

Fab binding to CD3 couples with weak antigen recognition to enhance T cell responses and immunotherapy.
Keywords: Antigen receptors, Co-potentiation, Synergy, T cell lymphocytes, T cell antigen recognition, T cell responses, Monovalent Fab fragments, cancer, melanoma, Immunotherapies
Abstract
Adaptive immunity is mediated by antigen receptors that can induce weak or strong immune responses depending on the nature of the antigen that is bound. In T lymphocytes, antigen recognition triggers signal transduction by clustering T cell receptor (TCR)/CD3 multiprotein complexes. In addition, it hypothesized that biophysical changes induced in TCR/CD3 that accompany receptor engagement may contribute to signal intensity. Nonclustering monovalent TCR/CD3 engagement is functionally inert despite the fact that it may induce changes in conformational arrangement or in the flexibility of receptor subunits. We report that the intrinsically inert monovalent engagement of TCR/CD3 can specifically enhance physiologic T cell responses to weak antigens in vitro and in vivo without stimulating antigen-unengaged T cells and without interrupting T cell responses to strong antigens, an effect that we term as “co-potentiation.” We identified Mono-7D6-Fab, which biophysically altered TCR/CD3 when bound and functionally enhanced immune reactivity to several weak antigens in vitro, including a gp100-derived peptide associated with melanoma. In vivo, Mono-7D6-Fab induced T cell antigen–dependent therapeutic responses against melanoma lung metastases, an effect that synergized with other anti-melanoma immunotherapies to significantly improve outcome and survival. We conclude that Mono-7D6-Fab directly co-potentiated TCR/CD3 engagement by weak antigens and that such concept can be translated into an immunotherapeutic design. The co-potentiation principle may be applicable to other receptors that could be regulated by otherwise inert compounds whose latent potency is only invoked in concert with specific physiologic ligands.
INTRODUCTION
Upon recognition of peptide–major histocompatibility complex (pMHC) antigens, T cell receptor (TCR)/CD3 triggering induces signal transduction that requires receptor clustering (1, 2) but may be influenced by biophysical changes proposed to occur in TCR (3–8) and CD3 (9–12). The structural nature of such ligand-induced receptor alteration is not completely understood, potentially representing allosteric movement (2, 6), relative movement between subunits (13, 14), or dynamic states of quaternary structure that respond to external torque and directional force induced by ligation (15–17). Although these and other (18–20) structural contributions to TCR/CD3 signaling lack complete biophysical and functional characterization, they tend to share the attribute that weak antigens poorly induce these effects in T cells whereas strong antigens readily induce them.
This generates the hypothesis that T cell responses to weak antigens might be enhanced under conditions where a putative “pro-activation” TCR/CD3 biophysical state could be exogenously provided. To test this hypothesis, we sought and identified a monovalent anti-TCR/CD3 Fab that could produce a biophysical alteration to the receptor without any intrinsic stimulatory capacity (because its monovalent binding does not induce receptor cross-linking). To ensure a lack of protein aggregation and preservation of monovalency, we subjected Fab to rigorous quality control by strictly following previously published procedures (21). As a marker of biophysical alteration upon Fab binding to TCR/CD3, a well-described CD3 conformational change (CD3Δc) was used; this CD3Δc involves the exposure of a cryptic proline-rich sequence (PRS) present in the cytoplasmic tail of CD3ε that is induced upon productive TCR triggering by antibody or pMHC-based ligands. (9, 10, 22–25). In mature T cells, weak pMHC ligands have poor efficacy for CD3Δc induction, TCR clustering, and downstream T cell signaling and activation (2, 26). In the current work, we accept CD3Δc induction as an indicator of Fab-induced TCR/CD3 biophysical alteration without seeking to fully characterize or account for all possible biophysical consequences of Fab binding. Selection of the anti-TCR/CD3 Fab on this basis allowed the present experiments to test the “co-potentiation” hypothesis by assessing the functional pro-immune consequences the Fab could provide when T cells respond to weak antigens.
A co-potentiation mechanism that could be immunopharmacologically harnessed may favorably contribute to therapeutic strategies against cancer. Stage IV melanoma is difficult to treat, but immunotherapy in recent years has significantly increased mean patient life span from 6.4 to 10.1 months after diagnosis (27). Thus, this cancer can be targeted by the immune system, while strategies to further improve outcome are of outstanding interest. We present experiments to test the extent to which Fab-based T cell co-potentiation could boost immune response in a mouse model of metastatic melanoma in vivo. We propose that the data support the co-potentiation model and its application to anti-metastatic melanoma immunotherapy, improving outcome as a lone immunotherapy and, even better, showing compatibility and synergy with other immunotherapies for best outcome and survival. The co-potentiation concept may be applicable to other receptor systems where latent potency of a drug might only be activated in the presence of specific physiologic ligands.
RESULTS
Mono-7D6-Fab binds T cells without interrupting pMHC:TCR interactions and is functionally inert
Mono-7D6-Fab was obtained by papain digestion of the monoclonal antibody (mAb) 7D6 [specific for the mouse CD3εγ heterodimer of TCR/CD3 (28)], followed by purification and storage in the presence of the osmolyte l-proline for long-term preservation of Fab monovalency (21). Mono-7D6-Fab bound to mouse CD4+ and CD8+ T cells (Fig. 1A) but did not prevent or enhance the binding of physiologic pMHC antigens to transgenic TCRs, Pmel-1, or OT-I [Fig. 1, B and C, and fig. S1 (21)]. Furthermore, Pmel-1 or OT-I T cells bound by Mono-7D6-Fab showed unaltered TCR down-regulation in response to signaling upon exposure to their respective antigenic peptides loaded in antigen-presenting cells (Fig. 1, D and E). As expected, because of its monovalency, Mono-7D6-Fab did not stimulate sensitive T cell responses such as CD69 or CD25 up-regulation, whereas cross-linking 2C11 anti-CD3ε mAb induced these responses (Fig. 1F).
