma membrane is a critical step in the Akt activation pathway. It is established that membrane binding of Akt is mediated by direct interactions between its pleckstrin homology domain (PHD) and phosphatidylinositol 3,4,5-trisphosphate (PI(3,4,5)P3). There is now evidence that Akt activation in many breast cancer cells is also modulated by the calcium-binding protein, calmodulin (CaM). Upon EGF stimulation of breast cancer cells, CaM co-localizes with Akt at the plasma membrane to enhance activation. However, the molecular details of Akt(PHD) interaction with CaM are not known. In this study, we employed NMR, biochemical, and biophysical techniques to characterize CaM binding to Akt(PHD). Our data show that CaM forms a tight complex with the PHD of Akt (dissociation constant = 100 nm). The interaction between CaM and Akt(PHD) is enthalpically driven, and the affinity is greatly dependent on salt concentration, indicating that electrostatic interactions are important for binding. The CaM-binding interface in Akt(PHD) was mapped to two loops adjacent to the PI(3,4,5)P3 binding site, which represents a rare CaM-binding motif and suggests a synergistic relationship between CaM and PI(3,4,5)P3 upon Akt activation. Elucidation of the mechanism by which Akt interacts with CaM will help in understanding the activation mechanism, which may provide insights for new potential targets to control the pathophysiological processes of cell survival.
Introduction
Induction of cell survival is often initiated at the cell surface and is triggered by a wide range of receptor-ligand interactions (1–5). One of the most frequent cell survival pathways involves activation of Akt, a serine/threonine kinase that is critical in regulating apoptosis and oncogenesis. Hyperactivation of Akt is a common tumorigenic event in many types of cancers, including breast cancer (1, 4, 5). Akt interferes with programmed cell death by phosphorylating and thereby inactivating a number of proteins involved in apoptosis. Phosphatidylinositol 3-kinase (PI3K), a lipid kinase and upstream effector of Akt, is implicated in numerous cellular functions, including survival (6), growth and proliferation (7), and differentiation (8). Upon growth factor stimulation, PI3K generates 3′-phosphorylated phosphoinositides, such as phosphatidylinositol 3,4,5-trisphosphate (PI(3,4,5)P3)2 and phosphatidylinositol 3,4-bisphosphate at the plasma membrane (PM) (4, 9–12). PI(3,4,5)P3 (and possibly phosphatidylinositol 3,4-bisphosphate) mediates membrane localization of Akt by binding to the pleckstrin homology domain (PHD) (1, 4, 13). The PHD is formed by two antiparallel β sheets, a C-terminal amphipathic helix, and three variable loops (Fig. 1). These structural elements are recognized as global features for PHDs in hundreds of proteins in mammals and bacteria (14–16). Interaction between the PHD of Akt and PI(3,4,5)P3 is considered a hallmark in the activation pathway and is required to initiate a cascade of events, including phosphorylation and downstream activation of effector proteins. The PM-anchored Akt becomes fully activated upon phosphorylation of residue Thr-308 in the kinase domain and Ser-473 in the hydrophobic motif through PDK1 (phosphoinositide-dependent kinase 1) and mTORC2 (mammalian target of rapamycin complex 2), respectively (12). It is believed that PI(3,4,5)P3 binding to a conserved basic patch on the PHD induces a conformational change, allowing for phosphorylation of residues Thr-308 and Ser-473. In addition to PI(3,4,5)P3, it has been recently shown that Akt translocation to the PM and subsequent activation are also modulated by phosphatidylserine (PS) (17). PS interacts with specific residues in the PHD and regulatory domain of Akt, promoting binding of Akt to PI(3,4,5)P3 (17). The synergy between PS and PI(3,4,5)P3 is thought to induce Akt interdomain conformational changes required for phosphorylation of Thr-308 and Ser-473 (17). Mutagenesis studies have also revealed that disruption of Akt-PS interaction impairs Akt signaling and increases susceptibility to cell death, suggesting that PS is critical for Akt activation and cell survival, particularly in conditions with limited PI(3,4,5)P3 availability (17). Taken together, these studies suggest that a synergy between membrane lipids is perhaps needed for Akt localization and activation.
FIGURE 1.
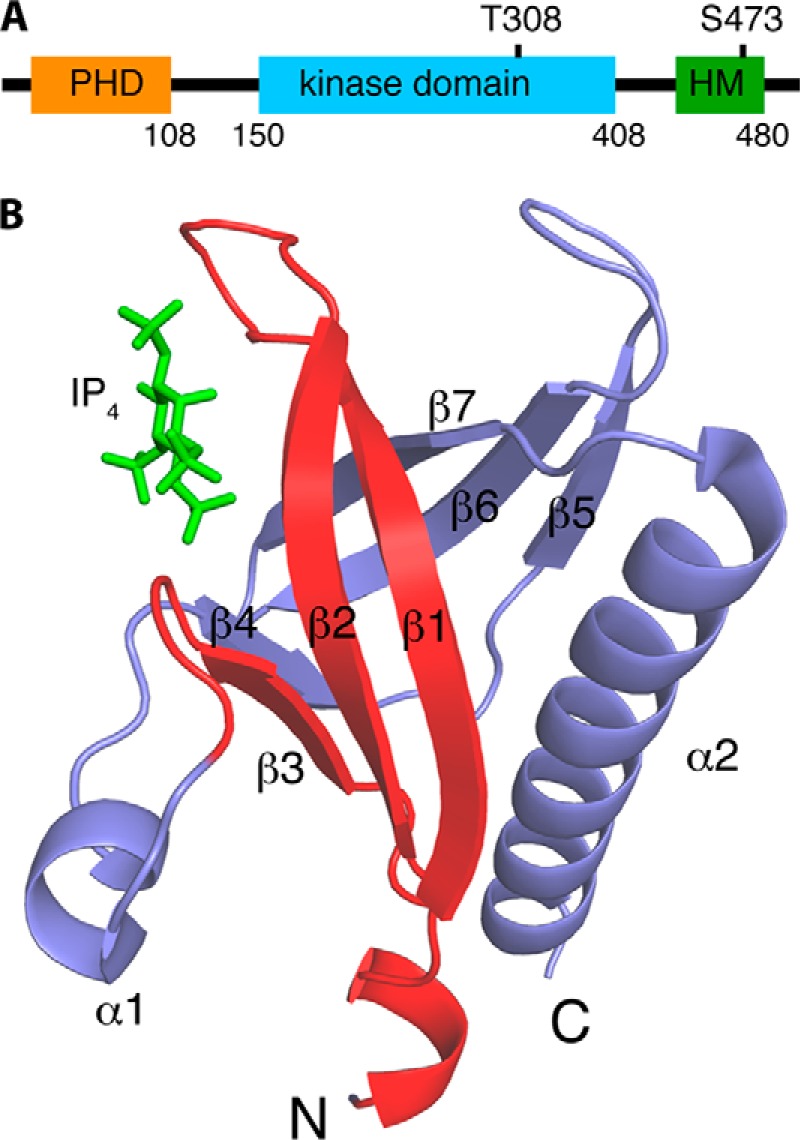
A, diagram of the human AKT1 protein. The protein domains and their length (indicated by number of limiting residues) are shown. B, schematic representation of the PHD of Akt1 (Protein Data Bank entry 1H10) (33). Residues 1–42, previously thought to be the CaM-binding region (31), are highlighted in red. Inositol 1,3,4,5-tetrakisphosphate (IP4) is shown in green sticks.
