

Abstract
Macroautophagy (autophagy hereafter) captures intracellular proteins and organelles and degrades them in lysosomes. The degradation breakdown products are released from lysosomes and recycled into metabolic and biosynthetic pathways. Basal autophagy provides protein and organelle quality control by eliminating damaged cellular components. Starvation-induced autophagy recycles intracellular components into metabolic pathways to sustain mitochondrial metabolic function and energy homeostasis. Recycling by autophagy is essential for yeast and mammals to survive starvation through intracellular nutrient scavenging. Autophagy suppresses degenerative diseases and has a context-dependent role in cancer. In some models, cancer initiation is suppressed by autophagy. By preventing the toxic accumulation of damaged protein and organelles, particularly mitochondria, autophagy limits oxidative stress, chronic tissue damage and oncogenic signaling, which suppresses cancer initiation. This suggests a role for autophagy stimulation in cancer prevention, although the role of autophagy in the suppression of human cancer is unclear. In contrast, some cancers induce autophagy and are dependent on autophagy for survival. Much in the way that autophagy promotes survival in starvation, cancers can use autophagy-mediated recycling to maintain mitochondrial function and energy homeostasis to meet the elevated metabolic demand of growth and proliferation. Thus autophagy inhibition may be beneficial for cancer therapy. Moreover, tumors are more autophagy-dependent than normal tissues, suggesting that there is a therapeutic window. Despite these insights, there remain many important, unanswered questions about the exact mechanisms of autophagy-mediated cancer suppression and promotion, how relevant these observations are to humans, and whether the autophagy pathway can be modulated therapeutically in cancer.
Introduction
Autophagy is intracellular lysosomal degradation and recycling of proteins and organelles. Autophagy-related genes (Atg) control the process of autophagy (1). The products of these Atg genes are regulated by nutrient (mammalian target of rapapmycin [mTOR]), energy (AMP-activated protein [AMPK]) and stress (hypoxia-inducible factors [HIFs]) sensing mechanisms in the cell that turn the pathway on and off (Figure 1). Once activated, a series of ATG protein complexes orchestrate the formation of double membrane vesicles called autophagosomes that capture cytoplasmic cargo (Figure 1). Cargo can be damaged or superfluous proteins, organelles, lipids, and glycogen that are tagged with ubiquitin and recognized by autophagy receptors such as Sequestasome1 (p62). Cargo receptors bind both cargo and the autophagosome membrane component LC3-II, facilitating cargo sequestration. Fusion between autophagosomes and lysosomes provides the hydrolases to degrade the cargo. The resulting amino acids, nucleosides, fatty acids and sugars are released into the cytoplasm for recycling (2) (Figure 1). Autophagy is essential to prevent the toxic accumulation of damaged proteins and organelles and to sustain metabolism, energy homeostasis and survival in starvation. Autophagy is also important to recycle ferritin, and autophagy defects cause perturbation of iron homeostasis that increases susceptibility to oxidative stress (3, 4).
Figure 1. Depiction of the process of autophagy.
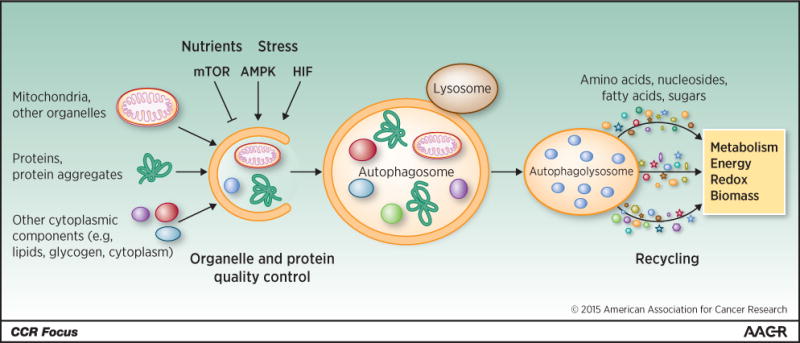
The cargo degraded by autophagy includes organelles, proteins and protein aggregates, and constituents of the cytoplasm. Autophagy is suppressed by nutrients and mTOR, and activated by stress, AMPK and HIF. Cargo is degraded when autophagosomes fuse with lysosomes that provide the hydrolytic enzymes. The breakdown products of autophagy are released into the cytoplasm where they are recycled into metabolic and biosynthetic pathways.
As autophagy activity depends of the level of activation of autophagosome initiation and the rate of cargo degradation in lysosomes, flux through the pathway is critical to assess. This typically involves blocking lysosome function and measuring the accumulation of autophagolysosomes and autophagy substrates. Alternatively, genetically engineered mouse models (GEMMs) of constitutive and conditional knockouts of essential Atg genes provide an accurate assessment of the functional requirement for autophagy. A great deal of what we know about the role of autophagy in normal tissues comes from these models, which also inform its role in cancer. This review will focus on what we know about the proposed tumor suppression and promotion roles for autophagy and how we may apply this knowledge to the treatment and prevention of human cancer.
Context-dependent role for autophagy in cancer
Autophagy has a major role in protein and organelle degradation and metabolism (2, 5). These activities influence the composition of half the proteome (6), prevent the buildup of toxic cellular waste products, maintain organelle function and host defense (7), and sustain the survival of cells and animals in the absence of nutrients (8, 9) (Figure 2). These diverse and wide-ranging cellular activities of autophagy result in a context-dependent role for autophagy in cancer.
Figure 2. Role of autophagy in starvation, tumor suppression and tumor promotion.
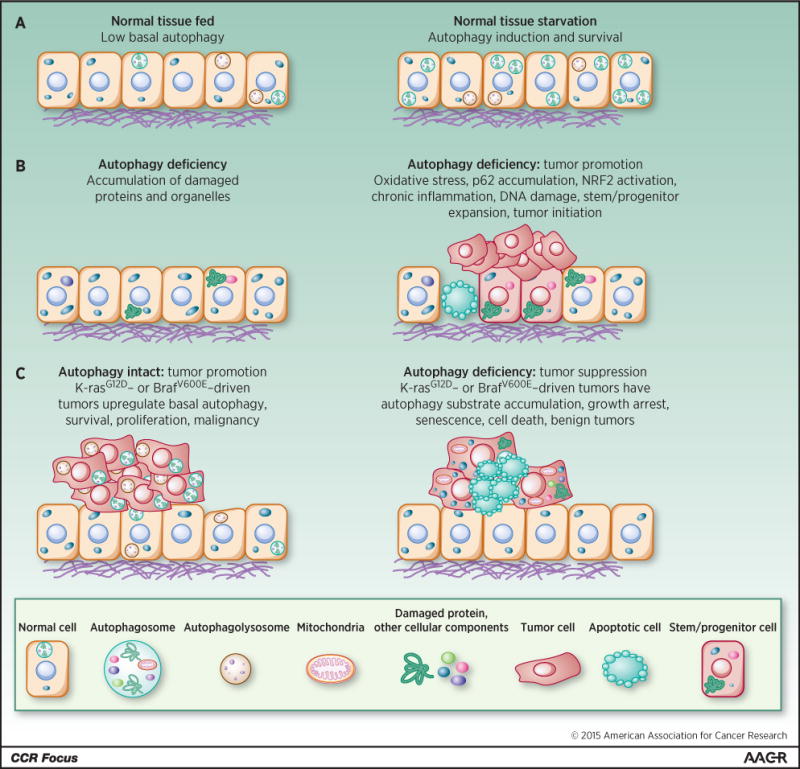
(A) Induction of autophagy-mediated survival in starvation. (B) Autophagy deficiency produces the accumulation of damaged proteins, particularly p62, and organelles. This causes cell death, tissue damage, DNA damage, oncogenic signaling, chronic inflammation, stem/progenitor expansion, and tumor initiation. (C) Autophagy is activated in tumors and promotes survival and growth, whereas loss of autophagy causes substrate accumulation, tumor cell growth arrest, senescence, and death, and restricts tumor progression to benign disease.
