

Abstract
The nicotine metabolite ratio (NMR), a stable measure of hepatic nicotine metabolism via the CYP2A6 pathway and total nicotine clearance, is a predictive biomarker of response to nicotine replacement therapy, with increased quit rates in slower metabolizers. Nicotine binds directly to nicotinic acetylcholine receptors (nAChRs) to exert its psychoactive effects. This study examined the relationship between NMR and nAChR availability (α4β2* subtype) using positron emission tomography (PET) imaging of the radiotracer 2-18F-FA-85380 (2-18F-FA).
Methods
Twenty four smokers, 12 slow metabolizers (NMR <0.26) and 12 normal metabolizers (NMR ≥0.26), underwent 2-18F-FA-PET brain imaging following overnight nicotine abstinence (18 hours prior to scanning), using a validated bolus plus infusion protocol. Availability of nAChRs was compared between NMR groups in a priori volumes of interest (VOIs), with total distribution volume (VT/fP) being the measure of nAChR availability. Cravings to smoke were assessed prior to and following the scans.
Results
Thalamic nAChR α4β2* availability was significantly reduced in slow (versus normal) nicotine metabolizers (P=0.04). Slow metabolizers exhibited greater reductions in craving than normal metabolizers from pre- to post-scanning; however, craving was unrelated to availability.
Conclusion
The rate of nicotine metabolism is associated with thalamic nAChR availability. Additional studies could examine whether altered nAChR availability underlies differences in treatment response between slow and normal metabolizers of nicotine.
Keywords: Positron Emission Tomography, Nicotine, Addiction, 2-18F-FA-85380, Nicotine Metabolite Ratio, α4β2* nAChR
INTRODUCTION
An estimated 43.8 million Americans and 1.1 billion people worldwide smoke cigarettes (1, 2). Tobacco use is the leading cause of preventable death, representing a significant medical and public health problem (3). Despite the burden of nicotine dependence on public health, available pharmacotherapies have long-term failure rates of 70-80% (4).
Individual differences in rate of nicotine metabolism via CYP2A6 have been linked to variation in smoking rates (i.e., cigarettes per day) (5, 6). The ratio of 3’-hydroxycotinine (3-HC) to cotinine, referred to as the nicotine metabolite ratio (NMR), has been validated as a measure of inherited differences in hepatic metabolism of nicotine, and a predictor of both total nicotine clearance and response to smoking cessation therapies (7, 8). Smokers who are normal metabolizers of nicotine are less likely to quit smoking with transdermal nicotine compared to slow metabolizers (7, 9). A recent multi-center randomized placebo-controlled trial of 1,246 smokers showed that varenicline was more efficacious than nicotine patch in normal metabolizers based on NMR; however, in slow metabolizers, these treatments had equivalent efficacy (10). Among pregnant women, who clear nicotine at higher rates compared to non-pregnant women, those with a higher NMR were also less likely to achieve cessation (11). Despite the validated clinical significance of the NMR for targeting smoking cessation therapy, the mechanisms underlying its association with treatment response are unknown. This study examined whether slow and normal metabolizers differ in nAChR availability following ~18 hours abstinence from nicotine. We focused on short-term abstinence because ability to refrain from nicotine during the first day of a quit attempt is highly predictive of long-term abstinence (12, 13).
Chronic nicotine exposure in both animal models and humans has been shown to increase nAChR density, based on immunohistochemistry and other in vitro molecular probes specific for the nAChR α4β2* subtype (14-18). Positron emission tomographic imaging (PET) and single photon emission computed tomography (SPECT) using 2-18F-fluoro-3-(2(S)-azetidinylmethoxy) pyridine (2-18F-FA) and 123I- 5-IA, respectively, have confirmed these results in vivo in humans (19-21). Specifically, chronic nicotine exposure leads to nAChR up-regulation (22), while quitting smoking leads to decreased receptor levels (i.e., normalization) (23). SPECT molecular imaging studies of the nAChR α4β2* subtype have also shown that nAChR availability in smokers peaks at one week following abstinence (22, 24), suggesting persistent occupation of nAChRs by nicotine. These studies have consistently shown the highest density of the nAChRs (α4β2* subtype) to be in the thalamus (21). Studies have also shown that smoking, exposure to secondhand smoke, and administration of the smoking cessation medication varenicline to displaces (2-18F-FA) and 123I-5-IA binding to the α4β2* receptor subtype (25-28). Based on these data, we hypothesized that following overnight nicotine abstinence, slow nicotine metabolizers based on the NMR would have reduced nAChR availability compared to normal metabolizers. Because changes in nAChR availability had been previously observed in the thalamus and cortex (25, 26, 29), our a priori analysis targeted the thalamus, frontal cortex, temporal cortex and whole brain.