Fig. 1. Mono-7D6-Fab binds T cells without interrupting pMHC:TCR interactions and is functionally inert.
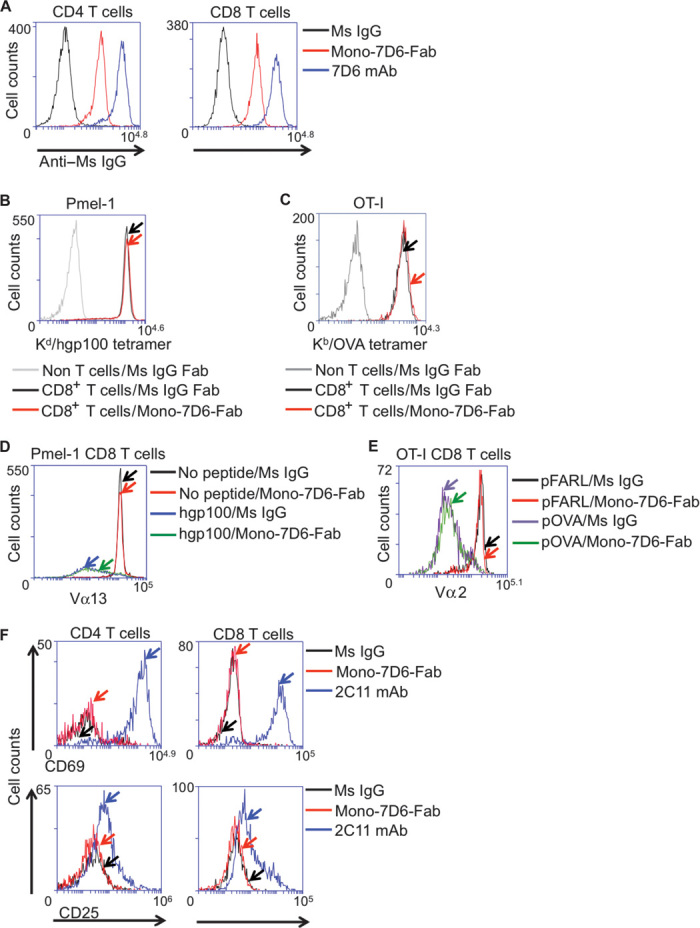
(A) Mono-7D6-Fab binds to T cells. Thy1.2+CD4+ or Thy1.2+CD8+ B6 cells were analyzed for binding irrelevant Ms IgG, Mono-7D6-Fab, or 7D6 mAb detected by anti–Ms IgG fluorescein isothiocyanate. (B and C) Mono-7D6-Fab does not block antigen binding to transgenic TCRs. (B) Pmel-1 TCR transgenic cells were preincubated with control Ms IgG Fab or Mono-7D6-Fab, and Thy1.1+CD8+ cells were analyzed for binding H-2KD/hgp100 tetramer. (C) OT-I TCR transgenic cells were preincubated with control Ms IgG Fab or Mono-7D6-Fab, and Thy1.2+CD8+ cells were analyzed for binding H-2KB-OVA tetramer. (D and E) Mono-7D6-Fab does not alter T cell response to stimulation of transgenic TCRs by strong pMHC ligands. (D) Pmel-1 cells were cultured with no peptide or hgp100 peptide (500 ng/ml) in the presence of control Ms IgG or Mono-7D6-Fab and analyzed at 24 hours for Pmel-1 Vβ13 TCR down-regulation. (E) OT-I T cells were cultured with no peptide or 0.2 nM pOVA in the presence of control Ms IgG or Mono-7D6-Fab and analyzed at 24 hours for OT-I Vα2 TCR down-regulation. (F) Mono-7D6-Fab binding does not stimulate T cells. B6 cells were incubated with Ms IgG Fab, Mono-7D6-Fab, or 2C11 mAb and analyzed for induction of CD69 or CD25 expression. From (A) to (F), one representative experiment out of three or more performed experiments is shown.
Mono-7D6-Fab does not stimulate early TCR/CD3 signaling events but induces CD3Δc
Mono-7D6-Fab did not stimulate early TCR/CD3 signaling products such as CD3-associated phosphotyrosine (Fig. 2A) or calcium flux (Fig. 2B). However, Mono-7D6-Fab induced open CD3 conformation (CD3Δc), as evidenced by exposure of the PRS in CD3ε (Fig. 2C). CD3Δc was assessed using the CD3 pull-down (CD3-PD) assay (see Materials and Methods), which utilized glutathione S-transferase (GST)–SH3.1Nck beads that bind to the PRS in CD3ε when it is accessible but not when it is cryptic (22). APA1/1 anti-CD3ε mAb binds the PRS and blocks CD3-PD assay, revealing the assay background (Fig. 2C, “Bckg.” lane), whereas in the absence of blockade, a moderate positive basal level of open CD3 is detectable [Fig. 2C, “Basal” lane (22)]. Upon ligation with either intact 2C11 anti-CD3ε mAb or Mono-7D6-Fab, CD3Δc was induced to a similar degree, which resulted in greater access of GST-SH3.1Nck to its PRS binding site and thus greater quantities of CD3 in the CD3-PD assay (Fig. 2C, “CD3Δc” lanes). In control lanes, total lysate content without CD3-PD assay contained similar amounts of CD3 (Fig. 2C, “TL”). Thus, Mono-7D6-Fab did not impede pMHC:TCR interactions and did not intrinsically induce TCR/CD3 cross-linking and signaling but induced a physical alteration to TCR/CD3, marked at least by CD3Δc.
Fig. 2. Mono-7D6-Fab does not stimulate early TCR/CD3 signaling events but induces CD3Δc.