Mechanism of tumor suppression
Autophagy was originally thought to be a tumor suppression mechanism due to the reported allelic loss of the essential autophagy gene BECN1 (ATG6) in 40–75% of human breast, ovarian and prostate cancers (10, 11). These reports are confounded by the location of BECN1 in close proximity to the known tumor suppressor breast cancer 1, early onset (BRCA1) on chromosome 17q21. Nonetheless, autophagy suppresses tissue damage, chronic inflammation, DNA damage response and genome instability, which are known to create an environment for cancer initiation (6, 12–16). Mosaic loss of ATG5 in mice produces chronic liver damage, inflammation and the development of benign liver tumors that fail to progress to carcinoma (17). Chronic liver damage is a known tumor promoter by inducing hepatocyte death and activation of Kupffer cells and cytokine production that stimulates compensatory proliferation (16). Similar stimulation of benign tumor development with ATG gene deletion is seen in the pancreas, another tissue where chronic inflammation is tumor-promoting (18, 19). These findings suggest that autophagy may suppress liver and pancreatic tumor initiation by protein and organelle quality control that limits tissue damage (Figure 2).
Further insight into the mechanism of tumor suppression by autophagy was provided by additional GEMMs. Accumulation of the autophagy substrate p62 induced by deletion of an essential Atg gene in mouse liver is critical for tumor promotion as compound p62 deficiency suppresses this tumorigenesis (20). p62 binds to and inhibits Kelch-like ECH-associated protein 1 (KEAP1), a Nuclear factor (erythroid-derived 2)-like 2, (NRF2) inhibitor, and thereby promotes the activity of NRF2. In humans, KEAP1 is a tumor suppressor and NRF2 is an oncogene. NRF2 is a transcription factor and master regulator of the antioxidant defense response that promotes cancer cell survival. Thus, inhibition of KEAP1 by p62 accumulation in autophagy-defective tumors and the resulting NRF2 activation may drive cancer cell growth and survival. Indeed, NRF2 induction in Atg7-deficient BRAFV600E-driven lung tumors is responsible for transient stimulation of tumor growth (21).
Allelic loss of Becn1 promotes mammary tumorigenesis following parity and when oncogenic wingless-type MMTV integration site family, member 1 (WNT1) is activated in the mammary gland (22). Becn1 heterozygosity causes immature mammary epithelial stem or progenitor cell expansion, considered to be the tumor-initiating cells of WNT1-driven mammary tumorigenesis. Interestingly, deletion of Atg7 in the mammary gland does not share this property (22). This suggests that allelic loss of Becn1 increases the pool of cells susceptible to malignant transformation by WTN1 activation (Figure 2). Whether this stem cell/progenitor expansion is due to loss of an autophagy-independent function of Becn1, is not known. It is important to note that Becn1 regulates growth factor receptor signaling independent of its role in autophagy that can contribute to cancer growth (23). There also may be different outcomes with suppression rather than complete loss of autophagy. If this tumor promotion is related to KEAP1/NRF2/p62 modulation as described above is also not known. Finally, if autophagy deficiency occurs in humans and promotes cancer by these mechanisms is not known.
Autophagy genetics in human cancer
Analysis of the genomic information available on human cancers has revealed that autophagy genes are not generally mutated in human cancer, with a few exceptions. Allelic loss of BECN1 was reported in a small number of human cancer cell lines (9 out of 22) (10). Since BECN1 resides at 17q21 in close proximity (211 Kb) to the known tumor suppressor BRCA1, this analysis did not resolve whether BECN1 copy number loss was a driver or passenger mutation. Moreover, no BECN1 coding region or splice site mutations characteristic of a tumor suppressor genes were found (10). Nonetheless, this gave rise to the notion that BECN1 is a candidate tumor suppressor gene that is monoallelically deleted in 40–75% of human breast, ovarian and prostate cancers (24). In support of that concept, mice with allelic loss of BECN1 are prone to liver tumors and lymphomas and mammary hyperplasia with long latency (25, 26). However, humans with germ line copy number loss of BECN1 have not been reported, thus the mouse models deficient for one copy of BECN1 do not represent a human condition. The vast majority of hereditary breast and ovarian cancers result from germ line missense mutations in BRCA1 with cancer arising following deletion of the remaining wild type allele along with hundreds of other genes, among them often is BECN1. Thus clear proof of the tumor suppressor status of BECN1 has been lacking.
Is there evidence of selection for BECN1 loss in human cancers? More recent analysis of the mutational status of BECN1 in The Cancer Genome Atlas (TCGA) data containing 10,000 human cancers with matched normal controls demonstrated no significant occurrence of copy number losses in BECN1 independent of co-deletion of BRCA1 in any cancer (27). Since BRCA1 deletions only occur significantly in breast and ovarian cancer, there are no significant copy number losses in BECN1 in prostate cancer as originally reported. This is consistent with BRCA1 mutations being rare in this disease (27). In the TCGA data set, there is also no significant occurrence of somatic mutations in BECN1 (27). Thus copy number losses in BECN1 rarely occur independent of deletion of BRCA1, and BECN1 is not otherwise mutated in human cancers.
Independent analysis of the TCGA breast cancer data confirmed the lack of significant copy number losses in BECN1 in the absence of co-deletion of BRCA1 (28). Interestingly, genomic analysis of another large human breast cancer data set, METABRIC, found significant BECN1-only deletions. The vast majority of the TCGA breast tumors are from untreated individuals, whereas the METABRIC data set is enriched in tumors from heavily pre-treated patients (28). This suggests that primary, untreated breast cancers lack BECN1-only copy number loss, but this is increased in heavily pretreated breast cancers in METABRIC. These results may reflect mutagenesis of homologous recombination-deficient BRCA1/2 and partner and localizer of BRCA2 (PALB2) mutant breast cancers, or possibly a role for BECN1 allelic loss in response to breast cancer treatment.
Going forward it will be interesting to generate a mouse model that resembles hereditary breast and ovarian cancer bearing a germ line pathogenic BRCA1 mutation with subsequent deletion of BRCA1 with or without deletion of BECN1. If loss of both BRCA1 and BECN1 promotes cancer to a greater extent than the loss of BRCA1 alone, that would support the concept that BECN1 is a tumor suppressor and would provide an approach to identify the underlying mechanism. Alternatively, the large deletions that arise with loss of heterozygosity of BRCA1 may have no functional consequence beyond deletion of the remaining BRCA1 allele, or may harbor additional tumor suppressors that could include BECN1 or other genes. It is also intriguing to speculate that the tumor suppression function of autophagy revealed in loss-of-function mouse models is due to mutations in genes other than those used to generate the mice. Thus the autophagy pathway can be tumor suppressive in some settings but the specific genes involved are different in humans from those used to demonstrate the phenotype in mice.
