

Abstract
Colloidal aggregation of organic molecules is the dominant mechanism for artifactual inhibition of proteins, and controls against it are widely deployed. Notwithstanding an increasingly detailed understanding of this phenomenon, a method to reliably predict aggregation has remained elusive. Correspondingly, active molecules that act via aggregation continue to be found in early discovery campaigns and remain common in the literature. Over the past decade, over 12 thousand aggregating organic molecules have been identified, potentially enabling a precedent-based approach to match known aggregators with new molecules that may be expected to aggregate and lead to artifacts. We investigate an approach that uses lipophilicity, affinity, and similarity to known aggregators to advise on the likelihood that a candidate compound is an aggregator. In prospective experimental testing, five of seven new molecules with Tanimoto coefficients (Tc’s) between 0.95 and 0.99 to known aggregators aggregated at relevant concentrations. Ten of 19 with Tc’s between 0.94 and 0.90 and three of seven with Tc’s between 0.89 and 0.85 also aggregated. Another three of the predicted compounds aggregated at higher concentrations. This method finds that 61 827 or 5.1% of the ligands acting in the 0.1 to 10 µM range in the medicinal chemistry literature are at least 85% similar to a known aggregator with these physical properties and may aggregate at relevant concentrations. Intriguingly, only 0.73% of all drug-like commercially available compounds resemble the known aggregators, suggesting that colloidal aggregators are enriched in the literature. As a percentage of the literature, aggregator-like compounds have increased 9-fold since 1995, partly reflecting the advent of high-throughput and virtual screens against molecular targets. Emerging from this study is an aggregator advisor database and tool (http://advisor.bkslab.org), free to the community, that may help distinguish between fruitful and artifactual screening hits acting by this mechanism.

INTRODUCTION
Colloidal aggregates, which are formed by many small organic molecules in aqueous solution, have long plagued early drug discovery.1,2 Ranging from 50 to over 800 nm in radius, these colloids form spontaneously and reversibly in aqueous buffer, undergoing a critical aggregation concentration (CAC) similar to a critical micelle concentration (CMC).3 When a colloid has formed, soluble and membrane4,5 proteins adsorb to its surface and are partially denatured, leading to nonspecific inhibition6,7 and occasionally activation.8,9 It is now well accepted that promiscuous inhibition caused by small molecule aggregation is a major source of false positive results in high-throughput and virtual screening.2,10,11 To mitigate this, use of a nonionic detergent such as Triton X-100 or Tween-80, which can disrupt aggregates, is now common in screening campaigns.10,12 However, detergent typically only right-shifts concentration-response curves, it does not fully eliminate aggregation,13–15 and it cannot always be tolerated by an assay. For this and other reasons, many early discovery efforts continue to be plagued with colloid-forming compounds.
The pervasiveness of aggregators16 has inspired efforts to predict them.17 Doman and co-workers investigated recursive partitioning, based on the physical properties of the fewer than 200 aggregators then known.18 This model successfully classified 94% of aggregators and nonaggregators retrospectively. However, in prospective testing,19 the model had a high false positive and a high false negative rate. Shelat and colleagues19 investigated a naive Bayesian model to predict aggregation. Against a set of 732 drug-like molecules, 40% of predicted aggregators were confirmed experimentally, while 7% of the predicted nonaggregators were aggregators (false negatives). A random forest version of the initial recursive partitioning model, optimized by inclusion of the new 732 compound data set, was also investigated, but this continued to both under-predict and overpredict new aggregators. Rao and co-workers20 used a support vector machine to classify aggregators and nonaggregators. Their model had a 71% success rate on 17 aggregators that were not used to build the model, but the rate of false positive prediction was not assessed, and prospective tests were not reported. Hsieh and colleagues used a k-nearest neighbor classification quantitative structure-activity relationship based approach to predict aggregation.21 A total of 342 predictive models were built based on 21 known aggregators and 80 compounds that had not been observed to aggregate under the same conditions. From among a library of 69 653 compounds, 15 compounds were predicted, and five compounds were tested for aggregation. All five were confirmed by experiment. Our own experience, with the subsequent development of much larger data sets, is that these models are good at classifying known aggregators but are less reliable at predicting aggregation prospectively.
Colloids have been described as a “fourth state of matter”, with particular physical properties. Colloidal aggregates of organic molecules undergo a critical-point transition22 from the soluble form and are sensitive to ionic strength and temperature,3 similar to micelle formation. Inhibition or occasionally activation8,23 of proteins by aggregates depends on their stoichiometry, since the colloid particles are present in the mid-femtomolar concentration range and become saturated with about 104 protein molecules. Preincubation with proteins such as serum albumin24 will attenuate the apparent activity of the colloids on the active target, by presaturation of the colloids with an inactive protein. These variable assay conditions can make colloid formers hard to identify. A colloid will form reproducibly under specified conditions of buffer, temperature, and concentration. However, its promiscuous inhibition will depend on other components of the buffer and the concentration of the target protein (increasing the concentration of the target protein can eliminate inhibition, owing to colloid saturation, even though the colloids themselves are still present). Formation of the colloid can occur over a small change in concentration, and they are temperature- and ionic strength-dependent. These factors have made it difficult for methods based on physical properties to predict aggregators, as is also true for methods that predict micelle or crystal formation, which also undergo such abrupt phase transitions. The problem is compounded by the use of protein inhibition as a proxy for whether an aggregator is present or not, rather than direct measurement of colloidal particles themselves.
Initial efforts to model aggregation were based on knowledge of fewer than 500 aggregating compounds. Subsequently, intentional screens for aggregation have been performed over libraries as large as 198 000 compounds,2,25 from which 12 600 compounds were identified as likely aggregators, because they caused inhibition that was reversible by detergent. For the 38 detergent-sensitive inhibitors that were followed up from these HTS campaigns, 37 were confirmed as particle formers by DLS. The aggregators that have emerged from these and other studies span a large range of properties and chemotypes. Drugs,18,26,27 probes, and natural products28 are now known to aggregate at screening-relevant concentrations, and aggregators are present in substantial numbers in every large compound library of which we are aware. It has become impossible for investigators, and certainly for ourselves, to intuit which molecules will aggregate at relevant concentrations. Conversely, the great expansion of known aggregators has made it possible to return to the question of prediction by integrating physical properties with better-powered pattern recognition.