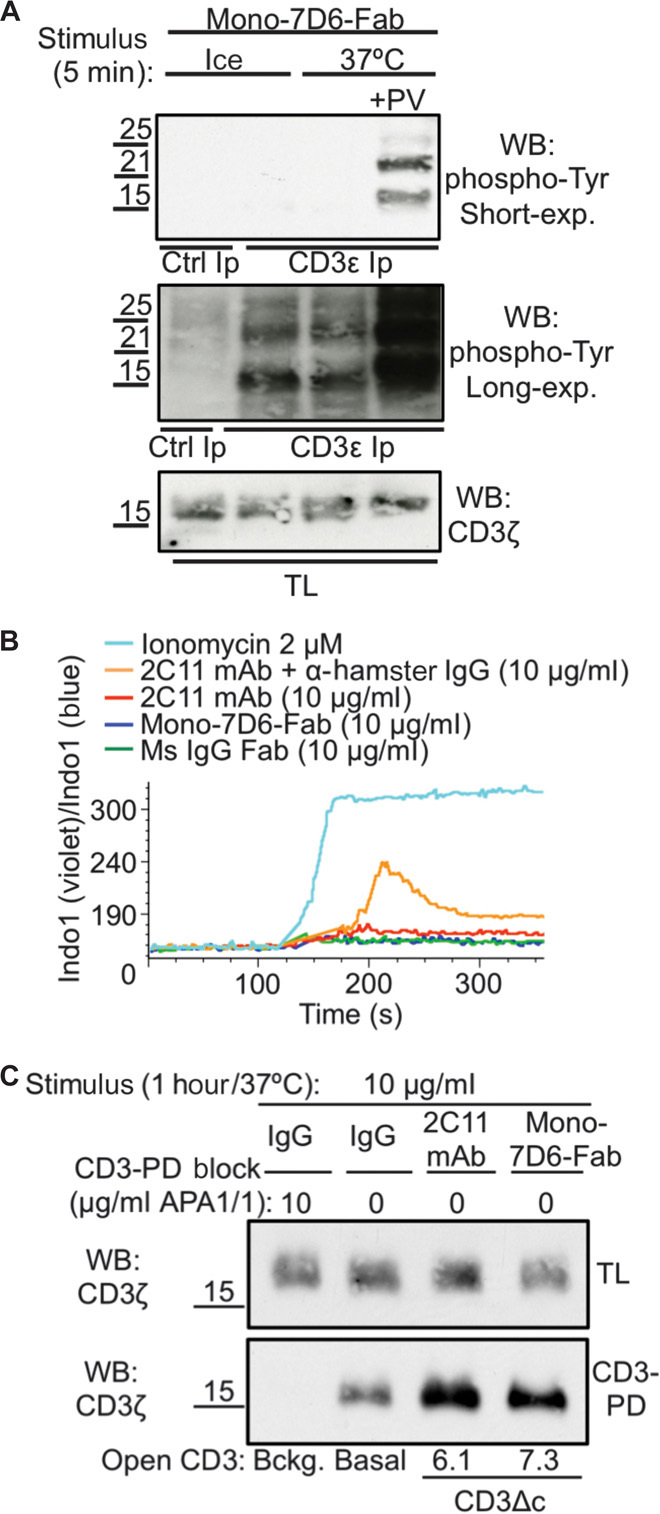
(A) B6 cells were treated with Mono-7D6-Fab on ice versus 37°C with or without pervanadate (PV) for 5 min. Lysates were immunoprecipitated with control IgG or anti-CD3ε (APA1/1). CD3-associated phosphotyrosine was detected by Western blot (WB) analysis using the anti-PY (phospho-tyrosine) mAb 4G10 (short and long exposures shown). TL, total lysate content before immunoprecipitation (Ip). (B) Indo1/AM-loaded B6 cells were stimulated with the indicated reagents for 5 min at 37°C. Calcium flux was measured as a ratio of 405-nm/495-nm fluorescence gated on Thy1.2+ T cells. (C) Pmel-1 lysate was incubated with APA1/1 (pull-down blocking condition), hamster IgG (basal condition), 2C11 (full induction condition), or Mono-7D6-Fab (test condition). Lysates were subjected to CD3-PD assay, and open CD3 conformation was determined by WB analysis of the CD3ζ subunit. Numbers represent a fold increase in PRS-accessible (open) CD3. TL, total lysate content before pull-down.
Mono-7D6-Fab increases T cell responses to weak antigens in vitro
We investigated whether Mono-7D6-Fab could affect T cell antigen sensitivity in vitro. In the OT-I TCR transgenic system, pOVA strongly stimulates OT-I T cells, whereas several altered peptide ligands (pE1, pG4, pQ7, pQ4H7, and pT4) are less stimulatory (26). At 24 hours, Mono-7D6-Fab had increased OT-I T cell responses to the weak peptides pQ7, pQ4H7, and pT4, observed through enhanced up-regulation of surface CD69 [Fig. 3A and fig. S2 (26)] and of the intracellular transcription factors interferon regulatory factor-4 (IRF4) and T-bet [Fig. 3, B and C, and fig. S3 (29)]. At 96 hours, Mono-7D6-Fab had increased the percentage of OT-I T cells that divided in response to the weak peptides pQ7 and pQ4H7 (Fig. 3D, “Cell ÷”). The effect of Mono-7D6-Fab on pT4 peptide stimulation involved enhancement of IRF4 and T-bet induction (Fig. 3, B and C) without concomitant enhancement of already positive cell division (Fig. 3D), suggesting that Mono-7D6-Fab can affect effector differentiation separately from expansion. Throughout these experiments, Mono-7D6-Fab was not intrinsically stimulatory under the null-peptide condition [no peptide, mouse IgG (Ms IgG), or pFARL; Figs. 1 to 3 and figs. S2 and S3], nor did Mono-7D6-Fab enhance responses to the weakest OT-I–specific peptides (Fig. 3 and figs. S2 and S3). Thus, Mono-7D6-Fab treatment enhanced T cell activation and effector differentiation only when weak pMHC ligands engaged OT-I TCRs with a specific minimal level of potency.
Fig. 3. Mono-7D6-Fab increases T cell responses to weak antigens in vitro.