MATERIALS AND METHODS
Participants
This study was conducted in accordance with the Declaration of Helsinki, and the University of Pennsylvania Institutional Review Board approved all procedures. Individuals who expressed interest in participating in smoking cessation research at the University of Pennsylvania completed an in-person intake session where they provided written informed consent and were screened for eligibility. Eligible participants were between the ages of 18-65 and reported smoking at least 10 cigarettes per day (CPD) for the past 6 months, confirmed with exhaled carbon monoxide (CO) reading greater than 10 parts per million (ppm), and were fluent English speakers. Persons with a history of DSM-IV Axis I psychiatric or substance disorders (except nicotine dependence), assessed using the Mini International Neuropsychiatric Interview (30), and those taking psychotropic medications were excluded. Other exclusion criteria included: current use of chewing tobacco, snuff, or smoking cessation products; pregnancy, planned pregnancy or breastfeeding; history of brain injury; or magnetic resonance imaging (MRI)-contraindicated material in the body.
The nicotine metabolite ratio was determined from a plasma sample provided at intake (6). Participants were classified as slow metabolizers (NMR < 0.26) or normal metabolizers (NMR ≥ 0.26) based on prior clinical data which show differences in smoking cessation outcomes using this cutoff (7, 9). Because slow metabolizers typically comprise one-quarter of the general population of smokers, we oversampled this group to obtain equal numbers of slow and normal metabolizers. Participants and research staff involved with the PET scans were blind to NMR. Of 35 eligible participants who scheduled a PET scan session, 5 voluntarily withdrew prior to the PET scan session and 6 were withdrawn from the study due to non-compliance with study requirements (breath alcohol concentration reading > 0.01 or exhaled CO >15 ppm at the PET scan session); or newly acquired MRI contraindication, leaving a final sample of 24 participants (12 SMs and 12 NMs). The 15ppm cut off for CO was used because overnight abstinence may not be sufficient to clear CO in smokers who may have compromised pulmonary function. The average CO reading at session initiation was 8.2 ppm (range: 4-15 ppm). A second CO reading, taken prior to scanning, averaged 3.5 ppm (range 2-7 ppm).
Scanning Procedures
Participants were instructed to cease smoking at 9 p.m. on the night prior to the scan visit. The visit started at 7:30 a.m. the following day and the scan data acquisition was initiated at 3:00 p.m. Thus, there was roughly 18 hours of abstinence prior to scanning. After overnight nicotine abstinence, participants underwent PET brain imaging of 2-18F-FA delivered by bolus plus infusion to shorten the required duration of the PET imaging session due to the relatively slow receptor uptake of 2-18F-FA(31). The time period of overnight abstinence was chosen because most smokers relapse in the first 24 hours of a quit attempt(12), and because our primary hypothesis concerned differences in receptor availability in early abstinence due to differences in rates of nicotine metabolism. At the start of the session, participants provided a urine sample for a drug screen, a breath alcohol sample (BrAC > 0.01 exclusionary), and a breath CO sample for biochemical confirmation of abstinence (CO <15ppm, or at least a 50% reduction from the intake session). During the PET scan session, 2-18F-FA (3.08-5.88 mCi) was administered as a 50% bolus initially with an additional 2-18F-FA (3.11-6.0 mCi) administered as a continuous infusion over 8 hours (50%). Participants completed 2 hours of brain scanning beginning 6 hours (±30 min) post-injection to allow for the radiotracer to achieve steady state (31). PET scanning was performed on a Gemini TruFlight (TF) Big Bore PET/CT scanner (Philips Healthcare, Eindhoven, The Netherlands). A low dose (120 kVp, 50 mAs), unenhanced CT scan of the head was performed before the emission acquisition in order to perform attenuation correction. The PET/CT images were reconstructed via an iterative LOR-RAMLA algorithm using 2 iterations and 4 subsets and the Sharp filter setting. Just prior to and immediately following the 2-hour PET scan, participants completed the Brief Questionnaire on Smoking Urges (QSU) (32), a validated measure of cravings to smoke.