Regardless of whether BECN1 is a tumor suppressor gene or not, another autophagy-related gene, PARK2 (Parkin), has been identified as a potential tumor suppressor on chromosome 6q25-q26 that is frequently deleted in human cancers (29, 30). Parkin is an E3 ubiquitin ligase that plays an important role in mitophagy (the autophagy of mitochondria) but also has autophagy-independent functions that regulate the cell cycle, potentially also explaining its role in cancer (30).
Another autophagy-related gene with a role in cancer is p62, the product of which is an autophagy cargo receptor and substrate that prominently accumulates with autophagy inhibition. In mouse models, p62 deficiency inhibits lung cancer development (31), and suppresses tumorigenesis triggered by autophagy deficiency in liver as discussed above (20) and in cell lines (32). Gain of p62 also promotes tumorigenesis (14) and the growth of FIP200-deficient tumors (32). Importantly, focal amplification of p62 on chromosome 5q occurs in renal cancer (33). P62 regulates NRF2 and also mTOR and NFκB, all of which are important in cancer signaling (34). Thus, deregulation of p62 by autophagy inhibition may be an important cancer–promoting mechanism suppressed by autophagy.
Mechanisms of tumor promotion
Autophagy promotes survival in starvation by recycling intracellular components, and this function is conserved from yeast to mammals (2). In normal, fed conditions, basal levels of autophagy are low. In starvation, autophagy is rapidly induced to high levels and the resulting degradation of intracellular components supplies substrates to support metabolism in the absence of extracellular nutrients (Figure 2). The autophagy substrates and metabolic pathways they support are not specifically known, but they might be expected to depend on the particular tissue and circumstances.
The metabolic survival-promoting function of autophagy is also activated in cancers. Some cancer cell lines have abnormally high levels of basal autophagy even under fed conditions. This suggests that oncogenic events create inherent metabolic stress necessitating autophagy activation to sustain tumor cell survival (19, 35–41). New evidence suggests that Ras-driven pancreatic cancers activate transcription programs for autophagy and lysosomal biogenesis by promoting the nuclear localization of the master regulatory MiT/TFE family (42). MiT/TFE family members are also activated by somatic mutation and other mechanisms in human cancers (43), suggesting the general importance of the autophagy-lysosome system in promoting tumor growth.
Knocking down the expression of essential autophagy genes or deleting them in cancer cell lines can reduce survival and tumorigenesis, establishing the functional importance of autophagy in tumor promotion. Autophagy is also upregulated in hypoxic tumor regions where it is required for tumor cell survival (35). Thus, both the activation of cancer pathways within tumor cells and stress in the tumor microenvironment can increase the requirement for autophagy to promote tumor growth and survival. But what is autophagy specifically doing and what are the circumstances?
Autophagy promotes tumorigenesis in GEMMs
To address the functional role of autophagy in physiological settings, essential autophagy genes were deleted in tumors of GEMMs for cancer. In these models, cancer initiation is driven by oncogene activation and/or tumor suppressor gene inactivation, which occur simultaneously with or without deletion of an Atg gene. Tumors emerge that are either wild type or deficient for autophagy while the host retains autophagy function. These models simply address the potential tumor cell-autonomous requirement for autophagy. Another characteristic of these models is that they address the role of autophagy in cancer as the tumor evolves spontaneously in its natural microenvironment in the presence of an intact immune system. This feature allows for a close preclinical approximation of human disease that is highly relevant given the emergence of new, effective immunotherapies with antitumor activity in multiple types of solid tumors (44). Other models such as xenografted human tumors and cancer cell lines grown in nude mice lack these important characteristics. This is specifically problematic given the known functional interactions between autophagy and the immune system. A limitation of these models is that autophagy deficiency is only in the tumor, so the selective role of autophagy in cancer versus normal tissue is not addressed.
Deletion of Atg7 in an autochthonous GEMM for K-rasG12D-driven lung cancer causes tumor cells to accumulate autophagy substrates, particularly defective mitochondria (37). Atg7-deficient tumors also activate the Trp53 tumor suppressor protein, and have less proliferation and more apoptosis, which likely contributes to the significant reduction in tumor burden. While Atg7 wild type tumors progress to adenocarcinomas, Atg7-deficiency instead produces benign oncocytomas, rare tumors characterized by the accumulation of defective mitochondria (37). This suggests that autophagy is required for development of aggressive cancer and that autophagy defects may be the underlying basis for the benign status of human oncocytomas. Mice bearing Atg7-deficient lung tumors die of pneumonia instead of cancer, consistent with a role for autophagy suppressing a lethal inflammatory response as seen in other settings (37). Compound deletion of Trp53 with activation of K-rasG12D in the lung alleviated some of the anti-tumor activity of Atg7 deficiency, however, tumor growth was still suppressed and limited to benign oncocytomas (37). Similar findings were obtained with deletion of Atg5 suggesting that these events are not due to an autophagy-independent function of Atg7 (45). Thus, complete autophagy ablation alters lung tumor fate from carcinomas to benign oncocytomas, activates Trp53, and suppresses tumorigenesis.
Interestingly, without Atg7, K-ras-driven, Trp53-deficient lung cancers accumulate lipid due to defective mitochondrial fatty acid oxidation (FAO). Therefore, in the absence of Trp53, K-ras-driven lung cancers require autophagy to maintain the functioning pool of mitochondria for lipid catabolism and homeostasis (37). Trp53 regulates metabolism and the metabolic changes that result from Trp53 absence in the tumors may alter the metabolic requirement for autophagy, increasing the requirement for FAO (46). Cell lines derived from these Atg7-deficient lung tumors are also more sensitive to starvation and dependent on glutamine than Atg7 wild type tumors, revealing another metabolic vulnerability (37). This suggests that autophagy not only maintains FAO but also supplies amino acids from protein degradation, particularly glutamine, to sustain mitochondrial metabolism and survival (47). It will be of great interest to map the changes in metabolism caused by defects in autophagy, and to identify the metabolic substrates produced by autophagy and the pathways they are used for to clearly define the role of autophagy in K-ras-driven lung cancer metabolism.
Are findings of autophagy-dependent cancers generalizable to tumors driven by distinct oncogenic events? To address this point, Atg7 was deleted in BrafV600E-driven lung tumor GEMM. Early in tumorigenesis, Atg7 deficiency accelerated tumor growth (see above) but later it suppressed it, generating oncocytomas and greatly extending mouse survival (48). The early acceleration of tumor growth by loss of Atg7 is due to hyperactivation of NRF2, since it is abolished by Nrf2 deficiency (48). Atg7 is similarly required for BrafV600E-driven lung tumorigenesis in the absence of Trp53 (48). Atg7-deficient tumor-derived cell lines from this model are sensitive to starvation and highly glutamine-dependent in comparison to the Atg7 wild type controls, although they do not display the lipid accumulation seen in the K-ras tumors (48). Thus lung cancers with different cancer drivers are autophagy-dependent, with BrafV600E tumors having a greater autophagy-dependence than K-rasG12D tumors. This may due to their increased respiration (21).
