

Abstract
Background
The myosin light chain kinase (MLCK) pathway controls intestinal epithelial barrier permeability by regulating the tight junction. 1,25-dihydroxyvitamin D (1,25(OH)2D3)-vitamin D receptor (VDR) signaling protects the epithelial barrier, but the molecular mechanism is incompletely understood.
Methods
MLCK activation and barrier permeability were studied using monolayers of HCT116, Caco-2 and SW480 cells treated with TNF-α ±1,25(OH)2D3. The MLCK pathway was analyzed in normal and inflamed colonic biopsies from ulcerative colitis patients. Colonic mucosal barrier permeability and MLCK activation were also investigated using TNBS-induced colitis models in vitamin D analog paricalcitol-treated wild-type mice and mice carrying VDR deletion in colonic epithelial cells.
Results
TNF-α increased cell monolayer permeability and induced long isoform of MLCK expression and myosin II regulatory light chain (MLC) phosphorylation, and 1,25(OH)2D3 blocked TNF-α-induced increases in monolayer permeability and MLCK-MLC pathway activation by a VDR-dependent fashion. 1,25(OH)2D3 directly suppressed long MLCK expression by attenuating NF-κB activation, and ChIP assays confirmed that 1,25(OH)2D3 disrupted p65 binding to three κB sites in long MLCK gene promoter. In human ulcerative colitis biopsies VDR reduction was associated with increases in long MLCK expression and MLC phosphorylation. In TNBS colitis models, paricalcitol ameliorated colitis, attenuated the increase in mucosal barrier permeability and inhibited long MLCK induction and MLC phosphorylation. In contrast, mice with colonic epithelial VDR deletion exhibited more robust increases in mucosal barrier permeability and MLCK activation compared with wild-type mice.
Conclusions
These data demonstrate that 1,25(OH)2D3-VDR signaling preserves the mucosal barrier integrity by abrogating MLCK-dependent tight junction dysregulation during colonic inflammation.
Keywords: Vitamin D, Intestinal epithelial barrier, Tight junction, Myosin light chain kinase, Colitis, Inflammatory bowel diseases
Introduction
The intestinal epithelial barrier consists of a monolayer of epithelial cells and intercellular junctions between adjacent cells that seal the paracellular space and regulate permeability of the barrier 1. This barrier separates harmful luminal substances such as microorganisms, toxins and antigens from the body, and thus plays a critical role in mucosal homeostasis. Impaired barrier function can lead to gut hyperpermeability and trigger mucosal inflammation 2; 3. Increased gut permeability usually results from aberrant apoptosis of intestinal epithelial cells and/or dysregulation of tight junction on the barrier, and recent studies suggest that these are two temporally and morphologically distinct mechanisms in colitis development 4. Increases in tight junction permeability occur in early disease stage and promotes disease initiation, whereas epithelial cell apoptosis causes tight junction-independent barrier loss and drives disease progression.
Myosin light chain kinase (MLCK) plays a central role in the regulation of tight junction permeability 5. MLCK has two splice variants derived from the same gene using different promoters. Short or smooth muscle MLCK is not expressed in intestinal epithelium, whereas long MLCK is highly expressed in intestinal epithelial cells and regulates tight junction permeability via inducing phosphorylation of myosin II regulatory light chain (MLC) 6; 7, which leads to remodeling of the tight junction structure 8. TNF-α, a pro-inflammatory cytokine central to the pathogenesis of inflammatory bowel diseases (IBD), particularly Crohn’s disease 9; 10, has been shown to promote tight junction dysregulation and induce epithelial barrier loss by up-regulating long MLCK expression and enzymatic activity 11; 12. This up-regulation is in part mediated by NF-κB 13; 14, and a few κB sites have been identified in the upstream promoter region that specifically drives long MLCK expression 15; 16.
1,25-dihydroxyvitamin D (1,25(OH)2D3) is a pleiotropic hormone with a broad range of physiological functions 17. The biological activity of 1,25(OH)2D3 is mediated by the vitamin D receptor (VDR), a nuclear hormone receptor 18 highly expressed in gut epithelial cells. Vitamin D-deficiency or - insufficiency (commonly defined as serum 25-hydroxyvitamin D < 30 ng/ml) is associated with increased risk of IBD 19, and a high prevalence of vitamin D deficiency or -insufficiency has been reported in patients with established as well as newly diagnosed IBD 20; 21; 22; 23. Conversely, high vitamin D intake lowers the risk of IBD 24. Thus, vitamin D is though to be an environmental risk factor for IBD 19. Recently we reported that the epithelial VDR signaling protects gut epithelial barrier by suppressing inflammation-induced epithelial cell apoptosis 25. Consistent with this notion, we found that epithelial VDR levels are >50% down-regulated in patients with IBD 25, partly due to TNF-α induction of miR-346, which targets VDR 26. Aside from inhibition of epithelial cell apoptosis, numerous studies have shown that 1,25(OH)2D3-VDR signaling also preserves epithelial barrier permeability by directly regulating tight junction 27; 28; 29. Apparently, vitamin D modulation of tight junction is an integral part of the mechanism whereby vitamin D protects the gut epithelial barrier; however, how vitamin D controls tight junction remains unclear. Therefore, in this study we aimed at elucidating the molecular mechanism whereby 1,25(OH)2D3 regulates tight junction. Our study demonstrates that 1,25(OH)2D3 suppresses the MLCK-MLC pathway via blocking NF-κB activation to maintain the mucosal barrier permeability.
Experimental procedures
Cell culture
HCT116, Caco-2 and SW480 cells were cultured in DMEM supplemented with 10% FBS at 37°C and 5% CO2. The cells were pre-treated with 1,25(OH)2D3 (20 nM) or ethanol vehicle for 24 hours followed by TNF-α (100 ng/ml) or saline treatment for 12 hours. In separate experiments, the cells were pre-treated with BAY 11-7082 (20 ng/ml), a NF-κB inhibitor, for 24 hours followed by 12-hour TNF-α or saline treatment. Cells were transfected with human (h)VDR-specific siRNA to reduce VDR levels, or with a plasmid expressing HA-IKKβ to increase IKKβ levels, both using Lipofectamine 2000 (Invitrogen).
Human biopsies
Colonic mucosal biopsies were obtained from patients with active ulcerative colitis at Shengjing Hospital of China Medical University. Biopsies were collected from the inflamed region and the adjacent normal tissue in each patient during endoscopic examination. Study subjects were recruited with written informed consent from the participants or their guardians. All human studies were approved by the Institutional Ethical Committee of Shengjing Hospital, China Medical University (Protocol # 2014PS145K). The collected biopsies were immediately processed for histology or for RNA and protein lysate preparations.