Here we investigate a method to predict aggregating molecules, implementing it as an open access tool to advise on the likelihood of aggregation. The method is simplified from the earlier physical property-based, trained approaches, and its ambitions are similarly reduced. It simply uses topological similarity to the over 12 600 precedented aggregators, calculated lipophilicity, and observed affinity range as criteria to classify molecules as more or less likely to aggregate. There is no training in this approach. This method is thus guaranteed to have false negatives and false positives: for instance, many known dye aggregators fall below our lipophilicity limits and would not be classified as aggregators, and the method will not predict aggregation for a molecule outside of precedented chemotypes. Still, we wondered whether it might be useful to capture many aggregating molecules with which investigators are commonly faced. We therefore tested the method’s ability to prospectively predict the aggregation of 40 molecules, each previously reported in the literature but not previously tested for aggregation, that scored as more or less similar to known aggregators. The method was then used to explore the prevalence of likely aggregators in the medicinal chemistry literature and in libraries of commercially available compounds from which screening decks are largely drawn. We caution from the outset that our goal is not to eliminate aggregating compounds from discovery libraries; they cover far too many chemotypes, and aggregation itself is a fundamental property of organic molecules from which no category of compound is entirely free.6 The purpose of this study is rather to identify plausible aggregators so that they can be tested for this mechanism, avoiding artifacts. A tool to do so is made freely available to the community (http://advisor.bkslab.org).
RESULTS
Properties and Chemotypes of Known Aggregators
We first investigated the diversity and the shared physical properties of the over 12 600 known2,19,25 aggregators (http://advisor.bkslab.org/rawdata/). These aggregators spanned a range of physical properties, with calculated LogP values from −5.3 to 9.8, molecular weights from 77 to 1702 Da, from 0 to 31 rotatable bonds, and exposed polar surface areas of 0 to 777 A2. However, 80% of these molecules had cLogP values of >3, and we adopted this as a criterion for predicting aggregation, consistent with an earlier model.18 The aggregators were chemically diverse. For instance, their average Tanimoto similarity, using default ChemAxon path-based fingerprints, was only 24%. Of the 9127 clusters of 70% self-similar molecules among 1.1 million compounds drawn from the medicinal chemistry literature, 12% of the clusters included at least one aggregator. Among FDA-approved drugs, 39, about 3.6%, are known aggregators and 71 are 80% or more similar to at least one aggregator. Known aggregators also included 217 natural products. It is apparent that aggregators span a broad range of biologically relevant chemotypes.
We therefore adopted chemical similarity as a second criterion for predicting aggregation, scanning the ChEMBL database29 for published compounds similar to one of the 12 600 known aggregators. Using ChemAxon axonpath fingerprints, we sought compounds with Tanimoto coefficients (Tc’s) of between 80% and 84%, 85% and 89%, 90% and 94%, and 95% and 99% to known aggregators, with calculated logP values >3, and with reported activities in the 0.1 to 10 µM range. From among the over 125 000 molecules that met these criteria (below), we chose 40 for prospective, experimental testing as colloidal aggregators, using two criteria: detergent-dependent inhibition of an established counter-screening enzyme, such as AmpC β-lactamase or malate dehydrogenase,2,6,18,19 and observation of colloidal particles by dynamic light scattering (DLS).3,15,22 To be considered an aggregator, a molecule had to inhibit AmpC β-lactamase or malate dehydrogenase with an IC50 value better than 100 µM, have that inhibition much diminished or eliminated by addition of 0.01% Triton X-100 (Figure 1) and form particles characteristic of aggregators observable by dynamic light scattering (DLS). For certain characteristic molecules, critical aggregation concentrations, where colloids form in a phase-transition like event, similar to a critical micelle concentration, were also measured by monitoring the dependence of particle formation on concentration (Figure 2). As is true of previously characterized aggregators, enzyme inhibition for the new aggregators were found to be exquisitely sensitive to nonionic detergent, formed readily measurable particles by DLS (Figure 1), and transitioned from a soluble to an apparently colloidal form over a small concentration range (Figure 2).
Figure 1.


Dynamic light scattering (DLS) curves (first and third columns) and β-lactamase inhibition concentration-response curves with (red) and without (black) Triton X-100 (second and fourth columns), for characteristic new aggregators identified here. For some compounds where inhibition was fully reversed by detergent addition, only points at the highest inhibitor concentration were determined and are shown (arrows with red points).
Figure 2.

Critical aggregation concentration (CAC) curves for selected compounds. Intensity of a compound in buffer is measured by dynamic light scattering (DLS) as concentration is raised. A characteristic of colloidal aggregators is that they transit through a critical point (CAC) beyond which all added compound contributes to the colloidal phase; this transition typically occurs over a brief concentration range.
The likelihood to form colloids for the new molecules closely tracked their similarity to precedented aggregators (Table 1 and Table 2, showing confirmed and falsified predictions, respectively). Five of seven of the newly predicted molecules that had Tc values 95% or greater to a known aggregator were found to be aggregators: they inhibited β-lactamase in a detergent dependent manner at a relevant concentration, forming particles by DLS at those concentrations (Figure 1), and, where measured, went through a sharp critical aggregation concentration (CAC) (Figure 2). Similarly, 10 of 19 that had Tc values between 90% and 94% also aggregated (two more, CHEMBL1508601 and CHEMBL1565380 were weak aggregators), while 3 of 7 with Tc values between 85% and 89% aggregated (another molecule in this Tc range, CHEMBL1333273, was a weak aggregator). Only 1 of 7 molecules with Tc values to a known aggregator between 80% and 84% was observed to aggregate. Based on these observations, we choose a Tc cutoff of 85% as the reliability limit of the method. More or less conservative cutoffs may be chosen, depending on the tolerance of the investigator to higher or lower levels of false positives and false negatives.
Table 1.