The three members of the Akt family, Akt1, Akt2, and Akt3, are closely related and highly conserved. However, functional isoform-specific differences concerning proliferation, apoptosis, and migration in human cancer cells have been identified (18–21). A role for Akt1 (which will be referred to as Akt throughout this work) in breast cancer cells has been proposed (3, 22). It has been found that epidermal growth factor (EGF)-induced membrane translocation and activation of Akt is modulated by calmodulin (CaM) (3, 22), a ubiquitous calcium-binding protein expressed in all eukaryotic cells (23–28). Perturbation of this targeting mechanism by CaM antagonists inhibited Akt-CaM translocation to the membrane and led to apoptotic cell death in tumorigenic mammary carcinoma cells (3). These studies are consistent with the findings that CaM is overexpressed in breast tumors and breast cancer cell lines (29, 30). Biochemical and mutagenesis studies have shown that Akt-CaM interaction is calcium-dependent and is mediated by the PHD (31). Based on biochemical assays, it was suggested that the CaM-binding region of Akt is located within the first 42 residues of the PHD (31). Interestingly, the three-dimensional structure of Akt(PHD) shows that these residues form a three-stranded β-sheet (Fig. 1). The requirement of the β-sheet region for CaM binding is unprecedented because CaM-binding regions often adopt helical structures (27, 32). Furthermore, this region is known to be important for PI(3,4,5)P3 and PS binding (17, 33, 34). The interplay between the roles of CaM, PI(3,4,5)P3, and PS in membrane translocation and activation of Akt is not clear. To understand these mechanisms, it is essential to characterize at the molecular level how CaM interacts with the PHD of Akt and whether this interaction synergizes with Akt binding to membrane lipids. Identification of the structural elements required for membrane translocation of Akt is critical for elucidation of the precise molecular mechanism of activation. Here, we employed NMR, biophysical, and biochemical methods to characterize how Akt(PHD) binds to CaM. We show that CaM binds to Akt(PHD) with a dissociation constant (Kd) of 100 nm and a 1:1 stoichiometry. We also show that both of the N- and C-terminal lobes of CaM are critical for Akt(PHD) binding. The CaM-binding interface in Akt(PHD) was mapped to two loops adjacent to the PI(3,4,5)P3 binding site, which represents a novel CaM-binding motif. Elucidation of the mechanism by which Akt interacts with CaM will help in the understanding of the activation mechanism, which may provide insights for new potential targets to control the pathophysiological processes of cell survival.
Experimental Procedures
Sample Preparation
A plasmid encoding full-length (amino acids 1–148) CaM was a generous gift from Dr. Madeline Shea (University of Iowa). Expression and purification of the CaM protein were conducted as described (35). CaM samples were stored in a buffer containing 50 mm HEPES or Tris at pH 7.0, 150 mm NaCl, and 5 mm CaCl2. CaM-N (residues 1–80) and CaM-C (residues 76–148) proteins were expressed and purified as described (36). CaM-N protein concentrations were measured using a bicinchoninic acid assay (BCA; Thermo Scientific). A plasmid encoding full-length Mus musculus (mouse) Akt (amino acids 1–541) was a kind gift from Dr. Yabing Chen (University of Alabama at Birmingham). M. musculus Akt protein sequence is 98% identical to that of human Akt1 (Swiss-Prot code P31749). The PHD of Akt encoding for amino acids 1–113 was inserted into pET28 vector, which was fused to a His6 SUMO tag gene on the 5′-end, at its XhoI and BamHI sites. The His6-SUMO-Akt(PHD) protein was overexpressed in Escherichia coli BL21 (DE3) codon plus RIL cells. Cells were grown at 37 °C until A600 was ∼0.6, induced with 0.3 mm isopropyl β-d-1-thigoalactopyranoside, and grown at 16 °C for 16 h. To make uniformly 15N- and 15N-,13C-labeled Akt(PHD) samples, cells were grown in 4L LB medium at 37 °C until A600 was ∼0.6. Cells were then spun down, washed with 1× M9 salt, and transferred into 1 liter of M9 minimal medium containing 15NH4Cl and 13C-glucose as the sole sources to produce 15N- and 13C-labeled proteins, respectively. Cells were then induced with 0.3 mm isopropyl β-d-1-thigoalactopyranoside, grown at 16 °C for ∼16 h, spun down, and stored overnight at −80 °C. The next day, the cell pellet was resuspended in 100 ml of lysis/binding buffer containing 50 mm phosphate (pH 8.0), 500 mm NaCl, 10 mm imidazole, and 5 mm 2-mercaptoethanol. Cells were sonicated, and cell lysate was spun down at 17,000 rpm for 30 min. The protein-containing supernatant was purified on nickel affinity resin. The Akt(PHD) protein was eluted with a buffer containing 50 mm phosphate (pH 8.0), 500 mm NaCl, 300 mm imidazole, and 5 mm 2-mercaptoethanol. The fractions were then pooled and dialyzed in a buffer containing 25 mm Tris (pH 8.0) and 300 mm NaCl. The His6-SUMO tag was cleaved via SUMO protease and purified via nickel affinity and gel filtration methods. The Akt(PHD) protein was stored in 50 mm Tris (pH 7.0), 150 mm NaCl, 2 mm tris(2-carboxyethyl)phosphine hydrochloride or 50 mm phosphate (pH 6.5), 300 mm NaCl, 50 mm glutamate, 50 mm arginine, and 2 mm tris(2-carboxyethyl)phosphine hydrochloride.
Gel Filtration Assay
The mobilities of Akt(PHD), CaM, and the Akt(PHD)-CaM complex were analyzed by a gel filtration assay. Briefly, 0.5 ml of ∼100 μm sample solutions of Akt(PHD), CaM, and the Akt(PHD)-CaM (1.5:1) complex were loaded onto a HiLoad Superdex 75 (10/300) column (GE Healthcare) in a buffer containing 50 mm Tris (pH 7.0), 150 mm NaCl, and 5 mm CaCl2. Protein fractions were analyzed by SDS-PAGE and stained by Coomassie Brilliant Blue. The approximate molecular weights of the loaded proteins were determined by low molecular weight calibration kits (GE Healthcare).
Isothermal Titration Calorimetry (ITC)
Thermodynamic parameters of CaM binding to Akt(PHD) were determined using an Auto-iTC200 microcalorimeter (Malvern Instruments). ITC experiments were performed on protein samples in 50 mm HEPES (pH 7.0), 50, 150, or 500 mm NaCl, and 5 mm CaCl2. CaM at 250 μm was titrated into the cell sample containing 28 μm Akt(PHD). Heat of reaction was measured at 20 °C for 19 injections. Heat of dilution was measured by titrating CaM into buffer. Data analysis was performed using the Microcal Origin package (version 8.1). Baseline corrections were performed by subtracting heat of dilution from the raw CaM-Akt(PHD) titration data. Binding curves were analyzed, and dissociation constants (Kd) were determined by nonlinear least square fitting of the baseline-corrected data. The formula used to fit the data as one binding site is as follows,
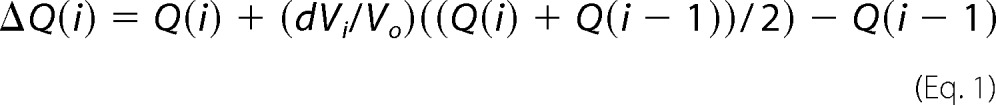 |
where ΔQ(i) is the heat released at the ith injection, Q(i) is the total heat content of the solution, dVi is injection volume, and Vo is the total volume.
Analytical Ultracentrifugation
Sedimentation velocity measurements were collected on a Beckman XL-I Optima system equipped with a 4-hole An-60-rotor (Beckman Coulter). CaM and Akt(PHD)-CaM complex were in a buffer containing 50 mm Tris (pH 7.0), 150 mm NaCl, and 5 mm CaCl2, whereas the Akt(PHD) sample was in a buffer containing 50 mm phosphate (pH 6.5), 400 mm NaCl, 50 mm glutamate, and 50 mm arginine. Akt(PHD), CaM, and Akt(PHD)-CaM complex were at 20, 60, and 20 μm, respectively. To ensure sample homogeneity, the Akt(PHD)-CaM complex was run through the gel filtration column. Rotor speed was set at 40,000 rpm, and scans were acquired at 280 nm and 20 °C. Partial specific volumes (v̄) and molar extinction coefficients were calculated by using the program SENDTERP, and buffer densities were measured pycnometrically. Sedimentation velocity data were analyzed using SEDFIT (37–40).
NMR Spectroscopy
NMR data were collected on Bruker Avance II and III (700 and 850 MHz 1H) spectrometers equipped with cryogenic triple-resonance probes, processed with NMRPIPE (41), and analyzed with NMRVIEW (42) or CCPN Analysis (43). Proton, carbon, and nitrogen NMR chemical shifts for CaM are reported elsewhere (44, 45). The backbone resonances of Akt(PHD) in the free state were assigned using standard triple resonance data (HNCA, HNCO, HNCOCA) and 15N-edited heteronuclear single quantum coherence (HSQC)-NOESY data collected at 20 or 30 °C on 200 μm samples in a buffer containing 50 mm phosphate (pH 6.5), 400 mm NaCl, 50 mm glutamate, 50 mm arginine, and 2 mm tris(2-carboxyethyl)phosphine hydrochloride. The backbone resonances of Akt(PHD) when bound to CaM were assigned using HNCA and 15N-edited HSQC-NOESY data collected at 30 °C on 200 μm samples in a buffer containing 50 mm phosphate (pH 6.5), 200 mm NaCl, 50 mm glutamate, 50 mm arginine, 2 mm tris(2-carboxyethyl)phosphine hydrochloride, and 1 mm CaCl2. NMR titrations were conducted in a buffer containing 50 mm Tris-d11 (pH 7.0), 150 mm NaCl and 5 mm CaCl2 (20 or 35 °C). Kd values were calculated by a non-linear least square fitting algorithm in the Origin software (OriginLab, Northampton, MA) using the equation,
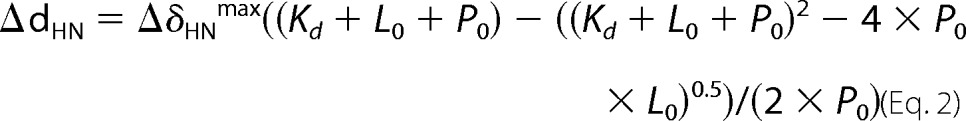 |
where ΔδHN is the chemical shift change between complex and free protein, L0 is the total concentration of lipid, and P0 is the total concentration of protein.