Is the autophagy-dependence of cancers related by the origin of their tissue type? Pancreatic cancer cell lines also have a high rate of basal autophagy and are autophagy-dependent (41), prompting functional assessment in GEMMs. In autochthonous models of pancreatic cancer driven by K-rasG12D with Trp53 intact or with the stochastic loss of heterozygosity of Trp53, genetic ablation of autophagy in the pancreas increases premalignant pancreatic intraepithelial neoplasia (PANIN) lesions (18, 19). Despite increased PANIN, these lesions are impaired for progression to invasive cancer, which increases mouse survival (18, 19). Thus autophagy suppresses PANIN but it is required for progression to aggressive cancer independent of Trp53 status, although the underlying mechanisms are unknown (49). It is interesting to note that, like liver cancer, pancreatic cancer is also triggered by chronic tissue damage and inflammation. We speculate that autophagy deficiency in mice may promote tumor initiation by the same mechanism. If this is also a mechanism of carcinogenesis in humans remains to be determined.
There is evidence that autophagy plays a role in promoting mammary tumors. Deficiency in FAK family-interacting protein of 200 kDa (FIP 200), which is part of the autophagosome initiation complex important for autophagy induction, impairs mammary tumorigenesis induced by PyMT (50). Tumor growth impairment is accompanied by induction of innate immunity genes and immune infiltration (50), as seen in other models (6, 8, 35, 37, 51). An important future goal is to determine if autophagy deficiency in tumors activates an anti-tumor immune response that contributes to suppression of tumorigenesis.
Germline mutations in BRCA1, BRCA2, and PALB2, which regulate DNA damage response, DNA repair by homologous recombination, and oxidative stress, cause hereditary breast cancer. Loss of Palb2, in the mouse mammary gland produces tumors with long latency that harbor mutations in Trp53 (52). Allelic loss of Becn1 reduces Palb2-associated mammary tumorigenesis in a Trp53-wild-type but not conditionally null genetic background (52). These findings suggest that augmentation of Trp53 activity upon allelic loss of Becn1 limits mammary cancer development (46). Consistent with the findings from lung and pancreas cancer models described above, autophagy promotes cancer by suppressing Trp53. As no impairment of mammary tumor development with allelic loss of Becn1 is observed in the absence of Trp53, this also suggests that complete rather than partial autophagy ablation may be necessary for anti-tumor activity without Trp53 as seen in the other models.
The findings that Becn1 allelic loss suppresses rather than promotes mammary tumorigenesis in a model of hereditary breast cancer are opposite to the proposed tumor suppression function of Becn1 described above. Since allelic loss of Becn1 promotes mammary tumorigenesis following parity and with oncogenic WNT1 (22), the context may influence whether mammary tumorigenesis is suppressed or stimulated.
What is the potential role of autophagy in a non-epithelial tumor type such as melanoma? Using an autochthonous GEMM for melanoma driven by conditional activation of oncogenic BrafV600E and deficiency in the Pten tumor suppressor gene in melanocytes, the functional consequence of loss of Atg7 was determined. Atg7 deficiency prevented melanoma development by BrafV600E activation and allelic Pten loss, and suppressed melanomagenesis induced by BrafV600E activation and Pten deficiency (51). Thus, autophagy is essential for melanoma development and melanomagenesis. In addition to accumulating autophagy substrates, BrafV600E-mutant, Pten- and Atg7-deficient melanomas display growth defects, and increased oxidative stress, senescence and animal survival in comparison to those that are Atg7 wild type (51). Thus, Atg7 promotes melanoma by suppressing oxidative stress and overcoming the senescence barrier, suggesting that autophagy inhibition may be of therapeutic value in melanoma (51). The autophagy-dependence of melanoma is also observed in human melanoma cell lines (40, 53) and with leucine deprivation (54). Moreover, the presence of autophagosomes potentially indicating active autophagy in patient melanomas is associated with poor outcome (38, 40). These findings suggest that melanomas are reliant on autophagy, perhaps reflecting the BrafV600E-mediated metabolic changes and the requirement for mitochondrial function (21, 48, 51, 55). Thus, deficient autophagy activates Trp53, induces growth arrest, senescence and apoptosis, and causes metabolic defects, limiting tumor progression to benign disease. The requirement for autophagy in cancers can be due to any or all of these activities, depending on the context. Autophagy has also been recently been shown to be required for the secretion of factors that promote invasion, and thus may have different cancer-promoting functions at specific stages of tumor progression (56).
Role of autophagy in adult mice
Are tumors more dependent on autophagy than normal tissues? The anti-tumor activity reported in the cancer GEMMs outlined above suggests that autophagy inhibition may provide a therapeutic advantage. Since autophagy also plays an important role in the function of some normal tissues (7), tumors would need to be even more autophagy-dependent to provide a therapeutic window. The first step to addressing this point is to switch off autophagy throughout an adult mouse similar to what would occur upon treatment with a highly selective and potent autophagy inhibitor, and determine the consequences. Indeed, constitutive Atg7 or Atg5 deficiency results in perinatal lethality by physiologic neonatal starvation, although nutrient supplementation is still insufficient to rescue survival (9, 57). It is clear from these results that newborn mice are exquisitely autophagy-dependent, but as they are physiologically very different from adult mice, what would happen upon conditional systemic autophagy deficiency in adult mice was unknown. Tissue-specific Atg knockouts indicated that autophagy would be important for some normal tissues including brain, liver, adipose and muscle (7).
In stark contrast to the perinatal lethality of mice with constitutive Atg deficiency, conditional, systemic deletion of Atg7 throughout adult mice is surprisingly well tolerated (8). While a small fraction of the mice succumb to Streptococcus infection early on, most mice remain reasonably healthy for more than a month (8). Autophagy deficiency in adult mice ultimately limits survival to no more than three months where the cause of death is predominantly neurodegeneration. Many tissues surprisingly retain function upon loss of Atg7, including lung (8), contrasting the critical requirement for autophagy for lung function in neonatal mice (58). It is clear that autophagy is far more important in the neonatal period, likely because of the unique metabolism and higher metabolic demand imposed by growth and differentiation. In adult mice with fully differentiated tissues, autophagy is substantially less important where autophagy deficiency causes selective tissue damage over time.
As switching off Atg7 in adult mice is not immediately lethal, and a conserved function of autophagy is enabling survival in starvation, what would happen if adult mice with Atg7 conditionally deleted were fasted? Mice tolerate fasting with robust activation of autophagy (59). In contrast to wild type adult mice, Atg7-ablated adult mice fail to survive fasting for 24 hours and die from hypoglycemia (8). Fasting adult mice with short-term Atg7 deficiency rapidly depletes adipose tissue and liver glycogen, and causes muscle atrophy, presumably to provide amino acid substrates for gluconeogenesis and ketogenesis (8). Degradation of muscle proteins is ultimately insufficient to sustain glucose homeostasis, and plummeting serum glucose level leads to death. Thus, like yeast, adult mice require autophagy to survive starvation. This suggests that there is a window in time where autophagy loss is tolerated with the important exception of the requirement for survival during fasting.