To facilitate automatic generation of brain volumes of interest (VOIs), all participants received a T1-weighted anatomic scan on a Siemens MR scanner (Siemens Medical Solutions USA, Knoxville, TN with 3 participants on a 3T Magnetom Sonata Syngo and 21 participants on a 1.5T Magnetom Trio A Tim System) acquired using magnetization-prepared 180° radio-frequency pulses and rapid gradient-echo (MP RAGE) sampling. Subjects underwent MRI scanning at a different session than PET imaging.
Measurement Of Unbound, Unmetabolized 2-[18F]FA in plasma
Baseline venous blood samples were obtained prior to injection for assessment of 2-18F-FA binding to plasma proteins, measurement of serum nicotine levels, and for preparation of standards for the solid phase extraction process. Average non-metabolized concentration of free 2-18F-FA in plasma during the scan period was determined from an average of 5.8 blood samples (range 3-6 samples, standard deviation +/− 0.7) for each participant from 5-7.5 hours after bolus injection 2-18F-FA binding to plasma proteins was evaluated by ultrafiltration using YM-10 filtration devices (Millipore Corporation, Bedford, MA). Additional venous blood samples (~3 mL each) were drawn at predetermined intervals during the infusion period for measurement of unbound, non-metabolized 2-18F-FA. Samples were centrifuged at 4000 × g for 15 min and plasma radioactivity was measured using an automated gamma counter. Radioactivity attributable to non-metabolized 2-18F-FA was determined using solid phase extraction with Clean Screen extraction columns (200 mg/10 ml, United Chemical Technologies, Inc., Bristol, PA) (33).
Data Analysis
PET Analysis Including Automatic Generation Of Brain VOIs And Partial Volume Correction
In order to standardize the PET imaging acquisition times following radiotracer administration across all subjects, we used only the 90 minutes of PET scanning that was contemporaneous for all participants acquired between 6.25 to 7.75 hours after bolus injection using six 15 min time bins (mean midpoint of scans was 7.0 hours +/− 0.1 hours after bolus injection). Each participant's PET image was co-registered to his or her T1-weighted MR scan and brain VOIs were automatically defined using the T1-MRI parcellation PNEURO tool in PMOD software (Pmod Technologies LTD, version 3.5, Zurich, Switzerland). Mean regional time activity curves (TAC) were output by Pmod software with partial volume correction based on a variant of the Rousset correction method, whereby only 15% of the pixels in the inner VOI were used for calculating the VOI average. Average activity concentrations for the 90-minute time period were decay corrected to the time of bolus injection for use in subsequent calculations.
Calculation Of Total Distribution Volume (VT/fP)
Participant total distribution volumes (designated as VT/fP) were calculated as the ratio of average partial volume corrected 2-18F-FA concentration in brain to unbound, non-metabolized 2-18F-FA concentration in plasma (31).
Generation Of Mean PET Images For Participant Cohorts
Using each individual participant's calculated distribution volumes (VT/fP), mean images of participant total distribution volumes (VT/fP) were created, with both PET image and plasma activity concentrations decay corrected to the time of 2-18F-FA bolus injection. All participants’ VT/fP images were spatially normalized to the same template followed by averaging the resulting VT/fP images for all participants with normal hepatic nicotine metabolism (n = 12) and averaging the resulting VT/fP images for all participants with slower rates of hepatic nicotine metabolism (n = 12).
Descriptive statistics were generated for demographic and smoking variables, with differences between NMR groups being tested using t-tests and χ2 tests. Group differences in VT/fP in the a priori VOIs were estimated by ANOVA including age, sex, and average cigarettes per day (CPD) as covariates; non-significant covariates were allowed to drop from the models at p > 0.1. Associations between VT/fP and craving were tested using regression models including mean QSU total score as the outcome and VT/fP in the a priori regions as the primary predictor. Alpha was adjusted on the basis of 4 VOIs with an average correlation of 0.95, resulting in an adjusted p value of 0.047. Twenty four smokers provided 80% power to detect an NMR group effect size of 1.2(34).
RESULTS
Demographic statistics for the study sample are summarized in Table 1. There were no significant differences in age, sex, race, CPD, or FTND score between NMR groups (all Ps > .05).
Table 1.