Results
PHDs are found in numerous membrane-associated proteins, and one of their most recognized functions is to mediate protein-phospholipid interactions (14, 46). Previous studies have shown that the PHDs of Akt and other proteins containing analogous PHD have a propensity to self-associate and form aggregates in solution at protein concentrations required for structural studies (34, 47–49). Although the oligomerization properties of Akt(PHD) have not been studied in detail, an NMR study revealed that the PHD of dynamin is present in a monomer-dimer equilibrium with a Kd of ∼1 mm (49). To minimize protein self-association and facilitate structural studies, the PHD of Akt2 has been solved by NMR methods only in the presence of inositol 1,4,5-trisphosphate, the polar head of phosphatidylinositol 4,5-bisphosphate (34). NMR data for structural determination were collected at high salt concentration (∼300 mm) and relatively low temperature (13 °C). In another study, the structure of the PHD of human Akt1 in complex with inositol 1,3,4,5-tetrakisphosphate was solved by x-ray crystallography (33). In other cases, mutations have been introduced in the PHD to reduce protein self-association (47, 50). Altogether, it is recognized that the PHD of Akt is amenable to self-association, a process that can be minimized but not fully eliminated at certain experimental conditions. Given those technical difficulties, we have expressed the PHD of Akt as a fusion protein with a SUMO tag attached to the N terminus. After SUMO cleavage, the Akt(PHD) protein was kept at concentrations of ∼50 μm at 4 °C. Because the oligomerization process is minimal at low protein concentration, all experiments described here were conducted with Akt(PHD) concentrations at 20–200 μm.
Mobility Characteristics of the Akt(PHD)-CaM Complex
To determine how CaM interacts with Akt(PHD) and to characterize the mobility and size of the complex, we first assessed the solution properties of Akt(PHD), CaM, and Akt(PHD)-CaM complex by a gel filtration mobility assay. Proteins were run on a size exclusion column (Superdex 75 GL) under the same buffer conditions at ∼100 μm. As shown in Fig. 2A, the elution volumes of Akt(PHD) and CaM were 14.3 and 11.3 ml, respectively. The Akt(PHD)-CaM complex was prepared with a 1.5:1 stoichiometry and yielded two peaks, the first at 10.9 ml and another at 14.3 ml, indicating excess Akt(PHD). Fractions of the complex were analyzed via SDS-PAGE. As shown in Fig. 2B, two bands representing Akt(PHD) and CaM with equal intensity were observed, suggesting formation of a 1:1 Akt(PHD)-CaM Complex. A gel filtration mobility assay with known protein standards revealed that the molecular mass of free Akt(PHD) migrates as a ∼13-kDa species, consistent with a monomeric form (Fig. 2C). As we and others have shown previously, the migration behavior of CaM (smaller than expected elution volume) is attributed to its elongated dumbbell shape (44, 51). Based on this mobility assay, the approximate molecular mass of the CaM-Akt(PHD) is ∼33 kDa, consistent with a 1:1 complex. Taken together, our gel filtration data establish a direct interaction between Akt(PHD) and CaM with a 1:1 stoichiometry. No differences in the mobility characteristics of the complex have been observed upon variation of the stoichiometry of proteins, excluding the formation of higher order ternary complexes.
FIGURE 2.
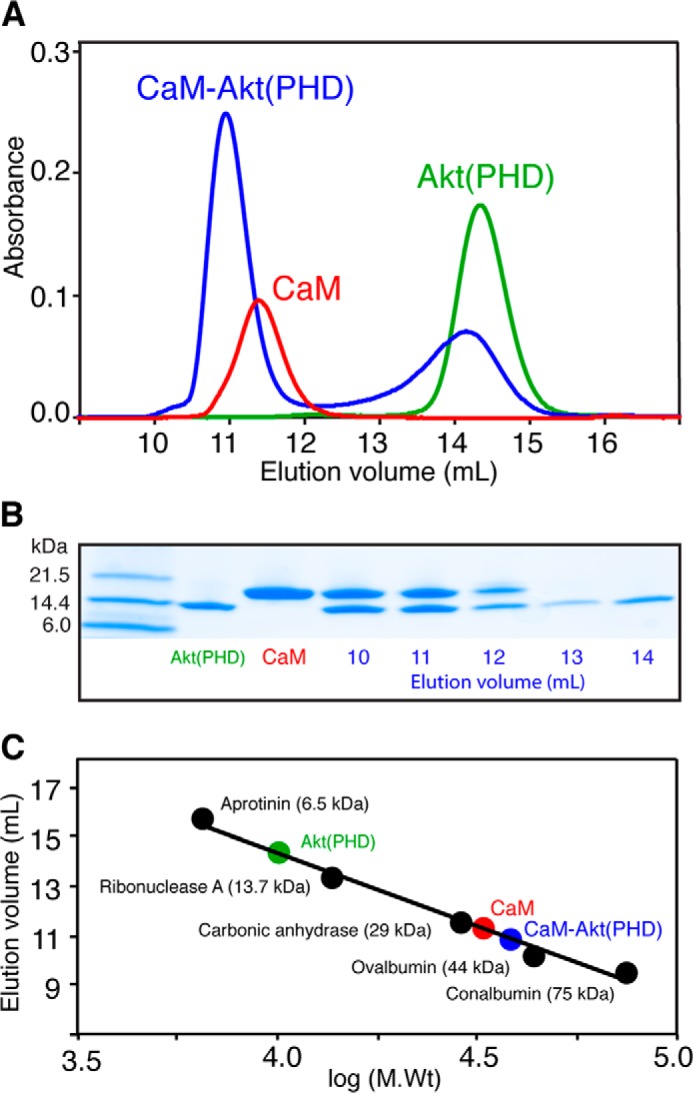
Interaction between CaM and Akt(PHD) as characterized by a gel filtration assay. A, elution profiles of Akt(PHD), CaM, and the Akt(PHD)/CaM (1.5:1) mixture on a HiLoad Superdex 75 (10/300) column. B, complex fractions from A were analyzed by SDS-PAGE. Gels were stained with Coomassie Brilliant Blue. C, a low molecular weight calibration kit was used to determine the approximate molecular weight of the Akt(PHD)-CaM complex and unbound proteins.
Sedimentation Studies of Akt(PHD)-CaM Interactions
We next employed analytical ultracentrifugation to examine the oligomerization properties of Akt(PHD) and its complex with CaM. We performed sedimentation velocity experiments on samples for Akt(PHD), CaM, and Akt(PHD)-CaM complex at 20, 60, and 20 μm, respectively. To ensure sample homogeneity, the Akt(PHD)-CaM complex was prepared by mixing equimolar amounts of CaM and Akt(PHD) and then run onto a gel filtration column. The fraction of the complex was used for the sedimentation velocity experiment. As shown in Fig. 3, sedimentation velocity profiles of all samples exhibit a single sedimentation boundary distribution. Analysis of the data using SEDFIT provided peaks at 1.3 and 1.9 S for Akt(PHD) and CaM, respectively. Molecular mass distribution analysis gave 13 and 17 kDa for Akt(PHD) and CaM, respectively, consistent with monomeric species. Based on this result, it is conceivable that the self-association of the Akt(PHD) protein at low concentration (20 μm) is minimal or absent. The sedimentation coefficient for Akt(PHD)-CaM complex is observed at 3 S, consistent with a homogenous complex. In agreement with the gel filtration data, the estimated molecular mass of the Akt(PHD)-CaM complex (∼30 kDa) indicates a 1:1 complex.
FIGURE 3.