Selective dependence of lung cancers on autophagy
Are cancers more autophagy-dependent than most normal tissues sufficient to provide a therapeutic window for an autophagy inhibition? Moreover, what would happen if autophagy were systemically ablated in the presence of established tumors? This contrasts previous work where tumors develop in the absence of autophagy. To answer these questions, mice were engineered to have lung cancer by conditionally activating K-rasG12D and deleting Trp53, controlled independently from deletion of Atg7. Systemic ablation of Atg7 throughout adult mice is induced either before or after tumorigenesis. Deleting Atg7 does not alter the efficiency by which K-rasG12D activation and Trp53 loss initiate lung tumorigenesis (8). In stark contrast, deleting Atg7 conditionally throughout adult mice with established lung tumors (deletion occurs simultaneously in established tumors and normal tissues) produces remarkable tumor cell death and regression, far more than expected from the tumor-specific knockouts of Atg7 (8). This suggests that conditional systemic autophagy ablation has potent anti-cancer activity, that host autophagy may contribute to tumor growth, that established tumors are more autophagy-dependent than tumors with constitutive Atg7 ablation, and that there may be compensation mechanisms in tumors evolving without Atg7 (8).
Role for mitochondrial quality control and function in cancer
What does autophagy do to promote the growth and aggressiveness of these cancers? The GEMMs for cancer illustrate the requirement for autophagy in many cancers. We know that autophagy recycles intracellular substrates to support metabolism during starvation. One hypothesis is that maintaining metabolic function through the supply of autophagy substrates is especially critical for tumor growth (46). Autophagy is also essential for the removal of damaged proteins and organelles, particularly mitochondria. A second overlapping hypothesis is that tumor cells need autophagy not only to supply substrates to mitochondria, but that autophagy is also needed to preserve the functioning pool of mitochondria by removing the defective ones, and that this is critical for sustaining metabolism and survival (46). To test these hypotheses, we have begun to identify metabolic vulnerabilities in autophagy-deficient tumor cells, the limiting substrates important for their growth and survival and the metabolic pathways they support.
Atg-deficient tumors accumulate abnormal mitochondria with impaired respiration, FAO, energy homeostasis, and likely other problems (2, 37). These tumor cells are also extremely sensitive to starvation-induced cell death. Thus autophagy is essential for maintenance of the functioning pool of mitochondria in tumor cells, the loss of which limits substrate utilization and metabolic fitness. These findings also point to the general importance of mitochondrial function to tumorigenesis.
To begin to address the nature of the substrates provided by the autophagy pathway that are important for tumor growth, substrates that rescue the survival of autophagy-deficient tumor cells in starvation were identified. In both K-ras- and Braf-driven lung cancer cells, supplementation with glutamine rescues respiration and cell death in starvation (21, 37, 46, 48). This suggests that autophagy replenishes tricarboxylic (TCA) cycle intermediates to sustain metabolism by generating glutamine and other amino acids from protein and organelle degradation (47, 48). Going forward, it will be important to determine specifically what metabolic pathways and products are supported by autophagy substrates. Since distinct oncogenic events impact metabolism in different ways, autophagy may have unique roles that are dependent on the cancer genes and also the tissue type involved.
Autophagy modulation to improve cancer treatment
Which human cancers are dependent on autophagy? Those with activated Ras or B-Raf, many autophagosomes and activated MiT/TFE is a good place to start. What is the best way to inhibit autophagy for cancer therapy? There are three concepts: inhibit the lysosome and autophagy cargo degradation, inhibit essential components of the autophagy machinery, and inhibit mitochondrial respiration.
The most advanced approach to inhibit autophagy for cancer therapy is the use of hydroxychloroquine (HCQ) to disrupt lysosome function. In numerous preclinical models HCQ blocks autophagic flux and cargo degradation and suppresses tumor growth (60). Since HCQ is in use in humans for malaria prophylaxis, clinical trials testing its utility for cancer treatment are ongoing. Recent clinical findings with HCQ are encouraging (60–67). HCQ can modulate autophagy in patients if the dose is high enough and is well tolerated even in combination therapies. Although the sample sizes are too small to establish efficacy, there are a subset of patients that respond to HCQ and others with stable disease. Whether HCQ will have sufficient potency to block autophagy in human tumors to be efficacious remains to be determined. This has prompted the development of more active derivatives of HCQ such as Lys05 (68). Note that targeting lysosomes has the added advantage of not only blocking intracellular protein scavenging by autophagy, but also blocking extracellular protein scavenging by macropinocytosis (69). Macropinocytosis and lysosomal degradation of albumin (and presumably other extracellular proteins) is an important survival mechanism for K-ras-driven pancreatic cancer (69, 70).
To target the autophagy machinery directly, initial approaches have focused on inhibiting enzymes necessary for autophagosome formation. The class III phosphatylinositol 3-kinase Vps34 is critical for generating the lipid phosphatidylinositol 3-phosphate used for autophagosome membrane formation. Specific Vps34 inhibitors have been recently developed, and they can suppress the growth and increase the death of cancer cells (3, 71, 72). Another potential target is the autophagy initiating kinases ULK1 and 2, and newly developed inhibitors block autophagy in vitro (73, 74). Other targetable enzymes include ATG7, the E1-like enzyme required for the conjugation steps and autophagososme formation, and ATG4b, which processes ATG8/LC3 essential for its autophagosome membrane association. There is genetic proof of principal that loss of ATG7 suppresses tumorigenesis (5), and an inhibitor might be expected to do the same. Over-expression of a dominant-negative ATG4b can either increase toxicity or promote resistance to cytotoxic drugs, so the situation where an inhibitor would be useful requires further investigation (75).
The dependence of cancers on autophagy and exploration of the underlying mechanism suggests that maintenance of mitochondrial function is important for cancer (5). Indeed, impairment of mitochondrial function by genetic inactivation of the essential mitochondrial transcription factor TFAM greatly suppresses growth of K-ras-driven lung cancers (76). Depletion of mitochondrial DNA also diminishes tumorigenesis and metastasis (77). Small molecule inhibitors of mitochondrial respiration suppress tumor growth, with growth only being restored upon recovery of normal mitochondrial function (78). Benign human oncocytomas may owe their non-aggressive status to their accumulation of mutations in the mitochondrial genome that inactivate respiration (79). Moreover many mitochondrial enzymes such as SHMT2 are upregulated in some cancers (80), and SHMT2 promotes glioma survival in hypoxia (81). Indeed, the diabetes drug and complex I poison, the biguanide metformin, has anti-cancer activity in the diabetic subpopulation (82). Tumors with increased glucose dependence due to mitochondrial mutations are more sensitive to biguanides (83). Defects in autophagy and the resulting inhibition of removal of defective mitochondria may act similarly to suppress tumorigenesis by allowing the functional deterioration of the pool of mitochondria. Collectively these findings suggest that targeting respiration or global function of mitochondria may be a new Achilles heel of cancer (Figure 3). Note that mitochondria regulate apoptosis, the loss of which may hinder cancer therapy, which is discussed elsewhere in this CCR Focus section (84, 85). Mitochondrial TCA cycle function is also important to control levels of metabolic intermediates, and their accumulation due to inactivating mutations in succinate dehydrogenase complex components (SDHA, SDHB, SDHC, SDHD, and SDHAF2), or isocitrate dehydrogenase or fumarate hydratase causes aberrant regulation of dioxygenases, producing epigenetic changes, HIF activation and cancer (86). Thus, targeting mitochondrial respiration, not the TCA cycle, may be the best approach for cancer therapy (Figure 3).
Figure 3. Role of mitochondria in tumorigenesis.