Demographic and smoking-related variables at baseline
| Group | Normal metabolizers (n =12) | Slow metabolizers (n =12) |
|---|---|---|
| Race | African American: 8 Caucasian: 4 |
African American: 9 Caucasian: 3 |
| Age, mean (SD, range) | 48.7 (9.2, 35-64) | 45.5 (11.6, 23-62) |
| Sex, n (%) | Male: 5 (42%) Female: 7 (58%) |
Male: 6 (50%) Female: 6 (50%) |
| CPD, mean (SD, range) | 16.9 (8.4,10-40) | 17.0 (3.4, 11-20) |
| FTND, mean (SD, range) | 5.9 (2.2, 2-9) | 5.4 (1.78, 2-8) |
| CO at intake session, mean (SD, range) | 21.2 (5.8, 12-29) | 25.0 (7.7, 12-35) |
| Plasma cotinine at intake session (ng/mL), mean (SD, range) | 219.7 (64.1, 119.8-329.2) | 323.9 (143.9, 154.1-604.5) |
| Plasma 3-hydroxycotinine at intake session (ng/mL), mean (SD, range) | 104.6 (50.8, 31.9-195.0) | 51.8 (27.8, 18.5-100.2) |
| Plasma NMR at intake session, mean (SD, range) | 0.467 (0.192, 0.266-0.908) | 0.170 (0.059, 0.08-0.259) |
Participant demographics and smoking variables by NMR group. CPD - cigarettes per day; FTND - Fagerström Test for Nicotine Dependence; CO – exhaled carbon monoxide.
No significant differences were present between groups (P= 0.67, 0.47, 0.7, 0.98, 0.62 and 0.18 for race, mean age, sex, CPD, FTD and CO, respectively). Serum nicotine values are not included as the time between last cigarette and sampling was not standardized.
There was significantly greater receptor availability (as measured by 2-18F-FA VT/fP) in normal metabolizers compared to slow metabolizers in the thalamus bilaterally, where there is the highest concentration of nAChRs (F = 4.92, p = 0.037). Significant associations were not observed in other tested regions: whole brain, temporal lobes and frontal lobes; however, trends in all regions were observed (Figure 1).
Figure 1. 2-[18F]FA VT/fP by region.
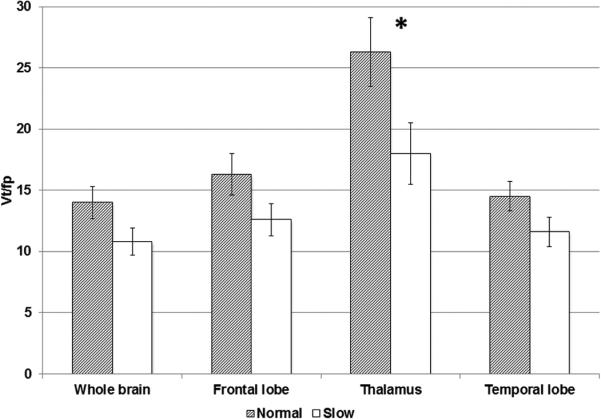
Partial volume corrected VT/fP values by NMR group: Smokers with slower nicotine metabolism (NMR <0.26) demonstrated decreased VT/fP in brain compared to those with normal metabolism including in the thalamus (p=0.037), the region of greatest nAChR density within the CNS. This relationship persisted and approached significance when comparing VT/fP between slow (bottom quartile) and normal NMR groups for the entire brain (p=0.07). P values were calculated using one-way ANOVA. “*” indicates P < 0.05. Error bars show standard error of the mean. For whole brain, frontal lobe, thalamus and temporal lobe, P=0.07, 0.1, 0.04 and 0.1, respectively).
There were no significant associations between VT/fP in the 4 VOIs and self-reported urges to smoke. Urges to smoke did not differ between slow and normal metabolizers prior to scanning; however, the groups differed with respect to changes in urges from pre- to post-scan, with slow metabolizers exhibiting a decrease (mean change in SMs -8.1 (SE=2.9) and normal metabolizers showing a slight increase (mean change of +1.8 (SE=2.2).
DISCUSSION
These data suggest that slow metabolizers of nicotine exhibit reduced nAChR availability in thalamus after 18 hours of abstinence from smoking, compared to normal nicotine metabolizers. Individual variation in hepatic metabolism influences nicotine's half-life in plasma from approximately 2 hours to 4 hours (35-37). In addition, nicotine levels in the brain can persist beyond nicotine's plasma half-life (22, 24). Therefore, differences in thalamic nAChR availability observed between slow and normal metabolizers may reflect differences in nicotine binding to nAChRs due to differences in elimination kinetics. Nonetheless, other possibilities should be considered, such as differences in nAChR expression or binding. For example, in non-human primates at baseline prior to any nicotine exposure, 2-18F-FA VT/fP binding is believed to be reflective of nAChR expression, since the radiotracer does not compete with nicotine, unlike the findings in smokers in the present study (38). However, definitive measurement of nAChR protein expression requires utilization of histopathological techniques (14, 39). Binding of the radiotracer with plasma proteins prior to crossing the blood brain barrier and interacting with nAChRs could occur, but the established bolus plus infusion technique used should account for such an interaction (31). Four of the 24 smokers had persistent nicotine in their blood following overnight abstinent; 3 slow metabolizers and 1 normal. These values measured less than 5 ng/mL. Although such levels could interfere with radiotracer binding, we intentionally timed this study in order that nicotine might persist in blood (and brain) of slow metabolizers. Not surprisingly because of its participation in numerous neuronal circuits, the thalamus has been implicated in nicotine dependence by both fMRI and PET studies (40, 41). Although in the present study significance was only detected in the thalamus, the region of greatest nAChR availability, the trend suggests a whole brain phenomenon. It is possible, however, that overnight abstinence was not sufficient to reveal other involved neuroanatomical locations.