Molecules Predicted and Experimentally Confirmed To Aggregatea
| ChEMBL ID |
Structure | xLogP | AmpC IC50 no Triton (µM) |
AmpC IC50 plus Triton (µM) |
DLS detected, µM (radius, nm) |
ChEMBL ID of most similar aggregator Tc% (ECFP4 %) |
|---|---|---|---|---|---|---|
| 57997 | 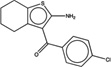 |
4.4 | 12.7 | >100 | 21 (270) |
1303983 90 (71) |
| 261275 | 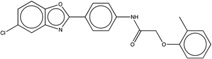 |
5.6 | 7.0 | >100 | 4 (2312) |
1329712 86 (73) |
| 1304694 |  |
5.5 | 8.0 | >100 | 20 (456) |
1416661 95 (78) |
| 1333273 |  |
4.0 | 146 | >400 | 100 (49) |
1328313 89 (80) |
| 1347854 | 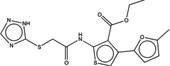 |
3.3* | 29 | >100 | 10 (115) |
1418209 85 (78) |
| 1349652 | 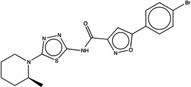 |
4.7 | 41 | >200 | 10 (484) |
6577804 96 (90) |
| 1359872 | 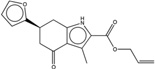 |
3.2* | 10 | >100 | 10 (405) |
326803 99 (91) |
| 1378476 | 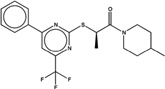 |
5.4 | 46 | >200 | 20 (163) |
1449091 96 (89) |
| 1388809 | 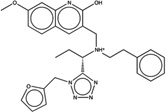 |
3.5 | 13 | >100 | 10 (177) |
1371567 90 (79) |
| 1420333 | 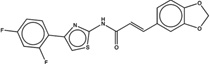 |
4.3 | 10.6 | >100 | 14 (3772) |
1310257 94 (89) |
| 1430688 | 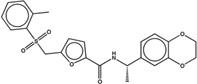 |
3.4 | 47 | >100 | 100 (240) |
1378455 86 (68) |
| 1486389 | 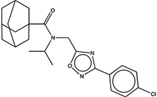 |
5.5 | 16 | >200 | 10 (248) |
1417302 96 (77) |
| 1508601 | 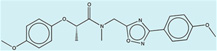 |
3.3 | 235 | >400 | 100 (62) |
1483036 91 (66) |
| 1515492 | 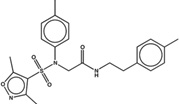 |
4.1 | 53 | >100 | 20 (142) |
1612324 92 (74) |
| 1560919 | 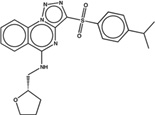 |
4.6 | 10.0 | >100 | 10 (410) |
1549087 91 (73) |
| 1565380 | 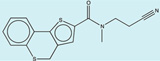 |
3.3 | 184 | >400 | 100 (71) |
1530398 91 (71) |
| 1572732 | 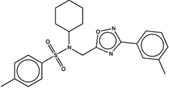 |
5.5 | 14.3 | >100 | 10 (238) |
1384931 90 (64) |
| 1607067 | 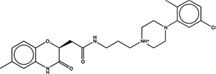 |
3.7 | 54 | >100 | 20 (291) |
1531142 90 (86) |
| 1778715 | 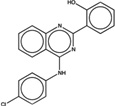 |
6.2 | 2.7 | >100 | 50 (1811) |
239009 92 (84) |
| 2165871 | 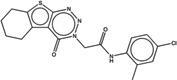 |
3.7 | 6.7 | >100 | 1.8 (2363) |
1474138 93 (85) |
| 3087539 | 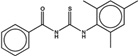 |
3.7 | 29 | >100 | 45 (383) |
1333224 93 (91) |
The criteria used to predict aggregation, xLogP and the Tanimoto coefficient to the closest known aggregators, are given, as are two experimental criteria of aggregator formation, detergent dependent inhibition of the counter-screening enzyme, AmpC, and the formation of particles detectable by DLS. Known aggregators are from refs 14 and 15.
LogP is derived from Molinspiration miLogP except those indicated by *, which are from Rdkit LogP. Weak aggregators are indicated in light blue
Table 2.
Molecules Predicted to Aggregate That Were Not Observed To Do so on Experimenta
| ChEMBL ID | Structure | xLogP | ChEMBL ID of most similar aggregator (Tc%) |
|---|---|---|---|
| 17052 | 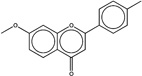 |
4.2 | 16751 (95) |
| 196585 | 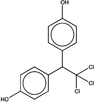 |
4.4 | 362919 (91) |
| 228369 | 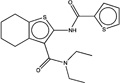 |
3.3 | 1605360 (93) |
| 239674 | 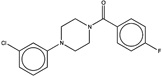 |
3.3 | 1531914 (91) |
| 276915 | 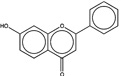 |
3.2 | 16761 (94) |
| 283196 | 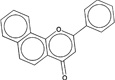 |
4.9 | 406835 (90) |
| 337993 | 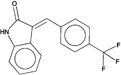 |
4.0 | 48760 (87) |
| 1492666 | 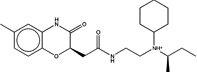 |
3.9 | 1608393 (93) |
| 1465049 | 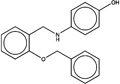 |
4.3 | 1084441 (89) |
| 1516338 | 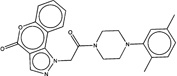 |
3.4 | 1572132 (86) |
| 1517395 | 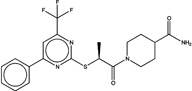 |
3.7 | 1449091 (92) |
| 1574158 |  |
4.1 | 1612523 (95) |
| 3113390 |  |
4.6 | 1492816 (89) |
These molecules did not form visible colloids by DLS up to concentrations of at least 200 µM, nor did they inhibit the counterscreening enzyme AmpC β-lactamase. These represent failed predictions of the model.
The newly predicted aggregators came from a diverse range of discovery modalities and were reported active on a diverse range of targets.30 Many came from high-throughput screening (HTS) campaigns, including CHEMBL1304694, CHEMBL1347854, and CHEMBL1359872 (Table 1), which emerged from screens targeting a calcium channel, 4′-phosphopantetheinyl transferase, and the microtubule-associated protein tau, respectively. This reflects the importance that HTS retains in early discovery.31 Other molecules came from medicinal chemistry optimization campaigns or from virtual screening. An example of the former is CHEMBL57997 (Table 1), a 6.8 µM positive allosteric modulator of the adenosine A1 G protein coupled receptor32 (GPCR). The compound is one of 13 in a synthetic series with EC50 values ranging from 4 to 32 µM. Intriguingly, none of the derivatives was found to be superior to the lead; it may be that the series represents both ligands that are acting classically on the A1 receptor and those like CHEMBL57997 that have transitioned to a colloidal mechanism, as has been previously observed.33 CHEMBL57997 resembles the known aggregator CHEMBL1303983,14 differing by the loss of two methyl groups and the addition of one chloride to the aromatic ring. It inhibited β-lactamase at an IC50 of 12.7 µM, while addition of 0.01% Triton X-100 diminished this activity to well above 100 µM, to the point that it could no longer be reliably detected within the solubility limits of the compound (Figure 1, top left). Correspondingly, CHEMBL57997 formed particles detectable by DLS at 21 µM (Figure 1, top left), with an average radius of 270 nm, and transitioned across its observable CAC at around the same concentration (Figure 2). A related example is CHEMBL261275, a 21 µM inhibitor of cholesterol ester transferase protein (CETP)34 that emerged from a medicinal chemistry study of several new compound series. Here too the compounds may represent a mixture of different mechanisms: the most potent series members had affinities in the 10 nM range; these are likely to behave by a classical 1:1 stoichiometry mechanism. Many others inhibit in the micromolar or tens of micromolar range, and like CHEMBL261275 some of these may be aggregators. CHEMBL261275 is 86% similar to the known aggregator CHEMBL1329712, differing by the gain of a chloride, the loss of a hydroxyl, and exchange of an ortho methyl for a para ethyl group. CHEMBL261275 inhibited β-lactamase at 7 µM, and this inhibition was fully abrogated by the addition of 0.01% Triton X-100. The molecule formed particles that strongly scattered light by DLS at 4 µM (Figure 1, top right), transiting through its observable CAC at around the same concentration (Figure 2).
