

Genome-environment associations and phenotypic analyses may reveal the basis of environmental adaptation.
Keywords: plant breeding, plasticity, quantitative trait loci, genotyping-by-sequencing, domestication, genomic selection, water stress, local adaptation, genome scan, climate
Abstract
Improving environmental adaptation in crops is essential for food security under global change, but phenotyping adaptive traits remains a major bottleneck. If associations between single-nucleotide polymorphism (SNP) alleles and environment of origin in crop landraces reflect adaptation, then these could be used to predict phenotypic variation for adaptive traits. We tested this proposition in the global food crop Sorghum bicolor, characterizing 1943 georeferenced landraces at 404,627 SNPs and quantifying allelic associations with bioclimatic and soil gradients. Environment explained a substantial portion of SNP variation, independent of geographical distance, and genic SNPs were enriched for environmental associations. Further, environment-associated SNPs predicted genotype-by-environment interactions under experimental drought stress and aluminum toxicity. Our results suggest that genomic signatures of environmental adaptation may be useful for crop improvement, enhancing germplasm identification and marker-assisted selection. Together, genome-environment associations and phenotypic analyses may reveal the basis of environmental adaptation.
INTRODUCTION
Many of the world’s staple crop species harbor great genetic diversity, having diffused to diverse environments and adapted locally under human and natural selection (1–3). The resulting diversity of crop landraces (that is, traditional varieties) is crucial to global food security, particularly for smallholder agriculture in Africa and Asia, where increasing temperature and more erratic precipitation due to climate change are major threats (4). Crop landraces originating from stressful environments are a major source of adaptive traits for crop improvement efforts (5, 6) and are often targeted at the initial steps of molecular breeding (7–10). Genomics-assisted breeding tools are rapidly improving (11), but accurate phenotyping in relevant field conditions remains a major limitation (12), particularly in the highly variable, low-fertility, and low-input conditions that are common in smallholder production (13). Thus, new strategies for germplasm screening and crop improvement are needed, especially for orphan food crops, which are less likely to see large-scale phenotyping efforts (14).
Recent genomic studies in wild populations have demonstrated that genome-environment associations [that is, associations between single-nucleotide polymorphism (SNP) alleles and accessions’ environment of origin] can be used to identify adaptive loci (15, 16) and predict phenotypic variation (17, 18). However, genome-environment associations in crop landraces have been largely uncharacterized and have not been exploited for crop improvement. Breeders have identified potential stress-adapted landraces based on their environment of origin (known as ecogeographic strategies), but this approach is limited to georeferenced germplasm and then only for a single generation before crossing. If genome-environment associations reflect local adaptation, then these associations could be used to predict genetic differences in fitness and yield across particular environments, that is, genotype-by-environment interactions (G×E). Thus, genome-environment associations may be useful for dissecting the genomic basis of crop adaptation and enhancing marker-assisted crop improvement.
The C4 cereal crop sorghum (Sorghum bicolor [L.] Moench) provides an excellent system to investigate local adaptation to environment, having diffused to diverse agroecological zones in Africa and Asia hundreds to thousands of years ago (19). Sorghum is a staple crop for about 500 million people and vital to smallholder farmer livelihoods in many areas that are too stressful for other major cereal crops (20). Here, we leverage environmental, genomic, and phenotypic data in a diverse panel of sorghum to characterize the genomic basis of local adaptation and genotype-by-environment interactions in response to abiotic stress.
RESULTS
Genotyping-by-sequencing provides a worldwide map of genome-environment associations in sorghum
Taking advantage of diversity found in global ex situ germplasm collections, we studied a diverse panel of 1943 georeferenced landrace accessions chosen to maximize geographic coverage. This panel includes 55 countries and all 12 of the tropical and temperate global agroecological zones (Fig. 1 and table S1) (21). In this large panel, we have high-density sampling of regional subsets of botanical races, allowing us to study regional variation in genomic associations (fig. S1). We used data on precipitation, temperature, soil, and length of growing season to characterize the environmental conditions and growing season length for each landrace in its site of origin (Supplementary Materials, section 1.2). These landraces originate from both temperate and tropical regions, and wet and dry climates. Many of these locations have short estimated growing seasons (<3 months), whereas many have favorable climate year round (Fig. 1). Estimated growing season was largely limited by precipitation in our panel, although in temperate locations, temperature also imposed limits.
Fig. 1. Diversity of sorghum landraces and environments.

(A to C) Map of georeferenced sorghum landraces genotyped in this study (A), with neighbor-joining tree (B and C) of landraces based on genome-wide SNP distance. Several landraces fall outside the plotted map region and are not shown. The monthly climates of four landraces representing diverse agroecological zones are shown in sets (i to iv; gray lines, temperature; black lines, precipitation) in addition to our estimate of each landrace’s local rainfed growing season length (brown to green colors). At right, the neighbor-joining trees show estimated growing season length (B) and botanical race classifications (C) of landraces, demonstrating variation in climate among related accessions.
We characterized genomic variation for these accessions at 404,627 SNPs using genotyping-by-sequencing (22, 23) and quantified allelic associations with environmental variables. The resulting data set was dominated by uncommon SNPs, with 23% having minor allele frequency (MAF) <0.01 and 65% of SNPs having MAF <0.1. We observed that many accessions that are similar based on genome-wide SNPs are often divergent in important environmental conditions, such as growing season length (Fig. 1B). This suggests that genome-environment associations may have power to detect loci underlying local adaptation, because environment is not completely confounded with genome-wide similarity.
Genome-environment associations reflect local adaptation at Maturity1 and Tannin1
To assess whether genome-environment associations can be used to reveal evidence of local adaptation at a gene level, we first investigated two cloned natural variants in the Maturity1 and Tannin1 genes that were tagged in our SNP data. These variants control two of the most important agroclimatic traits in this crop: photoperiod sensitivity and grain tannins (24). The first variant, in Maturity1, controls photoperiod-sensitive flowering, a trait that is common in tropical latitudes but prevents flowering in temperate latitudes, and has thus been selected against in higher latitudes (25). Ma1 functionality (null allele comprising 8% of allele copies among landraces) was most strongly associated with the minimum temperature of the coldest month (Spearman’s ρ = 0.24, P < 10−16), with the null allele (photoperiod-insensitive) being most common in cold locations, as predicted on the basis of its adaptation to temperate latitudes.
The second variant, in Tannin1, controls the presence of tannin in the grain testa (26) (the ancestral state). Tannins provide grain mold resistance (27) and reduced preharvest sprouting (28) under humid conditions, but are selected against where possible because of their astringency (24). We found that the tan1a SNP (29) (a null allele comprising 20% of allele copies) was strongly correlated with a number of bioclimatic gradients (table S4). Foremost of these was mean temperature of the warmest quarter (Spearman’s ρ = −0.28, P < 10−16), where the derived, null allele is nearly as common as the functional (ancestral) allele for temperatures >32°C. These findings suggest that the allelic distribution at Tannin1 is shaped by geographical gradients in human and natural selection. In cool, wet climates that favor mold and preharvest sprouting, natural selection may favor tannins. However, in hot, dry climates, the null allele is conditionally neutral with respect to natural selection (30), so human selection against tannins may dominate.
After accounting for genome-wide similarity among accessions using the mixed-model EMMA (31), associations between the SNPs at Tannin1 and Maturity1 and climate variables became much weaker and mostly nonsignificant (table S5), consistent with the strong relationship between kinship and allelic state at Tannin1 (Spearman’s ρ for the first three eigenvectors of kinship matrix = −0.13, 0.55, and 0.24) and Maturity1 (Spearman’s ρ for the first three eigenvectors of kinship matrix = −0.12, −0.11, and −0.32). These results highlight that controlling for population structure may limit power to detect true environmentally adaptive polymorphisms that are collinear with population structure (32).
If local adaptation to similar conditions in different parts of a species’ range occurs via convergent adaptation that generates similar phenotypes, then global genetic-environment associations may have little power to identify causal loci. Additionally, the confounding of population structure with environmental gradients might be reduced through a regional association approach. Thus, we also tested climate associations at Maturity1 and Tannin1 within each of four geographic regions where we had dense sampling of landraces: West Africa, East Africa, southern Africa, and South Asia. We found that within South Asia, where the null Tannin1 allele is most common, variation at Tannin1 was significantly and most strongly associated with growing season length, when accounting for population structure (EMMA, P = 0.0040, Fig. 2). The strongest associations for Maturity1 were found in southern Africa, where absolute latitude showed the strongest correlation (Spearman’s rank correlation, P < 10−16, Fig. 2). However, this was not significant after accounting for population structure (EMMA, P = 0.3831), which is expected for a causal allele that is correlated with population structure (32).
Fig. 2. Distribution of Tannin1 (top panels) and Maturity1 (bottom panels) alleles (represented by red/purple/blue) across space and along environmental gradients.

Gray boxes indicate regional subsets where the locus showed the strongest association with environment: Tannin1 was most strongly associated with growing season length in South Asia, and Maturity1 was most strongly associated with absolute latitude in southern Africa. The T allele at position 61667908 on chromosome 4 corresponds to the null Tannin1 allele, whereas the T allele at position 40286721 on chromosome 6 corresponds to the null Maturity1 allele.
A substantial proportion of genomic variation is shaped by environment
Next, we considered genome-wide allelic variation. Local adaptation to environment can generate correlations between environmental variables and allelic state (33) because of local selection for new favored variants or against deleterious variants. Here, we quantified these correlations using redundancy analysis (RDA), a form of eigenanalysis (34) that explains linear combinations of SNPs using linear combinations of geographic variables, such that SNP variation explained is maximized (35–37) (Fig. 3A). Overall, we found that 31% of total SNP variation among accessions can be explained by environmental variation (fig. S6), but that much of the environmentally correlated SNP variation was collinear with geographic spatial structure (23% of the total SNP variation). Thus, it is unclear how much environmentally structured SNP variation in sorghum is due to local adaptation versus dispersal limitation and isolation-by-distance.
