

Abstract
Fluorescence anisotropy imaging is a popular method to visualize changes in organization and conformation of biomolecules within cells and tissues. In such an experiment, depolarization effects resulting from differences in orientation, proximity and rotational mobility of fluorescently labeled molecules are probed with high spatial resolution. Fluorescence anisotropy is typically imaged using laser scanning and epifluorescence-based approaches. Unfortunately, those techniques are limited in either axial resolution, image acquisition speed, or by photobleaching. In the last decade, however, selective plane illumination microscopy has emerged as the preferred choice for three-dimensional time lapse imaging combining axial sectioning capability with fast, camera-based image acquisition, and minimal light exposure. We demonstrate how selective plane illumination microscopy can be utilized for three-dimensional fluorescence anisotropy imaging of live cells. We further examined the formation of focal adhesions by three-dimensional time lapse anisotropy imaging of CHO-K1 cells expressing an EGFP-paxillin fusion protein.
OCIS codes: (170.2520) Fluorescence microscopy, (180.6900) Three-dimensional microscopy
1. Introduction
As of today, many ligand binding, immuno-, and high throughput screening assays in the clinical and biomedical fields are based on fluorescence polarization [1]. Most of these applications are solution-based and performed with a spectrofluorimeter type of instrument; hence, the lack of spatial resolution is not relevant. However, with the development of advanced instrumentation, in particular confocal and two photon laser-scanning microscopy [2,3], fluorescence polarization imaging has become more and more popular over the last two decades [4,5]. Many polarization-based probes have been developed that can be used as biosensors in vitro as well as in living cells or tissues. Unfortunately, many commercial microscopes do not support polarization imaging as a standard feature. Often, the detection path contains prisms and gratings inducing a splitting by wavelength. Also, polarizing beam splitters in the form of a cube require special holders. Still, many instruments can be upgraded with additional components to support this type of fluorescence imaging. While laser-scanning microscopy provides optical sectioning, imaging speed is rather slow as the data is collected in a sequential, pixel-by-pixel fashion. Consequently, especially for time lapse imaging, laser-scanning microscopy is usually limited to a single plane. Widefield, camera-based image acquisition, on the other hand, is fast but, when combined with epi-illumination, lacks contrast in axial direction, or, when combined with evanescent excitation termed total internal reflection (TIRF) microscopy, is limited to the interface between coverslip and sample [6]. Still, polarized TIRF microscopy is very useful for surface studies [7,8]. Note that, in TIRF, the electric field has two components due to the elliptical polarization of the evanescent field with the intensity predominantly polarized normal to the interface. This can be exploited to study oriented systems [9]. One solution to combine the imaging speed of camera-based detection with axial sectioning capability is to rapidly move an array of focal spots across the observation plane, usually by the use of a rotating disc. Unfortunately, the photon collection efficiency of such a setup is poor leading to a lack in sensitivity and increased photobleaching. To overcome these limitations, a concept dating back more than one hundred years has been reintroduced recently; the basic idea is to illuminate the sample with a light beam perpendicular to the detection axis of the microscope [10–12]. Named the ultramicroscope by Siedentopf et al., the acronyms orthogonal plane fluorescence optical sectioning (OPFOS) and selective plane illumination microscopy (SPIM) are the most common ones these days. Based on the original concept, huge improvements in image quality were made by the development of long working distance objectives, combined with advances in camera and computer-based data processing technology. By injecting a thin sheet of light perpendicular to the detection axis, only the focal plane of the detection lens is illuminated, resulting in excellent optical sectioning and minimal light exposure of the sample. At the same time, wide field detection with a camera results in superior imaging speed. The SPIM method is also very versatile; it can be used to image larger specimens (~1 mm) such as tissues and whole organisms [13], or adopted for imaging of smaller samples such as single cells on a coverslip [14]. In this work, we demonstrate how polarization sensitive imaging can be done effectively in the SPIM configuration. We assessed the sensitivity of our instrument by measuring the fluorescence polarization of Rhodamine 110 dissolved in solutions of different water/glycerol content and comparing the results to measurements using a spectrofluorimeter. We further show how high quality, three-dimensional images of the fluorescence polarization exhibited by various genetically encoded fusion probes expressed in living CHO-K1 and HeLa cells can be obtained with SPIM. Finally, we visualized the aggregation of the EGFP-paxillin fusion probe in focal adhesions by 3D time lapse imaging of CHO-K1 cells.
