

New microscopy method reveals local variations of functional state of membrane protein associated with cancer cell growth.
Keywords: HER2 homodimers, breast cancer, SKBR3 cells, resting cells, membrane ruffles, correlative fluorescence- and electron microscopy, electron microscopy in liquid
Abstract
The formation of HER2 homodimers plays an important role in breast cancer aggressiveness and progression; however, little is known about its localization. We have studied the intra- and intercellular variation of HER2 at the single-molecule level in intact SKBR3 breast cancer cells. Whole cells were visualized in hydrated state with correlative fluorescence microscopy and environmental scanning electron microscopy (ESEM). The locations of individual HER2 receptors were detected using an anti-HER2 affibody in combination with a quantum dot (QD), a fluorescent nanoparticle. Fluorescence microscopy revealed considerable differences of HER2 membrane expression between individual cells and between different membrane regions of the same cell (that is, membrane ruffles and flat areas). Subsequent ESEM of the corresponding cellular regions provided images of individually labeled HER2 receptors. The high spatial resolution of 3 nm and the close proximity between the QD and the receptor allowed quantifying the stoichiometry of HER2 complexes, distinguishing between monomers, dimers, and higher-order clusters. Downstream data analysis based on calculating the pair correlation function from receptor positions showed that cellular regions exhibiting membrane ruffles contained a substantial fraction of HER2 in homodimeric state. Larger-order clusters were also present. Membrane areas with homogeneous membrane topography, on the contrary, displayed HER2 in random distribution. Second, HER2 homodimers appeared to be absent from a small subpopulation of cells exhibiting a flat membrane topography, possibly resting cells. Local differences in homodimer presence may point toward functional differences with possible relevance for studying metastasis and drug response.
INTRODUCTION
HER2, also known as ErbB2, is a transmembrane receptor tyrosine kinase belonging to the epidermal growth factor receptor (EGFR) family and is overexpressed in certain types of breast cancer (1) and other cancer types (2). In patients with HER2-overexpressing tumors, intratumoral heterogeneity of HER2 expression was found to be associated with therapeutic resistance and reduced disease-free survival times (3). HER2, being an orphan receptor, does not form homodimers in response to ligands, but forms ligand-independent homodimers in HER2-overexpressing cells (4). It also associates in heterodimers with other members of the receptor family (4), thereby contributing significantly to a dysregulation of intracellular signaling (5) and of cell growth (6). In principle, the amount of homodimers in a cell population can be quantified, for example with biochemical methods using pooled cellular material. However, this seems to be practically difficult for the HER2 homodimer because its extracellular domain does not homodimerize in solution (7, 8). Moreover, no information is available about the localization of HER2 homodimers. Of major interest is the question whether HER2 homodimers distribute homogeneously over the entire plasma membrane or whether distinct functional membrane regions are associated with a specific HER2 stoichiometry. Also, it is important to assess the intercellular variability of HER2 dimer abundance within a cancer cell population. Greater understanding of HER2’s spatial distribution may provide new insights into mechanisms controlling cancer cell responses toward HER2-targeting drugs.
Here, we describe a stoichiometric study that visualized HER2 at the molecular level in intact SKBR3 breast cancer cells, shedding light on its intra- and intercellular distribution patterns. The human breast cancer cell line SKBR3 overexpresses HER2 and has previously served as a model system of HER2+ breast cancer in numerous in vitro studies (9, 10). Whole cells were visualized in hydrated state with correlative fluorescence microscopy and environmental scanning electron microscopy (ESEM). Electron microscopy of labeled proteins in whole cells in liquid is a relatively new approach for studying protein localization and protein function (11–14). A protein label was developed to detect the HER2 position without interfering with its function, using an anti-HER2 affibody (15, 16) conjugated to a quantum dot (QD) (Fig. 1). Correlative fluorescence microscopy yielded information regarding the heterogeneity of HER2 membrane expression within the cancer cell population. Selected cellular areas were subsequently studied at nanoscale spatial resolution using ESEM combined with scanning transmission electron microscopy (STEM) detection, elucidating the organization of HER2 in the plasma membrane.
Fig. 1. Schematic representation of HER2 labeling with affibody-QD probes and correlative microscopy of whole cells in hydrated state.
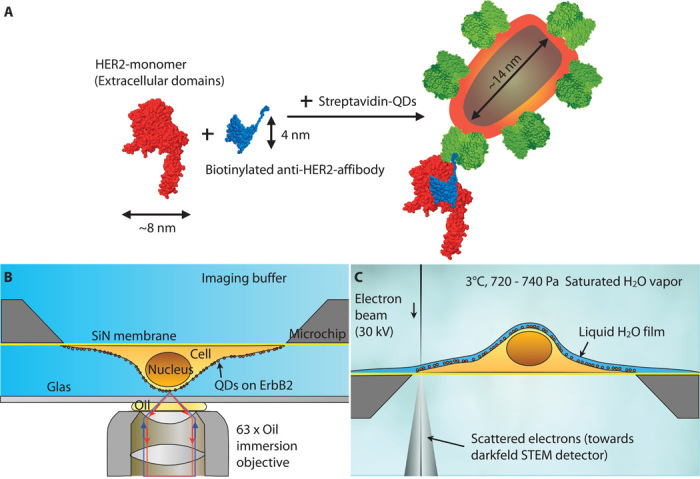
(A) The biotinylated affibody (blue) directed against HER2 (red) binds to a single epitope on the extracellular part of the membrane receptor. The single biotin moiety of the affibody binds a streptavidin (green)–conjugated QD. QDs can emit bright fluorescence signals, and their electron-dense core can be detected with electron microscopy. (B) Cells were grown on a silicon nitride (SiN) membrane supported by a silicon chip. A microchip with QD-labeled and fixed cells was positioned upside down in a saline-filled glass-bottom dish. Fluorescence imaging was performed with an oil immersion lens on an inverted bright-field microscope. (C) For ESEM, the same microchip was positioned upright on a cooled stage and kept in a saturated water vapor atmosphere. The hydrated cells were covered with a thin layer of purified water. Contrast was obtained on the QDs using the STEM detector located underneath the sample.
RESULTS
HER2 receptor membrane expression differs between cells and cellular regions as revealed by labeling of HER2 with QDs
To study the distribution of HER2 in the plasma membrane of SKBR3 cells at the single molecule level, we developed a two-step protocol. The label contained an affibody attached via a short biotin-streptavidin bond to a QD. Affibodies represent a new type of non–immunoglobulin-derived affinity protein with 10- to 20-fold lower molecular weight than antibodies (17). One of the main qualities of the affibody is its small dimension of 5 × 4 × 3 nm3 as determined from the x-ray structure in the Protein Data Bank (PDB entry: 3MZW). The affibody-QD label is bound to HER2 in a 1:1 ratio, on account of a single binding epitope at HER2 for the affibody (18) with a biotinylated C terminus. The anti-HER2 affibody binding site is at the side of the protein, at domain III and close to domain IV (18), distant from the dimerization side (7). After exposure to the anti-HER2 affibody, cells were subjected to fixation. Only then were the cells incubated with streptavidin-conjugated QDs, such that labeling-induced clustering (19) was avoided. Specific HER2 labeling was thus obtained (figs. S1 and S2).