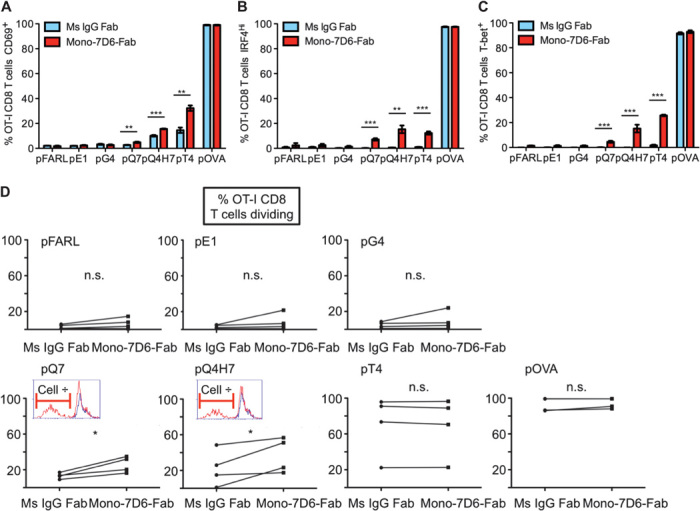
(A to E) OT-I cells were incubated with the indicated peptides in the presence of Ms IgG Fab or Mono-7D6-Fab for 24 hours (A to C) or 96 hours (D and E). For flow cytometry analysis, OT-I T cells were gated as Thy1.2+Vα2+CD8+. Representative experiments (expressed as percentage) are shown (mean ± SD from triplicate samples): (A) CD69 up-regulation assay (n ≥ 7), (B) IRF4 up-regulation assay (n = 4), (C) T-bet up-regulation assay (n = 2), and (D) CFSE cell division assay. For each peptide stimulation, graphs show paired Ms IgG Fab and Mono-7D6-Fab mean percentages from triplicate samples of dividing OT-I CD8 T cells found in each independent experiment (n = 4). Insets for pQ7 and pQ4H7 depict overlays of the CFSE profiles of Ms IgG Fab–treated (blue line) and Mono-7D6-Fab–treated (red line) samples from one representative experiment. (A to D) *P < 0.05, **P < 0.005, ***P < 0.0005, two-tailed unpaired Student’s t test.
Mono-7D6-Fab injection is inert in unimmunized mice in vivo
If the critical ligand potency level could be supplied by self-pMHC expressed systemically, then administration of Mono-7D6-Fab in vivo might cause overtly stimulatory or autoreactive effects. However, a single dose of Mono-7D6-Fab injected intravenously into C57BL/6 (B6) mice did not alter the T cell/B cell ratio in blood compared with equivalent injections of irrelevant Ms IgG Fab or Fc domain–containing complement-depleting 7D6 mAb or 2C11 mAb (Fig. 4A), indicating that Mono-7D6-Fab mediated neither T cell expansion nor depletion in mice. Mono-7D6-Fab also failed to produce overt signs of piloerection or diarrhea during 6 hours of observation, as would have been expected had cytokine release syndrome been induced (30). Mono-7D6-Fab administered four times per week failed to induce detectable levels of inflammatory cytokines unlike those induced by the superantigen staphylococcal enterotoxin B [Fig. 4B (31)]. Furthermore, the multi-injection regimen failed to induce detectable pathology in the liver, kidneys, or lungs (Fig. 4C). Therefore, in the absence of immune response, Mono-7D6-Fab was not intrinsically stimulatory, nor did it promote T cell responses against healthy tissues.
Fig. 4. Mono-7D6-Fab injection is inert in unimmunized mice in vivo.
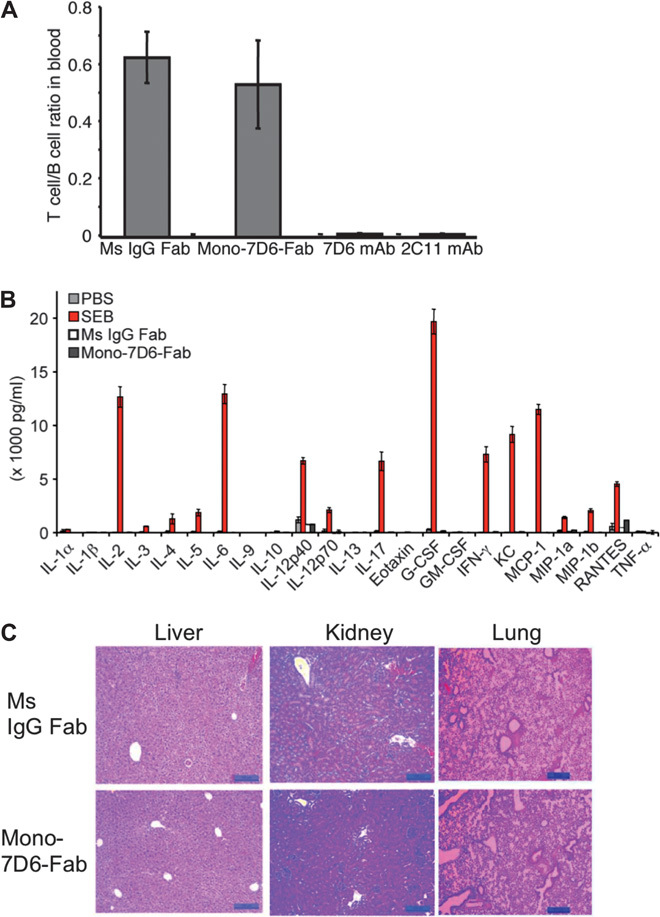
(A) Three B6 mice per experimental group were injected retro-orbitally with 10 μg of control Ms IgG Fab, Mono-7D6-Fab, 7D6 mAb, or 2C11 mAb. After 24 hours, blood samples were collected, and cells were counted in triplicate and analyzed by flow cytometry. The T cell/B cell ratio found in the blood is displayed (mean ± SEM). (B and C) Three B6 mice per experimental group were injected retro-orbitally with 10 μg of control Ms IgG Fab or Mono-7D6-Fab on days 0, 2, 4 and 6, and were sacrificed on day 7. (B) Serum cytokines found in plasma after 7 days. For comparison, serum cytokines from mice sacrificed 3 hours after a single injection of PBS or 50 μg of staphylococcal enterotoxin B (SEB) were quantified in parallel (mean ± SEM). (C) Liver, kidney, and lung tissues were collected in buffered formalin, paraffin-embedded, cut, and stained with hematoxylin and eosin (H&E) following standard procedures for histopathologic analysis. H&E-stained sections were evaluated by light microscopy. Scale bar, 100 μm. G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte-macrophage colony-stimulating factor; KC, keratinocyte chemoattractant; MCP-1, monocyte chemoattractant protein-1; MIP-1a, macrophage inflammatory protein-1α; MIP-1b, macrophage inflammatory protein-1β; TNF-α, tumor necrosis factor–α.