Fig. 3. Genome-wide, multivariate SNP-environment associations.
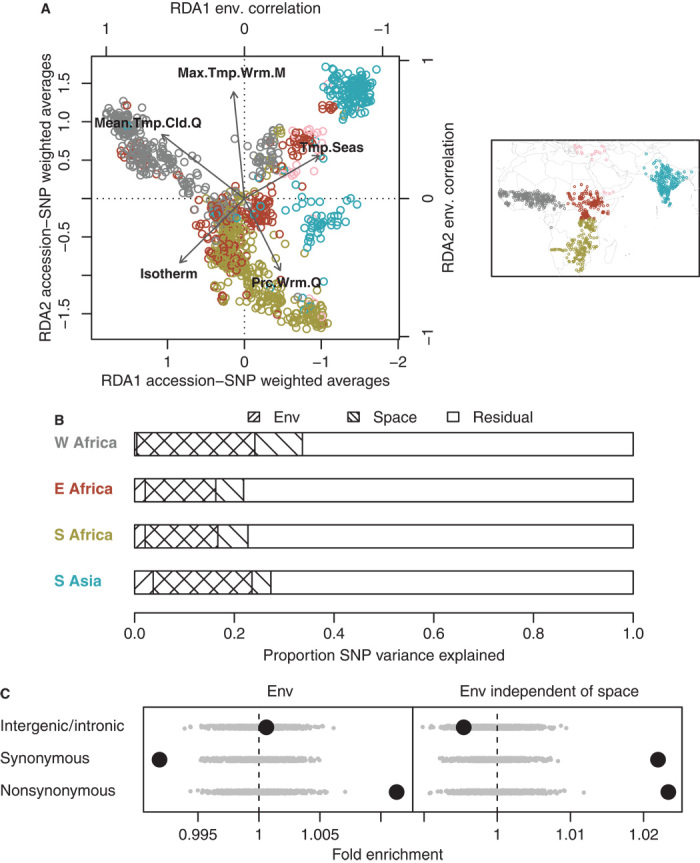
(A) Left panel shows the first two canonical axes (RDA1 and RDA2) of an RDA of variation in 871 SNPs among 1133 accessions (chosen to minimize missing SNP calls for accessions); inset map shows the geographic distribution of the accessions. Each canonical axis represents a linear combination of environmental variables (strongly loading variables shown as arrows) that explains variation in a linear combination of SNPs among accessions (colored points representing accessions from different regions: blue, South Asia; pink, Middle East; gray, West Africa; red, East Africa; green, South Africa). (B) Proportion of total SNP variation among accessions explained in RDA by environmental variables or spatial structure within each region (excluding Middle East). (C) Enrichment of three SNP categories for environmental structure, that is, the proportion of SNP variation among accessions (in all five regions) explained by environmental conditions. Gray dots represent 1000 circular permutations of SNP categories. The right panel shows enrichment for environmental structure after removing geographic spatial structure in SNP variation via partial RDA (akin to partial regression).
The greatest genomic differentiation along spatially structured environmental gradients occurred in West Africa and South Asia, potentially reflecting the rapid diffusion of genotypes to appropriate locations along shallow (that is, highly spatially autocorrelated) environmental gradients (Fig. 3B). By contrast, less genomic variation was explained by geography in East Africa, the center of sorghum domestication, potentially reflecting the accumulation of local diversity over time and more recombination events breaking linkage between adaptive and neighboring loci. Nevertheless, environmental structure in genomic variation among sorghum landraces is high in comparison to Arabidopsis (Supplementary Materials, section 2.1). Consistent with the hypothesis that genome-environment associations reflect adaptation (17, 37), we observed that amino acid changing (that is, nonsynonymous) SNPs exhibited greater environmental structure compared with the average SNP (Fig. 3C). These enrichments were small (~1 to 2%) but were highly significant and on the same order as what has been found in Arabidopsis (17).
Models derived from environment-associated SNPs can predict phenotypes
If environment-associated SNPs reflect adaptations to environmental gradients, then genotypic information at those SNPs should be predictive of phenotypic variation in traits related to environmental adaptation. We tested this hypothesis by growing two diverse panels of sorghum accessions in common garden experiments with treatments of well-watered and water-limited conditions. In Austin, United States (2013 growing season), we simulated drought throughout the growing season (figs. S2 to S4), whereas in Hyderabad, India (2010–2011 and 2011–2012 growing seasons), we simulated a terminal drought (fig. S5). Our panels consisted of diverse landraces in addition to improved varieties not in the georeferenced panel of 1943 landraces. Notably, many (191 of 342 in Austin and 110 of 242 in Hyderabad) accessions did not have environment-of-origin information that could be used directly to predict phenotype, either because the collection site was not recorded or because they represent admixed breeding lines, which have no single environment of origin.
To predict phenotypes using genotype and environmental data, we developed an approach that integrates genomic selection (GS) (11) and ecogeographic germplasm analysis (38). In marker-assisted selection (MAS) and GS, genotype to phenotype models are fit and then used to predict phenotype on new individuals using marker data (11). Here, we used the 1943 georeferenced landraces and first fit models for environmental associations with allelic state at individual SNPs (31). To generate a relative phenotypic prediction for a given accession, we used its allelic state at the SNPs with the strongest associations with landrace environmental conditions, filtered to SNPs with relatively low linkage disequilibrium (LD) to avoid making redundant predictions (17), and summed parameter estimates for the corresponding SNP-environment models. We generated a second set of phenotypic predictions based on environmental associations with estimated genome-wide kinship (39). Environment associations with kinship may result when local adaptation is driven by many loci of small effect, or when large effect variants underlying local adaptation are collinear with population structure (see Maturity1 and Tannin1 above).
We asked whether SNP associations with climate variables related to abiotic stress (annual aridity, growing season length, growing season aridity, precipitation seasonality, precipitation of the warmest quarter, and temperature seasonality) could be used to predict yield component phenotypes (biomass and grain yield) averaged across well-watered and drought treatments. Performance averaged across environments may be relevant to identifying genotypes that are high-yielding despite the spatiotemporal environmental heterogeneity common in the field. Environmental associations were good predictors of average phenotypes. For example, per-plant biomass for each accession averaged across treatments in Austin was best predicted by SNPs with the strongest associations with aridity, where genotypes with more alleles associated with arid environments exhibited higher biomass (top 5000 SNPs before filtering for LD, Pearson’s r = 0.33, P = 4.6 × 10−10, fig. S7). Per-plant grain weight averaged across wet and drought treatments in Hyderabad was best predicted by SNP associations with growing season aridity, with genotypes having more alleles associated with more arid environments exhibiting higher grain weight (top 10,000 SNPs before filtering for LD + kinship, Pearson’s r = 0.31, P = 6.1 × 10−7, fig. S8).
Environment-associated SNPs predict genotype-by-environment interactions
Characterizing genotype-by-environment interactions (G×E) presents a major challenge in crop improvement. Breeders often seek to minimize G×E (that is, increase yield stability) or at least ensure that G×E are favorable for the target population of environments (6, 40, 41). Because the environmental variables we studied are related to abiotic stress, we next asked whether SNP-environment associations could predict G×E. We studied phenotypic differences in yield components across well-watered and drought treatments, differences that may play a role in adaptation to local moisture conditions. We found that accession change in panicle (that is, seed head) weight from well-watered to drought treatments in Hyderabad was best predicted by SNPs with the strongest associations with growing season length (top 100 SNPs before filtering for LD + kinship). Accessions with alleles associated with long growing seasons showed larger reductions in panicle weight from well-watered to drought treatments compared with accessions with alleles associated with short growing seasons (Pearson’s r = 0.23, P = 0.0003, Fig. 4C). Our results suggest that alleles selected for in short growing seasons may confer drought tolerance (for example, if locations with short growing season frequently experience drought) or drought escape via shorter time to maturity (6, 42). (Note that to impose drought stress per se, we imposed stress by maturity classes in Hyderabad and used converted accessions with similar maturity in the Austin experiments.) Additionally, we found that accession change in harvest index (that is, grain yield/biomass) from well-watered to drought treatments in Austin was best predicted by SNPs with the strongest associations with precipitation in the warmest quarter (top 10,000 SNPs before filtering for LD). Accessions with alleles associated with wet warmest quarters tended to show large reductions in harvest index from well-watered to drought treatments, whereas accessions with alleles associated with dry warmest quarters often showed higher harvest index in drought than in well-watered treatments (Pearson’s r = 0.15, P = 0.018, Fig. 4F). This finding may indicate that alleles selected for in drought-prone production environments promote a shift of resource allocation from vegetative biomass to grain production when drought is sensed, a phenomenon that has been observed in some sorghum hybrids (43).
Fig. 4. Genome-wide associations with environment (A, D, and G) used to predict change in phenotypes across treatments for breeding lines and landraces (circles in C, F, and I), and comparisons of predictions using different numbers of predictor SNPs (B, E, and H).

The best prediction is shown for each trait in (C), (F), and (I). Note that phenotype data were not used in predictions. There were three experiments testing the effects of (i) drought treatment late in growing season in Hyderabad, India (A to C), (ii) drought treatment across growing season in Austin, United States (D to F), and (iii) aluminum toxicity in the laboratory (G to I). Horizontal dashed lines (A, D, and G) show P value thresholds delineating the nested subsets of SNPs with the strongest associations to environment tested in (B), (E), and (H). The solid horizontal line (A, D, and G) shows the set of SNPs giving the best predictions (C, F, and I; r = Pearson’s correlation coefficient). In (C) and (I), the best model combined the subset of SNPs indicated and genome-wide SNP similarity (“kinship”). SE bars (B, E, and H) were generated using nonparametric bootstraps of sampled accessions. Predictions were standardized to z scores (x axes of C, F, and I). Drought treatment data were generated here; aluminum toxicity response data are from (44). Because of skew in the data, the y axes in (F) and (I) are shown as proportions with log scaling.