Figure 2A shows an example of a confluent layer of SKBR3 cells grown on a silicon microchip. The rectangular area (highlighted with dashed line) outlines the viewing window in the microchip used for transmission electron microscopy. Several cells in this area were selected for a detailed analysis (nos. 1 to 7). The overlaid DIC image of the window in the viewing area provides a three-dimensional impression of the cell surfaces and reveals that most cells had a ruffled plasma membrane. All cells expressed HER2; however, individual cells exhibited discrete HER2 distribution patterns. Several cells showed an almost homogeneous membrane expression of HER2 over their entire surface, whereas other cells preferentially expressed HER2 on distinct, elongated plasma membrane regions extending toward the outer edges of cells. Likewise, the overall abundance of HER2 in the plasma membrane varied significantly between individual cells.
Fig. 2. Correlative light and electron microscopic overview images of affibody-QD–labeled HER2 on SKBR3 human breast cancer cells.
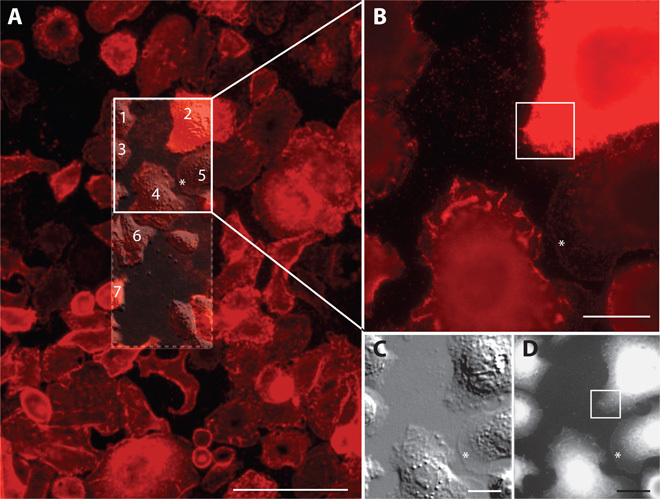
(A) Fluorescence overview image of the central region of a microchip, showing several dozen cells. Individual cells exhibit a high degree of heterogeneity in their morphology and HER2 membrane expression. The location of the SiN membrane window is indicated as a rectangular dashed outline. A differential interference contrast (DIC) image of the window area is overlaid. The DIC image provides an impression of the three-dimensional topography. Scale bar, 100 μm. Cells from which ESEM-STEM images were recorded are indicated by numbers. (B) Higher-resolution fluorescence image, recorded with a 63× oil immersion lens, of the cells in the upper half of the window area, marked as a solid line rectangle in (A). The fluorescence signal of cell #2 indicated a high level of HER2 membrane expression, whereas cell #5 showed only weak HER2 membrane expression. Cell #4 concentrated HER2 on membrane ruffles. The boxed area is shown in Fig. 3A. (C) DIC image of the same region as in (B), depicting the membrane topography in greater detail. (D) ESEM-STEM image from the same region as shown in (B) and (C) (×1000 magnification). Scale bars, 20 μm.
HER2 heterogeneity became even more apparent in the high-resolution fluorescence image shown in Fig. 2B. Here, the focus was adjusted to image peripheral membrane areas. Whereas the cell in the upper right corner (#2) displays very bright and almost saturated signals, the cell in the central part of the imaging area (#4) shows numerous elongated bright areas. The DIC image, shown in Fig. 2C, indicates that these areas represent membrane ruffles, suggesting that the HER2 receptor colocalized with membrane ruffles.
A small number of cells, presumably resting cells (~7% of all cells observed in the experiments), exhibited a different membrane topography, for example, cell #5 in Fig. 2A. This cell type had a weaker HER2 membrane expression level (fig. S3) compared to cells with membrane ruffles, and showed a mostly flat membrane topography with a 5- to 10-μm-wide skirt-like membrane flattening out toward the cell edge (marked with an asterisk in Fig. 2), in which membrane ruffles were absent in contrast to most of the cells exhibiting continuous and dynamic membrane ruffling (fig. S4). This type of cell was referred to as flat cell.
ESEM of hydrated SKBR3 cells enables study of HER2 distribution patterns on the single molecule level
To obtain quantitative information about the distribution of HER2 at the level of individual receptors, we studied intact SKBR3 cells using ESEM-STEM in liquid state. Several overview ESEM-STEM images were recorded for orientation. Figure 2D shows a low-resolution—in terms of electron microscopy—dark-field ESEM-STEM overview image of the selected region in Fig. 2A. The contours of cells are recognized, as well as membrane ruffles and vesicles. However, the structure cannot be resolved in the thicker regions around the nucleus on account of excessive electron scattering.
Figure 3A shows the fluorescence signal of the lower left tip of the strongly HER2-expressing cell (#2), indicated by a box in Fig. 2B. The heterogeneous pattern of signal intensities in the fluorescence image indicates the presence of membrane ruffles. Figure 3B represents the ESEM-STEM image recorded in the same region at ×15,000 magnification. The uneven pattern of electron-dense regions, emerging bright in the STEM dark-field contrast, along with less dense and hence dark-appearing regions, points toward an irregular membrane topography. Details of cell borders, protruding lamellipodia, and electron-dense areas of membrane ruffles are clearly discernible. At ×75,000 magnification (Fig. 3D), QD labels became visible as bright, bullet-shaped dots representing individual HER2 receptors. The achieved spatial resolution amounted to 3 nm for ESEM-STEM imaging (fig. S5) and was sufficient to resolve the QDs of dimensions of 6 × 14 nm2 (fig. S6). Most of the HER2-derived QD signals appeared to localize to electron-dense cellular structures, visible as bright areas in the dark-field STEM contrast and identified as membrane ruffles in the DIC image (Fig. 2C).
Fig. 3. Correlative light and electron microscopic images showing the distribution of QD-labeled HER2 on an SKBR3 cell.
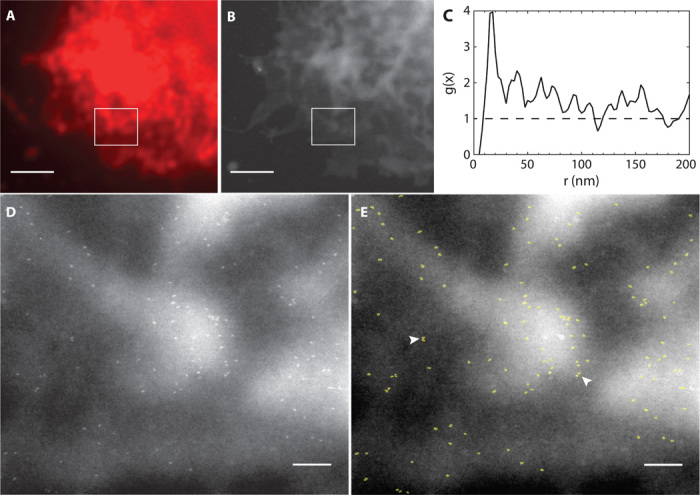
(A) Selected area of the fluorescence image in Fig. 2B. This region contained membrane ruffles as shown in the DIC image of Fig. 2C. (B) ESEM-STEM image at the same area, recorded at ×15,000 magnification. (C) Pair correlation function g(x) (line) determined from the 122 detected label positions in the rectangular region in (B). The dashed line at unity serves as a guide to the eye for a random distribution. (D) STEM image recorded of the boxed region shown in (B) at ×75,000 magnification. The localization of individual HER2 receptors became visible as the bright, bullet-shaped QDs. (E) Automatically detected labels were outlined in light green. Numerous pairs of HER2 were observed (two examples are indicated by arrowheads). Scale bars, 2 μm (A and B); 200 nm (D and E).