2. Methods
2.1 Definition and calculation of the fluorescence anisotropy
As our SPIM setup was designed for coverslip-based samples, such as a monolayer of single cells, the perpendicularly arranged excitation and detection lenses dip into the sample dish from the top at an angle of 45° with respect to the sample plane. To quantify the polarization of the emission, we calculated the fluorescence anisotropy, , defined as
| (1) |
with and the fluorescence intensities of vertical and horizontal polarization with respect to that of the vertically polarized excitation beam. A potential bias in the detection of the two polarizations due to polarization sensitive optics was accounted for by the factor defined as
| (2) |
To measure the factor, the polarization of the excitation beam is set in such a way that both detection channels are of perpendicular orientation with respect to the excitation. In this configuration, the emission intensities can be referred to as and . Alternatively, the sample could be detected at the magic angle (54.7°) [15]. An additional correction has to be considered when employing objective lenses of high numerical aperture (NA), which create a cone of light instead of a more or less parallel beam. At the periphery of the cone, there is a component of the electric field parallel to the optical axis causing partial depolarization of the light [15,16]. This depolarization can be accounted for by replacing the factor of two in Eq. (1) with a factor resulting in
| (3) |
This correction factor can be determined experimentally, e.g., by measuring a sample of high anisotropy with an objective lens of low NA () that has a negligible effect on the polarization [17,18]. This reference anisotropy value can then be used to calculate the correction factor that will match the result obtained with the high NA lens. For our SPIM setup, we determined by a reference measurement with a spectrofluorimeter, , using the following equation
| (4) |
2.2 Description of the SPIM setup used for anisotropy imaging
Figure 1 shows a scheme of our home built SPIM setup as used for fluorescence anisotropy imaging.
Fig. 1.

Schematic of the SPIM setup used for fluorescence anisotropy imaging, see main text for a description.
For fluorescence excitation, a 488-nm laser diode (488nm, ISS, Champaign, IL, USA), a 561-nm solid state laser (CL561-150, CrystaLaser, Reno, NV, USA) and a 635-nm laser diode (CPS180, Thorlabs, Newton, NJ, USA) were joined with long pass dichroic mirrors. The intensity of each laser line was controlled independently with an acousto optical tunable filter, AOTF, (AOTFnC-400.650, AA Opto-Electronic, Orsay, France). As a benefit, the AOTF acted as a polarizer, yielding linearly polarized light at the first order of diffraction. After passing a single mode polarization maintaining fiber, PMF, the orientation of the polarization was adjusted with a half wave plate (AQWP05M-600, Thorlabs). The beam was then scanned across the back focal plane of the excitation lens, OLE (CFI Plan Fluor 10XW, NA 0.3, Nikon, Melville, NY, USA), to form the light sheet. The excitation lobe was scanned across the field of view once per exposure cycle to ensure homogeneous illumination of the focal plane of the detection objective, OLD (Scaleview 25x, NA 1.05, Olympus, Center Valley, PA, USA). The scanning optics included a galvanometric mirror, GM (GVSM002, Thorlabs), a scan lens, SL (#49-356, Edmund Optics, Barrington, NJ, USA), and a tube lens, TLE (#49-362, Edmund Optics). A function generator (DS345, SRS, Sunnyvale, CA, USA) synchronized to the sCMOS camera (Zyla 4.2, Andor, Belfast, Ireland) was used to generate the proper voltage ramp. Fluorescence light was spectrally cleaned by a band pass filter, F (FF01-520/35, Semrock, Rochester, NY, USA), before splitting the beam into its parallel and perpendicular polarization components with a polarizing beam splitter cube, PBS (CM1-PBS251, Thorlabs). Via tube lenses, TLDs (#47-740, Edmund Optics), both beams were imaged onto the opposing sides of a knife-edge prism mirror, PM (MRAK25-P01, Thorlabs). With another lens, LD (#47-737, Edmund Optics), the two polarizations, and , were then re-imaged side-by-side onto the chip of the camera yielding a sample pixel size of 142 nm. A 3-axis stage (MAX343, Thorlabs) was used to move the sample. We used the freely available Micro-Manager open source microscopy software [19] to operate our SPIM system. The image acquisition process is described previously [20].