Animal treatment
C57BL/6 mice (6-8 week old) were treated with vitamin D analog paricalcitol (19-nor-1,25-dihydroxyvitamin D2, i.p. at 300 ng/kg, dissolved in 70% propylene glycol) or vehicle daily for one week. Experimental colitis was then induced in these mice using 2,4,6-trinitrobenzene sulfonic acid (TNBS) as previously reported 30; 25. Daily paricalcitol treatment continued after TNBS instillation. VDRΔCEC mice that carry VDR deletion in colonic epithelial cells were generated by crossing CDX2-Cre transgenic mice (Stock # 009350, Jackson Laboratory, Bar Harbor, Maine) and VDRflox/flox mice that contain LoxP sites flanking exon 4 of the Vdr gene 31. CDX2 promoter directs Cre recombinase expression in colonic epithelial cells and CDX2-Cre mice have been used to conditionally delete Apc gene from colonic epithelial cells 32. VDRΔCEC and VDRflox/flox mice were studied using TNBS-induced colitis model as reported before 25. Colons were collected immediately after sacrifice, and mucosa scraped to isolate total RNAs or proteins. All animal studies were approved by the Institutional Animal Care and Use Committee at the University of Chicago.
Permeability measurement
HCT116, Caco-2 and SW480 Cells were cultured in transwell inserts (0.4 µm pore size, Corning, Tewksbury, MA) until monolayers were formed. The cells were pre-treated with 1,25(OH)2D3 or BAY 11-7082 for 24 hours, followed by 12-hour TNF-α treatment placed on the apical side (no treatment compound was placed in the basolateral chamber). Transepithelial electrical resistance (TER) was measured using an Ussing chamber system. In separate experiments, monolayer paracellular permeability was also measured using FITC-conjugated dextran (MW 4000 Da, 10 mg/mL; Sigma-Aldrich, St. Louis, MO) as described previously 33. In TNBS models, colonic mucosal permeability was assessed by measuring colonic TER in freshly harvested colons using Ussing chamber as described 25, as well as by measuring FITC-dextran leak into the circulation as previously reported 34; 35. For the latter measurement, mice fasted for 6 hours were orally gavaged 4000 Da FITC-dextran at 200 mg/kg. After 2 hours, blood was collected from the tail, and serum FITC-dextran content was determined using a M200 TECAN microplate reader at 530 nm wavelength.
Histology and immunostaining
Freshly dissected colon or colon biopsies were fixed overnight with 4% formaldehyde in PBS (pH 7.2) and embedded in paraffin wax. Mouse colons were embedded as “Swiss roll” 36. Colonic morphology was examined by H&E staining, and histological scores were graded according to a previously published system 37; 38. Cell monolayers cultured on coverslips were fixed for 15 min in 4% formaldehyde and then incubated with anti-ZO-1 or anti-occludin antibodies (Invitrogen, Grand Island, NY), followed by FITC-conjugated anti-IgG antibodies (Santa Cruz Biotechnology, Dallas, Texas). Slides were examined under a Leica DFC425 fluorescence microscope.
Western blot
Western blot analyses were carried out as described previously 39. The following antibodies were used: anti-ZO-1, anti-occludin, anti-claudin-2 from Invitrogen; anti-MLC, anti-phospho-MLC and anti-IKKβ from Cell Signaling Technology (Beverly, MA); anti-TNF-α and anti-short MLCK from Sigma-Aldrich; anti-long MLCK from Abcam (Cambridge, MA) and anti-VDR from Santa Cruz Biotechnology.
RT-PCR
Total RNAs were extracted using TRIzol reagent (Invitrogen). First-strand cDNA templates were synthesized using PrimeScript RT reagent kit (TaKaRa, Mountain View, CA). Real-time PCR was carried out using SYBR Premix Ex kit (TaKaRa) in a Bio-RAD IQ5 real-time system. Relative amount of transcripts was calculated by the 2−ΔΔCt formula, using GAPDH as an internal control. PCR primers are provided in Table 1.
Table 1.
Nucleotide sequences of primers used in the study
| Primer name | Forward (5’-3’) | Revere (5’-3’) |
|---|---|---|
| hαB ChIP-1 | GCTGCCTCTGCTGCAGTTCA | ACACACAGCTCCCCTCTCTG |
| hαB ChIP-2 | CAAAGTGTCCCTCAAAGTGTC | TCACCCAGCCTCAGGTATTT |
| hαB ChIP-3 | CTGCTGACACTTCCTGTCCA | AGAGGGTGGAAACCCTGAGT |
| mTNF-α | TCAGCCTCTTCTCATTCCTG | CAGGCTTGTCACTCGAATTT |
| mIL-1β | CAGGATGAGGACATGAGCACC | CTCTGCAGACTCAAACTCCAC |
| mIL-6 | ATAGTCCTTCCTACCCCAATTTCC | CTGACCACAGTGAGGAATGTCCAC |
| mIFN-γ | CGGCACAGTCATTGAAAGCCTA | GTTGCTGATGGCCTGATTGTC |
| mIL-17 | TCCCTCTGTGATCTGGGAAG | AGCATCTTCTCGACCCTGAA |
| mIL-23p19 | AATAATGTGCCCCGTATCCA | CATGGGGCTATCAGGGAGTA |
| mMCP-1 | CAAGAAGGAATGGGTCCAGA | TGAGGTGGTTGTGGAAAAGG |
| mGAPDH | TGTGTCCGTCGTGGATCTGA | CCTGCTTCACCACCTTCTTGA |
Chromatin immunoprecipitation (ChIP) assays
Cells were treated with 1,25(OH)2D3 followed by TNF-α treatment for 4 hours. ChIP assays were performed as described previously 40, using anti-p65 antibodies. The assays were quantified by real time PCR using primers (Table 1) flanking the κB sites in the promoter of the MLCK gene 15; 16.
Myeloid peroxidase (MPO) activity assays
Colonic tissues were homogenized in 50 mM potassium phosphate and 50 mM hexadecyl trimethyl ammonium bromide (HTAB), sonicated, snap frozen and thawed twice, followed by addition of 50 mM potassium phosphate containing 0.167 mg/ml O-dianisidine dihydrochloride and 0.0005% hydrogen peroxide. Absorbance was read at 460 nm using M200 TECAN microplate system.
Statistical analysis
Data values were presented as means ± SD. Statistical comparisons were carried out using unpaired two-tailed Student’s t-test or one-way analysis of variance (ANOVA) as appropriate, with P< 0.05 being considered statistically significant.