To test our approach along edaphic gradients (rather than climate gradients), we considered adaptation to aluminum toxicity in acid soils, an important constraint for sorghum production in tropical regions (8, 44). We used SNP associations with soil conditions indicative of aluminum toxicity to predict phenotypic variation in a published experiment on variation in root response to soil aluminum toxicity in a diverse sorghum panel (44). We found that accessions carrying more alleles associated with high pH soils (that is, where Al cat ions are insoluble) had greater reductions in root growth rate when grown in aluminum toxicity treatments than accessions with alleles associated with low soil pH (that is, where Al cations are unbound and free). The best model combined the predictions based on soil pH with the top 1000 SNPs (before LD filtering) and kinship (Pearson’s r = 0.29, P = 0.0002, Fig. 4I).
Associations provide insight on genetic architecture of environmental adaptation
As in any marker-assisted selection (MAS) approach, the relationship between predictive ability and the number of loci used in predictions may reflect variation in the genetic architecture of traits (45). Note that the individual SNPs we used for predictions were thinned for LD and that environmental association models accounted for kinship, meaning that predictor SNPs represented relatively independent axes of genetic variation. Here, traits like average flowering time and biomass across treatments were best predicted by models including predictions based on environmental associations with 5000 SNPs, 10,000 SNPs, or all SNPs (that is, the kinship matrix), whereas models using small numbers of SNPs (for example, 100) had lower predictive ability (figs. S7 and S9). By contrast, response to aluminum toxicity was well predicted by associations between soil pH and small numbers of SNPs (for example, 100 SNP model: r = 0.20), suggesting the presence of more SNPs of large effect in comparison to average biomass and change in harvest index. Nevertheless, response to aluminum toxicity was also well predicted by kinship estimated from all SNPs, suggesting that loci of small effect also play an important role.
We next asked whether our identified putative signals of local adaptation (that is, genome-environment associations) overlap with genomic regions putatively under selection during domestication and subsequent landrace evolution. We used 682 candidate 10-kb regions (some of which were contiguous) identified by Mace et al. (46) that showed elevated differentiation between wild or weedy accessions and landraces, had low nucleotide diversity, and had highly skewed allele frequency spectra. These regions showed a significant enrichment with SNPs associated with precipitation of the warmest quarter (5th percentile of P values for SNP associations in candidate SNPs = 0.027, two-tailed permutation test z = −2.28, P = 0.0194). This enrichment in candidate regions for selection suggests that local adaptation to precipitation has played an important role in sorghum evolution. However, candidate regions (46) did not show enrichment for associations with growing season length (5th percentile of P values for SNP associations in candidate SNPs = 0.043, two-tailed permutation test z = −0.24, P = 0.8232) or topsoil pH (5th percentile of P values for SNP associations in candidate SNPs = 0.054, two-tailed permutation test z = 0.83, P = 0.39).
Combined environment and phenotype associations reveal putative adaptive loci
If much of the trait variation we observed is locally adapted to environmental stressors, variation at causal loci may be associated with both environmental conditions and phenotypes. Thus, SNP-environment and SNP-phenotype associations may carry complementary information that can be leveraged to identify loci underlying adaptive trait variation. To do so, we first tested SNP associations with phenotypic variables using the linear mixed model that accounts for kinship (EMMA) (31). We then used the strength of environmental associations for each SNP to develop a prior probability of association with phenotype for each SNP (47). Each SNP’s prior for the phenotype GWAS was equal to the −log(P) for the SNP’s association with the relevant environmental variable, scaled so that priors across SNPs summed to one. This prior was combined with the likelihood from the linear mixed model to generate an approximate posterior probability of association (APPA), following published methods (47–49). We used SNP associations with log precipitation in the warmest quarter as priors for SNP associations with harvest index plasticity in the U.S. experiment, SNP associations with growing season length as priors for SNP associations with panicle weight plasticity in the India experiment, and SNP associations with topsoil pH as priors for SNP associations with relative net root growth under Al toxicity in the experiment (44) (that is, the best phenotype predictors shown in Fig. 4).
We found a number of SNPs near genes of interest that were not among the strongest in phenotype associations (for example, in the top 100 SNPs) but were strongly associated with environmental associations and thus had high APPA (that is, posterior; tables S19 to S21). For example, S3_8974849 was ranked 302 among SNPs associated with harvest index plasticity (EMMA, P = 0.00108) but was also associated with precipitation of the warmest quarter (P = 0.00356) and, as a result, was the 58th ranked SNP by APPA for association with harvest index plasticity (table S19 and fig. S11). About 20 kb away is a NAC protein (Sb03g008470, similar to NAM-related protein 1) that is a putative co-ortholog of Arabidopsis genes ORE1 and ORS1, which are senescence-regulating transcription factors (50, 51). Related NAM/NAC genes are involved in yield under drought in rice (52) and senescence and grain fill in wheat (53).
Additionally, S10_58358066 was ranked 226 among SNPs associated with relative net root growth (EMMA, P = 0.00009) but was also associated with topsoil pH (P = 0.00869) and, as a result, was the 39th ranked SNP by APPA (table S20 and fig. S13). This SNP was found within the xyloglucan endotransglycosylase (XET) Sb10g028550. Several XETs in Arabidopsis are involved in Al tolerance (54), including one XTH17 that is 63% similar to this gene (the putative ortholog is 72% similar). Additionally, S10_58358066 is 20 kb (two genes away) from the candidate Sb10g028530, which is the putative ortholog of STAR1. Similarly, S3_45597499 was ranked 360 among SNPs associated with relative net root growth (EMMA, P = 0.00016) but was also associated with topsoil pH (P = 0.00084) and, as a result, was the 41st ranked SNP by APPA for association with relative net root growth. About 80 kb away is the candidate gene Sb03g022890, which is a paralog of the STAR1, which encodes part of an ABC (adenosine triphosphate–binding cassette) transporter conferring aluminum tolerance. S3_45597499 was also very close (~20 kb distant, two genes away) to a pectin methylesterase, which are involved in Al exclusion in rice (55).
Of additional note were the top SNPs associated with panicle weight plasticity. In particular, two nearby SNPs (S10_59797069 and S10_59797088) had the strongest associations with panicle weight plasticity, before (both EMMA P = 3.2 × 10−8) and after including SNP associations with growing season length as a prior (although the SNPs were not associated with growing season length, both EMMA P > 0.4, fig. S12). The two closest genes to these SNPs were a small auxin up-regulated RNA (7.5 kb away, Sb10g030050) and a calcium-dependent protein kinase (CDPK, 19 kb away, Sb10g030040), which is a homolog of the ZmCPK4 gene in maize that up-regulated ABA signaling and increased drought tolerance in transgenic Arabidopsis (56).
DISCUSSION
Predicting how individual organisms respond to a given set of environmental conditions is a shared challenge of agriculture, natural resources management, ecology, and evolutionary biology. Given the availability of de novo genotyping-by-sequencing, the approach we demonstrate can be applied in any species where local adaptation is prevalent and georeferenced genetic material is available. Thus, genome-environment associations have multiple potential applications. For example, selective breeding and managed gene flow may be applied to wild species to mitigate impacts of climate change (57, 58). In crop improvement, genome-environment associations may be integrated with genome-phenotype associations in selection for climate resilience traits (11), and in crop conservation, they may be integrated with ecogeographic germplasm identification strategies (38). Although knowledge of local adaptation has long been important in crop improvement (5, 6), here we provide a formalization and extension of this knowledge; genome-environment associations have not been previously leveraged to predict G×E. Models based on genome-environment associations may be used for any genotyped accession (for example, accessions of unknown origin and breeding lines) and integrated with phenotyping-based approaches to uncover the molecular basis of environmental adaptation.
MATERIALS AND METHODS
Data
Genotype data
Landraces were chosen to maximize geographic coverage. Landrace seeds were obtained from the NPGS-GRIN or ICRISAT gene banks. Seeds were grown at Cornell University, United States (NPGS-GRIN accessions), or at ICRISAT, India (ICRISAT accessions), and seedling tissue was used for DNA extraction. DNA preparation and genotyping-by-sequencing were carried out as described in (23). SNP calls were made using the TASSEL GBS pipeline version 3.0 (59) using the Sorghum bicolor genome v1.4 from Phytozome as a reference (60, 61) and BWA as the alignment algorithm (62). The GBS approach produces a large fraction of missing genotypes, so most missing genotypes were imputed on the basis of nearest neighbor searches using Viterbi algorithm in TASSEL version 4.0 (MinorWindowViterbiImputationPlugin) (http://tassel.bitbucket.org/TasselArchived.html).
Environmental data
Climate data
For each accession with a known collection location, we compiled data for multiple environmental parameters (37). All data were global, covering the period of the later 20th century, and publicly available (see Supplementary Materials, section 1.2). Although global environmental data sets are imperfect representations of the conditions at any given site, these data represent the best available high spatial or temporal resolution data for characterizing global environments.
The first climate data set, WorldClim (63), contained altitude and long-term averages of precipitation and mean monthly minimum, mean, and maximum temperatures. WorldClim additionally contains variables putatively of biological importance, derived from the monthly averages. We also included monthly and annual average potential evapotranspiration (PET), and a measure of aridity (mean annual precipitation divided by mean annual PET) derived from WorldClim data [data from Consultative Group for International Agricultural Research Consortium for Spatial Information (CGIAR-CSI) Global-Aridity database] (64). We obtained estimates of vapor pressure deficit using Climate Research Unit (CRU) data (65). The WorldClim (63) and CRU (65) authors estimated altitude and long-term averages of climate variables.
We used NCEP reanalysis data to estimate interannual variability in precipitation (data provided by the National Oceanic & Atmospheric Administration/Office of Oceanic and Atmospheric Research/Earth System Research Laboratory Physical Sciences Division, http://esrl.noaa.gov/psd/) (66). We extracted the monthly surface precipitation rate and then calculated each calendar month’s coefficient of variation (CV) across years. We used an additional data set to estimate photosynthetically active radiation (PAR) (data available at https://eosweb.larc.nasa.gov/project/srb/srb_table). We took SRB data and calculated average quarterly PAR across the years 1983 to 2007.