Although validation of these preliminary findings is necessary, the results suggest that normal metabolizers of nicotine, who have faster rates of nicotine elimination, also exhibit greater nAChR availability during the first day following a quit attempt. Other data show that smokers with higher specific binding volume of distribution (a measure of nAChR availability) are more likely to relapse following a quit attempt (42). Thus, future research could examine whether increased nAChR availability in normal metabolizers mediates the increased likelihood of relapse on nicotine patch or the greater efficacy of varenicline in this group (10).
Additionally, nAChR expression and availability are very dynamic and influenced by age, gender, and hormones, including estrogen and progesterone (43). However, age and gender were not associated with nAChR availability in our sample. Another limitation is the relatively small sample size; however, the relationship between NMR and nAChR availability was consistent with the a priori hypothesis. Prior 2-18F-FA-PET imaging studies have utilized abstinence periods of at least 36-48 hours to ensure complete depletion of nicotine. Future studies in slow and normal metabolizers that include a second scan at least one week later would be valuable to explore whether differences in availability are attributable to long-term occupancy or upregulation.
It is also important to note that the nAChR, a non-selective cation channel, may exist in multiple conformational states (44). Binding of an agonist, such as nicotine, may stabilize the receptor in either the open or desensitized state. 2-18F-FA-PET brain imaging cannot determine the current conformational state of unavailable receptors. The timing of nAChR densensitization, decreased response to nicotine exposure, has been studied in cell culture (45), but in vivo molecular imaging methods lack the temporal resolution and cannot distinguish between an nAChR that is densensitized to nicotine and one that is not. Despite this shortcoming, 2-18F-FA-PET brain imaging does have established validity for detecting differences in tobacco exposure and for predicting smoking cessation (19, 23, 26, 46). Lastly, it should be noted that although increases in cravings to smoke among normal (versus slow) metabolizers were noted, consistent with faster clearance of nicotine, levels of craving or changes in craving did not correlate with nAChR availability.
CONCLUSION
These results suggest that individual differences in the rates of metabolism of nicotine, based on the NMR, are associated with nAChR availability in smokers shortly after a quit attempt. Manipulation of nicotine hepatic metabolism could be valuable to improve smoking cessation rates (47, 48). This study further reinforces prior evidence that PET imaging of the α4β2* nAChR subtype represents a promising biomarker of nicotine exposure, and possibly smoking cessation success. Additionally, it raises the underappreciated issue of how psychoactive drug metabolism influences neuronal receptor expression and availability.
Figure 2. Mean 2-[18F]FA VT/fP Images.

Mean positron emission tomographic (PET) images demonstrating lower 2-[18F]FA binding in subgroup with slower hepatic metabolism of nicotine. Mean PET images for normal metabolizer cohort (left) and cohort with slower rates of hepatic nicotine metabolism (right) were spatially normalized to the same template. VT/fP indicates the total volume of distribution.