Naturally, not all predictions worked, even when similarity to a precedented aggregator was high. An example is CHEMBL17052, a 11.3 µM inhibitor of monoamine oxygenase B (MAOB),35 which despite its close resemblance (95%) to the known aggregator CHEMBL1675115 does not form colloids at accessible concentrations. This illustrates the capricious dependence of aggregation on physical molecular properties, something expected of a phase transition, and the difficulties of inferring these using similarity. Even in the high similarity bins, we expect to have false positives with this approach. The method’s inevitable false negatives are harder to quantify.
Analysis of ChEMBL and ZINC
We extended this analysis to all of the ChEMBL and ZINC36 databases, keeping to the higher similarity values where the approach was more successful. Of 1 121 615 eligible compounds (see Methods), 8753 compounds in ChEMBL Version 17 have been experimentally shown to be likely aggregators under biochemical assay conditions,2,25 about 0.7% fo the database. Of those followed up in detailed secondary assays, including DLS, all but one did aggregate (37 out of 38), strengthening our confidence 2,2 that these compounds are true aggregators. A further 4905 compounds in ChEMBL Version 17 are 96% or more similar (axonpath fingerprints) to a known aggregator with calculated logP values >3. Enlarging the Tanimoto circle to 85% or better, over 43 601 compounds in ChEMBL resemble known aggregators with calculated logP > 3. Meanwhile, of 17.9 million purchasable, “drug-like” compounds in ZINC, 14 967 known aggregators are commercially available. Two types of evidence are used to define what we consider “known aggregators” here. About 95% of the “known aggregators” were identified in high throughput screens against β-lactamase2 or cruzain25, where the goal was to find colloidal aggregators. Likely aggregators were those molecules that inhibited in the absence but not the presence of a small amount of nonionic detergent. Over a hundred examples of these were followed up in detailed assays, 14,15 and it was found that detergent sensitivity reliably predicted aggregation. A second, higher level of evidence came from those compounds that have been shown to aggregate by multiple lines of evidence, especially dynamic light scattering and occasionally transmission electron microscopy (about 5% of the 14 967). At the 85% Tc and calculated LogP > 3 cutoffs, 0.73% of purchasable drug-like37 compounds in ZINC are predicted to aggregate. Comparing ChEMBL Version 17, representing compounds tested in the medicinal chemistry literature, to ZINC, representing simply purchasable molecules, over 7% of compounds in the medicinal chemistry literature are predicted to aggregate and 1% are known to aggregate, while 0.73% of purchasable molecules are predicted to aggregate. This suggests that aggregators are actually being prioritized as ligands in medicinal chemistry.
One may extend this analysis to ask how the prevalence of likely aggregators has changed over time, and how the prevalence of aggregators compares with that of other artifact-inducing molecules. Investigating compounds that were published in or before 1995, that is, before the full advent of high-throughput and virtual screening in the literature, of 134 549 compounds in ChEMBL, only 0.83%, 1126 compounds, resemble what is now known to be an aggregator with Tc values of 85% or better and with calculated LogP values >3. Thus, since 1995 the prevalence of plausible aggregators has grown by more than 8-fold in the medicinal chemistry literature (we cannot exclude the effects of the different types of chemotypes that have been explored, for new targets, since 1995, in addition to the advent of target-based screening). By way of context, molecules containing PAINS chemotypes 38,39 comprise 5.1% of the molecules in ChEMBL, which though substantial is only slightly higher than their 4.2% presence among the 20.7 million purchasable in-stock compounds, though it is much enriched over their 1.4% occurrence rate among the 80.7 million make-on-demand compounds in ZINC. Of the 12 600 experimentally determined aggregators, 1071 (8.5%) are also PAINS, using the Rdkit version of the Guha translation40 of the original Sybyl Line Notation into the more widely standardized and widely used SMARTS patterns. Of the 8753 known and 43 601 predicted aggregators in ChEMBL, 342 and 1793, respectively, both 4%, are also PAINS. These figures should be interpreted with some caution, owing to the lack of consensus SMARTS for PAINS.
The Occurrence of Aggregators in Whole Organism Phenotypic Screens
Whereas colloidal aggregators may be the dominant single mechanism underlying false-positives in target-based HTS campaigns,2,25 their occurrence in phenotypic assays has not been well-investigated. We therefore investigated the occurrence of colloidal aggregators among three recent whole organism phenotypic screens, two on zebra fish embryos41,42 and one on worms.43 Of 93 active, machine readable compounds identified in the three screens, nine would be predicted to aggregate. This is a rate far below that expected for compounds emerging from biochemical, target-oriented HTS campaigns, at least from primary screens, consistent with the idea that at least whole organism screens are much less likely to enrich colloidal aggregators than are target-based screens (we have not compared with cell-based phenotypic screens, where aggregators may well be more prevalent, though the direction of their effects is hard to predict).27,44
A Web-Based Tool for Advising on Aggregation
To enable investigators to interrogate their own molecules for status as likely aggregators, we developed a web-based tool to query our database of known aggregators, both as a web page (http://advisor.bkslab.org) and by a command line script that may be downloaded from that site (http://advisor.bkslab.org/faq). The interface has a search feature and can browse historical data (http://advisor.bkslab.org/browse). The raw database files may be downloaded as text (http://advisor.bkslab.org/rawdata). An FAQ page answers the most common questions about aggregation and provides additional reference information.