Edaphic data
Water availability to plants is a function of both precipitation inputs and edaphic conditions. Thus, we included multiple data sets related to soil moisture. First, we used a global data set of estimated plant extractable water capacity of soil (67). Second, we used a global model of depth to water table, which represents potential groundwater availability to plants and is a strong predictor of wetlands (68). In addition to moisture stress, aluminum toxicity represents a major environmental stressor to tropical crops (69). We used a global aluminum soil toxicity data set (69). A major predictor of aluminum soil toxicity is soil pH; thus, we also used an additional higher-resolution data set on global soil pH (70).
Predicted growing seasons
We used long-term average data on monthly mean precipitation and temperature (63), monthly PET data (64), and local soil moisture capacity (67) to develop potential growing season windows for each accession. To determine when moisture permitted growing seasons, we followed Food and Agriculture Organization of the United Nations (FAO) criteria for rough growing season determination for dryland crops (http://fao.org/nr/climpag/cropfor/lgp_en.asp; http://www.fao.org/geonetwork/srv/en/metadata.show?id=73; see Supplementary Materials, section 1.2.3). Additionally, monthly mean temperature was required to be at least 15°C to begin the growing season, a criterion that was only important for sorghum grown in northern China. Temperature restrictions were placed on the end of growing seasons such that months where average minimum temperatures were less than 0°C were not part of the growing season, because sorghum performs poorly in cold conditions.
Phenotype data
Drought experiment in the United States
The study was conducted in Austin, TX [30°17′5.2″N, −97°46′52.6″W, elevation 133 m above sea level (asl)]. The planting was established under an existing 18.3 × 73.0–m rainout shelter (Windjammer Cold Frame, International Greenhouse Company). Irrigation was supplied to each planting bed via three rows of drip irrigation tape (Chapin Twin Wall BTF Drip Tape, Jain Irrigation Systems Ltd.). The beds supplied by each irrigation zone alternate down the length of the shelter, allowing for two interspersed and separate irrigation schemes.
Genetic material and experimental design
The diverse association panel consisted primarily of accessions from the U.S. Sorghum Association Panel (71) (table S2) with some accessions removed (for example, broomcorns) to avoid rare phenotypic covariates. From the accessions genotyped previously (23) and the landraces genotyped here, we chose accessions for the panel as those likely to be short statured, from diverse locations and botanical races, and for which we currently had seed stocks. Additionally, we selected lines of historical importance in sorghum breeding, such as Redland, Wheatland, and BTx623. Of the 408 accessions we planted, 344 accessions had at least some individuals survive, flower, and were in our SNP data. These 344 were used in phenotypic predictions (see below).
Planting and irrigation treatments
Plants were sown and maintained in the greenhouse until 12 May 2013. Plants were transplanted into the field by hand on 13 to 14 May. Differential irrigation was imposed by altering the run time of the irrigation drip tape to impose a persistent and progressive terminal drought in the stressed beds relative to the well-watered beds. Irrigation was applied at a ratio of 3:1 wet/dry by pulses that differed in both frequency and magnitude. Irrigation was triggered by evaluation of plant status with the goal of maintaining optimal growth for the well-watered treatment plants and preventing total crop failure of the drought stress plants.
Phenotype measurements
To examine genetic variation in phenotype plasticity under drought versus well-watered conditions, we measured days to flowering, plant height, chlorophyll content [via approximation by Soil-Plant Analysis Development (SPAD)], tiller number, above ground biomass, and grain mass (see Supplementary Materials, section 1.3.1). Days to flowering was scored as the number of days from emergence to the day when 50% of the florets on the head of the primary tiller had undergone anthesis. Plant height was assessed from the soil surface to the tip of the tallest panicle after anthesis. For genotypes that had not undergone panicle emergence at the time of harvest, height was assessed from the soil surface to the tip of the flag leaf of the tallest tiller. Aboveground biomass was determined by harvesting and drying plant material in paper bags (whole plants and panicles separately) at 50°C for 10 days and then weighing. Grain was threshed by hand.
Drought experiment in India
Two hundred forty-two germplasm accessions of the sorghum mini core collection (72) were grown at ICRISAT Patancheru, outside Hyderabad, India (78°12′E, 17°24′N, 545 m asl, table S3). The planting was in a split plot design using drought and control (fully irrigated) treatments as main plot and genotype as subplot, in five maturity groups [group 1, flowered <60 days after planting (DAP); group 2, flowered between 61 and 70 DAP; group 3, flowered between 71 and 80 DAP; group 4, flowered between 81 and 90 DAP; group 5, flowered >90 DAP], during the 2010–2011 and 2011–2012 post-rainy seasons. The experiment was sown in the second week of October each year and harvested in April of the following years. The seeds were sown at uniform depth, with standard agronomic and plant protection practices for sorghum. The irrigated environments received five irrigations (each time receiving about 7 cm of water). To impose drought at consistent phenological stages, we imposed drought at different stages depending on maturity group. Drought treatment plants received one to four irrigations (based on maturity groups) before the imposition of drought stress when the irrigation was stopped at 25 days in group 1, 37 days in group 2, 47 days in group 3, 57 days in group 4, and 66 days in group 5. Data were recorded on days to 50% flowering, plant height (cm), grain yield (g plant−1), and SPAD chlorophyll meter (SPAD-502, Minolta Corp.) reading (SPAD I) at 50% flowering and 30 days after flowering (SPAD II).
Aluminum toxicity experiment
We used published data on relative root growth under aluminum containing versus control media (44). We summarize their methods here. The authors’ panel included both landraces and breeding lines. Of their 167 accessions that we genotyped, 78 were in our georeferenced landrace panel. After 4 days of germination, seedlings were transferred to hydroponic growth chambers with a low pH (4.0) solution. After a day of acclimation, they were then exposed to a control solution or a solution with aluminum ions. The authors then measured net root growth (compared to root length at beginning of treatment) at 3 and 5 days after the treatment and calculated relative net root growth by dividing control net root growth by treatment for each time step. We took the average of relative net root growth at 3 and 5 days as our measure of accession response to aluminum toxicity (used in analyses below).
SNP-environment associations
Environmental associations for cloned natural variant Tannin1 and Maturity1
SNP S6_40286721 in our data set corresponds to the previously described K162N nonsynonymous polymorphism in the conserved pseudoresponse regulator domain of SbPRR37 (Maturity1) found in the Sbprr37-2 and Sbprr37-3 loss-of-function alleles (25). SNP S4_61667908 in our data set corresponds to the previously described synonymous polymorphism that is 218 base pairs upstream of the causative indel in Tannin1 and in perfect LD with the causative indel in a global sorghum diversity panel (n = 161) (26, 29).
Partitioning sorghum SNP variation
We sought to gain a broad perspective of the relationship between geography, environment, and genomic variation among landraces. Thus, we calculated the proportion of genome-wide SNP variation among accessions that could be explained by (i) global environmental gradients, (ii) geographic distance among accessions, and (iii) spatially structured environmental gradients [collinear portion of (i) and (ii)] (37). We used variance partitioning of RDA (34, 36), an eigenanalysis ordination that allows one to assess the explanatory power of multivariate predictors (environmental and geographical variables) for multivariate responses (SNPs). Because our sample of landraces drew heavily from Africa and South Asia, we did not include spatial outlier landraces from the Americas, China, and Southeast Asia/Oceania, which could heavily influence spatial patterns in SNP variation.
To understand regional variation in genome-environment associations, we also conducted variance partitioning on four subsets of accessions: (i) West Africa, defined as accessions west of 21°E and north of the equator; (ii) southern Africa, defined as accessions south of the equator (excluding a lone accession from the Congo River delta); (iii) East Africa, defined as accessions east of 21°E and north of the equator; and (iv) South Asia, defined as accessions between 60°E and 100°E.
We modeled spatial structure among accessions using principal components of neighborhood matrices (PCNMs) (35, 73). We calculated PCNM following (35). We calculated a distance matrix between accession locations of origin using great-circle distances calculated for the Vincenty Ellipsoid in the R “geosphere” package (74). We truncated the distance matrix at a threshold equal to the minimum distance required to form a minimum spanning tree, with distances greater than the threshold reassigned to four times the threshold. This threshold was chosen because it balances our ability to resolve fine and coarse-scale spatial structure (35). Finally, we calculated eigenvectors of the distance matrix, known as PCNM, to use as predictors of SNP variation. We kept only eigenvectors with positive eigenvalues. Additionally, we kept only as many eigenvectors as there were environmental variables in analyses.
For variance partitioning on all African and Eurasian landraces combined, we used all environmental variables (115 in total) described above in the “Environmental data” section, in addition to growing season length (in units of months). However, because regional variance partitioning was on a smaller number of accessions, we used a smaller number of variables (subset of 31), including only altitude, WorldClim-derived bioclimatic variables (“BioClim”), Aridity Index, PAR, edaphic, and growing season length variables.
Our purpose in Fig. 3A was to illustrate the basics of RDA and environmental structure in our SNP data. However, a single RDA requires no missing SNP data; thus, we chose the 871 SNPs that had missing calls for 15 or fewer accessions, and then culled any accession with missing SNP data, leaving 1133 accessions. We included altitude, Aridity Index, WorldClim-derived bioclimatic variables (BioClim), edaphic, and growing season length variables in the RDA used for Fig. 3A.
Because variance partitioning requires complete data sets, we analyzed repeated subsets of the accession by SNP data. We started by focusing on the 10% of SNPs with the fewest missing accession calls. Next, we selected 100 random SNPs, removed any accessions with missing data, and conducted variance partitioning. We then repeated this process, resampling 100 SNPs 10,000 times. Note that regions differed in the spatial distribution of samples. To control for effects of spatial distribution in regional comparisons, each time we resampled SNPs, we also took a subset of accessions, randomly sampling one accession from each degree cell.