Acknowledgments
RESEARCH SUPPORT
PNAT: Pharmacogenetics Research Network Grant (U01-DA20830), Abramson Cancer Center at the University of Pennsylvania, Institute for Translational Medicine and Therapeutics of the Perelman School of Medicine at the University of Pennsylvania (NIH), McCabe Pilot Award (UPENN), and the Commonwealth of Pennsylvania; the Pennsylvania Department of Health disclaims responsibility for analyses, interpretations or conclusions. Additional support was provided by Pfizer and National Center for Research Resources and the National Center for Advancing Translational Sciences, National Institutes of Health (UL1TR000003). Dr. Brody is supported by grants from the National Institute on Drug Abuse (R01 DA20872), the Department of Veterans Affairs, Office of Research and Development (CSR&D Merit Review Award I01 CX000412), and the Tobacco-Related Disease Research Program (#23XT-0002). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
ClinicalTrials.gov Identifier: NCT01788943
References
- 1.Organization WH. WHO Report on the Global Tobacco Epidemic, 2011. Warning about the Dangers of Tobacco. 2011 [Google Scholar]
- 2.Prevention CfDCa Current Cigarette Smoking Among Adults — United States, 2011. MMWR Morbidity and Mortality Weekly Report. 2012;61:889–894. [PubMed] [Google Scholar]
- 3.Prevention CfDCa Annual Smoking-Attributable Mortality, Years of Potential Life Lost, and Economic Costs --- United States, 1995--1999. MMWR Morbidity and Mortality Weekly Report 2002. 2002;51:300–303. [PubMed] [Google Scholar]
- 4.Phs Guideline Update Panel L, Staff Treating tobacco use and dependence: 2008 update U.S. Public Health Service Clinical Practice Guideline executive summary. Respir Care. 2008;53:1217–1222. [PubMed] [Google Scholar]
- 5.Benowitz NL, Pomerleau OF, Pomerleau CS, Jacob P., 3rd. Nicotine metabolite ratio as a predictor of cigarette consumption. Nicotine Tob Res. 2003;5:621–624. doi: 10.1080/1462220031000158717. [DOI] [PubMed] [Google Scholar]
- 6.Dempsey D, Tutka P, Jacob P, 3rd, et al. Nicotine metabolite ratio as an index of cytochrome P450 2A6 metabolic activity. Clin Pharmacol Ther. 2004;76:64–72. doi: 10.1016/j.clpt.2004.02.011. [DOI] [PubMed] [Google Scholar]
- 7.Lerman C, Tyndale R, Patterson F, et al. Nicotine metabolite ratio predicts efficacy of transdermal nicotine for smoking cessation. Clin Pharmacol Ther. 2006;79:600–608. doi: 10.1016/j.clpt.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 8.Ray R, Tyndale RF, Lerman C. Nicotine dependence pharmacogenetics: role of genetic variation in nicotine-metabolizing enzymes. J Neurogenet. 2009;23:252–261. doi: 10.1080/01677060802572887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schnoll RA, Patterson F, Wileyto EP, Tyndale RF, Benowitz N, Lerman C. Nicotine metabolic rate predicts successful smoking cessation with transdermal nicotine: a validation study. Pharmacol Biochem Behav. 2009;92:6–11. doi: 10.1016/j.pbb.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lerman C, Schnoll RA, Hawk LW, Jr., et al. Use of the nicotine metabolite ratio as a genetically informed biomarker of response to nicotine patch or varenicline for smoking cessation: a randomised, double-blind placebo-controlled trial. Lancet Respir Med. 2015;3(2):131–8. doi: 10.1016/S2213-2600(14)70294-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vaz LR, Coleman T, Cooper S, Aveyard P, Leonardi-Bee J. The nicotine metabolite ratio in pregnancy measured by trans-3′-Hydroxycotinine to Cotinine ratio: characteristics and relationship with smoking cessation. Nicotine Tob Res. 2015 doi: 10.1093/ntr/ntu342. doi: 10.1093/ntr/ntu342 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 12.Piasecki TM. Relapse to smoking. Clin Psychol Rev. 2006;26:196–215. doi: 10.1016/j.cpr.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 13.Westman EC, Behm FM, Simel DL, Rose JE. Smoking behavior on the first day of a quit attempt predicts long-term abstinence. Arch Intern Med. 1997;157:335–340. [PubMed] [Google Scholar]
- 14.Benwell ME, Balfour DJ, Anderson JM. Evidence that tobacco smoking increases the density of (-)-[3H]nicotine binding sites in human brain. J Neurochem. 1988;50:1243–1247. doi: 10.1111/j.1471-4159.1988.tb10600.x. [DOI] [PubMed] [Google Scholar]