Mono-7D6-Fab reduces tumor burden in a mouse model of melanoma lung metastasis
To determine whether Mono-7D6-Fab could enhance T cell responses against metastatic melanoma in vivo, we studied B16-F10, a subclone of the poorly immunogenic B16 melanoma, which, upon intravenous injection, produces melanoma nodules in the lungs (a recognized mouse model of melanoma lung metastasis) (32). Three weeks after intravenous tumor injection and a single 10-μg dose of Mono-7D6-Fab, we observed a significant decrease in lung tumor burden compared with mice receiving Ms IgG Fab control treatment (Fig. 5A), objectively quantified by lung weight (Fig. 5B) and pixel densitometry of digital lung images (Fig. 5C). Compared with control treatment, a higher percentage of CD4+ and CD8+ T cells in the mediastinal (lung-draining) lymph nodes of mice treated with Mono-7D6-Fab had up-regulated CD44 (Fig. 5D), indicating enhanced T cell activation. PD1+CD11a+ T cells were recently reported to mark tumor-reactive T cells in both mice and humans (33), and we observed this T cell subset to be increased in both CD4+ and CD8+ cells in the mediastinal lymph nodes of mice treated with Mono-7D6-Fab (Fig. 5E). These data suggest that Mono-7D6-Fab increased T cell reactivity against B16-F10 melanoma cells.
Fig. 5. Mono-7D6-Fab reduces tumor burden in a mouse model of melanoma lung metastasis.
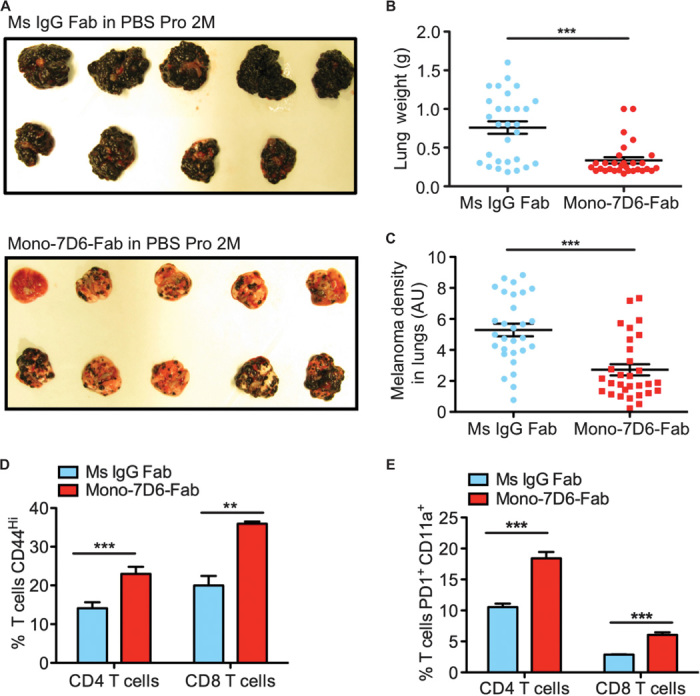
(A and C) B6 mice were intravenously injected with B16-F10 melanoma cells and then with control Ms IgG Fab or Mono-7D6-Fab. After 21 days, the lungs were photographed and weighed. (A) Lung photographs from one independent experiment. (B and C) Dots represent individual mice with data pooled from three independent experiments (mean ± SEM). (B) Lung weight. (C) Quantification of melanoma found in lungs (B and C; ***P < 0.0005, two-tailed unpaired Student’s t test). (D) Percentage of CD44+ and (E) percentage of PD1+CD11a+ T cells found in pooled mediastinal lymph nodes from mice engaged in one of three representative experiments, as shown in (A) to (C) (mean ± SD from triplicate samples; **P < 0.005, ***P < 0.0005, two-tailed unpaired Student’s t test).
Depletion of either the CD4+ T cell subset or the CD8+ T cell subset reduced the efficacy of Mono-7D6-Fab anti-melanoma effects to a similar degree, although an antitumor effect of decreased magnitude remained in either case (fig. S4), showing that both CD4+ and CD8+ T cells play a role in the anti-melanoma effects of Mono-7D6-Fab. In T cell–deficient mice [CD3ε−/−ζ−/− (34, 35)], Mono-7D6-Fab administration did not reduce lung melanoma burden compared with Ms IgG Fab control treatment (fig. S5, A and B). Mono-7D6-Fab also failed to reduce lung melanoma burden in OT-I/Rag2−/− mice, which express a T cell population bearing a single OT-I TCR that is melanoma-irrelevant (fig. S5, C and D). Finally, to assess pre-established metastases, we administered Mono-7D6-Fab (3 days after B16-F10 melanoma injection) to wild-type B6 mice, where we still observed a significant decrease in lung melanoma burden among those treated with Mono-7D6-Fab compared with those treated with control Ms IgG Fab (fig. S6). We conclude that the anti-melanoma effects of Mono-7D6-Fab are effective against immediate and pre-established metastases, requiring T cell function where antigenic specificity or clone identity is relevant.