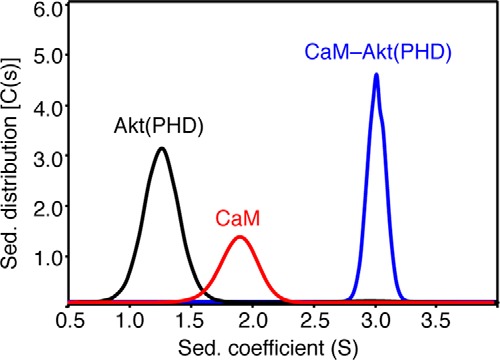
Sedimentation coefficient distributions, c(s), and sedimentation coefficient (s) obtained from the sedimentation profiles for Akt(PHD), CaM, and CaM-Akt(PHD) complex. Molecular mass distribution analysis afforded ∼13 and ∼17 kDa for Akt(PHD) and CaM, respectively, consistent with monomeric species. The estimated molecular mass of the Akt(PHD)-CaM complex (∼30 kDa) indicates a 1:1 complex.
Thermodynamic Properties of CaM Binding to Akt(PHD) as Detected by ITC
ITC measures heat absorbed or generated upon binding and provides values for the dissociation constant (Kd), stoichiometry of binding (n), enthalpy change (ΔH0) and the entropic term (TΔS0). The separation of the free energy of binding into entropic and enthalpic components reveals the nature of the forces that drive the binding reaction. Although it is recognized that the most frequent mechanism of CaM binding to target proteins involves the hydrophobic patches on the N- and C-terminal lobes (27), electrostatic interactions are also implicated in the stabilization of CaM-protein complexes, since CaM-binding motifs are often basic (52, 53). The ITC experiments can provide information on the importance and relative contribution of the hydrophobic versus electrostatic interactions in the Akt(PHD)-CaM interactions. We have obtained ITC data upon titration of CaM into Akt(PHD) under the same buffer conditions. As shown in Fig. 4, binding of CaM to Akt(PHD) is exothermic, as indicated by the sign of the heat of enthalpy. The binding data were fit into a one-site binding mode and yielded the following thermodynamic parameters: Kd =100 nm, n = 0.90 ± 0.02, ΔH0 = −3.87 kcal/mol, and ΔS0 = 19.3 calories/mol/degrees. Although the exothermic nature of the ITC thermogram suggests that electrostatic interactions contribute to the formation of the Akt(PHD)-CaM complex, the interaction also appears to be stabilized by favorable hydrophobic interactions, as indicated by the entropy factor (TΔS). To assess the importance of electrostatic interactions in stabilizing the Akt(PHD)-CaM complex, we obtained ITC data at variable salt concentrations (Fig. 4). As indicated by the Kd values, the binding affinity is reduced by 30-fold upon increasing the salt concentration from 50 to 500 mm (Table 1), indicating that ionic interactions contribute to the stabilization of the CaM-Akt(PHD) complex. Collectively, our ITC data show that CaM binds to Akt(PHD) in a 1:1 model and that ionic and hydrophobic interactions contribute to the formation of the CaM-Akt(PHD) complex.
FIGURE 4.
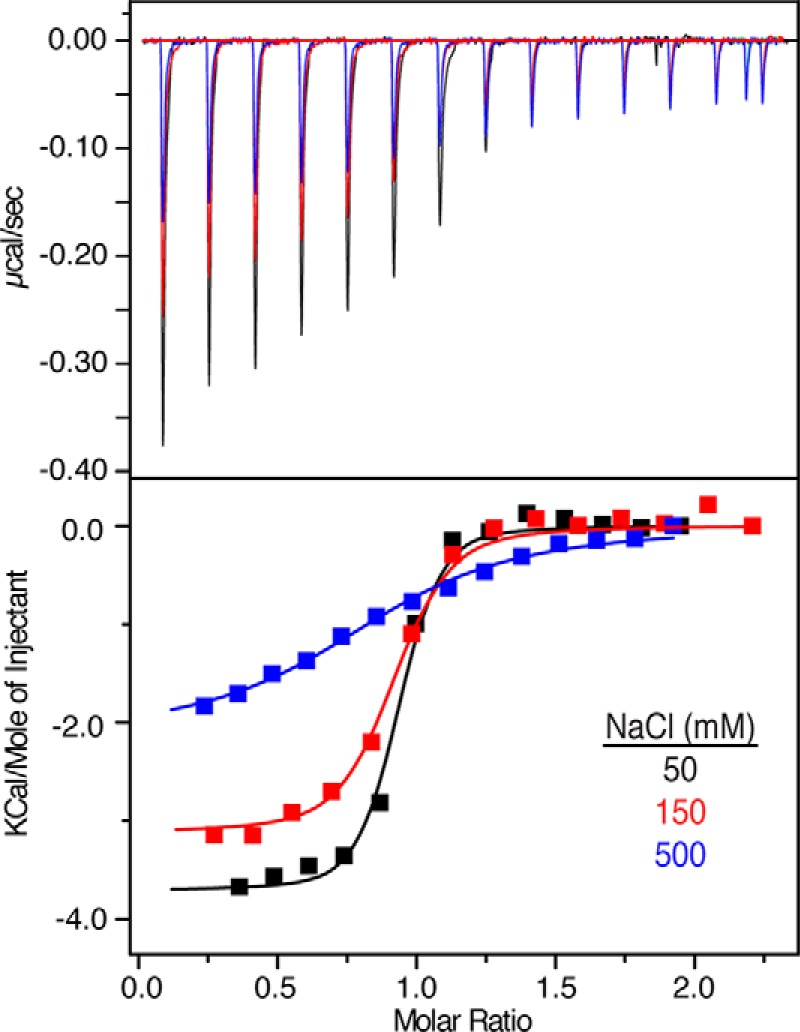
ITC data for binding of CaM to Akt(PHD) at 50, 150, and 500 mm NaCl. Data were best fit to one-site mode with Kd values of 100, 195, and 3 μm, respectively. The binding affinity is greatly reduced upon increasing salt concentration, indicating that complex formation is stabilized by electrostatic interactions.
TABLE 1.
Thermodynamic Parameters for CaM Binding to Akt(PHD) at different salt concentrations
| NaCl | Kd | ΔH | −TΔSa | ΔG | n |
|---|---|---|---|---|---|
| mm | nm | kcal/mol | kcal/mol | kcal/mol | |
| 50 | 100 | −3.87 ± 0.13 | −0.39 | −4.26 | 0.90 ± 0.02 |
| 100 | 195 | −3.12 ± 0.08 | −0.40 | −3.52 | 0.86 ± 0.01 |
| 500 | 3000 | −2.10 ± 0.10 | −0.36 | −2.46 | 0.85 ± 0.03 |
a Data were collected at 20 °C.
Characterization of the Interaction Interface of CaM-Akt(PHD) Complex by NMR Spectroscopy
The proton, carbon, and nitrogen resonances of Akt(PHD) were assigned using standard triple resonance methods (see “Experimental Procedures”). Approximately 90% of the signals of Akt(PHD) were assigned; signal broadening precluded assignment of the remaining ∼10% signals. To gain insights into the molecular elements of the CaM-Akt(PHD) interaction, we have utilized NMR resonance perturbations as detected in 1H-15N HSQC spectra. These experiments often allow for identification of residues involved in the interaction interface and/or accompanying conformational changes and provide an effective method for examining the folding of the protein. Because of the propensity of Akt(PHD) to self-associate, which often complicates the NMR studies, we conducted the two-dimensional HSQC NMR experiments at low protein concentration (60 μm). We first titrated CaM into a 15N-labeled Akt(PHD) and monitored chemical shift changes by two-dimensional HSQC experiments. The addition of a substoichiometric amount of CaM (0.5:1 CaM/PHD) led to a decrease in intensity for numerous 1H-15N resonances accompanied by the appearance of several new signals, consistent with a slow exchange on the NMR scale between the free and bound forms of Akt(PHD). A steady decrease in the intensity of 1H-15N signals and increase in intensity of the new signals was clearly observed with the further addition of CaM. Spectral changes ceased upon completion of CaM titration at 1:1 Akt(PHD)/CaM (Fig. 5). As indicated by the dispersion of signals, the Akt(PHD) protein remains folded upon binding to CaM. A subset of 1H-15N resonances exhibited significant chemical shift changes (>0.1 ppm), which correspond to residues Lys-14, Arg-15, Gly-16, Glu-17, Ile-19, Thr-21, Trp-22, Tyr-26, Leu-28, Thr-34, Gly-37, Asp-44, Val-45, Val-57, Gln-59, Lys-64, Ile-84, Val-90, Ile-103, Thr-105, Val-106, Asp-108, Gly-109, Leu-110, and Lys-111 (Figs. 5 and 6A). Moreover, several amide signals of residues located between positions 76 and 88 shifted significantly and became very broad, rendering signal assignment very difficult. Another spectral observation is the chemical shift perturbation of the side chain of Akt(PHD) tryptophan residues. As shown in Fig. 5, the side chain (ϵ1) 1H-15N resonances corresponding to Trp-80 and Trp-22 exhibited significant chemical shift changes upon binding of CaM. Aromatic residues, such as tryptophan, often have a high propensity to anchor to the hydrophobic patches of CaM (54, 55). To identify the interaction interface of Akt(PHD), the chemical shift perturbations were mapped on the structure of Akt(PHD) bound to inositol 1,3,4,5-tetrakisphosphate (33). As shown in Fig. 6B, the significantly perturbed residues are localized in relatively distant regions, but their presence on the same side of the molecule suggests a well defined interaction interface. The interaction interface involves the loops connecting strands β1/β2 (residues 13–23; red) and β6/β7 (residues 76–88; orange), and strand β3 (residues 40–45; yellow). Modest chemical shift changes are also observed for residues in the distant C-terminal helix (cyan). Interestingly, the binding interface is adjacent to the PI(3,4,5)P3 binding pocket and lacks any helical character, indicating that the binding region represents a rare CaM-binding motif.