Imaging individual HER2 receptors on intact SKBR3 cells reveals the presence of HER2 homodimers
The high resolution of ESEM-STEM allowed studying HER2 distribution patterns in individual cells of an SKBR3 cell population. The precise position of each individual QD label (Fig. 3E) was automatically detected and the distance between individual QD positions was recorded to determine the pair correlation function g(x) (20). Briefly, g(x) measures the likelihood of a particle being found within a certain radial distance with respect to a reference particle, whereby g(x) = 1 represents a random distribution, and a value >1 indicates clustering. Figure 3C shows g(x) for the total of 122 detected HER2 positions in Fig. 3E, with a peak above unity at 17.5 nm indicating the existence of a preferred QD pair distance. However, this number was calculated on a comparatively low number of events, and the peak position was therefore considered as a preliminary estimation of HER2 homodimer distance.
In measurements incorporating 14,043 HER2 positions in 11 cells (Fig. 4A), a center-to-center label distance of 20 nm was obtained for HER2 homodimers (see also Table 1). The peak of g(x) at x = 20 nm above the random level of g(x) = 1 indicates that a fraction of HER2 was clustered as homodimer. A control experiment involving QDs randomly attached to a SiN membrane (fig. S7), as well as a random simulation, verified g(x) = 1 (Fig. 4A) and ruled out the presence of preformed clustered labels. Because affibodies bind to their epitope on the HER2 receptor in a 1:1 stoichiometry (18), it thus seems likely that HER2 indeed formed homodimers on membrane ruffles. Further data supporting the notion of dimer formation are the measured most probable distance between the QD centers. Considering factors such as linker flexibility, a center-to-center QD distance of 20 nm seems plausible for QD-labeled HER2 homodimers. It is not possible to compare this value to the expected value from a molecular model because x-ray crystallography data are not available in the PDB for the HER2 homodimer. However, the corresponding data for the EGFR homodimer can be used instead as approximation because molecular modeling has shown that HER2 homodimers are of a size comparable to that reported for EGFR homodimers (7). The label distance of 20 nm is consistent with that of two 12-nm-diameter gold nanoparticle labels attached to an EGFR homodimer (11).
Fig. 4. Cluster analysis of HER2 proteins in 11 SKBR3 cells.
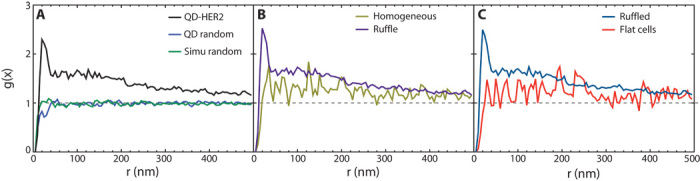
(A) The pair correlation function g(x) of a total of 14,171 labels exhibited a peak at 20 nm, indicating HER2 dimerization. Larger-sized clusters of up to several hundreds of nanometers were also observed. The curves of randomly dispersed QDs and a simulation (Simu) of random data were included as reference. (B) HER2 pairs were absent in cellular areas with a homogeneous membrane topography, contrasting g(x) in the ruffled areas. (C) HER2 does not appear clustered in the two analyzed flat cells. Clustering was only observable in cells with membrane ruffles.
Table 1. Density of HER2 labeling per morphological cell type.
A total of 11 SKBR3 cells were studied including two flat cells. Data of a test sample containing randomly distributed QDs are also indicated. The information includes the number of images in an experimental group Nimage, the number of all QD labels in a group Nlabel, and the average label density ρ.
| Cell group | Nimage | Nlabel | ρ (μm2) |
| All cells | 112 | 14,171 | 26.9 |
| Ruffled regions | 80 | 10,870 | 30.5 |
| Homogeneous regions | 32 | 3,307 | 19.3 |
| Ruffled cells | 90 | 10,513 | 28.8 |
| Flat cells | 22 | 3,664 | 22.6 |
| QD random | 24 | 4,506 | 41.2 |
QD label distances exceeding 20 nm and reaching up to several hundreds of nanometers were also observed with a higher likelihood than random. Hence, molecular control mechanisms driving HER2 receptor clustering on the cell surface possibly concentrate HER2 in lipid rafts, as was shown to occur for EGFR (21).
HER2 monodimers are preferentially found in membrane ruffles
HER2 distribution patterns were determined for two distinct cellular regions (that is, membrane ruffles and homogeneous areas). Figure 5A depicts the fluorescence signal of a cell with membrane ruffles and an intermediate level of HER2 membrane expression, similar to most cells in Fig. 2A. Two areas of this cell are selected in Fig. 5B: one with membrane ruffles and one with a homogeneously appearing topography. The corresponding g(x) curves were independently determined for both areas and are shown in Fig. 5, C and D, respectively. HER2 homodimers and larger HER2-containing clusters were identified as characteristic features of membrane ruffles, whereas homogeneous membrane areas exhibited a rather random HER2 distribution.
Fig. 5. Correlative light and electron microscopy of a cell with ruffles.
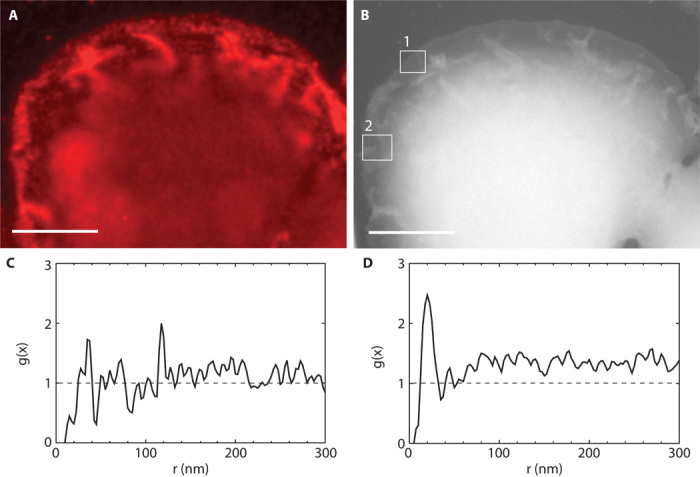
(A) Fluorescence image of a selected region at the edge of cell #11. (B) Same area as in (A) but imaged with ESEM-STEM. Several membrane ruffles are visible as structures of increased brightness. (C) An ESEM-STEM image was recorded at the location #1 in (B) of a region with homogeneous membrane topography, without ruffles. The g(x) function indicated a random distribution of 209 automatically detected QD label positions. (D) Region #2 in (B) contained membrane ruffles. The curve of g(x) determined from 417 label positions in the ESEM-STEM image in the area contained a peak at 20 nm and also pointed toward clustering at larger distances. Scale bars, 10 μm.