Results
1,25(OH)2D3 attenuates TNF-α-induced increases in monolayer permeability and blocks MLCK up-regulation
TNF-α plays a central role in the pathogenesis of IBD and has been shown to increase tight junction permeability. Therefore, we used TNF-α-treated epithelial cells as models to study effects of vitamin D on barrier permeability. As shown in Figure 1, consistent with previous findings 27; 28; 29, treatment with TNF-α markedly reduced the TER of monolayers formed by HCT116, Caco-2 or SW480 cells (Fig. 1A), as well as increased FITC-dextran paracellular passage through these monolayers (data not shown), but the TNFα-induced increase in monolayer permeability was largely corrected in the presence of 1,25(OH)2D3 co-treatment (Fig. 1A). Immunostaining of Caco-2 monolayer with anti-ZO-1 or anti-occludin antibodies confirmed that TNF-α disrupted the integrity of the tight junction, and the disruptive effect of TNF-α was attenuated by 1,25(OH)2D3 (Fig. 1B). As expected, TNF-α dramatically induced long MCLK (but not short MLCK) expression and MLC phosphorylation (but not MLC levels) in HCT116, Caco-2 and SW480 cells. These changes were accompanied by moderate claudin-2 induction and marked occludin inhibition. Claudin-2 is a tight junction protein that was found elevated in IBD patients 41; 42, and elevation of claudin-2 is associated with increased tight junction permeability 43. Remarkably, 1,25(OH)2D3 completely blocked the effect of TNF-α on MLCK up-regulation and MLC phosphorylation, as well as on claudin-2 and occludin expression in all three cell lines (Fig. 1C). Similar results were seen in cells treated with IFN-γ, another pro-inflammatory cytokine that can increase mucosal permeability 12 (Supplemental Figure 1A and B). Moreover, TNF-α and IFN-γ co-treatment caused more dramatic activation of the MLCK-MLC pathway and more dramatic loss of TER than IFN-γ alone, which can still be mostly reversed by 1,25(OH)2D3 (Supplemental Figure 1C and D). Therefore, 1,25(OH)2D3 is able to attenuate the ability of TNF-α or IFN-γ to activate the MLCK-MLC pathway and increase epithelial tight junction permeability in vitro.
Figure 1.

1,25(OH)2D3 counteracts the effects of TNF-α on paracellular permeability and MLCK activation. (A) Transepithelial resistance (TER) of monolayers formed by HCT-116, Caco-2 and SW480 cells under different treatment as indicated. The cells were grown on transwells and treated or untreated with TNF-α±1,25(OH)2D3 for 12 hours. (B) Caco-2 monolayer immunostained with antibodies against ZO-1 or occludin as indicated. Magnification, 200x. (C) Western blot analyses of HCT116, Caco-2 and SW480 cells under different treatments as indicated. The blots were analyzed with a number of antibodies against short MLCK, long MLCK, phospho-MLC, MLC, claudin-2, occludin and β-actin as shown. TER, transepithelial resistance; 1,25-VD, 1,25(OH)2D3; Ctrl, control. ** P<0.01, *** P<0.001 vs. the rest.
1,25(OH)2D3 inhibition of MLCK expression is VDR-dependent
To address whether the effect of 1,25(OH)2D3 on MLCK is dependent on VDR, we used hVDR-specific siRNA to knock down VDR in HCT116 cells (Fig. 2A). When VDR protein expression was reduced by >70% by siRNA, 1,25(OH)2D3 was unable to counteract the stimulatory effect of TNF-α on long MLCK expression and MLC phosphorylation (Fig. 2A). Consequently, 1,25(OH)2D3 failed to maintain the TER of the monolayer formed by hVDR-siRNA-treated HCT116 cells in the presence of TNF-α (Fig. 2B). Therefore, VDR is required to mediate the activity of 1,25(OH)2D3 to protect the monolayer barrier.
Figure 2.

1,25(OH)2D3 regulation of MLCK activation and paracellular permeability is dependent on VDR. (A) Western blot analyses of HCT116 cells transfected with scramble or hVDR-specific siRNA followed by various treatments as indicated. The blots were incubated with a number of antibodies as shown. (B) Transepithelial resistance (TER) of HCT116 monolayers transfected with scramble or hVDR-specific siRNA under different treatments as indicated. *P<0.05; ** P<0.01 vs. corresponding Ctrl or 1,25-VD; # P<0.05 vs. corresponding scramble.
1,25(OH)2D3 inhibits MLCK activation by counteracting TNF-α-induced NF-κB activation
Previous studies reported that TNF-α induces MLCK expression via activating NF-κB signaling pathway 15; 16. To confirm this finding, we treated HCT116, Caco-2 and SW480 cells with TNF-α in the presence of NF-κB inhibitor BAY 11-7082. BAY 11-7082 completely blocked the induction of long MLCK expression and MLC phosphorylation by TNF-α in these three cell lines (Fig. 3A, and Supplemental Figure 2A and 2B). BAY 11-7082 also prevented TNF-α-induced claudin-2 up-regulation and occludin reduction in these cells. Consistently, NF-κB inhibition abrogated the increase in cell monolayer permeability caused by TNF-α treatment (Fig. 3B).
Figure 3.

1,25(OH)2D3 blocks MLCK activation by targeting NF-κB activation. (A) Western blot analyses of HCT-116 cells treated or untreated with TNF-α ± BAY 11-7082 as indicated. (B) Transepithelial resistance (TER) of HCT116 cell monolayers under different treatments as indicated for 6 hours. *** P<0.001 vs. the rest. (C) HCT116 cells were transfected with empty vector or IKKβ-expressing plasmid, followed by 1,25(OH)2D3 or vehicle treatment as indicated. Cell lysates were analyzed by Western blotting. (D) HCT116 cells were transfected with increasing amount of IKKβ and then treated with 1,25(OH)2D3 as shown. Cell lysates were analyzed by Western blotting. (E) Schematic illustration of human MLCK gene promoter containing three putative κB sites (κB-1, κB-2 and κB-3) at −136, −1451 and −1584 as indicated. The PCR primer locations were also indicated. (F) ChIP assays using anti-p65 antibodies for HCT116 cells pre-treated with 1,25(OH)2D3 or vehicle followed by 4-hour TNF-α or saline treatment as indicated. The results for κB-1, κB-2 and κB-3 sites are shown. Similar results were seen for 10-hour treatment. **P<0.01 vs. the rest.
To directly confirm the involvement of NF-κB in MLCK pathway activation, we activated NF-κB by IKKβ transfection in HCT116 cells. IKKβ overexpression markedly induced the expression of long MLCK (but not short MLCK) and MLC phosphorylation, as well as claudin-2 up-regulation and occluding down-regulation in the absence of TNF-α, but all these changes were reversed when the cells were treated with 1,25(OH)2D3 (Fig. 3C). The inhibitory effect 1,25(OH)2D3 on long MLCK, however, was overcome when increasing amounts of IKKβ were expressed in the cells (Fig. 3D). These data provide compelling evidence that 1,25(OH)2D3 directly counteracts the NF-κB signaling pathway in the down-regulation of long MLCK. Three putative κB sites were identified previously in the long MLCK promoter region (Fig. 3E) that were thought to mediate the stimulatory effect of TNF-α on long MLCK expression 15; 16. By ChIP assays we found that all three sites at −136 (κB-1), −1415 ((κB-2) and −1584 (κB-3) were bound by NF-κB p65 after TNF-α treatment, but these TNF-α induced p65 bindings to these sites were completely disrupted in the presence 1,25(OH)2D3 (Fig. 3F). This observation provides a molecular basis to explain the inhibition of long MLCK gene expression by 1,25(OH)2D3.