FIGURE 5.

Two-dimensional 1H-15N HSQC spectra obtained on a 15N-labeled Akt(PHD) (60 μm) in the free (black) and CaM-bound (red) states. Amide signals that exhibited substantial chemical shift changes are highlighted in green. Tryptophan side chain signals are highlighted in blue (inset). Notice the significant perturbation observed for the side chain signals of Trp-80 and, to a lesser extent, Trp-22.
FIGURE 6.
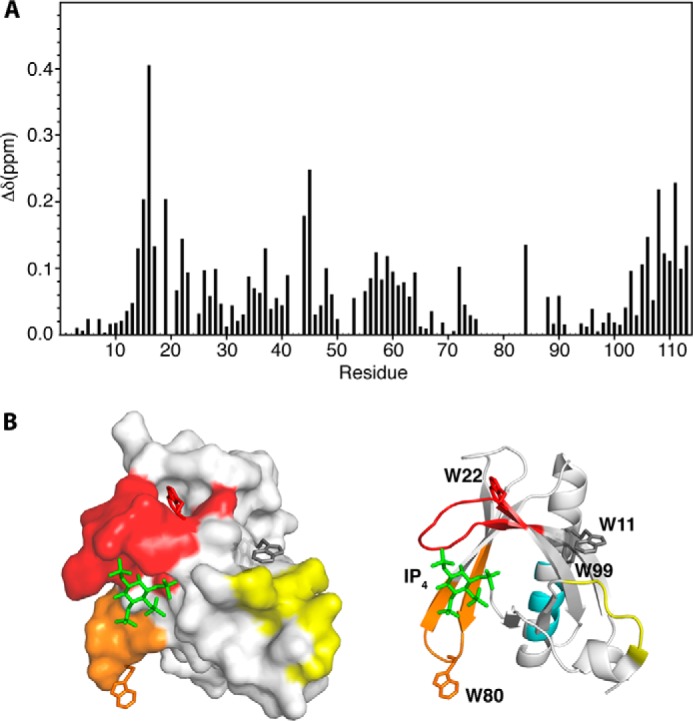
A, a histogram of normalized 1H-15N chemical shift changes versus residue number calculated from the HSQC spectra for Akt(PHD) upon titration with CaM. B, surface representation of the Akt(PHD) structure (Protein Data Bank entry 1H10) highlighting residues that exhibited substantial (>0.1 ppm) chemical shift changes. Red (residues 13–23), orange (residues 80–84), yellow (residues 31–43), and cyan (residues 103–110) highlight regions perturbed by CaM binding. IP4, 1,3,4,5-tetrakisphosphate.
To identify the interaction interface on the CaM protein, we have collected the reciprocal HSQC experiment with 15N-labeled CaM as titrated with unlabeled Akt(PHD). The addition of substoichiometric amounts of Akt(PHD) led to a decrease in intensity for numerous 1H-15N resonances accompanied by appearance of several new signals, consistent with a slow exchange on the NMR scale between the free and bound forms of CaM. Saturation was also achieved at 1:1 CaM/Akt(PHD). As shown in Fig. 7, the vast majority of 1H-15N resonances of CaM exhibited substantial chemical shift changes upon binding of Akt(PHD). These signals correspond to residues located on the N- and C-terminal lobes as well as the central linker of CaM, suggesting an engagement of a large interacting interface or induction of significant conformational changes in CaM upon binding to Akt(PHD). Altogether, the chemical shift perturbation data suggest that the binding mode of Akt(PHD) to CaM deviates from the typical mode that usually involves helical regions.
FIGURE 7.

Two-dimensional 1H-15N HSQC spectra obtained on a 15N-labeled CaM (60 μm) in the free (black) and Akt(PHD)-bound (red) states. The 1H-15N signals that exhibited significant chemical shift changes correspond to residues within the N- and C-terminal lobes as well as the central linker of CaM.
The N- and C-terminal Domains of CaM Are Required for Binding to the PHD of Akt
The chemical shift perturbation data shown above suggest that both of the N- and C-terminal lobes of CaM are probably involved in Akt(PHD) binding. One possible model to explain those findings is that Akt(PHD) anchors simultaneously to the N- and C-terminal lobes of CaM. Another scenario is that Akt(PHD) binds only to one lobe of CaM and induces a conformational change in the protein. We have devised two approaches to discern these two models and to identify the molecular elements of binding. In the first approach, we titrated isolated CaM-N and CaM-C domains into 15N-labeled samples of Akt(PHD), followed by acquisition of two-dimensional HSQC NMR data. As shown in Fig. 8, only a few 1H-15N signals exhibited significant chemical shift changes upon titration of CaM-N into Akt(PHD). These include residues Arg-16, Ile-19, Thr-21, Val-45, Asn-53, Thr-82, Ile-103, Thr-105, Val-106, Asp-108, and Lys-111. Because many signals corresponding to residues in the 76–87 region were too broad to assign, it was not possible to conclusively determine the extent of chemical shift perturbation. However, in addition to Thr-82, the 1H-15N signal corresponding to Trp-80 ϵ1 also exhibited a significant chemical shift change (Fig. 8A, inset), similar to that observed upon titration of Akt(PHD) with full-length CaM (Fig. 5). As opposed to what was observed with the full-length CaM, the chemical shift changes in the HSQC spectra indicate fast exchange on the NMR scale between free and bound forms. Titration data were fit by a one-site binding model with Kd of ∼112 ± 19 μm (Fig. 8), a value that is ∼103-fold weaker than that obtained for full-length CaM. This result demonstrates that CaM-N alone is not sufficient for binding of Akt(PHD).
FIGURE 8.

A, histograms of normalized 1H-15N chemical shift changes versus residue number calculated from the HSQC spectra for Akt(PHD) upon titration with CaM-N or CaM-C. Note the significant differences in chemical shift changes. For example, signals corresponding to residues 13–16 exhibited substantial chemical shift changes upon binding of CaM-C but were less sensitive to binding of CaM-N. Insets, selected regions of the 1H-15N HSQC titration data showing the chemical shift changes of tryptophan side chain signals. B, binding isotherms generated by plotting the change in 1H and 15N chemical shifts (Δδ) versus concentration as obtained from the HSQC titration data. Data were best fit into 1:1 binding isotherms. As indicated by the Kd values, binding of isolated CaM-N and CaM-C to Akt(PHD) is 103-fold weaker than that of full-length CaM.
The two-dimensional HSQC titration data obtained upon titration of CaM-C into Akt(PHD) also revealed a weaker binding than that of the intact CaM protein, as indicated by the chemical shift changes. The interaction between Akt(PHD) and CaM-C was undergoing fast exchange on the NMR scale, which allowed for calculation of the Kd value (34 ± 8 μm; Fig. 8). Binding of CaM-C to Akt(PHD) led to pronounced chemical shift changes for residues Lys-14, Arg-15, Gly-16, Ile-19, Thr-21, Trp-22, Arg-23, Val-45, Glu-49, and Asn-53 (Fig. 8). The 1H-15N signal corresponding to Trp-22 side chain (ϵ1) also exhibited a significant chemical shift change (Fig. 8A, inset), similar to that observed when Akt(PHD) is titrated with full-length CaM (Fig. 5). When comparing NMR data for CaM-N and CaM-C titration to Akt(PHD), distinct differences are observed. For example, the 1H-15N resonances for residues 14–23 (loop connecting β1/2) were more substantially perturbed upon binding of CaM-C, a result that is very similar to that observed for full-length CaM. The side chain 1H-15N signals for residues Trp-22 and Trp-80 are perturbed by CaM-C and CaM-N, respectively. Moreover, the C-terminal helix of Akt(PHD) appears to be perturbed only when CaM-N is bound. Altogether, these results indicate that the PHD of Akt simultaneously engages both lobes of CaM and that CaM-N appears to recognize the loop connecting β6 and β7, whereas CaM-C appears to bind to the loop connecting β1 and β2.