Nine of the 11 cells of interest exhibited characteristic fluorescence patterns similar to those shown in Figs. 3A and 5A. These cells contained regions with membrane ruffles and also homogeneous regions. Images of membrane ruffles were analyzed for all nine cells and the g(x) curve was calculated. An analogous procedure was carried out for images recorded from homogeneous membrane areas (that is, without ruffles visible with electron microscopy). Figure 4B (see also Table 1) summarizes data obtained from the analysis of 10,745 images and shows that homodimers were only present in regions with membrane ruffles. Note that the statistical fluctuations of the g(x) curve of homogeneous regions are larger than for the ruffled areas because a smaller number of QD labels was included in the analysis. However, the HER2 density determined for the two distinct topographical regions differed by less than a factor of 2.
HER2 receptors do not cluster in flat SKBR3 cells
Two cells were identified as flat cells, indicated as #5 in Fig. 2A and with asterisks in the fluorescence image in Fig. 6A, and both cells displayed a flat membrane topography in the DIC (Fig. 6B) and ESEM-STEM images (Fig. 6C). A total of 22 randomly selected areas of these cells were imaged with ESEM-STEM at a magnification sufficient to detect the QD labels attached to HER2. An example is shown in Fig. 6D. Analysis of the interlabel distances via the g(x) curve revealed a random distribution (Fig. 6E) of HER2. Figure 4C shows g(x) for all 3664 analyzed HER2 positions of these two cells (see Table 1) and compares the curve with g(x) obtained for the cells showing a ruffled membrane. The char??acteristic dimer peak at 20 nm was absent for flat cells. These data show that HER2 homodimerization was not detected in the so-called flat SKBR3 cells.
Fig. 6. Correlative light and electron microscopy of a typical flat cell.
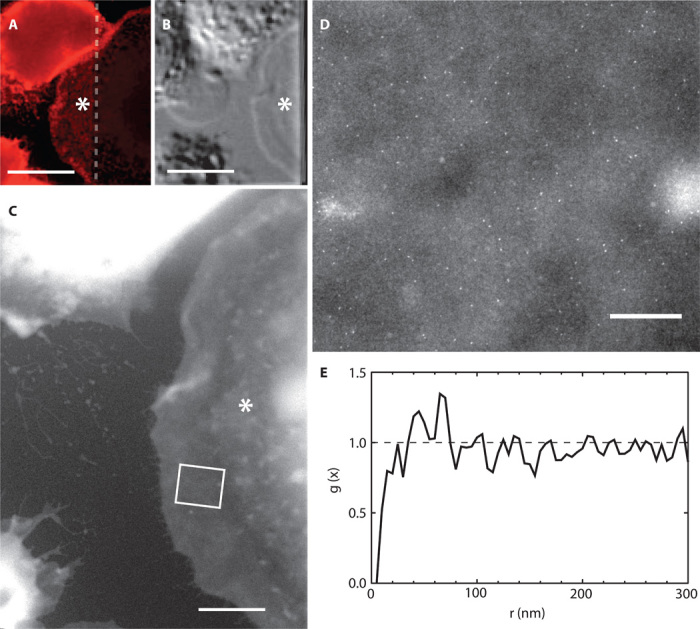
Flat cell is indicated with an asterisk. (A) A selected area of the fluorescence image showed a lower HER2 labeling density in this cell compared to adjacent cells. The dashed line indicated the edge of the SiN window. (B) DIC shows that the cell has a ~10-μm flat and smooth outer rim without membrane ruffles or protrusions. (C) Overview ESEM-STEM image in which the flat rim was clearly visible. (D) A selected region [indicated by the rectangle in (C)] of a high-resolution ESEM-STEM image showed scattering of QD labels. (E) The curve of g(x) obtained from the 188 QDs in this cellular region indicated a random distribution of HER2 around unity. Scale bars, 20 μm (A and B); 5 μm (C); 500 nm (D).
DISCUSSION
A protein labeling protocol was established combining the specificity of anti-HER2 affibody with the high spatial resolution achievable with QDs. It was used to map intra- and intercellular distribution patterns of HER2 on SKBR3 cells. The fluorescence images (for example, Fig. 2A) revealed significant differences of HER2 membrane expression on the SKBR3 population level and also between different functional regions of the same cell. This finding is in agreement with an earlier study reporting the preferential membrane expression of HER2 on membrane ruffles in SKBR3 cells (22). Heterogeneity of protein expression is known from tumors and has been closely linked with drug resistance mechanisms and the metastatic potential of tumors (23). In particular, heterogeneity in HER2-overexpressing tumors points toward an increased risk of therapeutic resistance (3). Moreover, because intratumoral heterogeneity of oncogenic receptors such as HER2 has been linked with the risk of cancer progression, understanding the molecular mechanisms behind the spatial heterogeneity of HER2 and in general of receptor tyrosine kinases might inspire new and more effective therapeutic concepts (24).
The small label used in this study and the high spatial resolution of ESEM-STEM allowed the detection of individual QD-labeled HER2 proteins not only as monomers but also when assembled into homodimers and higher-order clusters. It is feasible to label different protein species assembled into a protein complex (for example, HER2 and EGFR) with orthogonal labels in future studies. The analysis of cellular regions around the nucleus or cell types lacking thin edges would be possible using an electron microscope with a beam energy higher than 30 keV (14). Position information of 14,043 HER2 proteins in a total of 11 intact cells was obtained and analyzed. This approach can readily be used to investigate a wide range of membrane proteins in whole cells and merely requires access to specific labels and STEM.
Commonly used biochemical methods to detect and quantify membrane protein complexes rely on information from pooled cellular material, and as a consequence, exceptions deriving from rare cells remain hidden (see also Supplementary Discussion). Furthermore, these methods lack any spatial information about the cellular location of the occurring protein-protein interactions. It is practically impossible to obtain stoichiometric protein information and nanoscale localization information on membrane proteins within the context of tens of intact cells in liquid state using immunohistochemistry techniques, such as electron microscopy using antibody labeling (25). Light microscopy, on the other hand, does not exhibit sufficient spatial resolution (26) to directly image the individual constituents of small protein complexes such as the HER2 homodimer.
The key advantage of our method is its capability to measure distances, thus enabling us to distinguish regions containing randomly dispersed proteins from regions with a similar protein density but with a certain fraction of the proteins bound in complexes. In particular, for membrane regions containing endogenously expressed HER2, the density may be so high that many proteins are already found at a distance representative for a dimer by random chance (Fig. 6, D and E). As is evident from a cluster analysis comparing the flat and ruffled cells (see Supplementary Results), the fraction of label pairs at dimer distance differs only by a factor of 1.8 between these two cell types, whereas the labels are randomly distributed in flat cells. Cells and tissue can be examined with newly developed proximity methods (27, 28), but the measured fractions of clustered proteins contain both bound proteins and randomly occurring closely placed monomers, which could possibly lead to false-positive detection of, for example, the presence of HER2 homodimers. The high-content screening of ESEM-STEM of cells in liquid could thus possibly support the high-throughput screening capabilities of proximity methods.
Data obtained by analyzing all label positions revealed the presence of HER2 pairs at a characteristic homodimer distance of 20 nm. Figure 4B shows that these dimers are preferentially associated with ruffled membrane areas, whereby the ruffled areas revealed a twofold stronger labeling density than did flat areas of the same cells and corresponding areas of flat cells (Table 1). In cancer cells, the highly dynamic membrane ruffles, also referred to as invadopodia, are considered to serve as junctions for cellular signaling and drive motility, invasiveness, and metastasis of cancer cells (10, 29–31). Our findings of the preferential occurrence of HER2 homodimers in membrane ruffles could thus imply that HER2 homodimers play an important role in cancer cell spreading.