VDR reduction in human patient biopsies is associated with activation of MLCK pathway
We previously reported that colonic VDR levels in patients with ulcerative colitis and Crohn’s disease were reduced by about 50% 25 as a result of TNF-α-induced VDR down-regulation 26. Consistently, Figure 4 shows that in a new cohort of patients with ulcerative colitis, VDR protein levels in the majority of patients were markedly down-regulated in the inflamed lesions compared to the adjacent normal tissues from the same individuals (Fig. 4A), and the mean reduction for VDR protein is 46% in this cohort (Fig. 4B). Importantly, the VDR down-regulation was associated with dramatic and significant increases in long MLCK expression (by 110%) and MLC phosphorylation (by 370%) in the inflamed lesions in these patients (Fig. 4A and B). As expected, MPO activity in the lesions was also dramatically elevated (Fig. 4C), and so were pro-inflammatory cytokines (such as TNF-α, IFN-γ, IL-1β and IL-6) assessed by qPCR (data not shown), consistent with our early findings 26. These data are consistent with the notion that 1,25(OH)2D3–VDR signaling suppresses the MLCK-MLC pathway activation in humans.
Figure 4.

Reduction in VDR expression is associated with MLCK pathway activation in colonic biopsies from in ulcerative colitis patients. (A) Western blot analyses of inflamed lesion biopsies and adjacent normal tissues from seven ulcerative colitis patients using antibodies as indicated. (B) Relative protein levels in the lesions compared to the normal tissues, determined by densitometric quantitation of protein bands on the blots. (C) MPO activity in the normal and lesion biopsies. *P<0.05; ** P<0.01; ***P<0.001 vs. normal; n=7. N, normal adjacent tissue; D, diseased tissue.
Paricalcitol blocks MLCK activation, preserves mucosal barrier permeability and ameliorates colitis in colitismodel
To address whether vitamin D hormone regulates MLCK activation and mucosal barrier permeability in vivo, we investigated the effect of paricalcitol, a low-calcemic VDR agonist, in a TNBS-induced experimental colitis model, which develops TH1 cell-mediated mucosal inflammation 44; 45. As shown in Figure 5, paricalcitol treatment markedly impeded body weight loss following TNBS installation (Fig. 5A), ameliorated mucosal ulceration (Fig. 5B), reduced mucosal MPO activity (Fig. 5C) and attenuated mucosal production of pro-inflammatory cytokines and chemokines (TNF-α, IL-1β, IL-6, IFN-γ, IL-17, IL-23 and MCP-1) (Fig. 5D) in the TNBS-treated mice. Importantly, mucosal permeability of TNBS-treated mice increased one day after TNBS treatment when histological damage was not yet detectable in the colon, and paricalcitol treatment preserved colonic TER (Fig. 5E) and prevented FITC-dextran paracellular passage into the blood (Fig. 5F), indicating paricalcitol’s ability to protect against tight junction injury in vivo.
Figure 5.

Paricalcitol preserves mucosal barrier permeability, attenuates MLCK activation and ameliorates colitis in TNBS colitis model. (A) Mouse body weight changes in four treatment groups following TNBS instillation. *P<0.05, **P<0.01 vs. Ctrl and Paricalcitol; #P<0.05 vs. TNBS+Pari; n=5 in each group. (B) H&E staining of distal colon sections in four treatment groups of mice on day 3. (C) MPO activity in mucosal lysates from four groups of mice on day 3. **P<0.01 vs. the rest; n=5 in each group. (D) Real time RT-PCR quantitation of mucosal pro-inflammatory cytokines and chemokines in the four groups of mice on day 3. *P<0.05; ** P<0.01 vs. the rest; #P<0.05 vs. TNBS; n=5 in each genotype. (E) Transepithelial resistance (TER) of colon mucosa from control mice and mice treated with paricalcitol, TNBS and TNBS+Paricalcitol on day 1. (F) Quantitation of paracellular FITC-dextran passage across mucosal barrier in mice from the four treatment groups. ** P<0.01 vs. the rest. (G and H) Western blot analyses (G) and densitometric quantitation (H) of colonic mucosal lysates from the four treatment groups. *P<0.05, **P<0.01, ***P<0.001 vs. the rest for the same protein; #P<0.05 vs. TNBS; n=3.
Consistently, Western blot analyses of mucosal lysates obtained from these mice showed that, one day after TNBS treatment, mucosal VDR levels were already down-regulated, whereas long MLCK (not short MLCK) expression and MLC phosphorylation were markedly induced in the mucosa. Paricalcitol treatment reversed the decline in mucosal VDR levels and abrogated MCLK induction and MLC phosphorylation (Fig. 5G and H). Paricalcitol also reversed claudin-2 up-regulation and occludin down-regulation in TNBS-treated mice (Fig. 5G and H). Immunostaining showed that in TNBS-treated mice NF-κB p65 was translocated into the nuclei in colonic epithelial cells, and paricalcitol treatment blocked p65 nuclear translocation (Fig. 6), confirming that paricalcitol is able to inhibit NF-κB activation in vivo, which is required for MLCK up-regulation. Together, these data demonstrate that paricalcitol therapy is able to preserve mucosal barrier permeability in the early stage of the TNBS-induced colitis model by targeting MLCK activation pathway.
Figure 6.

Immunostaining of colon sections from control mice and mice treated with paricalcitol, TNBS and TNBS+Paricalcitol with antibody against NF-κB p65. The nuclei were stained with DAPI. Arrows indicate nuclei. Note that p65 is translocated in TNBS-treated colonocytes, and the p65 nuclear translocation is markedly attenuated by paricalcitol in colonocytes treated with TNBS+pariocalcitol.
Deletion of colonic epithelial VDR exacerbates MLCK activation and worsens mucosal leakin colitis model
To further evaluate the role of mucosal epithelial 1,25(OH)2D3-VDR signaling in the regulation of MLCK activation and tight junction in vivo, we specifically deleted VDR from colonic epithelial cells (Fig. 7C) by crossing CDX2-Cre and VDRflox/flox mice. The resultant VDRΔCEC mice, together with VDRflox/flox mice as controls, were subject to TNBS treatment. As shown in Figure 7, one day after TNBS instillation, mucosal paracellular barrier loss, as assayed by FITC-dextran leak, was much more robust in VDRΔCEC mice compared to VDRflox/flox mice (Fig. 7A). So was mucosal MPO activity (Fig. 7B). As expected, Western blot analyses confirmed that TNBS treatment reduced mucosal VDR and induced long MLCK expression and MLC phosphorylation in VDRflox/flox mice (Fig. 7C and D). In comparison, epithelial VDR deletion led to more robust induction of long MLCK (but not short MLCK) and MLC phosphorylation in VDRΔCEC mice (Fig. 7C and D). The increase of mucosal claudin-2 and decrease of mucosal occludin was also more robust in VDRΔIEC mice (Fig. 7C and D). Taken together, these genetic studies provide compelling evidence that epithelial VDR signaling regulates MLCK-dependent mucosal barrier permeability.