From each of the 10,000 resampled SNP sets, we calculated the portion of SNP variance explained by environmental variables, by spatial variables, and by their collinear portion. We then took the means of these values from the 10,000 resamples to estimate how much genome-wide SNP variation was explained by environmental and spatial variables.
Enrichment of SNP variation explained by climate in nonsynonymous SNPs
We tested whether three classes of SNPs differed from genome-wide expectations in terms of the portion of their variation explained by environment. SNPs were classified as nonsynonymous, synonymous, or intergenic. We generated a null expectation for the proportion of SNP variation explained by environment in each class using a permutation approach (37). For the observed statistic, we calculated how much variation in SNPs in each category was explained by environment, and environment independent of spatial variables (PCNM), selecting 100 SNPs from each category in the top 10% coverage SNPs and resampling 1000 times as described above for the previous analysis. To generate null distributions, we permuted SNP classifications along the genome by a random distance 1000 times, maintaining the order of SNP classifications. For each of the 1000 permutations, we then calculated proportion of variation in each SNP category explained by environment using the same resampling procedure used to calculate the observed.
Environmental associations with individual SNPs
We used the GAPIT implementation of a mixed linear association model (EMMA) (31) to test for associations between SNP alleles and climate of origin while controlling for genome-wide accession similarity (putative kinship). A kinship matrix was calculated following the method of (31) for genotype data with missing allele calls. For each environmental variable, we tested the null hypothesis that the environment of origin of the two alleles was equal (18, 75). Alleles were coded as numeric values such that homozygotes were 0 or 2 and heterozygotes were 1. To speed computation, missing allele calls were imputed as the average allele.
Predicting phenotypes based on environmental associations
We tested whether models based on SNPs associations with environment could predict genetic variation in (i) average phenotype across relatively benign and stressful environments and (ii) phenotypic change from relatively benign to stressful environments. We used two common approaches to predicting genotypic breeding value: MAS and genomic selection (GS). However, previous applications of MAS and GS have fit genotype-phenotype models and used them to predict phenotype. Here, we fit genotype-environment models and used them to predict phenotype. MAS uses a subset of strong marker associations with phenotype to predict phenotype of genotypes with unknown phenotype. GS uses associations between all markers and phenotype to predict phenotypes of genotypes with unknown phenotype. MAS is likely more accurate for oligogenic traits, whereas GS is likely more accurate for polygenic traits. A drawback to MAS is that it tends to overpredict phenotypes (that is, is biased) because effects of quantitative trait loci (QTL) are estimated independently of each other (45, 76). Furthermore, we developed a hybrid prediction model using combined information from both a subset of the strongest marker associations (MAS) and the genomic average association (GS).
Because this was our first attempt to implement predictions based on environmental associations, we used a relatively simple approach for MAS models. We started with the n SNPs with the lowest P values in mixed-model associations with a given environmental variable. We then selected a subset of those n SNPs, using the following criteria. We started with the lowest P value SNP and then sequentially added SNPs to the selected subset if the following SNPs (i) had allelic variation that was not correlated with any SNP already in the subset at ρ = 0.75 or greater (Spearman’s rank correlation) and (ii) at least five georeferenced landraces had the minor allele. The first criterion was used to avoid overestimating responses because of redundancy among SNPs. The second criterion was used to avoid including very rare alleles unlikely to predict variation in separate genotype panels. After implementing this procedure, typically 50 to 60% of the n original SNPs were selected for the predictive MAS model. To generate predictions for a given accession, the accession’s allelic state at each SNP in the model (coded as numeric where homozygotes were 0 or 2 and heterozygotes were 1) was multiplied by the corresponding estimated allelic effect (slope) from the environmental association model. Because SNP calls were often missing for a given accession, we took the average of the allelic state × allelic effect across SNPs in the model for each accession to get the total prediction for each accession. We tested predictions using six values of n: 100, 250, 500, 1000, 5000, and 10,000. These predictions, based on environment-SNP associations, were then compared to phenotypes that we hypothesized were locally adaptive along the environmental gradient (see below).
For GS models, we used a kinship matrix among accessions combined with environmental conditions to calculate genomic best linear unbiased predictors (gBLUP) using the “rrBLUP” package in R (39, 77). In this model, environment of origin was the response variable in a mixed model where genotype random effects had a correlation structure imposed by a kinship matrix among genotypes. The kinship matrix was constructed following (77). This model is analogous to the environmental association mixed model (described above) omitting any individual SNP effects. The model was fit using georeferenced landraces (as in the environmental association analyses described above). This model was then used to predict environment for a given genotype with known kinship relative to genotypes in the georeferenced set, using the “kin.blup” function in “rrBLUP” (39).
If phenotypes are affected by both many loci of small effect and a few loci of large effect, a combination of marker and kinship-based predictions may be most accurate. Thus, we tested additional predictions by combining MAS and GS predictions described in the previous two paragraphs. For each environmental variable tested, we calculated z scores of marker predictions and z scores of kinship predictions and then took the average of the two predictions for each genotype. Accession predictions based on genome-environment associations using each approach are given in tables S6 to S9.
For SPAD/chlorophyll, grain, panicle, harvest index, biomass, and height phenotypes from the two drought experiments, we tested predictions generated from associations with Aridity Index, growing season length, precipitation seasonality, precipitation of the warmest quarter, annual mean temperature, and temperature seasonality. Aridity Index as defined by (64) is greater in less arid environments. To make the interpretation more straightforward, we reversed the sign of predictions from Aridity Index such that larger predicted values correspond to more arid environments (that is, an intuitive interpretation of aridity). For flowering time, we tested predictions based on SNP associations with temperature seasonality (for reference, flowering time correlations with drought-related predictions are also included in tables S10 and S11). The environmental variables with the best prediction of each phenotype are shown in figs. S7 to S10 (see also tables S10 to S12 for correlations of phenotypes with all predictive models). For reference, phenotype correlations with environment of origin for landraces in each panel with known origin are included in tables S13 to S15.
For aluminum tolerance, we tested whether predictions generated from associations with soil aluminum toxicity and soil pH were associated with the change in root growth rate in non–aluminum-stressed versus aluminum-toxic treatments. Growth rate data were taken from (44) at two time points. The average of control/aluminum toxic relative growth rates at the two time points was used as the phenotype of interest for each accession. The best predictions of change in growth under aluminum toxicity tended to come from associations with topsoil pH (Fig. 4I). However, the single best predictive model of change in growth under aluminum toxicity came from associations with soil aluminum toxicity (based on kinship + 5000 SNP associations; r = −0.30, table S12). Because the best prediction from topsoil pH was nearly equivalent (based on kinship + 1000 SNP associations; r = 0.29), and topsoil pH predictions tended to be better across different prediction methods (table S12), we show topsoil pH in Fig. 4.
Acknowledgments
We thank E. Cooper, L. Gilbert, M. Kirkpatrick, J. Lovell, D. Lowry, J. McKay, R. Meyer, U. Mueller, and J. Schmutz for helpful comments on previous versions of this manuscript. We thank the staff at Beocat computing cluster at Kansas State University for technical support, E. Barona for making available the GIS data on global soil aluminum toxicity, and Brackenridge Field Laboratory (University of Texas at Austin) for support and access to field sites. Funding: We thank the National Science Foundation (NSF) for providing funds (ID: IOS-0965342) to carry out genotyping-by-sequencing under Basic Research to Enable Agricultural Development (BREAD) project to E.S.B., S.K., C.T.H., and S.E.M. NSF grant IOS-0922457 to T.E.J. supported the Austin, United States, field experiment. J.R.L. was funded by a fellowship from the Earth Institute at Columbia University. G.P.M. was funded by the U.S. Agency for International Development (USAID) under Cooperative Agreement No. AID-OAA-A-13-00047. This study is made possible through support by the American People provided to the Feed the Future Innovation Lab for Collaborative Research on Sorghum and Millet through USAID. The contents are the responsibility of the authors and do not necessarily reflect the views of USAID or the U.S. government. This work was undertaken as part of the CGIAR Research Program on Dryland Cereals. Competing interests: The authors declare that they have no competing interests. Data and materials availability: Phenotypes and genotypes have been deposited to the Dryad Digital Repository at doi:10.5061/dryad.jc3ht. Raw genotyping-by-sequencing data has been deposited to the NCBI Sequence Read Archive under accession SRP017504.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/1/6/e1400218/DC1
Fig. S1. Landrace accessions included in the study classified into botanical races based on morphological classification (five new world accessions not shown).
Fig. S2. Rainout shelter where plants were grown in Austin.
Fig. S3. Seedlings planted in the Austin experiment.
Fig. S4. Plants growing in Austin.
Fig. S5. A representative accession (IS 25836) under irrigated (left) and imposed terminal drought (right) conditions at experimental plot in India.
Fig. S6. Proportion of total SNP variation among accessions with known collection locations (excluding spatial outlier landraces from the Americas, China, and Southeast Asia/Oceania) explained by spatial structure or environmental variables.
Fig. S7. Predictions of phenotypes averaged across well-watered and drought conditions from drought treatment across growing season in Austin, United States.
Fig. S8. Predictions of phenotype change between well-watered and drought conditions from drought treatment across growing season in Austin, United States.
Fig. S9. Predictions of phenotypes averaged across well-watered and drought conditions from drought treatment late in growing season in Hyderabad, India.
Fig. S10. Predictions of phenotype change between well-watered and drought conditions from drought treatment late in growing season in Hyderabad, India.
Fig. S11. GWAS for harvest index plasticity (harvest index in wet/dry) in U.S. experiment, using SNP associations with precipitation in the warmest quarter as a prior.
Fig. S12. GWAS for panicle weight plasticity (panicle weight in wet − dry) in India experiment, using SNP associations with growing season length as a prior.
Fig. S13. GWAS for root growth plasticity (growth in control/Al toxic) in published aluminum toxicity experiment (44), using SNP associations with topsoil pH as a prior.