In the second approach, we assessed whether the hydrophobic patches on the N- and C-terminal lobes of CaM contribute to Akt(PHD) binding. These hydrophobic surfaces are widely implicated in the unique binding mode of CaM to target proteins (28) The methyl groups of Met residues are useful “NMR reporters” and often used to probe for target binding (35, 44, 56–58). Met residues 36, 51, 71, 72, and 76 are located in the N-terminal lobe, and residues 109, 124, 144, and 145 are located within the hydrophobic patch in the C-terminal lobe. Binding of target proteins or ligands to CaM often leads to substantial chemical shift changes in the 1H-13C methyl signals (35, 44, 56–58). To assess how the N- and C-terminal hydrophobic surfaces contribute to Akt(PHD) binding, we collected two-dimensional 1H-13C HMQC data on a uniformly 13C-labeled CaM sample upon titration with Akt(PHD). Binding of Akt(PHD) to CaM led to disappearance of the 1H-13C signals for Met-51, Met-71, Met-72, Met-109, and Met-144 (data not shown). Modest chemical shift changes have been observed for signals corresponding to Met-36 and Met-76, whereas no detectable changes occur for Met-124 and Met-145 signals. The HMQC NMR data indicate that Met residues from both the N- and C-terminal lobes contribute to Akt(PHD) binding. This result again demonstrates that both domains of CaM are critical for binding of Akt(PHD).
Discussion
Activation of Akt is critical in regulating apoptosis and oncogenesis and is a common tumorigenic event in many types of cancers, including breast cancer (1, 4, 5). The structural features of the PHD are shared by hundreds of analogs in mammals and bacteria (14–16). It is established that recruitment of Akt to the PM for activation is mediated by specific interactions between the PHD and PI(3,4,5)P3), a minor lipid localized on the inner leaflet of the PM (1, 4, 13). Additionally, it was shown that Akt translocation to the PM and subsequent activation are modulated by PS (17). PS is thought to play a synergistic effect by enhancing binding of PI(3,4,5)P3 to the PHD. It is now proposed that Akt activation in breast cancer cells is also modulated by CaM through interaction with the PHD (3, 22). Perturbation of this mechanism inhibited Akt-CaM translocation to the membrane and led to apoptotic cell death in tumorigenic mammary carcinoma cells (3). Here, we show that CaM binds tightly to Akt(PHD). The interaction between CaM and Akt(PHD) is enthalpically driven and dependent on salt concentration, suggesting that electrostatic interactions contribute into the formation of the complex. By using NMR chemical shift perturbation data, we mapped the CaM binding interface in Akt(PHD) to two loops adjacent to the PI(3,4,5)P3 binding site. We also showed that Akt(PHD) binding to the isolated N- and C-terminal lobes of CaM is 103-fold weaker than full-length CaM, suggesting that Akt(PHD) engages both domains simultaneously. Altogether, our findings provide the first mechanistic evidence for the mode of CaM binding to Akt(PHD).
Guided by genetic analysis and protein co-precipitation, it was previously suggested that the CaM-binding region of Akt is located within the first 42 residues (31). This region forms a three-stranded β-sheet (Fig. 1). The involvement of a β-sheet in CaM binding is, however, unprecedented because CaM-binding fragments often adopt helical structures (27, 32). The CaM-binding regions of target proteins are typically short (15–20 residues), hydrophobic-basic in nature, and have the propensity to form an α-helix (25, 32). Our NMR data, however, are not in agreement with the previous studies and strongly suggest that the CaM interaction interface on Akt(PHD) involves two loops connecting β strands 1 and 2 (residues 14–23), and β strands 6 and 7 (residues 80–84) (Figs. 5 and 9). Interestingly, this binding mode is similar to that observed when CaM binds to the PHD of a homologous protein from the Tec kinase family called interleukin-2-inducible tyrosine kinase (Itk) (47). The Tec family of tyrosine kinases is critical for the development and activation of immune cells (47, 59–61). NMR chemical shift perturbation data revealed that the Itk PHD interacts with CaM via the β3/β4 and β5/β6 loops (47). In both cases, Itk and Akt, the mode of binding of the PHD to CaM is rare. Sequence analysis of PHDs using a prediction algorithm based on known CaM-binding proteins (62) revealed that about half of the analyzed 236 PHDs were potential CaM-binding candidates (47). The ability for several PHDs to bind CaM was validated experimentally using co-precipitation assays (47). Our results along with the recent findings on CaM-Itk(PHD) complex suggest that the PHDs represent a novel class of CaM-binding proteins.
FIGURE 9.
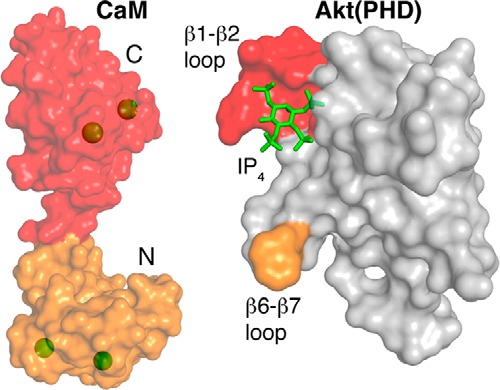
The NMR chemical shift perturbation data suggest that the N-terminal domain of CaM recognizes the Akt(PHD) loop connecting β6 and β7 (both highlighted in orange), whereas the C-terminal lobe of CaM appears to bind to the loop connecting β1 and β2 (both highlighted in red). The CaM interaction interface of Akt(PHD) is adjacent to the inositol 1,3,4,5-tetrakisphosphate (IP4) (green sticks) binding site. Calcium ions are shown as green spheres. Images were generated by PyMOL (PyMOL Molecular Graphics System version 1.7.4, Schrödinger, LLC).
The finding that EGF-induced membrane translocation and activation of Akt is modulated by CaM indicates a regulatory role of this protein in the membrane localization of Akt by binding directly to the PHD (3, 22). The confocal microscopy data show that stimulation of breast cancer cells with EGF caused translocation of both CaM and Akt to the PM, demonstrating that CaM and Akt localize on the same membrane compartment (3). Treatment of cells with a CaM antagonist blocked the translocation of both Akt and CaM to the PM (3). These results suggest that CaM is able to bind to the PHD of Akt without interfering with PI(3,4,5)P3 binding. It is likely that CaM plays a synergistic role in Akt activation by enhancing binding to PI(3,4,5)P3-enriched membranes. Indeed, this proposed role of CaM is in agreement with the recent findings on the role of CaM in Itk activation (47). It was shown that CaM enhances PI(3,4,5)P3 binding to the PHD of Itk and vice versa. Based on these findings, we propose that CaM binding to the Akt(PHD) loops connecting β1/β2 and β6/β7 strands possibly induces a conformational change that positively affects PI(3,4,5)P3 binding. Future studies will be focused on testing this hypothesis.
Elucidation of the structural details of how Akt binds to the PM and CaM may offer a better understanding of the molecular basis for Akt activation, which may help in the development of new therapeutic strategies against breast cancer and other types of cancer. Indeed, numerous Akt inhibitors are currently in clinical trials, and structure-based design and development of new inhibitors necessitates understanding, at the atomic level, the molecular mechanism of membrane translocation and activation of Akt.
Author Contributions
C. A. and R. H. G. designed and executed the experiments, analyzed the data, and compiled the figures. C. A. and J. S. S. wrote the manuscript.
Acknowledgments
We thank Yabing Chen (University of Alabama at Birmingham) for providing the Akt clone and Jiri Vlach (University of Alabama at Birmingham) for helping with the NMR experiments.
This work was supported by a pilot grant (to J. S. S.) from the comprehensive cancer center at the University of Alabama at Birmingham (supported by National Institutes of Health (NIH), NCI, Grant P30 CA013148). The high-field NMR facility and the x-ray core facility that houses the Auto-ITC200 (acquired through NIH Grant 1S10RR026478) are supported by the comprehensive cancer center at the University of Alabama at Birmingham. The authors declare that they have no conflicts of interest with the contents of this article.
The chemical shifts of Akt(PHD) in the free state and when bound to CaM have been deposited in the Biological Magnetic Resonance Bank with the accession codes 26651 and 26652, respectively.