2.3 Demonstration of depolarization effects induced by different fluorophore environments
To demonstrate how image contrast is obtained by fluorescence anisotropy imaging, we coated a glass coverslip with a ~100 µm thick layer of PMMA containing ~100 nM of Rhodamine 110. This sample was placed in a tissue culture dish (Falcon 353002, VWR, Radnor, PA, USA), filled with water, and subjected to SPIM imaging by exciting Rhodamine 110 fluorescence with 488-nm light. The immersion water was then supplemented with the same dye until the fluorescence intensity from the solution matched the fluorescence emanated by the PMMA-Rhodamine 110 layer, 3D images are shown in Fig. 2.
Fig. 2.
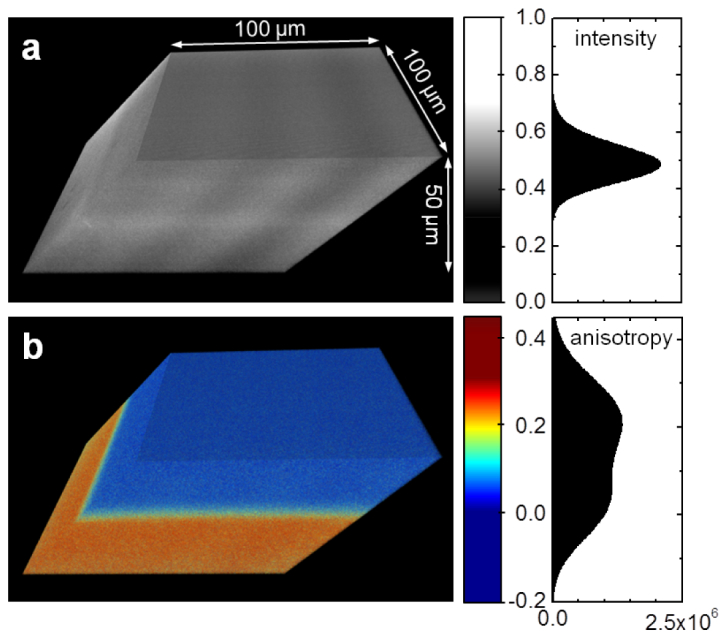
Three-dimensional projection of the fluorescence intensity (a) and anisotropy image (b) at the interface between a PMMA film covered with immersion water containing similar concentrations of Rhodamine 110. Fluorescence intensity and anisotropy histograms are shown to the side.
By means of the fluorescence intensity the PMMA layer cannot be distinguished from the immersion water anymore, see Fig. 2(a), while the two environments are clearly separated in the anisotropy image, see Fig. 2(b). The low anisotropy of Rhodamine in water is caused by fast rotational diffusion. This form of depolarization is inhibited in the PMMA matrix resulting in rather high anisotropy values.
3. Results
3.1 Evaluation of different water/glycerol mixtures containing Rhodamine 110
To evaluate the performance of our SPIM system, we prepared solutions of water and glycerol mixed a different ratios supplemented with ~100 nM of Rhodamine 110; fluorescence was excited with 488-nm light. Eleven solutions containing 0–100% v/v glycerol were measured at room temperature (24.3 ± 0.2°C). Subsequently, the solution containing 90% v/v glycerol was subjected to measurements at different temperatures between 27°C and 37°C. For each sample, we acquired two image series (2,048 × 2,048 pixels, 100 frames, 30 ms exposure time), one series with the half wave plate in a position yielding vertically polarized excitation light, and the other series with the half wave plate in a position yielding horizontally polarized excitation light. For each series, we calculated the temporal average followed by subtraction of the camera background. Both images were then split into their parallel and perpendicular components, a 64 pixel wide strip was removed for each detection channel to avoid artifacts introduced by the splitting edge of the dual view. Consequently, we obtained four images, , , , and of 2,048 × 960 pixels each. A factor image was calculated according to Eq. (2), followed by calculation of the final anisotropy image using Eq. (3). The relatively high NA of the detection lens used in our SPIM setup (NA 1.05) was also taken into account. The temperature series of the 90% v/v glycerol solution was used for this purpose, yielding a correction factor, . Each closed symbol in Fig. 3 represents the pixel average and standard deviation of the corresponding anisotropy image. For comparison, the fluorescence anisotropies of the same solutions were measured with a spectrofluorimeter (Fluoromax-4, Horiba Scientific, Edison, NJ, USA) maintaining the excitation wavelength and spectral detection window of the SPIM measurements.