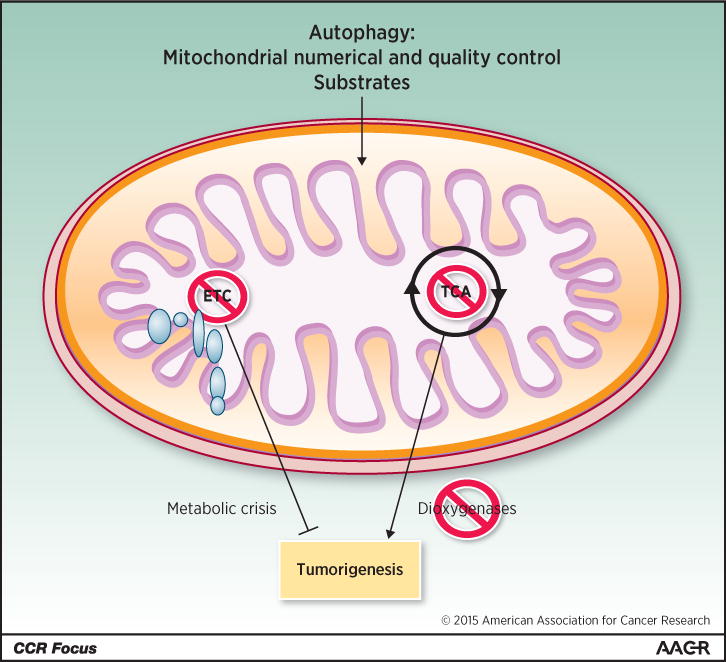
Autophagy supplies substrates for mitochondrial metabolic function. Inhibition of TCA cycle metabolism can alter the production of metabolites that, among other things, can alter regulation of dioxygenases, resulting in epigenetic changes that can promote tumorigenesis. In contrast, inhibition of mitochondrial respiratory function and the ETC might be selectively detrimental to tumorigenesis.
Future
Given that cancer represents approximately 200 different diseases, and autophagy has a global role in metabolism and protein and organelle quality control, it is not surprising that definition of the role of autophagy in cancer is still evolving. Autophagy stimulation may prevent disease, and although pharmacologic means to stimulate autophagy may have value, this can be accomplished with periodic fasting. The potential effectiveness of this is testable.
Autophagy inhibition can compromise tumorigenesis, but the underlying mechanisms require definition. Highly selective and potent autophagy inhibitors are becoming available, but how to identify the patients most likely to respond is not yet clear. The means to measure autophagy activity in human tumors would be useful. Optimal drug combinations are only beginning to emerge, with proteasome and BRAF inhibitor combinations with autophagy inactivation showing promise (35, 51, 55, 61, 66). Mechanisms of resistance to autophagy inhibition are unknown, although some human cancer cell lines are indifferent to ATG loss indicating the existence of compensation mechanisms (36, 41, 87). Other points to consider moving forward are the consequence of autophagy inhibition to the anti-tumor immune response and the ability of tumors versus normal tissue to recover from autophagy inactivation.
Acknowledgments
Grant Support: E. White is supported by the NIH under award numbers R01CA163591, R01CA130893, and R01 CA188096; the Val Skinner Foundation; and the Rutgers Cancer Institute of New Jersey (P30CA072720).
Footnotes
Disclosure of Potential Conflicts of Interest: E. White is a consultant/advisory board member for FORMA Therapeutics. No potential conflicts of interest were disclosed by the other authors.
References
- 1.Mizushima N, Yoshimori T, Ohsumi Y. The role of atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:107–32. doi: 10.1146/annurev-cellbio-092910-154005. [DOI] [PubMed] [Google Scholar]
- 2.Rabinowitz JD, White E. Autophagy and metabolism. Science. 2010;330:1344–8. doi: 10.1126/science.1193497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dowdle WE, Nyfeler B, Nagel J, Elling RA, Liu S, Triantafellow E, et al. Selective vps34 inhibitor blocks autophagy and uncovers a role for ncoa4 in ferritin degradation and iron homeostasis in vivo. Nat Cell Biol. 2014;16:1069–79. doi: 10.1038/ncb3053. [DOI] [PubMed] [Google Scholar]
- 4.Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC. Quantitative proteomics identifies ncoa4 as the cargo receptor mediating ferritinophagy. Nature. 2014;509:105–9. doi: 10.1038/nature13148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.White E. The role for autophagy in cancer. J Clin Invest. 2015;125:42–6. doi: 10.1172/JCI73941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mathew R, Khor S, Hackett SR, Rabinowitz JD, Perlman DH, White E. Functional role of autophagy-mediated proteome remodeling in cell survival signaling and innate immunity. Mol Cell. 2014;55:916–30. doi: 10.1016/j.molcel.2014.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–41. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 8.Karsli-Uzunbas G, Guo JY, Price S, Teng X, Laddha SV, Khor S, et al. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov. 2014;4:914–27. doi: 10.1158/2159-8290.CD-14-0363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, et al. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–6. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- 10.Aita VM, Liang XH, Murty VV, Pincus DL, Yu W, Cayanis E, et al. Cloning and genomic organization of beclin 1, a candidate tumor suppressor gene on chromosome 17q21. Genomics. 1999;59:59–65. doi: 10.1006/geno.1999.5851. [DOI] [PubMed] [Google Scholar]
- 11.Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–6. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- 12.Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013;13:722–37. doi: 10.1038/nri3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Karantza-Wadsworth V, Patel S, Kravchuk O, Chen G, Mathew R, Jin S, et al. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev. 2007;21:1621–35. doi: 10.1101/gad.1565707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137:1062–75. doi: 10.1016/j.cell.2009.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mathew R, Kongara S, Beaudoin B, Karp CM, Bray K, Degenhardt K, et al. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 2007;21:1367–81. doi: 10.1101/gad.1545107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer. 2012;12:401–10. doi: 10.1038/nrc3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, et al. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011;25:795–800. doi: 10.1101/gad.2016211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rosenfeldt MT, O’Prey J, Morton JP, Nixon C, MacKay G, Mrowinska A, et al. P53 status determines the role of autophagy in pancreatic tumour development. Nature. 2013;504:296–300. doi: 10.1038/nature12865. [DOI] [PubMed] [Google Scholar]
- 19.Yang A, Rajeshkumar NV, Wang X, Yabuuchi S, Alexander BM, Chu GC, et al. Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Discov. 2014;4:905–13. doi: 10.1158/2159-8290.CD-14-0362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131:1149–63. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- 21.Strohecker AM, White E. Targeting mitochondrial metabolism by inhibiting autophagy in braf-driven cancers. Cancer Discov. 2014;4:766–72. doi: 10.1158/2159-8290.CD-14-0196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cicchini M, Chakrabarti R, Kongara S, Price S, Nahar R, Lozy F, et al. Autophagy regulator becn1 suppresses mammary tumorigenesis driven by wnt1 activation and following parity. Autophagy. 2014;10:2036–52. doi: 10.4161/auto.34398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rohatgi RA, Janusis J, Leonard D, Bellve KD, Fogarty KE, Baehrecke EH, et al. Beclin 1 regulates growth factor receptor signaling in breast cancer. Oncogene. 2015 Feb 2; doi: 10.1038/onc.2014.454. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368:651–62. doi: 10.1056/NEJMra1205406. [DOI] [PubMed] [Google Scholar]