Mono-7D6-Fab promotes therapeutic antitumor T cell responses against pre-established melanoma lung metastases and synergizes with other immunotherapeutic modalities
We next assessed the extent to which Mono-7D6-Fab could amplify T cell responses to a specific melanoma-associated antigen. Mouse T cells expressing Pmel-1 TCR are more strongly stimulated by the human gp100 (hgp100) peptide than by the low-affinity self-peptide mouse gp100 (mgp100) (36) expressed by healthy melanocytes and melanoma cells (37). In vitro, we found mgp100 antigen to be stronger than weak antigens of the OT-I system such that CD69 up-regulation was already equally induced by both hgp100 and mgp100; however, Mono-7D6-Fab increased effector interferon-γ (IFN-γ) secretion by Pmel-1 T cells in response to mgp100 (fig. S7), promoting the hypothesis that Mono-7D6-Fab might enhance T cell responses directed against tumors expressing this antigen. To test this hypothesis in vivo, we combined Mono-7D6-Fab with a published anti–B16-F10 therapeutic approach involving adoptive transfer of Pmel-1 cytotoxic T lymphocytes (CTLs) and administration of human interleukin-2 (IL-2) 3 days after tumor injection (38). Control treatments involved injection of tumor-irrelevant OT-I CTLs and/or Ms IgG Fab, while all mice received IL-2. We observed that the Mono-7D6-Fab + Pmel-1 CTL treatment group combined to produce the most dramatic reduction of melanoma lung metastases; the Mono-7D6-Fab + control CTL group or the Pmel-1 CTL + control Ms IgG Fab group produced antitumor effects of decreased magnitude; and all targeted regimens reduced lung metastases compared with the control-only group (Fig. 6, A and B). Compared with control Ms IgG Fab, Mono-7D6-Fab combined with Pmel-1 CTLs to increase the number of Pmel-1 T cells found in both mediastinal lymph nodes and lungs (Fig. 6, C and D). In a separate longitudinal experiment, B6 mice that received Mono-7D6-Fab + Pmel-1 CTL survived significantly longer than those that received control Fab + Pmel-1 CTL (**P < 0.005; Fig. 6E). These data show that a single 10-μg injection of Mono-7D6-Fab was sufficient to enhance responses to the weak antigen mgp100 and to prolong survival in vivo.
Fig. 6. Mono-7D6-Fab promotes therapeutic antitumor T cell responses against pre-established melanoma lung metastases.
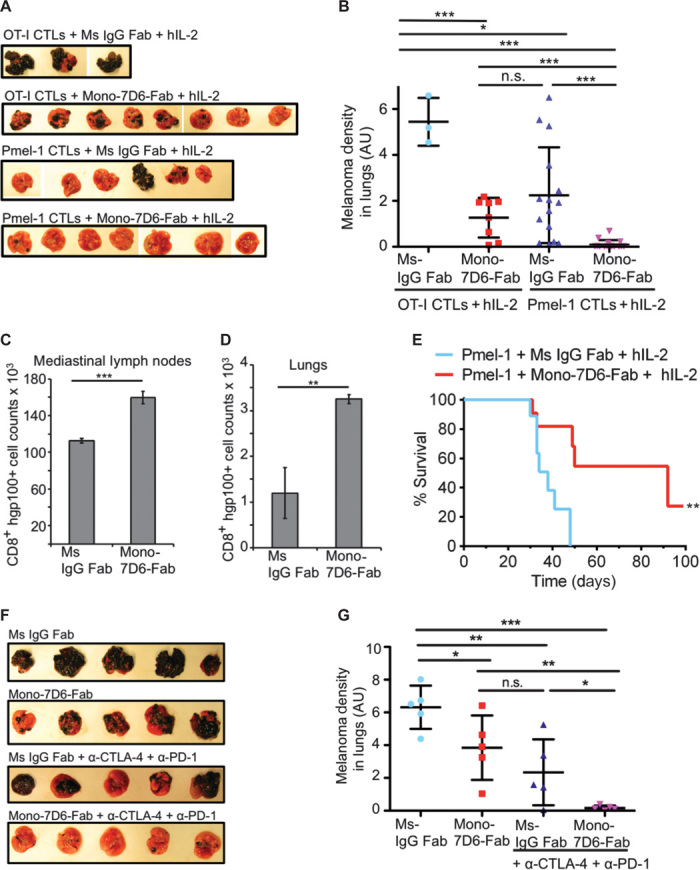
(A and B) B6 mice were intravenously injected with B16-F10 melanoma cells. Three days later, OT-I or Pmel-1 CTLs were adoptively transferred into tumor-bearing recipients. Subsequently, mice were injected with either control Ms IgG Fab or Mono-7D6-Fab. In addition, all mice were treated with 120,000 U of human IL-2 on day 3, followed by 60,000 U of human IL-2 on each of days 4 and 5. Mice were sacrificed after 28 days. (A) Lung photographs from one independent experiment. (B) Quantification of melanoma found in lungs. Dots represent individual mice with data pooled from two independent experiments (mean ± SEM; *P < 0.05, ***P < 0.0005, two-tailed unpaired Student’s t test). (C and D) Total CD8+ T cell counts positive for staining with Kd/hgp100 dextramer found in pooled (C) mediastinal lymph nodes or (D) lungs of mice adoptively transferred with Pmel-1 CTLs from (A) and (B) (mean ± SEM from triplicate samples; **P < 0.005, ***P < 0.0005, two-tailed unpaired Student’s t test). (E) Three days after B16-F10 injection, Pmel-1 CTLs were adoptively transferred into B6 recipients, together with Ms IgG Fab or Mono-7D6-Fab treatment [as described in (A) and (B)]. Survival was monitored (n = 10, **P < 0.005, Mantel-Cox log-rank test). (F and G) B6 mice were intravenously injected with B16-F10 melanoma cells. Three days later, mice were intraperitoneally injected with anti–CTLA-4 and anti-PD1, or control antibodies. In addition, on day 3, mice were intravenously injected with either Mono-7D6-Fab or control Ms IgG Fab. Subsequently, on days 5, 7, and 9, mice were re-treated with anti–CTLA-4 and anti-PD1 (or control) antibodies. After 24 days, the lungs were collected. (F) Lung photographs from one independent experiment. (G) Quantification of melanoma found in lungs. Dots represent individual mice (mean ± SEM; *P < 0.05, **P < 0.005, ***P < 0.0005, two-tailed unpaired Student’s t test).