Fig. 3.
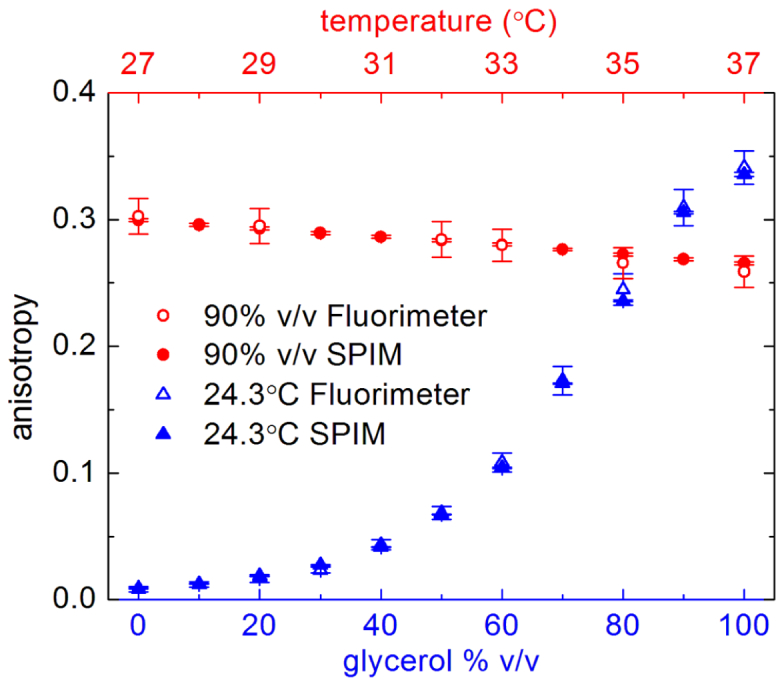
Fluorescence anisotropy measurements of Rhodamine 110 in solutions of different water/glycerol content (blue symbols) and varying temperature (red symbols) with a spectrofluorimeter (open symbols) and our SPIM setup (closed symbols).
At low glycerol content, the viscosity of the solution is low, allowing for fast rotational relaxation. Hence, the emitted fluorescence is almost completely depolarized, we obtained anisotropy values of with the SPIM and for the fluorimeter experiments, respectively. Dissolved in water at room temperature, these low values close to zero are typical for a small dye of rather long lifetime (~4 ns). Sometimes, such a sample is used to determine the factor assuming [4,17]. For the solution of highest glycerol content (99.6%), we obtained steady-state anisotropy values of 0.336 ± 0.002 with the SPIM and 0.341 ± 0.013 with the fluorimeter. With respect to the error, the anisotropy values obtained by SPIM perfectly match the corresponding fluorimeter values. The temperature series underlies the strong temperature dependence of the viscosity. We note the high precision of the anisotropy values obtained by SPIM, clearly resolving temperature differences as little as 1°C.
3.2 Three-dimensional live cell anisotropy imaging
There are three main mechanisms that modulate the anisotropy in fluorescence images of fluorescently labeled cells and tissues – rotational motion of the fluorophores, energy transfer between fluorophores, and their orientation relative to the polarization of the excitation light. In the first case, depolarization is caused by either passive, diffusional rotation or active rotation, e.g., by a motorprotein [21]. However, biomolecules are usually large compared to dye molecules resulting in a rather slow diffusional rotation with respect to the fluorescence lifetime. This is especially true for proteins tagged with genetically encoded markers such as green fluorescent protein (GFP). Consequently, their anisotropy is not significantly increased when bound to other cellular components compared to just free diffusion. To illustrate this point, we subjected enhanced (E)GFP expressing CHO-K1 cells to 3D SPIM anisotropy imaging, an exemplary cell is shown in Fig. 4(a). The anisotropy in each plane was calculated according to Eq. (3) with a global factor. Since background pixels yield arbitrary anisotropy values, an intensity threshold was applied. To get the correction factor, the same sample was imaged with horizontally polarized excitation. From this factor image, calculated according to Eq. (2), we used the position of the maximum of the resulting distribution instead of the average value in order to avoid any bias introduced by intensity thresholding. The resulting anisotropy of EGFP is high (0.29 ± 0.06) and relatively homogeneously distributed; note that the errors stated correspond to the pixel standard deviation, the standard error is negligible due to the large number of total pixels. In particular, there is no difference between the cell cytoplasm and the cell nucleus, which appears to be a little brighter in the intensity image displayed in Fig. 4(b). Nevertheless, we note an anisotropy decrease towards the top of the cell as depicted in Fig. 4(c). In the second case, depolarization is caused by energy transfer between differently orientated fluorophores. Many biosensors are based on that principle [17]. As an example, Fig. 4(d-f) depicts a HeLa cell expressing dimers of EGFP. Due to homotransfer between the two chromophores of the dimer, fluorescence