- PI(3,4,5)P3
- phosphatidylinositol 3,4,5-trisphosphate
- CaM
- calmodulin
- PHD
- pleckstrin homology domain
- HSQC
- heteronuclear single quantum coherence
- HMQC
- heteronuclear multiple quantum coherence
- ITC
- isothermal titration calorimetry
- Itk
- interleukin-2-inducible tyrosine kinase
- PM
- plasma membrane
- PS
- phosphatidylserine
- SUMO
- small ubiquitin-like modifier.
References
- 1.Altomare D. A., and Testa J. R. (2005) Perturbations of the AKT signaling pathway in human cancer. Oncogene 24, 7455–7464 [DOI] [PubMed] [Google Scholar]
- 2.Carpten J. D., Faber A. L., Horn C., Donoho G. P., Briggs S. L., Robbins C. M., Hostetter G., Boguslawski S., Moses T. Y., Savage S., Uhlik M., Lin A., Du J., Qian Y. W., Zeckner D. J., Tucker-Kellogg G., Touchman J., Patel K., Mousses S., Bittner M., Schevitz R., Lai M. H., Blanchard K. L., and Thomas J. E. (2007) A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature 448, 439–444 [DOI] [PubMed] [Google Scholar]
- 3.Coticchia C. M., Revankar C. M., Deb T. B., Dickson R. B., and Johnson M. D. (2009) Calmodulin modulates Akt activity in human breast cancer cell lines. Breast Cancer Res. Treat. 115, 545–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dillon R. L., White D. E., and Muller W. J. (2007) The phosphatidyl inositol 3-kinase signaling network: implications for human breast cancer. Oncogene 26, 1338–1345 [DOI] [PubMed] [Google Scholar]
- 5.Pérez-Tenorio G., and Stål O., Southeast Sweden Breast Cancer Group (2002) Activation of AKT/PKB in breast cancer predicts a worse outcome among endocrine treated patients. Br. J. Cancer 86, 540–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ahmed N. N., Grimes H. L., Bellacosa A., Chan T. O., and Tsichlis P. N. (1997) Transduction of interleukin-2 antiapoptotic and proliferative signals via Akt protein kinase. Proc. Natl. Acad. Sci. U.S.A. 94, 3627–3632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Valius M., and Kazlauskas A. (1993) Phospholipase C-γ1 and phosphatidylinositol 3 kinase are the downstream mediators of the PDGF receptor's mitogenic signal. Cell 73, 321–334 [DOI] [PubMed] [Google Scholar]
- 8.Kaliman P., Viñals F., Testar X., Palacín M., and Zorzano A. (1996) Phosphatidylinositol 3-kinase inhibitors block differentiation of skeletal muscle cells. J. Biol. Chem. 271, 19146–19151 [DOI] [PubMed] [Google Scholar]
- 9.Franke T. F., Yang S. I., Chan T. O., Datta K., Kazlauskas A., Morrison D. K., Kaplan D. R., and Tsichlis P. N. (1995) The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell 81, 727–736 [DOI] [PubMed] [Google Scholar]
- 10.Yap T. A., Garrett M. D., Walton M. I., Raynaud F., de Bono J. S., and Workman P. (2008) Targeting the PI3K-AKT-mTOR pathway: progress, pitfalls, and promises. Curr. Opin. Pharmacol. 8, 393–412 [DOI] [PubMed] [Google Scholar]
- 11.Miao B., and Degterev A. (2011) Targeting phospshatidylinositol 3-kinase signaling with novel phosphatidylinositol 3,4,5-triphosphate antagonists. Autophagy 7, 650–651 [DOI] [PubMed] [Google Scholar]
- 12.Franke T. F. (2008) Intracellular signaling by Akt: bound to be specific. Sci. Signal. 1, pe29. [DOI] [PubMed] [Google Scholar]
- 13.Cooray S. (2004) The pivotal role of phosphatidylinositol 3-kinase-Akt signal transduction in virus survival. J. Gen. Virol. 85, 1065–1076 [DOI] [PubMed] [Google Scholar]
- 14.Lemmon M. A. (2007) Pleckstrin homology (PH) domains and phosphoinositides. Biochem. Soc. Symp. 81–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu Q., Bateman A., Finn R. D., Abdubek P., Astakhova T., Axelrod H. L., Bakolitsa C., Carlton D., Chen C., Chiu H. J., Chiu M., Clayton T., Das D., Deller M. C., Duan L., Ellrott K., Ernst D., Farr C. L., Feuerhelm J., Grant J. C., Grzechnik A., Han G. W., Jaroszewski L., Jin K. K., Klock H. E., Knuth M. W., Kozbial P., Krishna S. S., Kumar A., Marciano D., McMullan D., Miller M. D., Morse A. T., Nigoghossian E., Nopakun A., Okach L., Puckett C., Reyes R., Rife C. L., Sefcovic N., Tien H. J., Trame C. B., van den Bedem H., Weekes D., Wooten T., Hodgson K. O., Wooley J., Elsliger M. A., Deacon A. M., Godzik A., Lesley S. A., and Wilson I. A. (2010) Bacterial pleckstrin homology domains: a prokaryotic origin for the PH domain. J. Mol. Biol. 396, 31–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu J. W., Mendrola J. M., Audhya A., Singh S., Keleti D., DeWald D. B., Murray D., Emr S. D., and Lemmon M. A. (2004) Genome-wide analysis of membrane targeting by S. cerevisiae pleckstrin homology domains. Mol. Cell 13, 677–688 [DOI] [PubMed] [Google Scholar]
- 17.Huang B. X., Akbar M., Kevala K., and Kim H. Y. (2011) Phosphatidylserine is a critical modulator for Akt activation. J. Cell Biol. 192, 979–992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cristiano B. E., Chan J. C., Hannan K. M., Lundie N. A., Marmy-Conus N. J., Campbell I. G., Phillips W. A., Robbie M., Hannan R. D., and Pearson R. B. (2006) A specific role for AKT3 in the genesis of ovarian cancer through modulation of G2-M phase transition. Cancer Res. 66, 11718–11725 [DOI] [PubMed] [Google Scholar]
- 19.Héron-Milhavet L., Franckhauser C., Rana V., Berthenet C., Fisher D., Hemmings B. A., Fernandez A., and Lamb N. J. (2006) Only Akt1 is required for proliferation, while Akt2 promotes cell cycle exit through p21 binding. Mol. Cell. Biol. 26, 8267–8280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Irie H. Y., Pearline R. V., Grueneberg D., Hsia M., Ravichandran P., Kothari N., Natesan S., and Brugge J. S. (2005) Distinct roles of Akt1 and Akt2 in regulating cell migration and epithelial-mesenchymal transition. J. Cell Biol. 171, 1023–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yun S. J., Tucker D. F., Kim E. K., Kim M. S., Do K. H., Ha J. M., Lee S. Y., Yun J., Kim C. D., Birnbaum M. J., and Bae S. S. (2009) Differential regulation of Akt/protein kinase B isoforms during cell cycle progression. FEBS Lett. 583, 685–690 [DOI] [PubMed] [Google Scholar]
- 22.Deb T. B., Coticchia C. M., and Dickson R. B. (2004) Calmodulin-mediated activation of Akt regulates survival of c-Myc-overexpressing mouse mammary carcinoma cells. J. Biol. Chem. 279, 38903–38911 [DOI] [PubMed] [Google Scholar]
- 23.Chin D., and Means A. R. (2000) Calmodulin: a prototypical calcium sensor. Trends Cell Biol. 10, 322–328 [DOI] [PubMed] [Google Scholar]
- 24.Hoeflich K. P., and Ikura M. (2002) Calmodulin in action: diversity in target recognition and activation mechanisms. Cell 108, 739–742 [DOI] [PubMed] [Google Scholar]
- 25.Ishida H., and Vogel H. J. (2006) Protein-peptide interaction studies demonstrate the versatility of calmodulin target protein binding. Protein Pept. Lett. 13, 455–465 [DOI] [PubMed] [Google Scholar]
- 26.Osawa M., Tokumitsu H., Swindells M. B., Kurihara H., Orita M., Shibanuma T., Furuya T., and Ikura M. (1999) A novel target recognition revealed by calmodulin in complex with Ca2+-calmodulin-dependent kinase kinase. Nat. Struct. Biol. 6, 819–824 [DOI] [PubMed] [Google Scholar]
- 27.Vetter S. W., and Leclerc E. (2003) Novel aspects of calmodulin target recognition and activation. Eur. J. Biochem. 270, 404–414 [DOI] [PubMed] [Google Scholar]
- 28.Yamniuk A. P., and Vogel H. J. (2004) Calmodulin's flexibility allows for promiscuity in its interactions with target proteins and peptides. Mol. Biotechnol. 27, 33–57 [DOI] [PubMed] [Google Scholar]