We demonstrate the interface with examples that illustrate the range of options available. Rottlerin is a known aggregator at micromolar concentrations and has been observed to modulate over 100 targets in over 70 papers in the scholarly literature. When the user puts in the SMILES string for rottlerin or draws it via the chemical drawing interface, its status as a known aggregator is returned, along with the papers where this is demonstrated by mechanistic experiment, its structure, and its physical properties. Meanwhile, the drug sorafenib is also an aggregator (CAC of 3.5 µM in biochemical buffer and in cell culture),27 and the papers illustrating this are returned, as are its physical properties and structures. ChEMBL1946170 closely resembles sorafenib (Tc value of 90%) and has a calculated LogP of 4.1. The advisor returns that this molecule is not known to aggregate but is a plausible aggregator given its close similarity to sorafenib and its lipophilicity. Finally, ZINC14007 (atenolol) is a polar molecule (calculated LogP 0.7) whose most related aggregator has a Tc value of only 72%. The advisor reports these physical properties and similarities, suggesting that this molecule is not a plausible aggregator.
Aggregation into colloidal particles reflects a phase transition, which is notoriously hard to predict. Also, molecules that do aggregate are not necessarily acting as artifacts in a particular assay. For instance, sorafenib is an aggregator at low micromolar concentrations, but at nanomolar concentrations it is a specific and well-behaved therapeutic. Thus, we do not pretend to accurately and reliably predict aggregators, rather the tool is meant to advise on plausibility and offer strategies to check for aggregation. Unlike many other molecules that can lead to artifacts in early discovery, colloidal aggregation acts by a specific, physical mechanisms of interference and can be controlled for by specific experiments. These include addition of nonionic detergent, centrifugation spin-down, increasing the stoichiometry of protein to putative aggregator, and others;13 these strategies are described in the advice from the tool.
DISCUSSION
Three principal observations emerge from this study. First, small molecule aggregators are chemically diverse, covering about 12% of the chemotype clusters that are represented in the medicinal chemistry literature. Over 8753 compounds in ChEMBL Version 17 are known to aggregate in vitro (over 0.7% of the annotated compounds), and a further 55 000 lipophilic ones are within a Tc of 85% of a known aggregator, making them plausible aggregators (about 7% of the compounds in ChEMBL Version 17). Intriguingly, known or plausible aggregators are 10-fold more common among literature-reported compounds than they are among purchasable compounds. Combined with the observation that they are over 8-fold more prevalent today than they were in 1995, before the full advent of combinatorial chemistry45 and high-throughput and virtual screening, this suggests that aggregators continue to be found in early discovery and that they are being found by target-based in vitro methods. Second, using conservative criteria of chemical similarity and lipophilicity, it is possible to predict new aggregators well enough to be pragmatic. Insisting on calculated LogP values of >3, 5 of 7 molecules with Tc values ≥95% to precedented aggregators aggregated, while 10 of 19 with Tc values between 90% and 94% did so, and 3 of 7 with Tc values between 0.85 and 0.89 aggregated; three others in these ranges were weak aggregators. Whereas this is an admittedly weak method, without a strong physical model, it is powered by the sheer number and diversity of colloidal aggregators now known. Third, the open access aggregator advisor tool allows for investigators to rapidly ask, at minimum, whether a molecule is already known to aggregate and offers guidance as to whether the molecule is likely to do so, and what controls may be undertaken (http://advisor.bkslab.org). This tool may be used interactively via its Web site, or the entire database may be downloaded for interrogation.
The caveats aired above merit re-emphasis. Just because a molecule aggregates, under some conditions, in the same concentration range as it is active, does not establish that its activity is artifactual. Differences in assay conditions could shift the compound’s CAC values to higher concentrations, the enzyme concentration could be high enough to be unaffected by the colloids, or the colloids could be presaturated with other proteins, such as serum albumin. What the observation of colloid formation does suggest is that further controls are warranted before interpreting inhibition by these molecules as a specific event. The nature of colloidal aggregation as a kinetically stable phase, balanced precariously between solubility and precipitation, also bears re-emphasis. We do not pretend that it can be captured by similarity and simple filters alone and certainly do not suggest that this method be used to filter aggregators from libraries. They are too prevalent, and whereas a molecule that aggregates is always, under those conditions, an aggregator, its aggregation at micromolar concentrations does not impugn its activity at nanomolar concentrations; sorafenib and fulvestrant are strong aggregators and bona fide drugs. The model advanced here is primitive and subject to false negatives and false positives. It only predicts positives, not negatives, and its domain of applicability is limited to Tc > 85% of known aggregators. We cannot exclude that the lower prediction rate simply reflects our inability to predict on compounds outside of the chemotypes covered by precedented aggregators, especially since we have not sought to characterize our false-negative rate. Even here, it has only been tested on several dozen compounds—a full test would involve a HTS campaign, such as for detergent sensitivity, as has been undertaken previously.2,25 The results of such a screen would undoubtedly refine the simple model described here. For now, chief recommendations for this model are its ease of use, the extensive library of chemotypes that have been observed to aggregate, powering the similarity criterion, and the simple, direct experimental methods that are available to investigators to control for aggregation.
These caveats should not obscure the central observations of this study: colloidal aggregators are perhaps the most widespread artifact in early drug discovery46 and likely comprise about 7% of the compounds in the medicinal chemistry literature, where they are 10-fold concentrated relative to their occurrence among purchasable compounds. They are far more common now than they were in the mid-1990s, before the phenomenon of colloidal aggregation had been described. Because colloid-formation reflects a physical event, many chemotypes are subject to it. The purpose of the aggregator advisor tool (http://advisor.bkslab.org) is thus not to filter out aggregators—this essentially cannot be done without removing many genuinely interesting molecules. Rather, its purpose is to rapidly identify those hits from early discovery, typically acting in the 100 nM range or higher, that are likely to aggregate, and to recommend rapid experimental controls to test if aggregation is in fact occurring and if so whether it is relevant to the activity being measured (sometimes it might not be). Used in this way, this tool, and more optimized versions that may be implemented by others using the data that underlies it, may be pragmatic to investigators in early discovery research.
METHODS
Data Collection
We assembled experimentally known aggregators from published sources.2,3,10,15,19,24,25,47 The data and data sources are available free to download at http://advisor.docking.org/rawdata/.