anisotropy is reduced (0.26 ± 0.06) compared to the cell expressing EGFP monomers [22]. Again, the average anisotropy decreases towards the top of the cell. A more interesting example is actin-GFP, a HeLa cells expressing this fusion protein is shown in Fig. 4(g-i). Monomeric actin (G-actin) does not show depolarization due to homotransfer until it is polymerized into strands (F-actin). Consequently, fluorescence anisotropy imaging can be used to visualize actin polymerization [23]. We observed that the concentration of F-actin is highest close to the cell membrane. Another example of aggregation leading to homotransfer is the EGFP-paxillin fusion protein. The paxillin protein is vital for cell adhesion, cytoskeletal reorganization and cell migration [24]. It is recruited to focal adhesions of a cell from a pool of monomeric paxillin residing in the cell cytoplasm [25,26]. Figure 4(j-l) shows the 3D projection of an EGFP-paxillin expressing CHO-K1 cell. The anisotropy is lowest close to the bottom of the cell, clearly identifying the focal adhesions as areas of highly compacted paxillin. We note that, in addition to monitoring fluorophore rotation and energy transfer, differences in fixed orientation, e.g., protein organization in a membrane [27], could also be revealed by SPIM fluorescence anisotropy imaging.
Fig. 4.

Measurements of EGFP expressing CHO-K1 cells (a-c), EGFP-EGFP expressing HeLa cells (d-f), actin-EGFP expressing HeLa cells (g-i), and EGFP-paxillin expressing CHO-K1 cells (j-l). 3D intensity projections of the anisotropy images are shown in the left column. The corresponding 3D intensity projections are displayed in the center column. The right column contains plots of the average anisotropy in each plane of the z stack, the error bars correspond to the pixel standard deviation. Anisotropy images of exemplary planes are shown as insets.
3.3 Three-dimensional time lapse anisotropy imaging
High imaging speed combined with low light exposure renders SPIM the ideal tool for 3D time lapse imaging. We took advantage of those properties to visualize paxillin aggregation in live CHO-K1 cells. A 3D series was acquired over 30 min at one minute intervals, the intensity projections of the fluorescence anisotropy are provided as Visualization 1 (1.4MB, AVI) , exemplary time points are shown in Fig. 5.
Fig. 5.

Three-dimensional time lapse anisotropy images of EGFP-paxillin expressing CHO-K1 cells (see Visualization 1 (1.4MB, AVI) ). As the cells shown in the 3D intensity projections travel along the surface, membrane protrusions are formed (arrows). At these sites, EGFP-paxillin is compacted forming new adhesions as indicated by the low anisotropy.
The high packing density of the EGFP-paxillin proteins at the adhesion sites facilitates homotransfer between chromophores resulting in a low anisotropy. Consequently, cellular adhesions present at the 2 min time point can be clearly identified in Fig. 5(a). The arrow, on the other hand, points to a part of a cell with no or little adhesions indicated, yet. Five minutes later, however, the cell has formed a fairly large membrane protrusion at the same spot, depicted in Fig. 5(b). The fluorescence anisotropy exhibited by this protrusion is reduced compared to cytoplasmic EGFP-paxillin, indicating that the protein is already forming aggregates. After another five minutes, which is the time point shown in Fig. 5(c), the protrusion has collapsed into smaller, stable adhesions. After yet another five minutes, this process is repeated on the other side of the cell, as depicted in Fig. 5(d-f).