Figure 7.

Epithelial hVDR deletion exacerbates MLCK activation and increases mucosal permeability in TNBS colitis model. (A) Paracellular FITC-dextran absorption across the barrier in the four groups of mice. **P<0.01 vs. two Ctrls; #P<0.05 vs. WT+TNBS. (B) Mucosal MPO activity. **P<0.01 vs. two Ctrls; #P<0.05 vs. WT+TNBS. (C) Western blot analyses of mucosal lysates in the four groups of mice on day 2. (D) Densitometric quantitation of Western blot data. *P<0.05, ** P<0.01; *** P<0.001 vs. two Ctrls; #P<0.05 vs. WT+TNBS; n=5 in each group.
Discussion
In this study we used three human colon cancer cell lines that possess colonic epithelial cell properties to investigate the effect of 1,25(OH)2D3 on MLCK-dependent tight junction permeability. Although these three cell lines have many differences such as tumor origins and karyotypes, the result is very consistent. Our data show that TNF-α strongly induces long MLCK expression and MLC phosphorylation and, consequently, increases the paracellular permeability in the cell monolayers. These observations are in agreement with early reports by other investigators 11; 12; 14; 46. Importantly, we demonstrate that the effect of TNF-α on tight junction permeability and MLCK activation is markedly attenuated by 1,25(OH)2D3 in a VDR-dependent fashion. 1,25(OH)2D3 treatment blocks long MLCK induction and MLC phosphorylation in all three cell lines. Moreover, we further show that robust MLCK activation and mucosal barrier loss are detectable in the early stage of TNBS-induced colitis, and paricalcitol, a low-calcemic VDR agonist, is able to disrupt MLCK activation, preserve mucosal barrier function, reduce colonic inflammation and alleviate colitis in TNBS treated mice. These observations demonstrate an ability of the VDR signaling to protect the mucosal epithelial tight junction in vivo. Consistent with this conclusion, a recent study demonstrates that vitamin D can mitigate the deleterious effects of adherent-invasive E. coli on intestinal mucosa by maintaining intestinal epithelial barrier homeostasis and preserving tight junction architecture 47. The diversity of the three cell lines used in this study makes the data compelling; however, giving the limitation of colon cancer cell lines, more compelling data might be obtained by studying organoids. Moreover, vitamin D inhibition of the MLCK-MLC pathway should be confirmed by human clinical trials in the future. Given the pleiotropic activity of vitamin D, however, it is possible that the inhibitory effect of vitamin D on colonic inflammation is mediated not only by epithelial cells, but also by immune cells.
We have reported that mucosal VDR levels are down-regulated by about 50% in disease biopsies from patients with IBD 25; 26, and this down-regulation largely results from TNF-α-induced increase of miR-346, which targets VDR translation in epithelial cells 26 (Fig. 8). In the current study we confirm our previous observation regarding mucosal VDR status in a new cohort of ulcerative colitis patients. Remarkably, we found that the ~50% VDR reduction is associated with an increase in long MLCK protein and MLC phosphorylation in the lesions. This observation establishes a pathological relevance of vitamin D regulation of the MLCK-MLC pathway. Furthermore, consistent with this human observation, we demonstrate, using mice with a genetic deletion of VDR specifically from colonic epithelial cells, that absence of epithelial VDR indeed enhances mucosal MLCK-MLC pathway activation and worsens mucosal barrier loss in experimental colitis. Although the impact of VDR deficiency or deletion may not be limited only to the MLCK-MLC pathway in the epithelial cells, these human and mouse data together confirm from another perspective a critical role of epithelial VDR signaling in the regulation of MLCK-dependent tight junction permeability.
Figure 8.
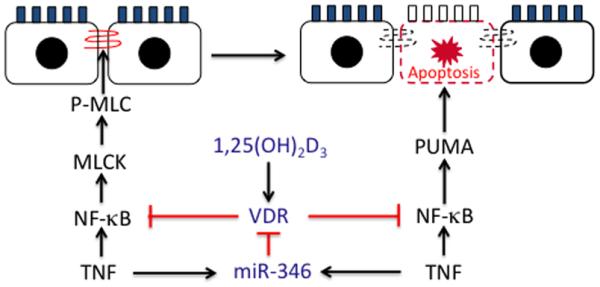
1,25(OH)2D3-VDR signaling protects the mucosal barrier. In early stage of colitis development, 1,25(OH)2D3-VDR signaling preserves tight junction barrier by down-regulating long MLCK (this study). With colitis progress, 1,25(OH)2D3-VDR inhibits MLCK-independent epithelial cell apoptosis by down-regulating PUMA (see ref. 25). The mechanism of both regulatory pathways is the blockade of NF-κB activation. Conversely, TNF-α, via induction of miR-346, down-regulates epithelial VDR to attenuate its protective effects (see ref. 26).
Down-regulation of long MLCK expression is the key for vitamin D protection against epithelial barrier loss. We show that the molecular basis for 1,25(OH)2D3 regulation of long MLCK expression is the blockade of NF-κB activation. There are two lines of crucial evidence to support this conclusion. One is 1,25(OH)2D3 suppression of IKKβ-induced long MLCK, and this suppression in turn can be overcome by increasing amount of IKKβ transfection. The other is the ChIP assays showing that 1,25(OH)2D3 disrupts TNF-α-induced p65 binding to the long MLCK promoter at three κB sites (Figure 3). It is worth pointing out that the κB sites at −1415 and −1584 were shown to interact with NF-κB by EMSA in an early study, but this interaction could not be confirmed by ChIP assays under TNF-α and IFN-γ co-treatment 15, whereas the κB site at −136 (previously at −75) was not analyzed by ChIP assays 16. Therefore, the ChIP data are novel and crucial to understand the regulatory mechanism. In fact, this mechanism is not surprising, as NF-κB is already a well established target for 1,25(OH)2D3-VDR action in the regulation of a variety of genes 48; 49; 50. We have recently shown that liganded VDR inhibits NF-κB activation by physical association with IKKβ 51. Relevant to the current work, induction of PUMA, a pro-apoptotic factor 52 driving inflammation-induced intestinal epithelial cell apoptosis, is also mediated by NF-κB 53, and we have reported that 1,25(OH)2D3-VDR signaling targets NF-κB to block PUMA expression, whereby suppressing epithelial cell apoptosis 25. NF-κB as a key transcription factor to mediate inflammatory response is central to colonic inflammation. It appears that NF-κB has emerged as a major vitamin D target in the protection of mucosal epithelial barrier (Fig. 8).