Database Design
We created a database in MySQL/JChemBase (ChemAxon, www.chemaxon.com). The database design follows the contents of the raw data files. One table contains data sources with three columns (data source ordinal number, DOI if available, publication or description of source) and one table for known aggregators with three columns (SMILES, original identifier, data source index).
Interface Design
We created a web-based graphical user interface using Code Igniter (http://ellislab.com/codeigniter). The interface has five page types: (1) a welcome page, which is also a search page incorporating the WebMe drawing tool (www.molinspiration.com); (2) a report (see Program Logic below) produced by clicking on “Run Query”, which searches the database and presents the report; (3) a help and frequently asked questions page to provide guidance to the user seeking to test compounds for aggregation; (4) a gallery of aggregators, enabling the user to view each known aggregator, follow links to references where aggregation was first reported, or search ZINC for analogs; (5) a contact page, where new aggregators may be reported.
Program Logic
The logic of the advisory function is as follows. The user enters the molecule as SMILES or uses the drawing tool, selects an affinity category among three choices (<0.1 µM; 0.1–10 µM, the default; >10 µM) and then clicks on Search. This generates a report based on three quantities: (A) the Tanimoto similarity, Tc, using standard axonpath fingerprints to the nearest known aggregator, (B) the calculated LogP using miLogP (mitools by Molinspiration), and (C) the affinity bin. The SMILES are standardized and canonicalized using the ChemAxon standardizer. The logic is as follows:
In the default affinity range of 100 nM to 10 µM, if calculated LogP > 3 and Tc ≥ 85%, the user is informed that this compound should be investigated as an aggregator. If either calculated LogP > 3 or Tc > 85%, the user is warned that one of these two contributing criteria are in effect and that controls should be run. If neither of these is true, this is reported, and the user is counseled that controls are always advised.
In the higher affinity range, we add to the logic above by stating that we are unaware of aggregators at such high affinity, but proper controls are always advised. In the lower affinity range, we remind the user that aggregators are particularly prevalent at high concentration and controls are strongly urged for all combinations of calculated LogP and Tc above 10 µM activity values.
Screening ChEMBL 17 against the Aggregator Advisor
We used ChEMBL version 17 containing 1 324 941 unique compounds released November 2013. We desalted the molecules using OEChem (OpenEye, Santa Fe, NM) and removed any molecules with strange valences using mitools (molinspiration.com) or that were over 1000 Da in weight, resulting in 1 121 615 eligible molecules. We calculated the most similar aggregator to each compound using the Tanimoto coefficient on the standard axonpath fingerprint in JChemBase (ChemAxon, Chemaxon.com) and tabulated compounds having a Tanimoto similarity of 80% or higher to a known aggregator.
Acquisition and Experimental Testing of Compounds by Tanimoto Range
To investigate whether compounds with high calculated LogP and high similarity to known aggregators were also aggregators, we picked representative commercially available compounds having Tanimoto similarity to a known aggregator between 80% and 99%, sampling as evenly as we could within that range. Although only 7% of ChEMBL is commercially available, there were many compounds we could have tested. We picked them at random, with our only biases being to avoid testing near duplicates, too many having the same ring or chemotype, and covering a wide range in LogP and molecular weight. Compounds were purchased from Molport (molport.com), Sigma (sigmaaldrich.com), ChemBridge (chembridge.com), Enamine (enamine.net), and eMolecules (emolecules.com).
Enzyme Inhibition Assays
Inhibition of AmpC β-lactamase was measured as previously described.18,22,26 The final concentration of DMSO was 1% for all samples. Values reported are the average of duplicate samples run in two independent experiments.
Dynamic Light Scattering
Particle formation was measured using a DynaPro MS/X (Wyatt Technology) as previously described.22 Compounds were measured both in the absence and in the presence of 0.01% Triton X-100. Critical aggregation concentrations values were calculated from three independent experiments. Each histogram shows a single representative sample.
How To Use the Tool
Once the page http://advisor.bkslab.org is opened, the user may specify a molecule by drawing it using the WebME tool (left) or by pasting a SMILES string into the text box (right). The affinity range of 0.1 −10 µM may be left unchanged or set to a higher (<0.1 µM) or lower (>10 µM) range. The user clicks on the “Run Query” button to run the calculation. The report page provides advice based on two calculated properties, the LogP and the similarity to previously known aggregators, as well as the affinity range; this calculation is typically completed in seconds.
Two further features of the Web site merit mention. The user may also browse the database of compounds observed to aggregate using the “Gallery” menu item (top left). Within the gallery, the user may view original citations in which the aggregator was reported. The “Help” menu item (top left) provides general advice about what to look for in one’s own work or when reading the work of others, answers to some frequently asked questions, literature references to original papers investigating aggregation, data files, and a command line program that may be downloaded and used in batch mode.
The PAINS calculations were done using 480 PAINS from the Rdkit version of the Guha translation40 of the original Sybyl Line Notation into the more widely standardized and widely used SMARTS patterns. This conversion resembles that of the publicly available reference implementation in the cApp program.48
ACKNOWLEDGMENTS
This work was supported by Grant GM71630. We thank Greg Landrum of Novartis and Rdkit for curated PAINS SMARTS. We are grateful to ChemAxon for JChemBase, Molinspiration for a license for their molecule properties (mib) and molecule depiction tool, and OpenEye Scientific Software for a free academic license for OEChem. We thank Cassidy Kelly and Pascal Wassam for programming support, and Drs. Trent Balius and Marcus Fischer for reading the manuscript.
ABBREVIATIONS
- DLS
dynamic light scattering
- Tc
Tanimoto coefficient
Footnotes
The authors declare no competing financial interest.