A key result of our study is the observation that HER2 homodimers are apparently absent from a small subpopulation of cells (Fig. 6) characterized by a flat topography and an almost complete lack of membrane ruffling. Already the difference in cell morphology compared to the typical SKBR3 cell could point toward a functional difference of these cells with respect to cells representing the major population. Possibly, these flat cells are resting cells (fig. S4). The absence of HER2 homodimers from flat cells could indicate that these cells display a different intercellular signaling mechanism than the average SKBR3 cell enlaced by membrane ruffles. Future studies of cancer cells that include the examination of selected rare cell subpopulations (for example, cancer stem cells) (32), while also providing an overview on the cell population, may provide additional information about the effectiveness of HER2-targeting drugs inhibiting HER2 homodimer formation (33), considering that cells with HER2 homodimers might respond differently to HER2-targeting drugs compared to cells lacking dimers. In conclusion, our study revealed a preferred localization of HER2 homodimers in membrane ruffles, whereas these dimers appear to be absent from flat cells.
MATERIALS AND METHODS
Experimental design
The main goal of the experiments was to study the localized distribution of the HER2 stoichiometry in the plasma membranes of SKBR3 breast cancer cells. The following objectives were considered.
(i) Imaging close to the native state
It was our aim to analyze the location of HER2 in a state as close as possible to that of the native cell, thus avoiding sectioning, membrane rupture, or drying effects. Therefore, correlative light microscopy and ESEM-STEM, a new imaging technology, were used to visualize the membrane proteins in intact cells kept in liquid state.
(ii) Obtaining representative data of a cell population
Considering the large morphological differences between individual cells of a cancer cell population, the ESEM-STEM–based analysis was applied to many cells, resulting in accumulating the location of more than 10,000 proteins. Imaging included correlative light microscopy, which was used to select suitable cells from the cancer cell population, and ESEM-STEM to obtain nanoscale information on proteins’ positions. This study was carried out for 11 whole cells. QDs were used as label, providing a fluorescence signal, while being made of a heavy material such that contrast was obtained in STEM.
(iii) Labeling individual HER2 in the plasma membrane
To study the stoichiometry of HER2 as assembled in homodimers, or larger-order protein complexes or clusters, it was crucial to optimize the spatial accuracy of the receptor localization. Therefore, both the label-to-target distance and the label were designed to be as small as possible so that all individual HER2 proteins in a complex could be labeled. A HER2-specific affibody was used as a small specific label that also allowed a one-to-one coupling to a QD. Linkage between affibody and QD was based on the strong and specific streptavidin-biotin interaction. Various control experiments were carried out to ensure that the labeling protocol was specific for HER2 without inducing HER2 uptake and without impact on cell viability.
(iv) Studying localized HER2 distribution
Our aims were to study in which membrane regions HER2 homodimers occurred and whether differences between cells existed. Numerous light microscopic images were recorded to characterize cellular differences. A total of 112 regions of 11 selected cells were studied with a spatial resolution of 3 nm using ESEM-STEM. A statistic analysis was performed including information on 14,171 HER2 positions via the calculation of the pair correlation function.
Materials
SKBR3 cells were obtained from the American Type Culture Collection. Biotin-conjugated anti-HER2 affibody molecule [HER2–AFF-B, termed: (ZHER2:477)2] was purchased from Affibody AB. Dulbecco’s phosphate-buffered saline (DPBS) was from Lonza Cologne GmbH. Dulbecco’s modified Eagle’s medium (DMEM) GlutaMAX with high glucose and pyruvate, fetal bovine serum (FBS), minimum essential medium nonessential amino acids (NEAA) 100× solution, QD Qdot 655 streptavidin conjugate (strep-QD), and FBS were from Life Technologies. CellStripper was from Mediatech. Normal goat serum (GS) was from Rockland Immunochemicals Inc. High-performance liquid chromatography (HPLC)–grade acetone and ethanol, 10× PBS solution, 25% electron microscopy–grade glutaraldehyde (GA) solution, d-glucose, d-saccharose, glycine, biotin-free and molecular biology–grade albumin Fraktion V [bovine serum albumin (BSA)], and sodium cacodylate trihydrate were from Carl Roth GmbH + Co. KG. Electron microscopy–grade formaldehyde (FA) 16% solution was from Electron Microscopy Sciences. HPLC-grade deionized water, poly-l-lysine (PLL) hydrobromide (molecular weight, 70,000 to 150,000), sodium tetraborate, boric acid, and gelatin from cold water fish skin (GEL) were from Sigma-Aldrich Chemie GmbH. Glass-bottom cell culture dishes (uncoated, 35-mm diameter) were from MatTek Corp. Sample support microchips (14, 34) with a central SiN membrane window of a dimension of 50 × 400 μm2 and a thickness of 50 nm were custom-made by Protochips. The microchips had diced edges so that the edges were flat, which was convenient for handling with wide-beak and Teflon-coated tweezers. The chips were of dimensions 2.00 × 2.60 × 0.30 mm3, and they fitted in the wells of a standard 96-well plate.
SiN membrane microchip preparation for cell settlement
SiN membrane microchips were prepared for SKBR3 cells by rinsing the microchips for 2 min in acetone, followed by a 2-min rinse in 100 ml of ethanol (both solvents were of HPLC grade). The dried microchips were cleaned with ArO2 plasma for 5 min and immediately placed in 0.01% PLL for 5 min at room temperature. Subsequently, the microchips were rinsed in water twice and kept in water until the cell suspension was ready (within 10 to 15 min).
Cell culture and cell seeding on microchips
SKBR3 cells (human breast cancer cell line overexpressing HER2) were cultured in flasks (25 cm2) with cell medium (DMEM, supplemented with NEAA and 10% FBS), in a 5% CO2 atmosphere, at 37 °C. The cells were passaged twice a week and experiments were carried out with cells between passages 4 and 10. The cells were harvested at half confluency by rinsing the attached cell layer in DPBS and subsequent dissociation with CellStripper (15 to 20 min at 37 °C), followed by a quench in the cell medium. The previously prepared microchips were each placed in 200 μl of cell medium at the bottom of a well in a 96-well plate. A 20 μl of harvested cells in suspension was added to each well. After 5 to 10 min, the microchips, with usually 5 to 12 cells adhering on the SiN window, were transferred into new wells containing 200 μl of cell medium. The microchips were then incubated for another 2 to 4 hours and finally placed into wells filled with cell medium without FBS for 12 to 24 hours of incubation under serum starvation; these incubations were done in a 5% CO2 atmosphere at 37 °C.