To test whether Mono-7D6-Fab could synergize with yet another immunotherapeutic modality, we combined Mono-7D6-Fab treatment with blockade of the T cell inhibitory receptors CTLA-4 and PD1 (39). Again, we observed the most dramatic reduction of melanoma lung metastases in the combined Mono-7D6-Fab + anti–CTLA-4/anti-PD1 treatment group compared with separate treatment groups or the control group (Fig. 6, F and G). Note that a single 10-μg dose of Mono-7D6-Fab exerted its effects while 100-μg doses of anti–CTLA-4 and 50-μg doses of anti-PD1 were co-injected on days 3, 5, 7, and 9. Therefore, Mono-7D6-Fab can increase T cell sensitivity against weak tumor antigens and can be combined with alternative complementary strategies targeting T cell function to achieve a superior anti-metastatic tumor effect.
DISCUSSION
Mono-7D6-Fab co-potentiated TCR/CD3 signaling in response to weak pMHC antigens that possessed a critical minimal antigenic strength. Here, we present the main model of receptor co-potentiation by an otherwise intrinsically inert monovalent Fab (fig. S8). Ligation of TCR/CD3 by weak antigens induces weak signal transduction and weak T cell responses (fig. S8A). In contrast, ligation by anti-TCR/CD3 monovalent Fab is inert, inducing no classical signal transduction and producing no functional T cell response even though such Fab binding may induce biophysical alteration to the receptor (fig. S8B). The co-potentiation principle states that weak antigenic ligation plus intrinsically inert monovalent Fab ligation can synergize to enhance TCR signaling and T cell responses (fig. S8C).
The identification of specific properties displayed by Mono-7D6-Fab permitted an investigation of the co-potentiation concept. Mono-7D6-Fab did not impede pMHC:TCR interactions and did not intrinsically induce TCR/CD3 cross-linking and signaling but induced a biophysical alteration to TCR/CD3 marked by CD3Δc (Figs. 1 and 2). We accepted CD3Δc as a marker indicating that Fab binding caused a biophysical alteration to TCR/CD3. Because there is no accepted means of fully characterizing the biophysical state of TCR/CD3, a limitation of this and other studies is that it has not been possible to experimentally isolate as a variable a specific biophysical parameter of TCR/CD3 (such as conformation, heat capacity, and rigidity/flexibility) to assign specific signaling functions to that parameter, having kept all others constant. Therefore, although the present work succeeded in observing and applying the co-potentiation effect, future studies will be required to comprehend the specific biophysical property (or properties) that explains its structural basis. Furthermore, the precise mechanisms by which CD3 signaling and downstream pathways are enhanced when TCRs engage weak antigens remain to be elucidated. Here, the present experiments were designed to directly demonstrate mechanisms in terms of functional consequences on TCR signaling and T cell responses caused by Fab complementation of weak TCR recognition. Thus, we conclude that Mono-7D6-Fab binding to TCR/CD3 co-potentiated signaling and T cell responses to weak pMHC ligands in vitro and in vivo, and we propose that, even without full structural characterization, enough knowledge exists to harness this co-potentiation principle for application to immunotherapy against metastatic melanoma.
One critical observation was that Mono-7D6-Fab treatment enhanced T cell activation and effector differentiation only when weak pMHC ligands engaged TCRs with a specific minimal level of potency (Fig. 3). We speculate that this is the reason that, in the absence of an immune response, Mono-7D6-Fab was not intrinsically stimulatory and failed to promote T cell responses against healthy tissues (Fig. 4). These observations also make the formal possibility that a low level of Fab aggregation induced TCR/CD3 cross-linking and mediated the co-potentiation response unlikely. Although our controls and practices were sufficient to ensure Fab monovalency up to the point of administration (21), we must still accept the possibility that, after addition to tissue culture or injection in vivo, changes in Fab valency could potentially occur in situ, which we could not directly control or monitor. However, if this had been the case, the clear prediction is that pro-signaling effects would have been observed under the null conditions in vitro and in the negative control experiments in vivo, but this was not observed. This argues that monovalent Fab mediated the co-potentiation effect, which occurred specifically for pMHC ligands of requisite minimal stimulatory capacity, selectively augmenting specific T cell responses without pan T cell activation.
Mono-7D6-Fab increased T cell reactivity against B16-F10 melanoma cells, with efficacy against immediate and pre-established metastases requiring T cell function wherein antigenic specificity or clone identity played a role (Figs. 5 and 6). It is important to note that Mono-7D6-Fab was compatible and synergistic in combination with alternative complementary strategies targeting T cell function to achieve an anti-metastatic tumor effect that was superior to any of the lone therapeutic modalities (Fig. 6). This compatibility with therapeutic combination makes sense considering that co-potentiation involves “signal 1” (the TCR/CD3 signal), checkpoint inhibition regimens (anti–CTLA-4/anti-PD1) are related to costimulatory/coinhibitory “signal 2” (40), and ex vivo CTL regimens utilize signals 1, 2, and 3 (including innate immune and cytokine signals) to maximize tumor-specific T cell responses (36). In those scenarios, co-potentiation to increase responsiveness to weak T cell antigens is predicted to be helpful, without foreseeable reasons a priori to hypothesize conflicts or untoward outcomes from combination immunotherapy. In this regard, the effects of Mono-7D6-Fab on B16-F10 metastasis were observed with a single injection of 10 μg, which, though a solo regimen, induced results comparable to those of four injections each of anti–CTLA-4 (100-μg doses) + anti-PD1 (50-μg doses; Fig. 6). Future experiments must determine the effectiveness of Mono-7D6-Fab in other tumor models. On the whole, the present data cast a good light on co-potentiation as a mechanism that has a significant potential for therapeutic development.