REFERENCES
- 1.Roche O, Schneider P, Zuegge J, Guba W, Kansy M, Alanine A, Bleicher K, Danel F, Gutknecht EM, Rogers-Evans M, Neidhart W, Stalder H, Dillon M, Sjogren E, Fotouhi N, Gillespie P, Goodnow R, Harris W, Jones P, Taniguchi M, Tsujii S, von Der Saal W, Zimmermann G, Schneider G. Development of a Virtual Screening Method for Identification of “Frequent Hitters” in Compound Libraries. J. Med. Chem. 2002;45:137–142. doi: 10.1021/jm010934d. [DOI] [PubMed] [Google Scholar]
- 2.Feng BY, Simeonov A, Jadhav A, Babaoglu K, Inglese J, Shoichet BK, Austin CP. A high-throughput screen for aggregation-based inhibition in a large compound library. J. Med. Chem. 2007;50:2385–2390. doi: 10.1021/jm061317y. [DOI] [PubMed] [Google Scholar]
- 3.McGovern SL, Helfand BT, Feng B, Shoichet BK. A specific mechanism of nonspecific inhibition. J. Med. Chem. 2003;46:4265–4272. doi: 10.1021/jm030266r. [DOI] [PubMed] [Google Scholar]
- 4.Sassano MF, Doak AK, Roth BL, Shoichet BK. Colloidal aggregation causes inhibition of G protein-coupled receptors. J. Med. Chem. 2013;56:2406–2414. doi: 10.1021/jm301749y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lin H, Sassano MF, Roth BL, Shoichet BK. A pharmacological organization of G protein-coupled receptors. Nat. Methods. 2013;10:140–146. doi: 10.1038/nmeth.2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McGovern SL, Caselli E, Grigorieff N, Shoichet BK. A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening. J. Med. Chem. 2002;45:1712–1722. doi: 10.1021/jm010533y. [DOI] [PubMed] [Google Scholar]
- 7.Coan KE, Maltby DA, Burlingame AL, Shoichet BK. Promiscuous aggregate-based inhibitors promote enzyme unfolding. J. Med. Chem. 2009;52:2067–2075. doi: 10.1021/jm801605r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wolan DW, Zorn JA, Gray DC, Wells JA. Small-molecule activators of a proenzyme. Science. 2009;326:853–858. doi: 10.1126/science.1177585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zorn JA, Wolan DW, Agard NJ, Wells JA. Fibrils colocalize caspase-3 with procaspase-3 to foster maturation. J. Biol. Chem. 2012;287:33781–33795. doi: 10.1074/jbc.M112.386128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feng BY, Shoichet BK. A detergent-based assay for the detection of promiscuous inhibitors. Nat. Protoc. 2006;1:550–553. doi: 10.1038/nprot.2006.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thorne N, Inglese J, Auld DS. Illuminating insights into firefly luciferase and other bioluminescent reporters used in chemical biology. Chem. Biol. 2010;17:646–657. doi: 10.1016/j.chembiol.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ryan AJ, Gray NM, Lowe PN, Chung C. Effect of Detergent on “Promiscuous” Inhibitors. J. Med. Chem. 2003;46:3448–3451. doi: 10.1021/jm0340896. [DOI] [PubMed] [Google Scholar]
- 13.Shoichet BK. Interpreting steep dose-response curves in early inhibitor discovery. J. Med. Chem. 2006;49:7274–7277. doi: 10.1021/jm061103g. [DOI] [PubMed] [Google Scholar]
- 14.Ferreira RS, Simeonov A, Jadhav A, Eidam O, Mott BT, Keiser MJ, McKerrow JH, Maloney DJ, Irwin JJ, Shoichet BK. Complementarity between a docking and a high-throughput screen in discovering new cruzain inhibitors. J. Med. Chem. 2010;53:4891–4905. doi: 10.1021/jm100488w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Babaoglu K, Simeonov A, Irwin JJ, Nelson ME, Feng B, Thomas CJ, Cancian L, Costi MP, Maltby DA, Jadhav A, Inglese J, Austin CP, Shoichet BK. Comprehensive mechanistic analysis of hits from high-throughput and docking screens against beta-lactamase. J. Med. Chem. 2008;51:2502–2511. doi: 10.1021/jm701500e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Giannetti AM, Koch BD, Browner MF. Surface plasmon resonance based assay for the detection and characterization of promiscuous inhibitors. J. Med. Chem. 2008;51:574–580. doi: 10.1021/jm700952v. [DOI] [PubMed] [Google Scholar]
- 17.Horobin RW, Rashid-Doubell F, Pediani JD, Milligan G. Predicting small molecule fluorescent probe localization in living cells using QSAR modeling 1. Overview and models for probes of structure, properties and function in single cells. Biotech. Histochem. 2013;88:440–460. doi: 10.3109/10520295.2013.780634. [DOI] [PubMed] [Google Scholar]
- 18.Seidler J, McGovern SL, Doman TN, Shoichet BK. Identification and prediction of promiscuous aggregating inhibitors among known drugs. J. Med. Chem. 2003;46:4477–4486. doi: 10.1021/jm030191r. [DOI] [PubMed] [Google Scholar]
- 19.Feng BY, Shelat A, Doman TN, Guy RK, Shoichet BK. High-throughput assays for promiscuous inhibitors. Nat. Chem. Biol. 2005;1:146–148. doi: 10.1038/nchembio718. [DOI] [PubMed] [Google Scholar]
- 20.Rao H, Li Z, Li X, Ma X, Ung C, Li H, Liu X, Chen Y. Identification of small molecule aggregators from large compound libraries by support vector machines. J. Comput. Chem. 2010;31:752–763. doi: 10.1002/jcc.21347. [DOI] [PubMed] [Google Scholar]
- 21.Hsieh JH, Wang XS, Teotico D, Golbraikh A, Tropsha A. Differentiation of AmpC beta-lactamase binders vs. decoys using classification kNN QSAR modeling and application of the QSAR classifier to virtual screening. J. Comput.-Aided Mol. Des. 2008;22:593–609. doi: 10.1007/s10822-008-9199-2. [DOI] [PubMed] [Google Scholar]
- 22.Coan KE, Shoichet BK. Stoichiometry and physical chemistry of promiscuous aggregate-based inhibitors. J. Am. Chem. Soc. 2008;130:9606–9612. doi: 10.1021/ja802977h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wells JA, McClendon CL. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature. 2007;450:1001–1009. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- 24.Coan KE, Shoichet BK. Stability and equilibria of promiscuous aggregates in high protein milieus. Mol. BioSyst. 2007;3:208–213. doi: 10.1039/b616314a. [DOI] [PubMed] [Google Scholar]