Labeling of HER2 on SKBR3 cells
HER2–AFF-B stock solution (20 μM) was adjusted to a final concentration of 200 nM in PBS supplemented with 1% GS, 0.5% BSA, and 0.1% GEL (GS-BSA-GEL-PBS). Strep-QD stock solution (1 μM) was diluted 1:20 in 40 mM borate buffer (sodium tetraborate, boric acid, pH 8.3) and then brought to a final concentration of 5 nM by dilution in TB, supplemented with 1% BSA. After 12 to 24 hours of serum starvation, the cells on the microchips were rinsed once with GS-BSA-GEL-PBS and then incubated for 10 min at room temperature in GS-BSA-GEL-PBS to inhibit unspecific binding of HER2–AFF-B. Subsequently, the chips were incubated for 15 min at 37 °C with the HER2–AFF-B labeling solution. After rinsing four times with PBS and once with CB (0.1 M cacodylate buffer, 0.1 M saccharose, pH 7.4), the cells were fixed with 3% FA in CB for 10 min. Cells were rinsed once with CB and three times with PBS, followed by incubation in 0.1 M glycine in PBS (pH 7.4) (GLY-PBS) for 2 min and rinsing twice with PBS, before incubation in strep-QD labeling solution for 10 min at room temperature. After being rinsed three times with PBS and once with PBS supplemented with 1% BSA (BSA-PBS), the chips were imaged in BSA-PBS in a glass-bottom dish with fluorescence microscopy. Thereafter, the cells were rinsed once with CB and fixed for 12 min at room temperature with 2% GA in CB for electron microscopy. Cells were then rinsed once with CB and three times with BSA-PBS and stored in BSA-PBS at 4 °C until electron microscopic investigation.
DIC and fluorescence microscopy
The FA-fixed and labeled cells were imaged using an inverted light microscope (DMI6000B, Leica). Cells on microchips were imaged in BSA-PBS and placed upside down in a plasma-cleaned cell culture glass-bottom dish, first with a 20× air objective and then with a 63× oil immersion objective. Images were recorded with bright light (that is, DIC) to yield information about membrane limits and topography, such as membrane ruffles. To detect bound strep-QDs, a filter cube with a 340- to 380-nm excitation and >420-nm emission window was used. To ensure that all HER2-bound QDs were detected, taking into account the stochastic blinking behavior of QD-emitted fluorescence signals, high-resolution fluorescence images were created from multiple images. For this purpose, the flat cell areas were brought into focus and a time series, consisting of 25 images, was recorded using an exposure time of 62 ms and a repetition rate of 6.5 Hz. Afterward, the cells were fixed with 2% GA to increase the stability of the cellular material under electron beam irradiation. A typical experiment encompassed four to six samples and required a total preparation time of 3 to 4 hours, including the seeding of cells on the microchips, the actual labeling protocol, and the time necessary to record the light microscopic images.
Loading a sample into the ESEM and preparation for imaging
After fixation with GA, the labeled cells on the SiN support membranes were imaged in hydrated state with ESEM (Quanta 400 FEG, FEI) using the STEM detector. The setup of the ESEM included a stage with a Peltier cooling element and a two-segment solid-state detector mounted underneath the sample serving as STEM detector; a detailed description of the setup for STEM in ESEM can be found elsewhere (35). Only the dark-field contrast mode was used for this study. A gaseous secondary electron detector mounted at the pole piece (above the sample) was used to check for the presence of the thin water film on the cell. Loading was done by taking a microchip out of the cooled BSA-PBS and rinsing four times in cooled water to remove the salt. After blotting the backside of the microchip, it was placed into the precooled Peltier stage (3 °C). Cooled water (3 μl) was pipetted onto the sample surface to provide sufficient water coverage for starting the air evacuation in the microscope vacuum chamber. Three additional 3-μl water droplets were placed close to the sample on the Peltier stage as an additional water source. The pressure in the specimen chamber was then cycled five times between 800 and 1500 Pa to fill the specimen chamber with saturated water vapor (35, 36). Afterward, a controlled thinning of the water film was initiated by lowering the pressure stepwise to 740 Pa; in some cases, the pressure was lowered to 720 Pa. ESEM-STEM analysis was started after the water layer was sufficiently thin to allow high-resolution imaging.
Wet ESEM-STEM imaging
We used an electron beam energy of 30 kV, a spot size of 1 nm, a probe current of 600 pA, and a working distance of 6.2 mm. For every sample, an overall overview ESEM-STEM image was recorded showing the whole membrane window area with all cells; these images were also used to provide a spatial correlation of the fluorescence images with those obtained with ESEM-STEM. Then, overview images were recorded from all individual cells at a higher magnification. To discern HER2-bound QDs on the plasma membrane, the magnification was set to ×50,000 to ×75,000, and pixel dwell times between 30 and 50 μs were used. The image size was usually 1024 × 884 pixels, but several images with 2048 × 1886 pixels were also recorded. For these large images, the magnification was set to ×25,000 to ×37,500. The stage temperature was kept at 3 °C, and the pressure was set to values ranging between 720 and 740 Pa during the imaging, except for the biotinylated control chips with the randomly immobilized strep-QDs, which were imaged at 700 Pa. These pressure and temperature settings created 100% relative humidity in the ESEM chamber as needed to ensure the constant coverage of the cells with a thin film of water. Depending on the chosen pixel dwell time, the frame time needed to record a high-resolution image usually ranged between 40 and 80 s (except for the large size images); it was therefore possible to record >100 images from several cells within a half day.
Correlative image analysis
Image processing was accomplished with ImageJ (National Institutes of Health). All images were adjusted in brightness and contrast. Cropping was applied for certain images as indicated in the figure captions. High-resolution fluorescence images were constructed from the maximum intensity overlay of the 25 images from a time series at a certain cellular region. The correlation of the ESEM images acquired at ×75,000 magnification with their corresponding fluorescence images was done in two steps. The respective overview ESEM image served as the main orientation image. An ESEM image recorded at ×50,000 or ×75,000 magnification was first reduced in size until it fitted the scale of the corresponding overview image. The reduced image was then positioned into its corresponding position in the overview ESEM image using the cellular features as reference objects for positioning, and the borders of this area were marked. Next, the respective high-resolution fluorescence image was adjusted in size to fit the scale of the ESEM overview image. Both images were then overlaid using the cellular features as reference (cellular features were recognizable in both the fluorescence image and the ESEM-STEM image), and the previously marked area was pasted in the fluorescence image.
Particle detection
The positions of the QD nanoparticle protein labels were determined in an automated procedure programmed in ImageJ. The particle positions in an image were measured as follows. An image was first noise-filtered by applying a Gaussian filter with a radius of 1 pixel. Background variations in the image were then filtered by applying a Fourier filter removing spatial frequencies a factor of 3 smaller and a factor of 3 higher than those of the expected particle diameter. The image was then binarized using an automated threshold with maximum entropy setting. Finally, the particles were selected using the Analyze Particles tool selecting for particles with an area within a factor of 5 of the expected area. These settings provided automated detection of the nanoparticles, where typically only a few particles were not detected.
Statistical analysis using the pair correlation function
The pair correlation function g(r) is defined as follows (20):
| (1) |
where r is the radial distance, ρ is the labeling density in the image, γ is the covariance function, and k is the kernel. The distance between two points i and j is indicated by the modulus |xi − xj|, where x is the two-dimensional position (x, y) of a particle in the image. The label positions were assumed to be planar. The covariance function γ corrects for edge effects of the rectangular image and is defined as follows (37):
| (2) |
where h and w are the height and width of the image, respectively. The kernel is written as el (38):
| (3) |
the parameter ε is called the bandwidth. Locally designed software in C++ was used to calculate g(r) of the particle positions in image. A histogram of r with a bin width of 2.5 nm was defined, and the value of g(r) was calculated for each bin r. The bandwidth was adjusted to 5 nm to obtain an optimal balance between a sharp response and the lowest fluctuation level of the obtained curves. The data of two or more images were averaged, whereby the average was weighted by the particle density. Values smaller than 10 nm were not allowed, taking the size of the nanoparticles into account.