- 25.Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112:1809–20. doi: 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A. 2003;100:15077–82. doi: 10.1073/pnas.2436255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Laddha SV, Ganesan S, Chan CS, White E. Mutational landscape of the essential autophagy gene becn1 in human cancers. Mol Cancer Res. 2014;12:485–90. doi: 10.1158/1541-7786.MCR-13-0614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tang H, Sebti S, Titone R, Zhou Y, Isidoro C, Ross TS, et al. Decreased mrna expression in human breast cancer is associated with estrogen receptor-negative subtypes and poor prognosis. EBioMedicine. 2015;2:255–63. doi: 10.1016/j.ebiom.2015.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cesari R, Martin ES, Calin GA, Pentimalli F, Bichi R, McAdams H, et al. Parkin, a gene implicated in autosomal recessive juvenile parkinsonism, is a candidate tumor suppressor gene on chromosome 6q25-q27. Proc Natl Acad Sci U S A. 2003;100:5956–61. doi: 10.1073/pnas.0931262100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gong Y, Zack TI, Morris LG, Lin K, Hukkelhoven E, Raheja R, et al. Pan-cancer genetic analysis identifies park2 as a master regulator of g1/s cyclins. Nat Genet. 2014;46:588–94. doi: 10.1038/ng.2981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Duran A, Linares JF, Galvez AS, Wikenheiser K, Flores JM, Diaz-Meco MT, et al. The signaling adaptor p62 is an important nf-kappab mediator in tumorigenesis. Cancer Cell. 2008;13:343–54. doi: 10.1016/j.ccr.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 32.Wei H, Wang C, Croce CM, Guan JL. P62/sqstm1 synergizes with autophagy for tumor growth in vivo. Genes Dev. 2014;28:1204–16. doi: 10.1101/gad.237354.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li L, Shen C, Nakamura E, Ando K, Signoretti S, Beroukhim R, et al. Sqstm1 is a pathogenic target of 5q copy number gains in kidney cancer. Cancer Cell. 2013;24:738–50. doi: 10.1016/j.ccr.2013.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moscat J, Diaz-Meco MT. P62: A versatile multitasker takes on cancer. Trends Biochem Sci. 2012;37:230–6. doi: 10.1016/j.tibs.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10:51–64. doi: 10.1016/j.ccr.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, et al. Activated ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011;25:460–70. doi: 10.1101/gad.2016311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guo JY, Karsli-Uzunbas G, Mathew R, Aisner SC, Kamphorst JJ, Strohecker AM, et al. Autophagy suppresses progression of k-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev. 2013;27:1447–61. doi: 10.1101/gad.219642.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lazova R, Camp RL, Klump V, Siddiqui SF, Amaravadi RK, Pawelek JM. Punctate lc3b expression is a common feature of solid tumors and associated with proliferation, metastasis, and poor outcome. Clin Cancer Res. 2012;18:370–9. doi: 10.1158/1078-0432.CCR-11-1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lock R, Roy S, Kenific CM, Su JS, Salas E, Ronen SM, et al. Autophagy facilitates glycolysis during ras-mediated oncogenic transformation. Mol Biol Cell. 2011;22:165–78. doi: 10.1091/mbc.E10-06-0500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ma XH, Piao S, Wang D, McAfee QW, Nathanson KL, Lum JJ, et al. Measurements of tumor cell autophagy predict invasiveness, resistance to chemotherapy, and survival in melanoma. Clin Cancer Res. 2011;17:3478–89. doi: 10.1158/1078-0432.CCR-10-2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011;25:717–29. doi: 10.1101/gad.2016111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Perera RM, Stoykova S, Nicolay BN, Ross KN, Fitamant J, Boukhali M, et al. Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature. 2015;524:361–5. doi: 10.1038/nature14587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ferguson SM. Beyond indigestion: Emerging roles for lysosome-based signaling in human disease. Curr Opin Cell Biol. 2015;35:59–68. doi: 10.1016/j.ceb.2015.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Drake CG, Lipson EJ, Brahmer JR. Breathing new life into immunotherapy: Review of melanoma, lung and kidney cancer. Nat Rev Clin Oncol. 2014;11:24–37. doi: 10.1038/nrclinonc.2013.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rao S, Tortola L, Perlot T, Wirnsberger G, Novatchkova M, Nitsch R, et al. A dual role for autophagy in a murine model of lung cancer. Nat Commun. 2014;5:3056. doi: 10.1038/ncomms4056. [DOI] [PubMed] [Google Scholar]
- 46.Guo JY, Xia B, White E. Autophagy-mediated tumor promotion. Cell. 2013;155:1216–9. doi: 10.1016/j.cell.2013.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guo JY, White E. Autophagy is required for mitochondrial function, lipid metabolism, growth, and fate of kras(g12d)-driven lung tumors. Autophagy. 2013;9:1636–8. doi: 10.4161/auto.26123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Strohecker AM, White E. Autophagy promotes brafv600e-driven lung tumorigenesis by preserving mitochondrial metabolism. Autophagy. 2014;10:384–5. doi: 10.4161/auto.27320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Amaravadi R, Debnath J. Mouse models address key concerns regarding autophagy inhibition in cancer therapy. Cancer Discov. 2014;4:873–5. doi: 10.1158/2159-8290.CD-14-0618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wei H, Wei S, Gan B, Peng X, Zou W, Guan JL. Suppression of autophagy by fip200 deletion inhibits mammary tumorigenesis. Genes Dev. 2011;25:1510–27. doi: 10.1101/gad.2051011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xie X, Koh JY, Price S, White E, Mehnert JM. Atg7 overcomes senescence and promotes growth of brafv600e-driven melanoma. Cancer Discov. 2015;5:410–23. doi: 10.1158/2159-8290.CD-14-1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huo Y, Cai H, Teplova I, Bowman-Colin C, Chen G, Price S, et al. Autophagy opposes p53-mediated tumor barrier to facilitate tumorigenesis in a model of palb2-associated hereditary breast cancer. Cancer Discov. 2013;3:894–907. doi: 10.1158/2159-8290.CD-13-0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xie X, White EP, Mehnert JM. Coordinate autophagy and mtor pathway inhibition enhances cell death in melanoma. PLoS One. 2013;8:e55096. doi: 10.1371/journal.pone.0055096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sheen JH, Zoncu R, Kim D, Sabatini DM. Defective regulation of autophagy upon leucine deprivation reveals a targetable liability of human melanoma cells in vitro and in vivo. Cancer Cell. 2011;19:613–28. doi: 10.1016/j.ccr.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ma XH, Piao SF, Dey S, McAfee Q, Karakousis G, Villanueva J, et al. Targeting er stress-induced autophagy overcomes braf inhibitor resistance in melanoma. J Clin Invest. 2014;124:1406–17. doi: 10.1172/JCI70454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lock R, Kenific CM, Leidal AM, Salas E, Debnath J. Autophagy-dependent production of secreted factors facilitates oncogenic ras-driven invasion. Cancer Discov. 2014;4:466–79. doi: 10.1158/2159-8290.CD-13-0841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, et al. Impairment of starvation-induced and constitutive autophagy in atg7-deficient mice. J Cell Biol. 2005;169:425–34. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cheong H, Wu J, Gonzales LK, Guttentag SH, Thompson CB, Lindsten T. Analysis of a lung defect in autophagy-deficient mouse strains. Autophagy. 2014;10:45–56. doi: 10.4161/auto.26505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15:1101–11. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rebecca VW, Amaravadi RK. Emerging strategies to effectively target autophagy in cancer. Oncogene. 