- 15.Breese CR, Marks MJ, Logel J, et al. Effect of smoking history on [3H]nicotine binding in human postmortem brain. J Pharmacol Exp Ther. 1997;282:7–13. [PubMed] [Google Scholar]
- 16.Flores CM, Rogers SW, Pabreza LA, Wolfe BB, Kellar KJ. A subtype of nicotinic cholinergic receptor in rat brain is composed of alpha 4 and beta 2 subunits and is up-regulated by chronic nicotine treatment. Mol Pharmacol. 1992;41:31–37. [PubMed] [Google Scholar]
- 17.Perry DC, Davila-Garcia MI, Stockmeier CA, Kellar KJ. Increased nicotinic receptors in brains from smokers: membrane binding and autoradiography studies. J Pharmacol Exp Ther. 1999;289:1545–1552. [PubMed] [Google Scholar]
- 18.Whiting PJ, Lindstrom JM. Characterization of bovine and human neuronal nicotinic acetylcholine receptors using monoclonal antibodies. J Neurosci. 1988;8:3395–3404. doi: 10.1523/JNEUROSCI.08-09-03395.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brody AL, Mukhin AG, La Charite J, et al. Up-regulation of nicotinic acetylcholine receptors in menthol cigarette smokers. Int J Neuropsychopharmacol. 2013;16:957–966. doi: 10.1017/S1461145712001022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ding YS, Molina PE, Fowler JS, et al. Comparative studies of epibatidine derivatives [18F]NFEP and [18F]N-methyl-NFEP: kinetics, nicotine effect, and toxicity. Nucl Med Biol. 1999;26:139–148. doi: 10.1016/s0969-8051(98)00070-5. [DOI] [PubMed] [Google Scholar]
- 21.Gallezot JD, Bottlaender M, Gregoire MC, et al. In vivo imaging of human cerebral nicotinic acetylcholine receptors with 2-18F-fluoro-A-85380 and PET. J Nucl Med. 2005;46:240–247. [PubMed] [Google Scholar]
- 22.Staley JK, Krishnan-Sarin S, Cosgrove KP, et al. Human tobacco smokers in early abstinence have higher levels of beta2* nicotinic acetylcholine receptors than nonsmokers. J Neurosci. 2006;26:8707–8714. doi: 10.1523/JNEUROSCI.0546-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brody AL, Mukhin AG, Shulenberger S, et al. Treatment for tobacco dependence: effect on brain nicotinic acetylcholine receptor density. Neuropsychopharmacology. 2013;38:1548–1556. doi: 10.1038/npp.2013.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cosgrove KP, Batis J, Bois F, et al. beta2-Nicotinic acetylcholine receptor availability during acute and prolonged abstinence from tobacco smoking. Arch Gen Psychiatry. 2009;66:666–676. doi: 10.1001/archgenpsychiatry.2009.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brody AL, Mandelkern MA, London ED, et al. Effect of secondhand smoke on occupancy of nicotinic acetylcholine receptors in brain. Arch Gen Psychiatry. 2011;68:953–960. doi: 10.1001/archgenpsychiatry.2011.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brody AL, Mandelkern MA, London ED, et al. Cigarette smoking saturates brain alpha 4 beta 2 nicotinic acetylcholine receptors. Arch Gen Psychiatry. 2006;63:907–915. doi: 10.1001/archpsyc.63.8.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lotfipour S, Mandelkern M, Alvarez-Estrada M, Brody AL. A single administration of low-dose varenicline saturates alpha4beta2* nicotinic acetylcholine receptors in the human brain. Neuropsychopharmacology. 2012;37:1738–1748. doi: 10.1038/npp.2012.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lotfipour S, Mandelkern M, Brody AL. Quantitative molecular imaging of neuronal nicotinic acetylcholine receptors in the human brain with A-85380 radiotracers. Curr Med Imaging Rev. 2011;7:107–112. doi: 10.2174/157340511795445676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brody AL, Mandelkern MA, Costello MR, et al. Brain nicotinic acetylcholine receptor occupancy: effect of smoking a denicotinized cigarette. Int J Neuropsychopharmacol. 2009;12:305–316. doi: 10.1017/S146114570800922X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sheehan DV, Lecrubier Y, Sheehan KH, et al. The Mini-International Neuropsychiatric Interview (M.I.N.I.): the development and validation of a structured diagnostic psychiatric interview for DSM-IV and ICD-10. J Clin Psychiatry. 1998;59(Suppl 20):22–33. quiz 34-57. [PubMed] [Google Scholar]