Table S1. Landraces studied and environment of origin data.
Table S2. Accession phenotypes from experiment in Austin, United States.
Table S3. Mean phenotypes across the 2 years of the experiment in Hyderabad, India.
Table S4. Spearman’s rank correlation test results for two SNPs that tag known candidate genes potentially involved in local adaptation.
Table S5. EMMA t test results for two SNPs that tag known candidate genes potentially involved in local adaptation.
Table S6. Predictions for each accession in the Austin experiment based on SNP-environment associations in the landrace panel.
Table S7. Predictions for each accession in the Hyderabad experiment based on SNP-environment associations in the landrace panel.
Table S8. Predictions for each accession in the Caniato et al. (44) experiment based on SNP-environment associations in the landrace panel.
Table S9. Predicted environments for accession in the three experiments based on kinship associations with environment of landraces (gBLUP).
Table S10. Pearson’s correlations between predictions based on environment-genome associations and phenotypes in Austin.
Table S11. Pearson’s correlations between predictions based on environment-genome associations and phenotypes in Hyderabad.
Table S12. Pearson’s correlations between predictions based on environment-genome associations and phenotypes in the Caniato et al. (44) Al toxicity experiment.
Table S13. Pearson’s correlations between phenotypes and environment of origin (where known) for landraces in the Austin experiment.
Table S14. Pearson’s correlations between phenotypes and environment of origin (where known) for landraces in the Hyderabad experiment.
Table S15. Pearson’s correlations between relative net root growth and environment of origin (where known) for landraces in the Caniato et al. (44) experiment.
Table S16. Pearson’s correlation coefficients between predicted phenotypes in the Austin experiment and observed, where predictions based on genome associations with phenotypes in fivefold cross-validation.
Table S17. Pearson’s correlation coefficients between predicted phenotypes in the Hyderabad experiment and observed, where predictions based on genome associations with phenotypes in fivefold cross-validation.
Table S18. Pearson’s correlation coefficients between predicted phenotypes in the Caniato et al. (44) experiment and observed, where predictions based on genome associations with phenotypes in fivefold cross-validation.
Table S19. Top 1000 SNPs associated with harvest index plasticity in Austin using SNP associations with precipitation of the warmest quarter as priors.
Table S20. Top 1000 SNPs associated with relative net root growth (comparing control treatment with Al toxic treatment) in Caniato et al. (44) experiment, using SNP associations with topsoil pH as priors.
Table S21. Top 1000 SNPs associated with panicle weight plasticity in Hyderabad, using SNP associations with growing season length as priors.
REFERENCES AND NOTES
- 1.Huang X., Zhao Y., Wei X., Li C., Wang A., Zhao Q., Li W., Guo Y., Deng L., Zhu C., Fan D., Lu Y., Weng Q., Liu K., Zhou T., Jing Y., Si L., Dong G., Huang T., Lu T., Feng Q., Qian Q., Li J., Han B., Genome-wide association study of flowering time and grain yield traits in a worldwide collection of rice germplasm. Nat. Genet. 44, 32–39 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Hung H. Y., Shannon L. M., Tian F., Bradbury P. J., Chen C., Flint-Garcia S. A., McMullen M. D., Ware D., Buckler E. S., Doebley J. F., Holland J. B., ZmCCT and the genetic basis of day-length adaptation underlying the postdomestication spread of maize. Proc. Natl. Acad. Sci. U.S.A. 109, E1913–E1921 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pallotta M., Schnurbusch T., Hayes J., Hay A., Baumann U., Paull J., Langridge P., Sutton T., Molecular basis of adaptation to high soil boron in wheat landraces and elite cultivars. Nature 514, 88–91 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Lobell D. B., Burke M. B., Tebaldi C., Mastrandrea M. D., Falcon W. P., Naylor R. L., Prioritizing climate change adaptation needs for food security in 2030. Science 319, 607–610 (2008). [DOI] [PubMed] [Google Scholar]
- 5.N. I. Vavilov, Origin and Geography of Cultivated Plants (Cambridge Univ. Press, Cambridge, ed. 1, 2009). [Google Scholar]
- 6.A. Blum, Plant Breeding for Water-Limited Environments (Springer Publishing, New York, 2010). [Google Scholar]
- 7.Xu K., Xu X., Fukao T., Canlas P., Maghirang-Rodriguez R., Heuer S., Ismail A. M., Bailey-Serres J., Ronald P. C., Mackill D. J., Sub1A is an ethylene-response-factor-like gene that confers submergence tolerance to rice. Nature 442, 705–708 (2006). [DOI] [PubMed] [Google Scholar]
- 8.Magalhaes J. V., Liu J., Guimarães C. T., Lana U. G., Alves V. M., Wang Y. H., Schaffert R. E., Hoekenga O. A., Piñeros M. A., Shaff J. E., Klein P. E., Carneiro N. P., Coelho C. M., Trick H. N., Kochian L. V., A gene in the multidrug and toxic compound extrusion (MATE) family confers aluminum tolerance in sorghum. Nat. Genet. 39, 1156–1161 (2007). [DOI] [PubMed] [Google Scholar]
- 9.Magalhaes J. V., Liu J., Guimarães C. T., Lana U. G., Alves V. M., Wang Y. H., Schaffert R. E., Hoekenga O. A., Piñeros M. A., Shaff J. E., Klein P. E., Carneiro N. P., Coelho C. M., Trick H. N., Kochian L. V., Sorghum stay-green QTL individually reduce post-flowering drought-induced leaf senescence. J. Exp. Bot. 58, 327–338 (2007). [DOI] [PubMed] [Google Scholar]
- 10.Gamuyao R., Chin J. H., Pariasca-Tanaka J., Pesaresi P., Catausan S., Dalid C., Slamet-Loedin I., Tecson-Mendoza E. M., Wissuwa M., Heuer S., The protein kinase Pstol1 from traditional rice confers tolerance of phosphorus deficiency. Nature 488, 535–539 (2012). [DOI] [PubMed] [Google Scholar]
- 11.Heffner E. L., Sorrells M. E., Jannink J.-L., Genomic selection for crop improvement. Crop. Sci. 49, 1–12 (2009). [Google Scholar]
- 12.Norton G. J., Douglas A., Lahner B., Yakubova E., Guerinot M. L., Pinson S. R., Tarpley L., Eizenga G. C., McGrath S. P., Zhao F. J., Islam M. R., Islam S., Duan G., Zhu Y., Salt D. E., Meharg A. A., Price A. H., Genome wide association mapping of grain arsenic, copper, molybdenum and zinc in rice (Oryza sativa L.) grown at four international field sites. PLOS One 9, e89685 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haussmann B. I. G., Fred Rattunde H., Weltzien-Rattunde E., Traoré P. S. C., vom Brocke K., Parzies H. K., Breeding strategies for adaptation of pearl millet and sorghum to climate variability and change in West Africa. J. Agron. Crop Sci. 198, 327–339 (2012). [Google Scholar]
- 14.Varshney R. K., Ribaut J. M., Buckler E. S., Tuberosa R., Rafalski J. A., Langridge P., Can genomics boost productivity of orphan crops? Nat. Biotechnol. 30, 1172–1176 (2012). [DOI] [PubMed] [Google Scholar]
- 15.Turner T. L., Bourne E. C., Von Wettberg E. J., Hu T. T., Nuzhdin S. V., Population resequencing reveals local adaptation of Arabidopsis lyrata to serpentine soils. Nat. Genet. 42, 260–263 (2010). [DOI] [PubMed] [Google Scholar]
- 16.Jones F. C., Grabherr M. G., Chan Y. F., Russell P., Mauceli E., Johnson J., Swofford R., Pirun M., Zody M. C., White S., Birney E., Searle S., Schmutz J., Grimwood J., Dickson M. C., Myers R. M., Miller C. T., Summers B. R., Knecht A. K., Brady S. D., Zhang H., Pollen A. A., Howes T., Amemiya C. Broad Institute Genome Sequencing Platform & Whole Genome Assembly Team Baldwin J., Bloom T., Jaffe D. B., Nicol R., Wilkinson J., Lander E. S., Di Palma F., Lindblad-Toh K., Kingsley D. M., The genomic basis of adaptive evolution in threespine sticklebacks. Nature 484, 55–61 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hancock A. M., Brachi B., Faure N., Horton M. W., Jarymowycz L. B., Sperone F. G., Toomajian C., Roux F., Bergelson J., Adaptation to climate across the Arabidopsis thaliana genome. Science 334, 83–86 (2011). [DOI] [PubMed] [Google Scholar]
- 18.Yoder J. B., Stanton-Geddes J., Zhou P., Briskine R., Young N. D., Tiffin P., Genomic signature of adaptation to climate in Medicago truncatula. Genetics 196, 1263–1275 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.C. T. Kimber, in Sorghum: Origin, History, Technology, and Production, C. W. Smith, R. A. Frederiksen, Eds. (John Wiley and Sons, New York, 2000), pp. 3–98. [Google Scholar]
- 20.National Research Council, Lost Crops of Africa: Volume I: Grains (National Academy Press, Washington, DC, 1996). [Google Scholar]
- 21.N. Ramankutty, T. Hertel, H.-L. Lee, S. K. Rose, Global agricultural land use data for integrated assessment modeling, in Human-Induced Climate Change: An Interdisciplinary Assessment, M. E. Schlesinger, H. S. Kheshgi, J. Smith, F. C. de la Chesnaye, J. M. Reilly, T. Wilson, C. Kolstad, Eds. (Cambridge Univ. Press, Cambridge, 2007), pp. 252–265. [Google Scholar]
- 22.Elshire R. J., J. C. Glaubitz, Q. Sun, J. A. Poland, K. Kawamoto, E. S. Buckler, S. E. Mitchell, A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLOS One 6, e19379 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morris G. P., Ramu P., Deshpande S. P., Thomas Hash C., Shah T., Upadhyaya H. D., Riera-Lizarazu O., Brown P. J., Acharya C. B., Mitchell S. E., Harriman J., Glaubitz J. C., Buckler E. S., Kresovich S., Population genomic and genome-wide association studies of agroclimatic traits in sorghum. Proc. Natl. Acad. Sci. U.S.A. 110, 453–458 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.H. Doggett, Sorghum (Longman Scientific & Technical, Harlow, UK, 1988). [Google Scholar]
- 25.Murphy R. L., Klein R. R., Morishige D. T., Brady J. A., Rooney W. L., Miller F. R., Dugas D. V., Klein P. E., Mullet J. E., Coincident light and clock regulation of pseudoresponse regulator protein 37 (PRR37) controls photoperiodic flowering in sorghum. Proc. Natl. Acad. Sci. U.S.A. 108, 16469–16474 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu Y., Li X., Xiang W., Zhu C., Lin Z., Wu Y., Li J., Pandravada S., Ridder D. D., Bai G., Wang M. L., Trick H. N., Bean S. R., Tuinstra M. R., Tesso T. T., Yu J., Presence of tannins in sorghum grains is conditioned by different natural alleles of Tannin1. Proc. Natl. Acad. Sci. U.S.A. 109, 10281–10286 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Esele J. P., Frederiksen R. A., Miller F. R., The association of genes controlling caryopsis traits with grain mold resistance in sorghum. Phytopathology 83, 490–495 (1993). [Google Scholar]