2015 Apr 20; doi: 10.1038/onc.2015.99. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Levy JM, Thompson JC, Griesinger AM, Amani V, Donson AM, Birks DK, et al. Autophagy inhibition improves chemosensitivity in braf(v600e) brain tumors. Cancer Discov. 2014;4:773–80. doi: 10.1158/2159-8290.CD-14-0049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mahalingam D, Mita M, Sarantopoulos J, Wood L, Amaravadi RK, Davis LE, et al. Combined autophagy and hdac inhibition: a phase i safety, tolerability, pharmacokinetic, and pharmacodynamic analysis of hydroxychloroquine in combination with the hdac inhibitor vorinostat in patients with advanced solid tumors. Autophagy. 2014;10:1403–14. doi: 10.4161/auto.29231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rangwala R, Chang YC, Hu J, Algazy KM, Evans TL, Fecher LA, et al. Combined mtor and autophagy inhibition: Phase i trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma. Autophagy. 2014;10:1391–402. doi: 10.4161/auto.29119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rangwala R, Leone R, Chang YC, Fecher LA, Schuchter LM, Kramer A, et al. Phase i trial of hydroxychloroquine with dose-intense temozolomide in patients with advanced solid tumors and melanoma. Autophagy. 2014;10:1369–79. doi: 10.4161/auto.29118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rosenfeld MR, Ye X, Supko JG, Desideri S, Grossman SA, Brem S, et al. A phase i/ii trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy. 2014;10:1359–68. doi: 10.4161/auto.28984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vogl DT, Stadtmauer EA, Tan KS, Heitjan DF, Davis LE, Pontiggia L, et al. Combined autophagy and proteasome inhibition: a phase 1 trial of hydroxychloroquine and bortezomib in patients with relapsed/refractory myeloma. Autophagy. 2014;10:1380–90. doi: 10.4161/auto.29264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rebecca VW, Massaro RR, Fedorenko IV, Sondak VK, Anderson AR, Kim E, et al. Inhibition of autophagy enhances the effects of the akt inhibitor mk-2206 when combined with paclitaxel and carboplatin in braf wild-type melanoma. Pigment Cell Melanoma Res. 2014;27:465–78. doi: 10.1111/pcmr.12227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McAfee Q, Zhang Z, Samanta A, Levi SM, Ma XH, Piao S, et al. Autophagy inhibitor lys05 has single-agent antitumor activity and reproduces the phenotype of a genetic autophagy deficiency. Proc Natl Acad Sci U S A. 2012;109:8253–8. doi: 10.1073/pnas.1118193109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.White E. Exploiting the bad eating habits of ras-driven cancers. Genes Dev. 2013;27:2065–71. doi: 10.1101/gad.228122.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S, et al. Macropinocytosis of protein is an amino acid supply route in ras-transformed cells. Nature. 2013;497:633–7. doi: 10.1038/nature12138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pasquier B, El-Ahmad Y, Filoche-Romme B, Dureuil C, Fassy F, Abecassis PY, et al. Discovery of (2s)-8-[(3r)-3-methylmorpholin-4-yl]-1-(3-methyl-2-oxobutyl)-2-(trifluoromethyl)- 3,4-dihydro-2h-pyrimido[1,2-a]pyrimidin-6-one: a novel potent and selective inhibitor of vps34 for the treatment of solid tumors. J Med Chem. 2015;58:376–400. doi: 10.1021/jm5013352. [DOI] [PubMed] [Google Scholar]
- 72.Ronan B, Flamand O, Vescovi L, Dureuil C, Durand L, Fassy F, et al. A highly potent and selective vps34 inhibitor alters vesicle trafficking and autophagy. Nat Chem Biol. 2014;10:1013–9. doi: 10.1038/nchembio.1681. [DOI] [PubMed] [Google Scholar]
- 73.Petherick KJ, Conway OJ, Mpamhanga C, Osborne SA, Kamal A, Saxty B, et al. Pharmacological inhibition of ulk1 kinase blocks mammalian target of rapamycin (mtor)-dependent autophagy. J Biol Chem. 2015;290:11376–83. doi: 10.1074/jbc.C114.627778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Egan DF, Chun MG, Vamos M, Zou H, Rong J, Miller CJ, et al. Small molecule inhibition of the autophagy kinase ulk1 and identification of ulk1 substrates. Mol Cell. 2015;59:285–97. doi: 10.1016/j.molcel.2015.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tran E, Chow A, Goda T, Wong A, Blakely K, Rocha M, et al. Context-dependent role of atg4b as target for autophagy inhibition in prostate cancer therapy. Biochem Biophys Res Commun. 2013;441:726–31. doi: 10.1016/j.bbrc.2013.10.117. [DOI] [PubMed] [Google Scholar]
- 76.Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, et al. Mitochondrial metabolism and ros generation are essential for kras-mediated tumorigenicity. Proc Natl Acad Sci U S A. 2010;107:8788–93. doi: 10.1073/pnas.1003428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tan AS, Baty JW, Dong LF, Bezawork-Geleta A, Endaya B, Goodwin J, et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial. DNA Cell Metab. 2015;21:81–94. doi: 10.1016/j.cmet.2014.12.003. [DOI] [PubMed] [Google Scholar]
- 78.Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sanchez N, Marchesini M, et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature. 2014;514:628–32. doi: 10.1038/nature13611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gasparre G, Romeo G, Rugolo M, Porcelli AM. Learning from oncocytic tumors: why choose inefficient mitochondria? Biochim Biophys Acta. 2011;1807:633–42. doi: 10.1016/j.bbabio.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 80.Hu J, Locasale JW, Bielas JH, O’Sullivan J, Sheahan K, Cantley LC, et al. Heterogeneity of tumor-induced gene expression changes in the human metabolic network. Nat Biotechnol. 2013;31:522–9. doi: 10.1038/nbt.2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kim D, Fiske BP, Birsoy K, Freinkman E, Kami K, Possemato RL, et al. Shmt2 drives glioma cell survival in ischaemia but imposes a dependence on glycine clearance. Nature. 2015;520:363–7. doi: 10.1038/nature14363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pollak M. Potential applications for biguanides in oncology. J Clin Invest. 2013;123:3693–700. doi: 10.1172/JCI67232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Birsoy K, Possemato R, Lorbeer FK, Bayraktar EC, Thiru P, Yucel B, et al. Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature. 2014;508:108–12. doi: 10.1038/nature13110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fulda S. Promises and challenges of smac mimetics as cancer therapeutics. Clin Cancer Res. 2015;21:xxxx–xxxx. doi: 10.1158/1078-0432.CCR-15-0365. [DOI] [PubMed] [Google Scholar]
- 85.Gibson CJ, Davids MS. Bcl-2 antagonism to target the intrinsic mitochondrial pathway of apoptosis. Clin Cancer Res. 2015;21:xxxx–xxxx. doi: 10.1158/1078-0432.CCR-15-0364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gaude E, Frezza C. Defects in mitochondrial metabolism and cancer. Cancer Metab. 2014;2:10. doi: 10.1186/2049-3002-2-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mandelbaum J, Rollins N, Shah P, Bowman D, Lee JY, Tayber O, et al. Identification of a lung cancer cell line deficient in atg7-dependent autophagy. Autophagy. 2015 Jun 19; doi: 10.1080/15548627.2015.1056966. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]