- 31.Kimes AS, Chefer SI, Matochik JA, et al. Quantification of nicotinic acetylcholine receptors in the human brain with PET: bolus plus infusion administration of 2-[18F]F-A85380. Neuroimage. 2008;39:717–727. doi: 10.1016/j.neuroimage.2007.09.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cox LS, Tiffany ST, Christen AG. Evaluation of the brief questionnaire of smoking urges (QSU-brief) in laboratory and clinical settings. Nicotine Tob Res. 2001;3:7–16. doi: 10.1080/14622200020032051. [DOI] [PubMed] [Google Scholar]
- 33.Shumway DA, Pavlova OA, Mukhin AG. A simplified method for the measurement of nonmetabolized 2-[18F]F-A-85380 in blood plasma using solid-phase extraction. Nucl Med Biol. 2007;34:221–228. doi: 10.1016/j.nucmedbio.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 34.Faul F, Erdfelder E, Lang AG, Buchner A. G*Power 3: a flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav Res Methods. 2007;39:175–191. doi: 10.3758/bf03193146. [DOI] [PubMed] [Google Scholar]
- 35.Benowitz NL, Perez-Stable EJ, Herrera B, Jacob P., 3rd. Slower metabolism and reduced intake of nicotine from cigarette smoking in Chinese-Americans. J Natl Cancer Inst. 2002;94:108–115. doi: 10.1093/jnci/94.2.108. [DOI] [PubMed] [Google Scholar]
- 36.Benowitz NL, Swan GE, Jacob P, 3rd, Lessov-Schlaggar CN, Tyndale RF. CYP2A6 genotype and the metabolism and disposition kinetics of nicotine. Clin Pharmacol Ther. 2006;80:457–467. doi: 10.1016/j.clpt.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 37.Levi M, Dempsey DA, Benowitz NL, Sheiner LB. Population pharmacokinetics of nicotine and its metabolites I. Model development. J Pharmacokinet Pharmacodyn. 2007;34:5–21. doi: 10.1007/s10928-006-9027-z. [DOI] [PubMed] [Google Scholar]
- 38.Le Foll B, Chefer SI, Kimes AS, et al. Baseline expression of alpha4beta2* nicotinic acetylcholine receptors predicts motivation to self-administer nicotine. Biol Psychiatry. 2009;65:714–716. doi: 10.1016/j.biopsych.2008.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marks MJ, McClure-Begley TD, Whiteaker P, et al. Increased nicotinic acetylcholine receptor protein underlies chronic nicotine-induced up-regulation of nicotinic agonist binding sites in mouse brain. J Pharmacol Exp Ther. 2011;337:187–200. doi: 10.1124/jpet.110.178236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Menossi HS, Goudriaan AE, de Azevedo-Marques Perico C, et al. Neural bases of pharmacological treatment of nicotine dependence - insights from functional brain imaging: a systematic review. CNS Drugs. 2013;27:921–941. doi: 10.1007/s40263-013-0092-8. [DOI] [PubMed] [Google Scholar]
- 41.Sharma A, Brody AL. In vivo brain imaging of human exposure to nicotine and tobacco. Handb Exp Pharmacol. 2009:145–171. doi: 10.1007/978-3-540-69248-5_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brody AL, Mukhin AG, Mamoun MS, et al. Brain nicotinic acetylcholine receptor availability and response to smoking cessation treatment: a randomized trial. JAMA Psychiatry. 2014;71(7):797–805. doi: 10.1001/jamapsychiatry.2014.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cosgrove KP, Esterlis I, McKee SA, et al. Sex differences in availability of beta2*-nicotinic acetylcholine receptors in recently abstinent tobacco smokers. Arch Gen Psychiatry. 2012;69:418–427. doi: 10.1001/archgenpsychiatry.2011.1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cooper JR, Bloom FE, Roth RH. The Biochemical Basis of Neuropharmacology. Oxford University Press; New York: 2003. pp. 151–179. [Google Scholar]
- 45.Campling BG, Kuryatov A, Lindstrom J. Acute activation, desensitization and smoldering activation of human acetylcholine receptors. PLoS One. 2013;8:e79653. doi: 10.1371/journal.pone.0079653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mukhin AG, Kimes AS, Chefer SI, et al. Greater nicotinic acetylcholine receptor density in smokers than in nonsmokers: a PET study with 2-18F-FA-85380. J Nucl Med. 2008;49:1628–1635. doi: 10.2967/jnumed.108.050716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sellers EM, Ramamoorthy Y, Zeman MV, Djordjevic MV, Tyndale RF. The effect of methoxsalen on nicotine and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) metabolism in vivo. Nicotine Tob Res. 2003;5:891–899. doi: 10.1080/14622200310001615231. [DOI] [PubMed] [Google Scholar]
- 48.Siu EC, Tyndale RF. Selegiline is a mechanism-based inactivator of CYP2A6 inhibiting nicotine metabolism in humans and mice. J Pharmacol Exp Ther. 2008;324:992–999. doi: 10.1124/jpet.107.133900. [DOI] [PubMed] [Google Scholar]