Acknowledgments
We thank E. Arzt for his support through INM, J. Hermannsdörfer for experimental help, and Protochips Inc. for providing the microchips. Funding: Research in part supported by the Leibniz Competition 2014. U.K. acknowledges support by the German Federal Ministry of Education and Research via the program of Medical Systems Biology “BreastSys” (grant 0315396) and the e:BIO project “MetastaSys” (grant 0316173). Author contributions: D.B.P. conducted the experiments. N.d.J. analyzed the data. All authors designed the experiments and wrote the paper. Competing interests: The authors declare that they have no competing interests.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/1/6/e1500165/DC1
Fig. S1. Micrographs of control experiments on SKBR3 cells.
Fig. S2. Overlay of DIC and fluorescence microscopic images of typical live or fixed SKBR3 cells.
Fig. S3. Selection of flat cells.
Fig. S4. Micrographs (DIC) showing examples of live flat SKBR3 cells.
Fig. S5. ESEM-STEM image used to determine the resolution on QDs of labeled HER2 on SKBR3 cells.
Fig. S6. High-resolution TEM image of QDs.
Fig. S7. ESEM-STEM image of immobilized streptavidin-conjugated QDs.
Table S1. Cluster analysis of HER2 labeling.
REFERENCES AND NOTES
- 1.Ross J. S., Fletcher J. A., The HER-2/neu oncogene in breast cancer: Prognostic factor, predictive factor, and target for therapy. Stem Cells 16, 413–428 (1998). [DOI] [PubMed] [Google Scholar]
- 2.Yu D. H., Hung M. C., Overexpression of ErbB2 in cancer and ErbB2-targeting strategies. Oncogene 19, 6115–6121 (2000). [DOI] [PubMed] [Google Scholar]
- 3.Seol H., Lee H. J., Choi Y., Lee H. E., Kim Y. J., Kim J. H., Kang E., Kim S. W., Park S. Y., Intratumoral heterogeneity of HER2 gene amplification in breast cancer: Its clinicopathological significance. Mod. Pathol. 25, 938–948 (2012). [DOI] [PubMed] [Google Scholar]
- 4.Yarden Y., Sliwkowski M. X., Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2, 127–137 (2001). [DOI] [PubMed] [Google Scholar]
- 5.Schmick M., Bastiaens P. I., The interdependence of membrane shape and cellular signal processing. Cell 156, 1132–1138 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Brennan P. J., Kumagai T., Berezov A., Murali R., Greene M. I., HER2/Neu: Mechanisms of dimerization/oligomerization. Oncogene 21, 328 (2002). [DOI] [PubMed] [Google Scholar]
- 7.Arkhipov A., Shan Y. B., Kim E. T., Dror R. O., Shaw D. E., Her2 activation mechanism reflects evolutionary preservation of asymmetric ectodomain dimers in the human EGFR family. eLife 2 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Badache A., Hynes N. E., A new therapeutic antibody masks ErbB2 to its partners. Cancer Cell 5, 299–301 (2004). [DOI] [PubMed] [Google Scholar]
- 9.Hynes N. E., Dey J. H., PI3K inhibition overcomes trastuzumab resistance: Blockade of ErbB2/ErbB3 is not always enough. Cancer Cell 15, 353–355 (2009). [DOI] [PubMed] [Google Scholar]
- 10.Henjes F., Bender C., von der Heyde S., Braun L., Mannsperger H. A., Schmidt C., Wiemann S., Hasmann M., Aulmann S., Beissbarth T., Korf U., Strong EGFR signaling in cell line models of ERBB2-amplified breast cancer attenuates response towards ERBB2-targeting drugs. Oncogenesis 1, e16 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peckys D. B., Baudoin J. P., Eder M., Werner U., de Jonge N., Epidermal growth factor receptor subunit locations determined in hydrated cells with environmental scanning electron microscopy. Sci. Rep. 3, 1–6 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dukes M. J., Peckys D. B., de Jonge N., Correlative fluorescence microscopy and scanning transmission electron microscopy of quantum-dot-labeled proteins in whole cells in liquid. ACS Nano 4, 4110–4116 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nishiyama H., Suga M., Ogura T., Maruyama Y., Koizumi M., Mio K., Kitamura S., Sato C., Atmospheric scanning electron microscope observes cells and tissues in open medium through silicon nitride film. J. Struct. Biol. 169, 438–449 (2010). [DOI] [PubMed] [Google Scholar]
- 14.de Jonge N., Peckys D. B., Kremers G. J., Piston D. W., Electron microscopy of whole cells in liquid with nanometer resolution. Proc. Natl. Acad. Sci. U.S.A. 106, 2159–2164 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Steffen A. C., Wikman M., Tolmachev V., Adams G. P., Nilsson F. Y., Stahl S., Carlsson J., In vitro characterization of a bivalent anti-HER-2 affibody with potential for radionuclide-based diagnostics. Cancer Biother. Radiopharm. 20, 239–248 (2005). [DOI] [PubMed] [Google Scholar]
- 16.Orlova A., Nilsson F. Y., Wikman M., Widstrom C., Stahl S., Carlsson J., Tolmachev V., Comparative in vivo evaluation of technetium and iodine labels on an anti-HER2 affibody for single-photon imaging of HER2 expression in tumors. J. Nucl. Med. 47, 512–519 (2006). [PubMed] [Google Scholar]
- 17.Lofblom J., Feldwisch J., Tolmachev V., Carlsson J., Stahl S., Frejd F. Y., Affibody molecules: Engineered proteins for therapeutic, diagnostic and biotechnological applications. FEBS Lett. 584, 2670–2680 (2010). [DOI] [PubMed] [Google Scholar]
- 18.Eigenbrot C., Ultsch M., Dubnovitsky A., Abrahmsen L., Hard T., Structural basis for high-affinity HER2 receptor binding by an engineered protein. Proc. Natl. Acad. Sci. U.S.A. 107, 15039–15044 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brown E., Verkade P., The use of markers for correlative light electron microscopy. Protoplasma 244, 91–97 (2010). [DOI] [PubMed] [Google Scholar]
- 20.Stoyan D., Stoyan H., Estimating pair correlation functions of planar cluster processes. Biom. J. 38, 259–271 (1996). [Google Scholar]
- 21.Nagy P., Vereb G., Sebestyen Z., Horvath G., Lockett S. J., Damjanovich S., Park J. W., Jovin T. M., Szollosi J., Lipid rafts and the local density of ErbB proteins influence the biological role of homo- and heteroassociations of ErbB2. J. Cell Sci. 115, 4251–4262 (2002). [DOI] [PubMed] [Google Scholar]