- 25.Jadhav A, Ferreira RS, Klumpp C, Mott BT, Austin CP, Inglese J, Thomas CJ, Maloney DJ, Shoichet BK, Simeonov A. Quantitative analyses of aggregation, autofluorescence, and reactivity artifacts in a screen for inhibitors of a thiol protease. J. Med. Chem. 2010;53:37–51. doi: 10.1021/jm901070c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Doak AK, Wille H, Prusiner SB, Shoichet BK. Colloid formation by drugs in simulated intestinal fluid. J. Med. Chem. 2010;53:4259–4265. doi: 10.1021/jm100254w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Owen SC, Doak AK, Wassam P, Shoichet MS, Shoichet BK. Colloidal aggregation affects the efficacy of anticancer drugs in cell culture. ACS Chem. Biol. 2012;7:1429–1435. doi: 10.1021/cb300189b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Duan D, Doak AK, Nedyalkova L, Shoichet BK. Colloidal Aggregation the in vitro Activity of Traditional Chinese Medicines. ACS Chem. Biol. 2015;10:978. doi: 10.1021/cb5009487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gaulton A, Bellis LJ, Bento AP, Chambers J, Davies M, Hersey A, Light Y, McGlinchey S, Michalovich D, Al-Lazikani B, Overington JP. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012;40:D1100–D1107. doi: 10.1093/nar/gkr777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Overington JP, Al-Lazikani B, Hopkins AL. How many drug targets are there? Nat. Rev. Drug Discovery. 2006;5:993–996. doi: 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- 31.Zhu PJ, Hobson JP, Southall N, Qiu C, Thomas CJ, Lu J, Inglese J, Zheng W, Leppla SH, Bugge TH, Austin CP, Liu S. Quantitative high-throughput screening identifies inhibitors of anthrax-induced cell death. Bioorg. Med. Chem. 2009;17:5139–5145. doi: 10.1016/j.bmc.2009.05.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van der Klein PAM, Kourounakis AP, IJzerman AP. Allosteric modulation of the adenosine A(1) receptor. Synthesis and biological evaluation of novel 2-amino-3-benzoylthiophenes as allosteric enhancers of agonist binding. J. Med. Chem. 1999;42:3629–3635. doi: 10.1021/jm991051d. [DOI] [PubMed] [Google Scholar]
- 33.Ferreira RS, Bryant C, Ang KK, McKerrow JH, Shoichet BK, Renslo A. R Divergent modes of enzyme inhibition in a homologous structure-activity series. J. Med. Chem. 2009;52:5005–5008. doi: 10.1021/jm9009229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harikrishnan LS, Kamau MG, Herpin TF, Morton GC, Liu Y, Cooper CB, Salvati ME, Qiao JX, Wang TC, Adam LP, Taylor DS, Chen AY, Yin X, Seethala R, Peterson TL, Nirschl DS, Miller AV, Weigelt CA, Appiah KK, O’Connell JC. Michael Lawrence, R 2-Arylbenzoxazoles as novel cholesteryl ester transfer protein inhibitors: optimization via array synthesis. Bioorg. Med. Chem. Lett. 2008;18:2640–2644. doi: 10.1016/j.bmcl.2008.03.030. [DOI] [PubMed] [Google Scholar]
- 35.Chimenti F, Fioravanti R, Bolasco A, Chimenti P, Secci D, Rossi F, Yanez M, Orallo F, Ortuso F, Alcaro S, Cirilli R, Ferretti R, Sanna ML. A new series of flavones, thioflavones, and flavanones as selective monoamine oxidase-B inhibitors. Bioorg. Med. Chem. 2010;18:1273–1279. doi: 10.1016/j.bmc.2009.12.029. [DOI] [PubMed] [Google Scholar]
- 36.Irwin JJ, Sterling T, Mysinger MM, Bolstad ES, Coleman RG. ZINC - A free tool to discover chemistry for biology. J. Chem. Inf. Model. 2012;52:1757. doi: 10.1021/ci3001277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery development settings. Adv. Drug Delivery Rev. 1997;23:3. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 38.Baell JB, Holloway G. A New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010;53:2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 39.Erlanson DA. Learning from PAINful lessons. J. Med. Chem. 2015;58:2088–2090. doi: 10.1021/acs.jmedchem.5b00294. [DOI] [PubMed] [Google Scholar]
- 40.Saubern S, Guha R, Baell JB. KNIME Workflow to Assess PAINS Filters in SMARTS Format. Comparison of RDKit and Indigo Chemoinformatics Libraries. Mol. Inf. 2011;30:847–850. doi: 10.1002/minf.201100076. [DOI] [PubMed] [Google Scholar]
- 41.Kokel D, Bryan J, Laggner C, White R, Cheung CY, Mateus R, Healey D, Kim S, Werdich AA, Haggarty SJ, Macrae CA, Shoichet B, Peterson RT. Rapid behavior-based identification of neuroactive small molecules in the zebrafish. Nat. Chem. Biol. 2010;6:231–237. doi: 10.1038/nchembio.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Laggner C, Kokel D, Setola V, Tolia A, Lin H, Irwin JJ, Keiser MJ, Cheung CY, Minor DL, Jr, Roth BL, Peterson RT, Shoichet BK. Chemical informatics and target identification in a zebrafish phenotypic screen. Nat. Chem. Biol. 2012;8:144–146. doi: 10.1038/nchembio.732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lemieux GA, Keiser MJ, Sassano MF, Laggner C, Mayer F, Bainton RJ, Werb Z, Roth BL, Shoichet BK, Ashrafi K. In silico molecular comparisons of C. elegans and mammalian pharmacology identify distinct targets that regulate feeding. PLoS Biol. 2013;11:e1001712. doi: 10.1371/journal.pbio.1001712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Owen SC, Doak AK, Ganesh AN, Nedyalkova L, McLaughlin CK, Shoichet BK, Shoichet MS. Colloidal drug formulations can explain “bell-shaped” concentration-response curves. ACS Chem. Biol. 2014;9:777–784. doi: 10.1021/cb4007584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lowe DB. Drug discovery: Combichem all over again. Nat. Chem. 2014;6:851–852. doi: 10.1038/nchem.2074. [DOI] [PubMed] [Google Scholar]
- 46.Edwards AM, Bountra C, Kerr DJ, Willson TM. Open access chemical and clinical probes to support drug discovery. Nat. Chem. Biol. 2009;5:436–440. doi: 10.1038/nchembio0709-436. [DOI] [PubMed] [Google Scholar]
- 47.Feng BY, Shoichet BK. Synergy and antagonism of promiscuous inhibition in multiple-compound mixtures. J. Med. Chem. 2006;49:2151–2154. doi: 10.1021/jm060029z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Amani P, Sneyd T, Preston S, Young ND, Mason L, Bailey UM, Baell J, Camp D, Gasser RB, Gorse AD, Taylor P, Hofmann A. A practical Java tool for small-molecule compound appraisal. J. Cheminf. 2015;7:28. doi: 10.1186/s13321-015-0079-1. [DOI] [PMC free article] [PubMed] [Google Scholar]