- 29.Desai K. V., Xiao N., Wang W., Gangi L., Greene J., Powell J. I., Dickson R., Furth P., Hunter K., Kucherlapati R., Simon R., Liu E. T., and Green J. E. (2002) Initiating oncogenic event determines gene-expression patterns of human breast cancer models. Proc. Natl. Acad. Sci. U.S.A. 99, 6967–6972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krishnaraju K., Murugesan K., Vij U., Kapur B. M., and Farooq A. (1991) Calmodulin levels in oestrogen receptor positive and negative human breast tumours. Br. J. Cancer 63, 346–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dong B., Valencia C. A., and Liu R. (2007) Ca2+/calmodulin directly interacts with the pleckstrin homology domain of AKT1. J. Biol. Chem. 282, 25131–25140 [DOI] [PubMed] [Google Scholar]
- 32.Villarroel A., Taglialatela M., Bernardo-Seisdedos G., Alaimo A., Agirre J., Alberdi A., Gomis-Perez C., Soldovieri M. V., Ambrosino P., Malo C., and Areso P. (2014) The ever changing moods of calmodulin: how structural plasticity entails transductional adaptability. J. Mol. Biol. 426, 2717–2735 [DOI] [PubMed] [Google Scholar]
- 33.Thomas C. C., Deak M., Alessi D. R., and van Aalten D. M. (2002) High-resolution structure of the pleckstrin homology domain of protein kinase b/akt bound to phosphatidylinositol (3,4,5)-trisphosphate. Curr. Biol. 12, 1256–1262 [DOI] [PubMed] [Google Scholar]
- 34.Auguin D., Barthe P., Augé-Sénégas M. T., Stern M. H., Noguchi M., and Roumestand C. (2004) Solution structure and backbone dynamics of the pleckstrin homology domain of the human protein kinase B (PKB/Akt). Interaction with inositol phosphates. J. Biomol. NMR 28, 137–155 [DOI] [PubMed] [Google Scholar]
- 35.Samal A. B., Ghanam R. H., Fernandez T. F., Monroe E. B., and Saad J. S. (2011) NMR, biophysical, and biochemical studies reveal the minimal calmodulin-binding domain of the HIV-1 matrix protein. J. Biol. Chem. 286, 33533–33543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fernandez T. F., Samal A. B., Bedwell G. J., Chen Y., and Saad J. S. (2013) Structural and biophysical characterization of the interactions between the death domain of Fas receptor and calmodulin. J. Biol. Chem. 288, 21898–21908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lebowitz J., Lewis M. S., and Schuck P. (2002) Modern analytical ultracentrifugation in protein science: a tutorial review. Protein Sci. 11, 2067–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schuck P. (2000) Size distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys. J. 78, 1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schuck P. (2003) On the analysis of protein self-association by sedimentation velocity analytical ultracentrifugation. Anal. Biochem. 320, 104–124 [DOI] [PubMed] [Google Scholar]
- 40.Schuck P., Perugini M. A., Gonzales N. R., Howlett G. J., and Schubert D. (2002) Size-distribution analysis of proteins by analytical ultracentrifugation: strategies and application to model systems. Biophys. J. 82, 1096–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., and Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 42.Johnson B. A., and Blevins R. A. (1994) NMRview: a computer program for the visualization and analysis of NMR data. J. Biomol. NMR 4, 603–614 [DOI] [PubMed] [Google Scholar]
- 43.Vranken W. F., Boucher W., Stevens T. J., Fogh R. H., Pajon A., Llinas M., Ulrich E. L., Markley J. L., Ionides J., and Laue E. D. (2005) The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins 59, 687–696 [DOI] [PubMed] [Google Scholar]
- 44.Ghanam R. H., Fernandez T. F., Fledderman E. L., and Saad J. S. (2010) Binding of calmodulin to the HIV-1 matrix protein triggers myristate exposure. J. Biol. Chem. 285, 41911–41920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ikura M., Kay L. E., and Bax A. (1990) A novel approach for sequential assignment of 1H, 13C, and 15N spectra of larger proteins: heteronuclear triple-resonance three-dimensional NMR spectroscopy: application to calmodulin. Biochemistry 29, 4659–4667 [DOI] [PubMed] [Google Scholar]
- 46.Lemmon M. A. (2003) Phosphoinositide recognition domains. Traffic 4, 201–213 [DOI] [PubMed] [Google Scholar]
- 47.Wang X., Boyken S. E., Hu J., Xu X., Rimer R. P., Shea M. A., Shaw A. S., Andreotti A. H., and Huang Y. H. (2014) Calmodulin and PI(3,4,5)P(3) cooperatively bind to the Itk pleckstrin homology domain to promote efficient calcium signaling and IL-17A production. Sci. Signal. 7, ra74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Meuillet E. J. (2011) Novel inhibitors of AKT: assessment of a different approach targeting the pleckstrin homology domain. Curr. Med. Chem. 18, 2727–2742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fushman D., Cahill S., and Cowburn D. (1997) The main-chain dynamics of the dynamin pleckstrin homology (PH) domain in solution: analysis of 15N relaxation with monomer/dimer equilibration. J. Mol. Biol. 266, 173–194 [DOI] [PubMed] [Google Scholar]
- 50.Boyken S. E., Fulton D. B., and Andreotti A. H. (2012) Rescue of the aggregation prone Itk pleckstrin homology domain by two mutations derived from the related kinases, Btk and Tec. Protein Sci. 21, 1288–1297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Majava V., and Kursula P. (2009) Domain swapping and different oligomeric states for the complex between calmodulin and the calmodulin-binding domain of calcineurin A. PLoS One 4, e5402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hayashi N., Matsubara M., Jinbo Y., Titani K., Izumi Y., and Matsushima N. (2002) Nef of HIV-1 interacts directly with calcium-bound calmodulin. Protein Sci. 11, 529–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Matsubara M., Nakatsu T., Kato H., and Taniguchi H. (2004) Crystal structure of a myristoylated CAP-23/NAP-22 N-terminal domain complexed with Ca2+/calmodulin. EMBO J. 23, 712–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mruk K., Farley B. M., Ritacco A. W., and Kobertz W. R. (2014) Calmodulation meta-analysis: predicting calmodulin binding via canonical motif clustering. J. Gen. Physiol. 144, 105–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tidow H., and Nissen P. (2013) Structural diversity of calmodulin binding to its target sites. FEBS J. 280, 5551–5565 [DOI] [PubMed] [Google Scholar]
- 56.Siivari K., Zhang M., Palmer A. G. 3rd, and Vogel H. J. (1995) NMR studies of the methionine methyl groups in calmodulin. FEBS Lett. 366, 104–108 [DOI] [PubMed] [Google Scholar]
- 57.Yuan T., Ouyang H., and Vogel H. J. (1999) Surface exposure of the methionine side chains of calmodulin in solution: a nitroxide spin label and two-dimensional NMR study. J. Biol. Chem. 274, 8411–8420 [DOI] [PubMed] [Google Scholar]
- 58.Osawa M., Swindells M. B., Tanikawa J., Tanaka T., Mase T., Furuya T., and Ikura M. (1998) Solution structure of calmodulin-W-7 complex: the basis of diversity in molecular recognition. J. Mol. Biol. 276, 165–176 [DOI] [PubMed] [Google Scholar]
- 59.Rawlings D. J., Saffran D. C., Tsukada S., Largaespada D. A., Grimaldi J. C., Cohen L., Mohr R. N., Bazan J. F., Howard M., and Copeland N. G. (1993) Mutation of unique region of Bruton's tyrosine kinase in immunodeficient XID mice. Science 261, 358–361 [DOI] [PubMed] [Google Scholar]
- 60.Thomas J. D., Sideras P., Smith C. I., Vorechovský I., Chapman V., and Paul W. E. (1993) Colocalization of X-linked agammaglobulinemia and X-linked immunodeficiency genes. Science 261, 355–358 [DOI] [PubMed] [Google Scholar]
- 61.Vanhaesebroeck B., Stephens L., and Hawkins P. (2012) PI3K signalling: the path to discovery and understanding. Nat. Rev. Mol. Cell Biol. 13, 195–203 [DOI] [PubMed] [Google Scholar]
- 62.Yap K. L., Kim J., Truong K., Sherman M., Yuan T., and Ikura M. (2000) Calmodulin target database. J. Struct. Funct. Genomics 1, 8–14 [DOI] [PubMed] [Google Scholar]