- 22.Hommelgaard A. M., Lerdrup M., van Deurs B., Association with membrane protrusions makes ErbB2 an internalization-resistant receptor. Mol. Biol. Cell 15, 1557–1567 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meacham C. E., Morrison S. J., Tumour heterogeneity and cancer cell plasticity. Nature 501, 328–337 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Casaletto J. B., McClatchey A. I., Spatial regulation of receptor tyrosine kinases in development and cancer. Nat. Rev. Cancer 12, 387–400 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cambi A., Lidke D. S., Nanoscale membrane organization: Where biochemistry meets advanced microscopy. ACS Chem. Biol. 7, 139–149 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lippincott-Schwartz J., Manley S., Putting super-resolution fluorescence microscopy to work. Nat. Methods 6, 21–23 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leuchowius K. J., Clausson C. M., Grannas K., Erbilgin Y., Botling J., Zieba A., Landegren U., Soderberg O., Parallel visualization of multiple protein complexes in individual cells in tumor tissue. Mol. Cell. Proteomics 12, 1563–1571 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Citri A., Yarden Y., EGF–ERBB signalling: Towards the systems level. Nat. Rev. Mol. Cell Biol. 7, 505–516 (2006). [DOI] [PubMed] [Google Scholar]
- 29.Weaver A. M., Invadopodia: Specialized cell structures for cancer invasion. Clin. Exp. Metastasis 23, 97–105 (2006). [DOI] [PubMed] [Google Scholar]
- 30.Feldner J. C., Brandt B. H., Cancer cell motility—On the road from c-erbB-2 receptor steered signaling to actin reorganization. Exp. Cell Res. 272, 93–108 (2002). [DOI] [PubMed] [Google Scholar]
- 31.Brix D., Clemmensen K., Kallunki T., When good turns bad: Regulation of invasion and metastasis by ErbB2 receptor tyrosine kinase. Cells 3, 53–78 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.O. D. Wiestler, B. Haendler, D. Mumberg, Cancer Stem Cells. Springer Series on Biofilms (Springer, Berlin, 2007), vol. 2006/5. [Google Scholar]
- 33.Ghosh R., Narasanna A., Wang S. E., Liu S., Chakrabarty A., Balko J. M., González-Angulo A. M., Mills G. B., Penuel E., Winslow J., Sperinde J., Dua R., Pidaparthi S., Mukherjee A., Leitzel K., Kostler W. J., Lipton A., Bates M., Arteaga C. L., Trastuzumab has preferential activity against breast cancers driven by HER2 homodimers. Cancer Res. 71, 1871–1882 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ring E. A., Peckys D. B., Dukes M. J., Baudoin J. P., de Jonge N., Silicon nitride windows for electron microscopy of whole cells. J. Microsc. 243, 273–283 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bogner A., Thollet G., Basset D., Jouneau P. H., Gauthier C., Wet STEM: A new development in environmental SEM for imaging nano-objects included in a liquid phase. Ultramicroscopy 104, 290–301 (2005). [DOI] [PubMed] [Google Scholar]
- 36.Kirk S., Skepper J., Donald A. M., Application of environmental scanning electron microscopy to determine biological surface structure. J. Microsc. 233, 205–224 (2009). [DOI] [PubMed] [Google Scholar]
- 37.Stoyan D., Bertram U., Wendrock H., Estimation variances for estimators of product densities and pair correlation functions of planar point processes. Ann. Inst. Stat. Math. 45, 211–221 (1993). [Google Scholar]
- 38.Fiksel T., Edge-corrected density estimators for points processes. Statistics 19, 67–75 (1988). [Google Scholar]
- 39.Chung I., Akita R., Vandlen R., Toomre D., Schlessinger J., Mellman I., Spatial control of EGF receptor activation by reversible dimerization on living cells. Nature 464, 783–787 (2010). [DOI] [PubMed] [Google Scholar]
- 40.L. Reimer, Scanning Electron Microscopy: Physics of Image Formation and Microanalysis (Springer, Berlin, 1998). [Google Scholar]
- 41.Gavin A. C., Bösche M., Krause R., Grandi P., Marzioch M., Bauer A., Schultz J., Rick J. M., Michon A. M., Cruciat C. M., Remor M., Höfert C., Schelder M., Brajenovic M., Ruffner H., Merino A., Klein K., Hudak M., Dickson D., Rudi T., Gnau V., Bauch A., Bastuck S., Huhse B., Leutwein C., Heurtier M. A., Copley R. R., Edelmann A., Querfurth E., Rybin V., Drewes G., Raida M., Bouwmeester T., Bork P., Seraphin B., Kuster B., Neubauer G., Superti-Furga G., Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature 415, 141–147 (2002). [DOI] [PubMed] [Google Scholar]
- 42.Desmedt C., Sperinde J., Piette F., Huang W., Jin X., Tan Y., Durbecq V., Larsimont D., Giuliani R., Chappey C., Buyse M., Winslow J., Piccart M., Sotiriou C., Petropoulos C., Bates M., Quantitation of HER2 expression or HER2:HER2 dimers and differential survival in a cohort of metastatic breast cancer patients carefully selected for trastuzumab treatment primarily by FISH. Diagn. Mol. Pathol. 18, 22–29 (2009). [DOI] [PubMed] [Google Scholar]
- 43.Bergersen L. H., Storm-Mathisen J., Gundersen V., Immunogold quantification of amino acids and proteins in complex subcellular compartments. Nat. Protoc. 3, 144–152 (2008). [DOI] [PubMed] [Google Scholar]
- 44.Ilgen P., Stoldt S., Conradi L. C., Wurm C. A., Rüschoff J., Ghadimi B. M., Liersch T., Jakobs S., STED super-resolution microscopy of clinical paraffin-embedded human rectal cancer tissue. PLOS One 9, e101563 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Piston D. W., Kremers G. J., Fluorescent protein FRET: The good, the bad and the ugly. Trends Biochem. Sci. 32, 407–414 (2007). [DOI] [PubMed] [Google Scholar]
- 46.Warren C. M., Landgraf R., Signaling through ERBB receptors: Multiple layers of diversity and control. Cell. Signal. 18, 923–933 (2006). [DOI] [PubMed] [Google Scholar]
- 47.Needham S. R., Hirsch M., Rolfe D. J., Clarke D. T., Zanetti-Domingues L. C., Wareham R., Martin-Fernandez M. L., Measuring EGFR separations on cells with ~10 nm resolution via fluorophore localization imaging with photobleaching. PLOS One 8, e62331 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu P., Sudhaharan T., Koh R. M., Hwang L. C., Ahmed S., Maruyama I. N., Wohland T., Investigation of the dimerization of proteins from the epidermal growth factor receptor family by single wavelength fluorescence cross-correlation spectroscopy. Biophys. J. 93, 684–698 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Moreira J. M., Thorsen S. B., Brunner N., Stenvang J., Proximity probing assays for simultaneous visualization of protein complexes in situ. Expert Rev. Proteomics 10, 219–221 (2013). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/1/6/e1500165/DC1
Fig. S1. Micrographs of control experiments on SKBR3 cells.
Fig. S2. Overlay of DIC and fluorescence microscopic images of typical live or fixed SKBR3 cells.
Fig. S3. Selection of flat cells.
Fig. S4. Micrographs (DIC) showing examples of live flat SKBR3 cells.
Fig. S5. ESEM-STEM image used to determine the resolution on QDs of labeled HER2 on SKBR3 cells.
Fig. S6. High-resolution TEM image of QDs.
Fig. S7. ESEM-STEM image of immobilized streptavidin-conjugated QDs.
Table S1. Cluster analysis of HER2 labeling.