4. Discussion
Due to higher image acquisition speed, reduced light exposure and cost-effectiveness, SPIM has started to replace laser scanning microscopy (LSM) for three-dimensional imaging applications. Therefore, it is important to transfer all the methodologies developed for the LSM platform to the SPIM platform. Anisotropy imaging is a powerful tool to study the organization, composition and dynamics of molecules in biological systems and anisotropy studies can greatly benefit from the advantages SPIM has to offer. We first verified the accuracy of our SPIM system by measuring and comparing the anisotropy of dye solutions of different water/glycerol content with spectrofluorimeter measurements. Subsequently, we visualized the aggregations state of various fusion proteins in live cells, and, finally, by 3D time lapse anisotropy imaging, we were able to show that focal adhesion assembly is preceded by the formation of membrane protrusions including the formation of paxillin aggregates. Detection of the two perpendicular polarizations was facilitated by a custom dual view. However, anisotropy imaging is also possible by sequential imaging with a rotating polarizer, which might be easier to add to a commercial setup. With the exception of TIRF microscopy, another quality unique to the SPIM configuration is to be able to excite the fluorophores with light that is polarized parallel to the detection axis. In this study, we have been looking exclusively at isotropic systems without particular molecular orientations. We excited with horizontally polarized light only to measure the factor. However, the perpendicular orientation of the detection axis with respect to the excitation axis in SPIM could allow detailed characterization of oriented systems, such as proteins in cellular membranes; multiple angles could thus be probed by rotating the polarization of the excitation. With further establishment of the SPIM technology, we believe that 3D (time lapse) anisotropy imaging will find a wide range of applications in biological/biomedical research.
Supplementary Material
Acknowledgment
This work was supported by the National Institutes of Health (NIH)-P41 GM103540 and the NIH-P50 GM076516 grants. Also, we would like to thank Milka Stakic for cell culturing.
References and links
- 1.Jameson D. M., Ross J. A., “Fluorescence polarization/anisotropy in diagnostics and imaging,” Chem. Rev. 110(5), 2685–2708 (2010). 10.1021/cr900267p [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Davidovits P., Egger M. D., “Scanning laser microscope,” Nature 223(5208), 831 (1969). 10.1038/223831a0 [DOI] [PubMed] [Google Scholar]
- 3.Denk W., Strickler J. H., Webb W. W., “Two-photon laser scanning fluorescence microscopy,” Science 248(4951), 73–76 (1990). 10.1126/science.2321027 [DOI] [PubMed] [Google Scholar]
- 4.Bigelow C. E., Conover D. L., Foster T. H., “Confocal fluorescence spectroscopy and anisotropy imaging system,” Opt. Lett. 28(9), 695–697 (2003). 10.1364/OL.28.000695 [DOI] [PubMed] [Google Scholar]
- 5.Koskinen M., Hotulainen P., “Measuring F-actin properties in dendritic spines,” Front. Neuroanat. 8(74), 74 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Axelrod D., “Total internal reflection fluorescence microscopy,” Methods Cell Biol. 30, 245–270 (1989). 10.1016/S0091-679X(08)60982-6 [DOI] [PubMed] [Google Scholar]
- 7.Bos M. A., Kleijn J. M., “Determination of the orientation distribution of adsorbed fluorophores using TIRF. I. Theory,” Biophys. J. 68(6), 2566–2572 (1995). 10.1016/S0006-3495(95)80439-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bos M. A., Kleijn J. M., “Determination of the orientation distribution of adsorbed fluorophores using TIRF. II. Measurements on porphyrin and cytochrome c,” Biophys. J. 68(6), 2573–2579 (1995). 10.1016/S0006-3495(95)80440-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burghardt T. P., Ajtai K., Chan D. K., Halstead M. F., Li J., Zheng Y., “GFP-tagged regulatory light chain monitors single myosin lever-arm orientation in a muscle fiber,” Biophys. J. 93(6), 2226–2239 (2007). 10.1529/biophysj.107.107433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Siedentopf H., Zsigmondy R., “Ueber Sichtbarmachung und Groessenbestimmung ultramikroskopischer Teilchen, mit besonderer Anwendung auf Goldrubinglaeser,” Ann. Phys. 10, 1–39 (1903). [Google Scholar]