Claudin-2 and occludin are proteins involved in the formation of tight junctions. It has been reported that claudin-2 is elevated in ulcerative colitis and Crohn’s disease 42 and induced by IL-13 in cell cultures 43. In the latter report, TNF-α was shown to induce claudin-2 mRNA, but it was not able to induce claudin-2 protein 43. Here we report induction of claudin-2 protein by TNF-α in three human epithelial cell lines. Consistently, claudin-2 protein levels were also increased in the colonic biopsies from the human IBD cohort (Fig. 4). The discrepancy between our observations and the previous finding43 is unclear but could be due to difference in cell lines used. Additionally, occludin was previously reported to be increased in mRNA levels in active ulcerative colitis patients 54, but here in this cohort of human biopsies we found little changes in occludin protein levels, whereas in the three cell lines under TNF-α treatment occludin protein was down-regulated. These discrepancies suggest that the occludin status in colonic inflammation requires further investigation.
Our previous studies have demonstrated that epithelial VDR signaling protects the mucosal epithelial barrier during colonic inflammation by blocking epithelial cell apoptosis 25. In this study we provide evidence from cells, human biopsies and animal models that 1,25(OH)2D3-VDR signaling controls MLCK-dependent tight junction permeability by regulating the MLCK-MLC pathway (Fig. 8). It is thought that tight junction dysregulation occurs in the early stage of colitis development and is involved in disease initiation, whereas excess epithelial cell apoptosis causes tight junction-independent barrier loss and promote disease progression 4. The dual protective mechanisms of 1,25(OH)2D3-VDR signaling against tight junction loss and epithelial apoptosis explain at least in part the potent anti-colitic activity of the epithelial VDR that we observed previously in various experimental colitis models 25.
Long MLCK as a central regulator to control tight junction permeability makes it an excellent drug target to prevent Crohn’s disease initiation, maintain remission and reduce disease activity 5. However, because of the high similarity between short and long MLCKs, which share the same catalytic domain, current MLCK pharmacological inhibitors cannot distinguish between these two MLCKs and are mostly toxic and not therapeutically useful, as inhibition of short MLCK that is expressed in the smooth muscle can cause adverse consequence. Therefore, isoform-specific drugs that only target long MLCK are highly desirable. In this study we show that active vitamin D and a low-calcemic vitamin D analog strongly down-regulate long MLCK expression but have no effect at all on short MLCK, which makes vitamin D therapy a very promising new method to target long MLCK. In fact, we have shown here that, paricalcitol, a FDA-approved drug, can indeed preserve mucosal barrier and ameliorate colitis in an experimental model. Therefore, developing new vitamin D analog drugs to target MLCK and block epithelial cell apoptosis for the management of IBD might be a useful strategy that warrants further exploration. In fact, in the clinical trials published in recent years, nutritional vitamin D supplementation has shown variable effects in IBD patients, ranging from clear clinical benefits to little beneficial effects on colonic inflammation 55; 56; 57, and active vitamin D metabolite appears to have better efficacy in reducing disease activity in patients with Cohn’s disease 56. Thus, active vitamin D analogs with low hypercalcemic side effects should be tested in more clinical studies in the future.
Supplementary Material
Acknowledgements
We thank Dr. David Gardner (University of California at San Francesco) for providing the VDRflox/flox mice, and Dr. Jerrod Turner (University of Chicago) for critical reading of the manuscript.
Grant support: This work was supported in part by NIH grant R01CA180087, and a research fund from Liaoning Provincial Government, China.
Abbreviations
- 1,25(OH)2D3
1,25-dihydroxyvitamin D3
- VDR
vitamin D receptor
- IBD
inflammatory bowel diseases
- MLC
myosin II regulatory light chain
- MCLK
myosin light chain kinase
- TNF-α
tissue necrosis factor-α
- IFN-γ
interferon-γ
Footnotes
Disclosure: The authors have declared that no conflict of interest exists in this work.
References
- 1.Laukoetter MG, Bruewer M, Nusrat A. Regulation of the intestinal epithelial barrier by the apical junctional complex. Curr Opin Gastroenterol. 2006;22:85–89. doi: 10.1097/01.mog.0000203864.48255.4f. [DOI] [PubMed] [Google Scholar]
- 2.Gibson PR. Increased gut permeability in Crohn's disease: is TNF the link? Gut. 2004;53:1724–1725. doi: 10.1136/gut.2004.047092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fasano A, Shea-Donohue T. Mechanisms of disease: the role of intestinal barrier function in the pathogenesis of gastrointestinal autoimmune diseases. Nat Clin Pract Gastroenterol Hepatol. 2005;2:416–422. doi: 10.1038/ncpgasthep0259. [DOI] [PubMed] [Google Scholar]
- 4.Su L, Nalle SC, Shen L, et al. TNFR2 activates MLCK-dependent tight junction dysregulation to cause apoptosis-mediated barrier loss and experimental colitis. Gastroenterology. 2013;145:407–415. doi: 10.1053/j.gastro.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cunningham KE, Turner JR. Myosin light chain kinase: pulling the strings of epithelial tight junction function. Ann N Y Acad Sci. 2012;1258:34–42. doi: 10.1111/j.1749-6632.2012.06526.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kamm KE, Stull JT. Dedicated myosin light chain kinases with diverse cellular functions. J Biol Chem. 2001;276:4527–4530. doi: 10.1074/jbc.R000028200. [DOI] [PubMed] [Google Scholar]
- 7.Clayburgh DR, Rosen S, Witkowski ED, et al. A differentiation-dependent splice variant of myosin light chain kinase, MLCK1, regulates epithelial tight junction permeability. J Biol Chem. 2004;279:55506–55513. doi: 10.1074/jbc.M408822200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shen L, Black ED, Witkowski ED, et al. Myosin light chain phosphorylation regulates barrier function by remodeling tight junction structure. J Cell Sci. 2006;119:2095–2106. doi: 10.1242/jcs.02915. [DOI] [PubMed] [Google Scholar]