- 28.Harris H. B., Burns R. E., Influence of tannin content on preharvest seed germination in sorghum. Agron. J. 62, 835 (1970). [Google Scholar]
- 29.Morris G. P., Rhodes D. H., Brenton Z., Ramu P., Thayil V. M., Deshpande S., Hash C. T., Acharya C., Mitchell S. E., Buckler E. S., Yu J., Kresovich S., Dissecting genome-wide association signals for loss-of-function phenotypes in sorghum flavonoid pigmentation traits. G3 3, 2085–2094 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fournier-Level A., Korte A., Cooper M. D., Nordborg M., Schmitt J., Wilczek A. M., A map of local adaptation in Arabidopsis thaliana. Science 334, 86–89 (2011). [DOI] [PubMed] [Google Scholar]
- 31.Kang H. M., Zaitlen N. A., Wade C. M., Kirby A., Heckerman D., Daly M. J., Eskin E., Efficient control of population structure in model organism association mapping. Genetics 178, 1709–1723 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bergelson J., Roux F., Towards identifying genes underlying ecologically relevant traits in Arabidopsis thaliana. Nat. Rev. Genet. 11, 867–879 (2010). [DOI] [PubMed] [Google Scholar]
- 33.J. A. Endler, Natural Selection in the Wild (Princeton Univ. Press, Princeton, NJ, 1986). [Google Scholar]
- 34.van den Wollenberg A. L., Redundancy analysis an alternative for canonical correlation analysis. Psychometrika 42, 207–219 (1977). [Google Scholar]
- 35.Borcard D., Legendre P., All-scale spatial analysis of ecological data by means of principal coordinates of neighbour matrices. Ecol. Model. 153, 51–68 (2002). [Google Scholar]
- 36.Peres-Neto P. R., Legendre P., Dray S., Borcard D., Variation partitioning of species data matrices: Estimation and comparison of fractions. Ecology 87, 2614–2625 (2006). [DOI] [PubMed] [Google Scholar]
- 37.Lasky J. R., Des Marais D. L., McKay J. K., Richards J. H., Juenger T. E., Keitt T. H., Characterizing genomic variation of Arabidopsis thaliana: The roles of geography and climate. Mol. Ecol. 21, 5512–5529 (2012). [DOI] [PubMed] [Google Scholar]
- 38.Endresen D. T. F., Street K., Mackay M., Bari A., De Pauw E., Predictive association between biotic stress traits and eco-geographic data for wheat and barley landraces. Crop. Sci. 51, 2036 (2011). [Google Scholar]
- 39.Endelman J. B., Ridge regression and other kernels for genomic selection with R package rrBLUP. Plant Genome J. 4, 250 (2011). [Google Scholar]
- 40.Des Marais D. L., Hernandez K. M., Juenger T. E., Genotype-by-environment interaction and plasticity: Exploring genomic responses of plants to the abiotic environment. Annu. Rev. Ecol. Evol. Syst. 44, 5–29 (2013). [Google Scholar]
- 41.Chenu K., Cooper M., Hammer G. L., Mathews K. L., Dreccer M. F., Chapman S. C., Environment characterization as an aid to wheat improvement: Interpreting genotype–environment interactions by modelling water-deficit patterns in North-Eastern Australia. J. Exp. Bot. 62, 1743–1755 (2011). [DOI] [PubMed] [Google Scholar]
- 42.Sabadin P. K., Malosetti M., Boer M. P., Tardin F. D., Santos F. G., Guimarães C. T., Gomide R. L., Andrade C. L., Albuquerque P. E., Caniato F. F., Mollinari M., Margarido G. R., Oliveira B. F., Schaffert R. E., Garcia A. A., van Eeuwijk F. A., Magalhaes J. V., Studying the genetic basis of drought tolerance in sorghum by managed stress trials and adjustments for phenological and plant height differences. Theor. Appl. Genet. 124, 1389–1402 (2012). [DOI] [PubMed] [Google Scholar]
- 43.Borrell A. K., Hammer G. L., Henzell R. G., Does maintaining green leaf area in sorghum improve yield under drought? II. Dry matter production and yield. Crop. Sci. 40, 1037 (2000). [Google Scholar]
- 44.Caniato F. F., Guimarães C. T., Hamblin M., Billot C., Rami J. F., Hufnagel B., Kochian L. V., Liu J., Garcia A. A., Hash C. T., Ramu P., Mitchell S., Kresovich S., Oliveira A. C., de Avellar G., Borém A., Glaszmann J. C., Schaffert R. E., Magalhaes J. V., The relationship between population structure and aluminum tolerance in cultivated sorghum. PLOS One 6, e20830 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jannink J.-L., Lorenz A. J., Iwata H., Genomic selection in plant breeding: From theory to practice. Brief. Funct. Genomics 9, 166–177 (2010). [DOI] [PubMed] [Google Scholar]
- 46.Mace E. S., Tai S., Gilding E. K., Li Y., Prentis P. J., Bian L., Campbell B. C., Hu W., Innes D. J., Han X., Cruickshank A., Dai C., Frère C., Zhang H., Hunt C. H., Wang X., Shatte T., Wang M., Su Z., Li J., Lin X., Godwin I. D., Jordan D. R., Wang J., Whole-genome sequencing reveals untapped genetic potential in Africa’s indigenous cereal crop sorghum. Nat. Commun. 4, 2320 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Segura V., Vilhjálmsson B. J., Platt A., Korte A., Seren Ü., Long Q., Nordborg M., An efficient multi-locus mixed-model approach for genome-wide association studies in structured populations. Nat. Genet. 44, 825–830 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kass R. E., Raftery A. E., Bayes factors. J. Am. Stat. Assoc. 90, 773–795 (1995). [Google Scholar]
- 49.Stephens M., Balding D. J., Bayesian statistical methods for genetic association studies. Nat. Rev. Genet. 10, 681–690 (2009). [DOI] [PubMed] [Google Scholar]
- 50.Rauf M., Arif M., Dortay H., Matallana-Ramírez L. P., Waters M. T., Gil Nam H., Lim P. O., Mueller-Roeber B., Balazadeh S., ORE1 balances leaf senescence against maintenance by antagonizing G2-like-mediated transcription. EMBO Rep. 14, 382–388 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Balazadeh S., Kwasniewski M., Caldana C., Mehrnia M., Zanor M. I., Xue G. P., Mueller-Roeber B., ORS1, an H2O2-responsive NAC transcription factor, controls senescence in Arabidopsis thaliana. Mol. Plant 4, 346–360 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Redillas M. C., Jeong J. S., Kim Y. S., Jung H., Bang S. W., Choi Y. D., Ha S. H., Reuzeau C., Kim J. K., The overexpression of OsNAC9 alters the root architecture of rice plants enhancing drought resistance and grain yield under field conditions. Plant Biotechnol. J. 10, 792–805 (2012). [DOI] [PubMed] [Google Scholar]
- 53.Distelfeld A., Avni R., Fischer A. M., Senescence, nutrient remobilization, and yield in wheat and barley. J. Exp. Bot. 65, 3783–3798 (2014). [DOI] [PubMed] [Google Scholar]
- 54.Zhu X. F., Wan J. X., Sun Y., Shi Y. Z., Braam J., Li G. X., Zheng S. J., Xyloglucan endotransglucosylase-hydrolase17 interacts with xyloglucan endotransglucosylase-hydrolase31 to confer xyloglucan endotransglucosylase action and affect aluminum sensitivity in Arabidopsis. Plant Physiol. 165, 1566–1574 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang X. Y., Zeng Z. H., Yan J. Y., Fan W., Bian H. W., Zhu M. Y., Yang J. L., Zheng S. J., Association of specific pectin methylesterases with Al-induced root elongation inhibition in rice. Physiol. Plant. 148, 502–511 (2013). [DOI] [PubMed] [Google Scholar]
- 56.Jiang S., Zhang D., Wang L., Pan J., Liu Y., Kong X., Zhou Y., Li D., A maize calcium-dependent protein kinase gene, ZmCPK4, positively regulated abscisic acid signaling and enhanced drought stress tolerance in transgenic Arabidopsis. Plant Physiol. Biochem. 71, 112–120 (2013). [DOI] [PubMed] [Google Scholar]
- 57.Aitken S. N., Whitlock M. C., Assisted gene flow to facilitate local adaptation to climate change. Annu. Rev. Ecol. Evol. Syst. 44, 367–388 (2013). [Google Scholar]
- 58.van Oppen M. J. H., Oliver J. K., Putnam H. M., Gates R. D., Building coral reef resilience through assisted evolution. Proc. Natl. Acad. Sci. U.S.A. 112, 2307–2313 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Glaubitz J. C., Casstevens T. M., Lu F., Harriman J., Elshire R. J., Sun Q., Buckler E. S., TASSEL-GBS: A high capacity genotyping by sequencing analysis pipeline. PLOS One 9, e90346 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Paterson A. H., Bowers J. E., Bruggmann R., Dubchak I., Grimwood J., Gundlach H., Haberer G., Hellsten U., Mitros T., Poliakov A., Schmutz J., Spannagl M., Tang H., Wang X., Wicker T., Bharti A. K., Chapman J., Feltus F. A., Gowik U., Grigoriev I. V., Lyons E., Maher C. A., Martis M., Narechania A., Otillar R. P., Penning B. W., Salamov A. A., Wang Y., Zhang L., Carpita N. C., Freeling M., Gingle A. R., Hash C. T., Keller B., Klein P., Kresovich S., McCann M. C., Ming R., Peterson D. G., Mehboob-ur-Rahman , Ware D, Westhoff P, Mayer KF, Messing J, Rokhsar DS, The Sorghum bicolor genome and the diversification of grasses. Nature 457, 551–556 (2009). [DOI] [PubMed] [Google Scholar]
- 61.Goodstein D. M., Shu S., Howson R., Neupane R., Hayes R. D., Fazo J., Mitros T., Dirks W., Hellsten U., Putnam N., Rokhsar D. S., Phytozome: A comparative platform for green plant genomics. Nucleic Acids Res. 40, D1178–D1186 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li H., Durbin R., Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hijmans R. J., Cameron S. E., Parra J. L., Jones P. G., Jarvis A., Very high resolution interpolated climate surfaces for global land areas. Int. J. Climatol. 25, 1965–1978 (2005). [Google Scholar]
- 64.Zomer R. J., Trabucco A., Bossio D. A., Verchot L. V., Climate change mitigation: A spatial analysis of global land suitability for clean development mechanism afforestation and reforestation. Agric. Ecosyst. Environ. 126, 67–80 (2008). [Google Scholar]
- 65.New M., Lister D., Hulme M., Makin I., A high-resolution data set of surface climate over global land areas. Clim. Res. 21, 1–25 (2002). [Google Scholar]
- 66.Kalnay E., Kanamitsu M., Kistler R., Collins W., Deaven D., Gandin L., Iredell M., Saha S., White G., Woollen J., Zhu Y., Leetmaa A., Reynolds R., Chelliah M., Ebisuzaki W., Higgins W., Janowiak J., Mo K. C., Ropelewski C., Wang J., Jenne Roy, Joseph Dennis, The NCEP/NCAR 40-year reanalysis project. Bull. Am. Meteorol. Soc. 77, 437–471 (1996). [Google Scholar]
- 67.K. A. Dunne, C. J. Willmott, Global distribution of plant-extractable water capacity of soil (Dunne); http://daac.ornl.gov/SOILS/guides/DunneSoil.html.