- 11.Voie A. H., Burns D. H., Spelman F. A., “Orthogonal-plane fluorescence optical sectioning: three-dimensional imaging of macroscopic biological specimens,” J. Microsc. 170(3), 229–236 (1993). 10.1111/j.1365-2818.1993.tb03346.x [DOI] [PubMed] [Google Scholar]
- 12.Huisken J., Swoger J., Del Bene F., Wittbrodt J., Stelzer E. H. K., “Optical sectioning deep inside live embryos by selective plane illumination microscopy,” Science 305(5686), 1007–1009 (2004). 10.1126/science.1100035 [DOI] [PubMed] [Google Scholar]
- 13.Krzic U., Gunther S., Saunders T. E., Streichan S. J., Hufnagel L., “Multiview light-sheet microscope for rapid in toto imaging,” Nat. Methods 9(7), 730–733 (2012). 10.1038/nmeth.2064 [DOI] [PubMed] [Google Scholar]
- 14.Hedde P. N., Stakic M., Gratton E., “Rapid measurement of molecular transport and interaction inside living cells using single plane illumination,” Sci. Rep. 4(2), 7048 (2014). 10.1038/srep07048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fisz J. J., “Another look at magic-angle-detected fluorescence and emission anisotropy decays in fluorescence microscopy,” J. Phys. Chem. A 111(50), 12867–12870 (2007). 10.1021/jp0775855 [DOI] [PubMed] [Google Scholar]
- 16.Fisz J. J., “Fluorescence polarization spectroscopy at combined high-aperture excitation and detection: application to one-photon-excitation fluorescence microscopy,” J. Phys. Chem. A 111(35), 8606–8621 (2007). 10.1021/jp072113b [DOI] [PubMed] [Google Scholar]
- 17.Devauges V., Matthews D. R., Aluko J., Nedbal J., Levitt J. A., Poland S. P., Coban O., Weitsman G., Monypenny J., Ng T., Ameer-Beg S. M., “Steady-state acceptor fluorescence anisotropy imaging under evanescent excitation for visualisation of FRET at the plasma membrane,” PLoS One 9(10), e110695 (2014). 10.1371/journal.pone.0110695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Devauges V., Marquer C., Lécart S., Cossec J.-C., Potier M.-C., Fort E., Suhling K., Lévêque-Fort S., “Homodimerization of amyloid precursor protein at the plasma membrane: A homoFRET study by time-resolved fluorescence anisotropy imaging,” PLoS One 7(9), e44434 (2012). 10.1371/journal.pone.0044434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Edelstein A. D., Tsuchida M. A., Amodaj N., Pinkard H., Vale R. D., Stuurman N., “Advanced methods of microscope control using μManager software,” J. Biol. Methods 1(2), e10 (2014). 10.14440/jbm.2014.36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hedde P. N., Gratton E., “Active focus stabilization for upright selective plane illumination microscopy,” Opt. Express 23(11), 14707–14714 (2015). 10.1364/OE.23.014707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peterman E. J., Sosa H., Goldstein L. S., Moerner W. E., “Polarized fluorescence microscopy of individual and many kinesin motors bound to axonemal microtubules,” Biophys. J. 81(5), 2851–2863 (2001). 10.1016/S0006-3495(01)75926-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gautier I., Tramier M., Durieux C., Coppey J., Pansu R. B., Nicolas J.-C., Kemnitz K., Coppey-Moisan M., “Homo-FRET microscopy in living cells to measure monomer-dimer transition of GFP-tagged proteins,” Biophys. J. 80(6), 3000–3008 (2001). 10.1016/S0006-3495(01)76265-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vishwasrao H. D., Trifilieff P., Kandel E. R., “In vivo imaging of the actin polymerization state with two-photon fluorescence anisotropy,” Biophys. J. 102(5), 1204–1214 (2012). 10.1016/j.bpj.2012.01.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deakin N. O., Turner C. E., “Paxillin comes of age,” J. Cell Sci. 121(Pt 15), 2435–2444 (2008). 10.1242/jcs.018044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Digman M. A., Wiseman P. W., Horwitz A. R., Gratton E., “Detecting protein complexes in living cells from laser scanning confocal image sequences by the cross correlation raster image spectroscopy method,” Biophys. J. 96(2), 707–716 (2009). 10.1016/j.bpj.2008.09.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Digman M. A., Dalal R., Horwitz A. F., Gratton E., “Mapping the number of molecules and brightness in the laser scanning microscope,” Biophys. J. 94(6), 2320–2332 (2008). 10.1529/biophysj.107.114645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kampmann M., Atkinson C. E., Mattheyses A. L., Simon S. M., “Mapping the orientation of nuclear pore proteins in living cells with polarized fluorescence microscopy,” Nat. Struct. Mol. Biol. 18(6), 643–649 (2011). 10.1038/nsmb.2056 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.