- 9.Rutgeerts P, Sandborn WJ, Feagan BG, et al. Infliximab for induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2005;353:2462–2476. doi: 10.1056/NEJMoa050516. [DOI] [PubMed] [Google Scholar]
- 10.Colombel JF, Sandborn WJ, Rutgeerts P, et al. Adalimumab for maintenance of clinical response and remission in patients with Crohn's disease: the CHARM trial. Gastroenterology. 2007;132:52–65. doi: 10.1053/j.gastro.2006.11.041. [DOI] [PubMed] [Google Scholar]
- 11.Taylor CT, Dzus AL, Colgan SP. Autocrine regulation of epithelial permeability by hypoxia: role for polarized release of tumor necrosis factor alpha. Gastroenterology. 1998;114:657–668. doi: 10.1016/s0016-5085(98)70579-7. [DOI] [PubMed] [Google Scholar]
- 12.Wang F, Graham WV, Wang Y, et al. Interferon-gamma and tumor necrosis factor-alpha synergize to induce intestinal epithelial barrier dysfunction by up-regulating myosin light chain kinase expression. Am J Pathol. 2005;166:409–419. doi: 10.1016/s0002-9440(10)62264-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ma TY, Iwamoto GK, Hoa NT, et al. TNF-alpha-induced increase in intestinal epithelial tight junction permeability requires NF-kappa B activation. Am J Physiol Gastrointest Liver Physiol. 2004;286:G367–376. doi: 10.1152/ajpgi.00173.2003. [DOI] [PubMed] [Google Scholar]
- 14.He F, Peng J, Deng XL, et al. Mechanisms of tumor necrosis factor-alpha-induced leaks in intestine epithelial barrier. Cytokine. 2012;59:264–272. doi: 10.1016/j.cyto.2012.04.008. [DOI] [PubMed] [Google Scholar]
- 15.Graham WV, Wang F, Clayburgh DR, et al. Tumor necrosis factor-induced long myosin light chain kinase transcription is regulated by differentiation-dependent signaling events. Characterization of the human long myosin light chain kinase promoter. J Biol Chem. 2006;281:26205–26215. doi: 10.1074/jbc.M602164200. [DOI] [PubMed] [Google Scholar]
- 16.Ye D, Ma I, Ma TY. Molecular mechanism of tumor necrosis factor-alpha modulation of intestinal epithelial tight junction barrier. Am J Physiol Gastrointest Liver Physiol. 2006;290:G496–504. doi: 10.1152/ajpgi.00318.2005. [DOI] [PubMed] [Google Scholar]
- 17.Bouillon R, Carmeliet G, Verlinden L, et al. Vitamin D and human health: lessons from vitamin D receptor null mice. Endocr Rev. 2008;29:726–776. doi: 10.1210/er.2008-0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haussler MR, Whitfield GK, Haussler CA, et al. The nuclear vitamin D receptor: biological and molecular regulatory properties revealed. J Bone Miner Res. 1998;13:325–349. doi: 10.1359/jbmr.1998.13.3.325. [DOI] [PubMed] [Google Scholar]
- 19.Lim WC, Hanauer SB, Li YC. Mechanisms of Disease: vitamin D and inflammatory bowel disease. Nature Clinical Practice Gastroenterology & Hepatology. 2005;2:308–315. doi: 10.1038/ncpgasthep0215. [DOI] [PubMed] [Google Scholar]
- 20.Driscoll RH, Jr., Meredith SC, Sitrin M, et al. Vitamin D deficiency and bone disease in patients with Crohn's disease. Gastroenterology. 1982;83:1252–1258. [PubMed] [Google Scholar]
- 21.Harries AD, Brown R, Heatley RV, et al. Vitamin D status in Crohn's disease: association with nutrition and disease activity. Gut. 1985;26:1197–1203. doi: 10.1136/gut.26.11.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vogelsang H, Ferenci P, Woloszczuk W, et al. Bone disease in vitamin D-deficient patients with Crohn's disease. Dig Dis Sci. 1989;34:1094–1099. doi: 10.1007/BF01536381. [DOI] [PubMed] [Google Scholar]
- 23.Pappa HM, Gordon CM, Saslowsky TM, et al. Vitamin D status in children and young adults with inflammatory bowel disease. Pediatrics. 2006;118:1950–1961. doi: 10.1542/peds.2006-0841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ananthakrishnan AN, Khalili H, Higuchi LM, et al. Higher predicted vitamin D status is associated with reduced risk of Crohn's disease. Gastroenterology. 2012;142:482–489. doi: 10.1053/j.gastro.2011.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu W, Chen Y, Golan MA, et al. Intestinal epithelial vitamin D receptor signaling inhibits experimental colitis. J Clin Invest. 2013;123:3983–3996. doi: 10.1172/JCI65842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen Y, Du J, Zhang Z, et al. MicroRNA-346 mediates tumor necrosis factor alpha-induced downregulation of gut epithelial vitamin D receptor in inflammatory bowel diseases. Inflamm Bowel Dis. 2014;20:1910–1918. doi: 10.1097/MIB.0000000000000158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kong J, Zhang Z, Musch MW, et al. Novel role of the vitamin D receptor in maintaining the integrity of the intestinal mucosal barrier. Am J Physiol Gastrointest Liver Physiol. 2008;294:G208–216. doi: 10.1152/ajpgi.00398.2007. [DOI] [PubMed] [Google Scholar]
- 28.Assa A, Vong L, Pinnell LJ, et al. Vitamin D deficiency promotes epithelial barrier dysfunction and intestinal inflammation. J Infect Dis. 2014;210:1296–1305. doi: 10.1093/infdis/jiu235. [DOI] [PubMed] [Google Scholar]
- 29.Chen SW, Wang PY, Zhu J, et al. Protective Effect of 1,25-Dihydroxyvitamin D3 on Lipopolysaccharide-Induced Intestinal Epithelial Tight Junction Injury in Caco-2 Cell Monolayers. Inflammation. 2014 doi: 10.1007/s10753-014-0041-9. [DOI] [PubMed] [Google Scholar]
- 30.Wirtz S, Neufert C, Weigmann B, et al. Chemically induced mouse models of intestinal inflammation. Nat Protoc. 2007;2:541–546. doi: 10.1038/nprot.2007.41. [DOI] [PubMed] [Google Scholar]
- 31.Chen S, Law CS, Grigsby CL, et al. Cardiomyocyte-specific deletion of the vitamin D receptor gene results in cardiac hypertrophy. Circulation. 2011;124:1838–1847. doi: 10.1161/CIRCULATIONAHA.111.032680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hinoi T, Akyol A, Theisen BK, et al. Mouse model of colonic adenoma-carcinoma progression based on somatic Apc inactivation. Cancer Res. 2007;67:9721–9730. doi: 10.1158/0008-5472.CAN-07-2735. [DOI] [PubMed] [Google Scholar]