- 68.Fan Y., Li H., Miguez-Macho G., Global patterns of groundwater table depth. Science 339, 940–943 (2013). [DOI] [PubMed] [Google Scholar]
- 69.Sanchez P. A., Palm C. A., Buol S. W., Fertility capability soil classification: A tool to help assess soil quality in the tropics. Geoderma 114, 157–185 (2003). [Google Scholar]
- 70.F. Nachtergaele, N. Batjes, Harmonized World Soil Database (FAO, Rome, 2012); http://www.fao.org/nr/water/docs/harm-world-soil-dbv7cv.Pdf.
- 71.Casa A. M., Pressoir G., Brown P. J., Mitchell S. E., Rooney W. L., Tuinstra M. R., Franks C. D., Kresovich S., Community resources and strategies for association mapping in sorghum. Crop. Sci. 48, 30–40 (2008). [Google Scholar]
- 72.Upadhyaya H. D., Pundir R. P. S., Dwivedi S. L., Gowda C. L. L., Reddy V. G., Singh S., Developing a mini core collection of sorghum for diversified utilization of germplasm. Crop. Sci. 49, 1769–1780 (2009). [Google Scholar]
- 73.Manel S., Poncet B. N., Legendre P., Gugerli F., Holderegger R., Common factors drive adaptive genetic variation at different spatial scales in Arabis alpina. Mol. Ecol. 19, 3824–3835 (2010). [DOI] [PubMed] [Google Scholar]
- 74.R. J. Hijmans, E. Williams, C. Vennes, geosphere: Spherical trigonometry; http://cran.r-project.org/web/packages/geosphere/index.html.
- 75.Lasky J. R., Des Marais D. L., Lowry D. B., Povolotskaya I., McKay J. K., Richards J. H., Keitt T. H., Juenger T. E., Natural variation in abiotic stress responsive gene expression and local adaptation to climate in Arabidopsis thaliana. Mol. Biol. Evol. 31, 2283–2296 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lande R., Thompson R., Efficiency of marker-assisted selection in the improvement of quantitative traits. Genetics 124, 743–756 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Endelman J. B., Jannink J.-L., Shrinkage estimation of the realized relationship matrix. G3 2, 1405–1413 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Murray F. W., On the computation of saturation vapor pressure. J. Appl. Meteorol. 6, 203–204 (1967). [Google Scholar]
- 79.Cerovic Z. G., Masdoumier G., Ghozlen N. B., Latouche G., A new optical leaf-clip meter for simultaneous non-destructive assessment of leaf chlorophyll and epidermal flavonoids. Physiol. Plant. 146, 251–260 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.D. Bates, M. Maechler, B. M. Bolker, S. Walker, lme4: Linear mixed-effects models using Eigen and S4; http://cran.r-project.org/web/packages/lme4/index.html.
- 81.Shimizu K. K., Purugganan M. D., Evolutionary and ecological genomics of Arabidopsis. Plant Physiol. 138, 578–584 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/1/6/e1400218/DC1
Fig. S1. Landrace accessions included in the study classified into botanical races based on morphological classification (five new world accessions not shown).
Fig. S2. Rainout shelter where plants were grown in Austin.
Fig. S3. Seedlings planted in the Austin experiment.
Fig. S4. Plants growing in Austin.
Fig. S5. A representative accession (IS 25836) under irrigated (left) and imposed terminal drought (right) conditions at experimental plot in India.
Fig. S6. Proportion of total SNP variation among accessions with known collection locations (excluding spatial outlier landraces from the Americas, China, and Southeast Asia/Oceania) explained by spatial structure or environmental variables.
Fig. S7. Predictions of phenotypes averaged across well-watered and drought conditions from drought treatment across growing season in Austin, United States.
Fig. S8. Predictions of phenotype change between well-watered and drought conditions from drought treatment across growing season in Austin, United States.
Fig. S9. Predictions of phenotypes averaged across well-watered and drought conditions from drought treatment late in growing season in Hyderabad, India.
Fig. S10. Predictions of phenotype change between well-watered and drought conditions from drought treatment late in growing season in Hyderabad, India.
Fig. S11. GWAS for harvest index plasticity (harvest index in wet/dry) in U.S. experiment, using SNP associations with precipitation in the warmest quarter as a prior.
Fig. S12. GWAS for panicle weight plasticity (panicle weight in wet − dry) in India experiment, using SNP associations with growing season length as a prior.
Fig. S13. GWAS for root growth plasticity (growth in control/Al toxic) in published aluminum toxicity experiment (44), using SNP associations with topsoil pH as a prior.
Table S1. Landraces studied and environment of origin data.
Table S2. Accession phenotypes from experiment in Austin, United States.
Table S3. Mean phenotypes across the 2 years of the experiment in Hyderabad, India.
Table S4. Spearman’s rank correlation test results for two SNPs that tag known candidate genes potentially involved in local adaptation.
Table S5. EMMA t test results for two SNPs that tag known candidate genes potentially involved in local adaptation.
Table S6. Predictions for each accession in the Austin experiment based on SNP-environment associations in the landrace panel.
Table S7. Predictions for each accession in the Hyderabad experiment based on SNP-environment associations in the landrace panel.
Table S8. Predictions for each accession in the Caniato et al. (44) experiment based on SNP-environment associations in the landrace panel.
Table S9. Predicted environments for accession in the three experiments based on kinship associations with environment of landraces (gBLUP).
Table S10. Pearson’s correlations between predictions based on environment-genome associations and phenotypes in Austin.
Table S11. Pearson’s correlations between predictions based on environment-genome associations and phenotypes in Hyderabad.
Table S12. Pearson’s correlations between predictions based on environment-genome associations and phenotypes in the Caniato et al. (44) Al toxicity experiment.
Table S13. Pearson’s correlations between phenotypes and environment of origin (where known) for landraces in the Austin experiment.
Table S14. Pearson’s correlations between phenotypes and environment of origin (where known) for landraces in the Hyderabad experiment.
Table S15. Pearson’s correlations between relative net root growth and environment of origin (where known) for landraces in the Caniato et al. (44) experiment.
Table S16. Pearson’s correlation coefficients between predicted phenotypes in the Austin experiment and observed, where predictions based on genome associations with phenotypes in fivefold cross-validation.
Table S17. Pearson’s correlation coefficients between predicted phenotypes in the Hyderabad experiment and observed, where predictions based on genome associations with phenotypes in fivefold cross-validation.
Table S18. Pearson’s correlation coefficients between predicted phenotypes in the Caniato et al. (44) experiment and observed, where predictions based on genome associations with phenotypes in fivefold cross-validation.
Table S19. Top 1000 SNPs associated with harvest index plasticity in Austin using SNP associations with precipitation of the warmest quarter as priors.
Table S20. Top 1000 SNPs associated with relative net root growth (comparing control treatment with Al toxic treatment) in Caniato et al. (44) experiment, using SNP associations with topsoil pH as priors.
Table S21. Top 1000 SNPs associated with panicle weight plasticity in Hyderabad, using SNP associations with growing season length as priors.