- 33.Cheadle GA, Costantini TW, Lopez N, et al. Enteric glia cells attenuate cytomix-induced intestinal epithelial barrier breakdown. PLoS One. 2013;8:e69042. doi: 10.1371/journal.pone.0069042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cani PD, Possemiers S, Van de Wiele T, et al. Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut. 2009;58:1091–1103. doi: 10.1136/gut.2008.165886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yan F, Cao H, Cover TL, et al. Colon-specific delivery of a probiotic-derived soluble protein ameliorates intestinal inflammation in mice through an EGFR-dependent mechanism. J Clin Invest. 2011;121:2242–2253. doi: 10.1172/JCI44031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Park CM, Reid PE, Walker DC, et al. A simple, practical 'swiss roll' method of preparing tissues for paraffin or methacrylate embedding. J Microsc. 1987;145:115–120. doi: 10.1111/j.1365-2818.1987.tb01321.x. Pt 1. [DOI] [PubMed] [Google Scholar]
- 37.Appleyard CB, Wallace JL. Reactivation of hapten-induced colitis and its prevention by anti-inflammatory drugs. Am J Physiol. 1995;269:G119–125. doi: 10.1152/ajpgi.1995.269.1.G119. [DOI] [PubMed] [Google Scholar]
- 38.Hyland NP, Chambers AP, Keenan CM, et al. Differential adipokine response in genetically predisposed lean and obese rats during inflammation: a role in modulating experimental colitis? Am J Physiol Gastrointest Liver Physiol. 2009;297:G869–877. doi: 10.1152/ajpgi.00164.2009. [DOI] [PubMed] [Google Scholar]
- 39.Li YC, Bolt MJG, Cao L-P, et al. Effects of vitamin D receptor inactivation on the expression of calbindins and calcium metabolism. Am J Physiol Endocrinol Metab. 2001;281:E558–E564. doi: 10.1152/ajpendo.2001.281.3.E558. [DOI] [PubMed] [Google Scholar]
- 40.Yuan W, Pan W, Kong J, et al. 1,25-Dihydroxyvitamin D3 Suppresses Renin Gene Transcription by Blocking the Activity of the Cyclic AMP Response Element in the Renin Gene Promoter. J Biol Chem. 2007;282:29821–29830. doi: 10.1074/jbc.M705495200. [DOI] [PubMed] [Google Scholar]
- 41.Prasad S, Mingrino R, Kaukinen K, et al. Inflammatory processes have differential effects on claudins 2, 3 and 4 in colonic epithelial cells. Lab Invest. 2005;85:1139–1162. doi: 10.1038/labinvest.3700316. [DOI] [PubMed] [Google Scholar]
- 42.Weber CR, Nalle SC, Tretiakova M, et al. Claudin-1 and claudin-2 expression is elevated in inflammatory bowel disease and may contribute to early neoplastic transformation. Lab Invest. 2008;88:1110–1120. doi: 10.1038/labinvest.2008.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Weber CR, Raleigh DR, Su L, et al. Epithelial myosin light chain kinase activation induces mucosal interleukin-13 expression to alter tight junction ion selectivity. J Biol Chem. 2010;285:12037–12046. doi: 10.1074/jbc.M109.064808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Neurath MF, Fuss I, Kelsall BL, et al. Antibodies to interleukin 12 abrogate established experimental colitis in mice. J Exp Med. 1995;182:1281–1290. doi: 10.1084/jem.182.5.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.te Velde AA, Verstege MI, Hommes DW. Critical appraisal of the current practice in murine TNBS-induced colitis. Inflamm Bowel Dis. 2006;12:995–999. doi: 10.1097/01.mib.0000227817.54969.5e. [DOI] [PubMed] [Google Scholar]
- 46.Al-Sadi R, Guo S, Ye D, et al. TNF-alpha modulation of intestinal epithelial tight junction barrier is regulated by ERK1/2 activation of Elk-1. Am J Pathol. 2013;183:1871–1884. doi: 10.1016/j.ajpath.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Assa A, Vong L, Pinnell LJ, et al. Vitamin D deficiency predisposes to adherent-invasive Escherichia coli-induced barrier dysfunction and experimental colonic injury. Inflamm Bowel Dis. 2015;21:297–306. doi: 10.1097/MIB.0000000000000282. [DOI] [PubMed] [Google Scholar]
- 48.Zhang Z, Yuan W, Sun L, et al. 1,25-Dihydroxyvitamin D(3) targeting of NF-kappaB suppresses high glucose-induced MCP-1 expression in mesangial cells. Kidney Int. 2007;72:193–201. doi: 10.1038/sj.ki.5002296. [DOI] [PubMed] [Google Scholar]
- 49.Chen Y, Kong J, Sun T, et al. 1,25-Dihydroxyvitamin D(3) suppresses inflammation-induced expression of plasminogen activator inhibitor-1 by blocking nuclear factor-kappaB activation. Arch Biochem Biophys. 2011;507:241–247. doi: 10.1016/j.abb.2010.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen Y, Liu W, Sun T, et al. 1,25-Dihydroxyvitamin D Promotes Negative Feedback Regulation of TLR Signaling via Targeting MicroRNA-155-SOCS1 in Macrophages. J Immunol. 2013;190:3687–3695. doi: 10.4049/jimmunol.1203273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen Y, Zhang J, Ge X, et al. Vitamin D Receptor Inhibits Nuclear Factor kappaB Activation by Interacting with IkappaB Kinase beta Protein. J Biol Chem. 2013;288:19450–19458. doi: 10.1074/jbc.M113.467670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Qiu W, Wu B, Wang X, et al. PUMA-mediated intestinal epithelial apoptosis contributes to ulcerative colitis in humans and mice. J Clin Invest. 2011;121:1722–1732. doi: 10.1172/JCI42917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang P, Qiu W, Dudgeon C, et al. PUMA is directly activated by NF-kappaB and contributes to TNF-alpha-induced apoptosis. Cell Death Differ. 2009;16:1192–1202. doi: 10.1038/cdd.2009.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yamamoto-Furusho JK, Mendivil-Rangel EJ, Fonseca-Camarillo G. Differential expression of occludin in patients with ulcerative colitis and healthy controls. Inflamm Bowel Dis. 2012;18:E1999. doi: 10.1002/ibd.22835. [DOI] [PubMed] [Google Scholar]
- 55.Benchimol EI, Ward LM, Gallagher JC, et al. Effect of calcium and vitamin D supplementation on bone mineral density in children with inflammatory bowel disease. J Pediatr Gastroenterol Nutr. 2007;45:538–545. doi: 10.1097/MPG.0b013e3180dca0cc. [DOI] [PubMed] [Google Scholar]
- 56.Miheller P, Muzes G, Hritz I, et al. Comparison of the effects of 1,25 dihydroxyvitamin D and 25 hydroxyvitamin D on bone pathology and disease activity in Crohn's disease patients. Inflamm Bowel Dis. 2009;15:1656–1662. doi: 10.1002/ibd.20947. [DOI] [PubMed] [Google Scholar]
- 57.Jorgensen SP, Agnholt J, Glerup H, et al. Clinical trial: vitamin D3 treatment in Crohn's disease -a randomized double-blind placebo-controlled study. Aliment Pharmacol Ther. 2010;32:377–383. doi: 10.1111/j.1365-2036.2010.04355.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.