One wonders whether all of the specific biophysical features required to enhance TCR/CD3 responsiveness upon binding Mono-7D6-Fab might occur naturally when TCRs bind physiologically strong pMHC ligands. Regardless, the co-potentiation described here is a surprising observation given that monovalent TCR ligands administered alone have long been accepted as being intrinsically inert (41, 42), a point that is in agreement with the negative controls of the present experiments. Furthermore, co-potentiation appears to be a readily applicable concept, occurring at the level of the surface receptor and available to be targeted and harnessed for immunotherapy. It is possible that the co-potentiation principle may also apply to other receptor systems, where creating latent potency by design (invoked only in combination with specific physiologic ligands) might produce additional biological insights or biomedical applications.
MATERIALS AND METHODS
Experimental design
Sample size was determined by published precedent data
All in vitro assays were performed in triplicate, which allowed for calculation of central values, proportions, SD, and SEM. In addition, interexperimental n was decided to be at least 3 a priori. For in vivo experiments, the minimum was five mice per experimental group (43). We routinely prepared up to 10 mice per experimental group when age/sex matching and cost permitted. For tumor data, we included all data from multiple experiments. Statistical analysis is presented unpaired (that is, data from the same experimental group but different independent experiments are reported together on a single scale); thus, we expect that the statistical significance we report is conservative.
Rules for stopping data collection
All assay endpoints were decided in advance, with one exception. We allowed the endpoint for in vivo tumor experiments to be flexible such that if positive control mice (those with predicted maximal tumor burden) in a particular experiment did not show outward signs of sickness by 21 days after tumor inoculation, then the experiment could be extended to 28 days before sacrificing and tissue harvest.
Inclusion and exclusion criteria
All criteria were established prospectively. All data were included in the analyses, with one exception. For in vivo tumor experiments, if a mouse died within 3 hours of intravenous tumor injection, it was considered a technical failure of injection and was excluded from the experiment. There was no identification or exclusion of outliers from any data set.
Research objectives
The main prespecified hypothesis was that Mono-Fab alone would not induce T cell activation but would enhance T cell stimulation by weak pMHC ligands. We did not hypothesize or pre-estimate the magnitude of a putative co-potentiation effect, which is why we chose not to pre-estimate or rely on a power calculation to decide experimental n.
Research subjects
B6 mice were used in the experiments. Transgenic and knockout mice are specified in the corresponding experiments and in the following section.
This study used controlled laboratory experiments, with specific details outlined in the corresponding sections of the text, in bibliographic references, and in the following sections. Individual mice were chosen without subjective or objective precharacterization. Single blinding was used at one specific step during the in vivo tumor experiments: (i) one coauthor (M.J.H.) injected all tumors through the tail vein and (ii) passed all mice to another coauthor (M.M.H.) who, 3 to 72 hours later, sorted tumor recipients into treatment groups and injected treatments retro-orbitally and/or intraperitoneally (as specified for each treatment). This was performed to prevent the treatment injector from experiencing any intuition about the quality of tumor injections in individual mice before assigning them to treatment groups.
Mice
B6 mice were bred as needed in-house at Mayo Clinic and used between 6 and 12 weeks of age. Female B6 mice were purchased from The Jackson Laboratory. OT-I and Pmel-1 mice were provided by R. Vile (Mayo Clinic). CD3ε−/−ζ−/− mice (T cell–deficient mice lacking all four CD3 subunits CD3γδεζ) (34, 35) were originally provided by D. Vignali (St. Jude Children’s Research Hospital) with permission from Cox Terhorst (Beth Israel Deaconess Medical Center, Harvard Medical School). The cells analyzed and represented in each mouse were pooled whole splenocytes plus peripheral lymph node cells, unless otherwise noted. Mouse procedures were approved by the Mayo Institutional Care and Use Committee, consistent with National Institutes of Health guidelines for the care and use of animals.
Cell lines
B16-F10 melanoma cells were a gift from R. Vile (Mayo Clinic). B16-F10 cells were grown in Dulbecco’s modified Eagle’s medium (Gibco Life Technologies) supplemented with 10% (v/v) cosmic calf serum (HyClone), 1× Gibco tissue culture additives (l-glutamine, penicillin/streptomycin, nonessential amino acids, and sodium pyruvate), and 50 μM β-mercaptoethanol (Sigma).
Peptides, antibodies, and other reagents
The peptides pFARL (SSIEFARL; negative control without specificity for OT-I TCR), pE1 (EIINFEKL), pG4 (SIIGFEKL), pQ7 (SIINFEQL), pQ4H7 (SIIQFEHL), pT4 (SIITFEKL), pOVA (SIINFEKL), mgp100 (EGSRNQDWL), and hgp100 (KVPRNQDWL) were purchased from Elim Biopharmaceuticals. Allophycocyanin (APC)–labeled H2-Kb/OVA and APC-labeled H2-Kb/Q7 tetramers were prepared in the laboratory as previously described (44). Phycoerythrin (PE)–labeled H2-Kd/hgp100 tetramer was purchased from Beckman Coulter. PE-labeled H2-Kd/hgp100 dextramer was purchased from Immudex. The following mAbs were purified in-house from hybridoma supernatant: 7D6 (Ms IgG2a, specific for mouse CD3εγ), 2C11 (hamster IgG1, specific for mouse CD3ε), and H146 (hamster IgG, specific for mouse CD3ζ). 7D6 hybridoma was provided by B. Alarcón (Centro de Biología Molecular Severo Ochoa, Universidad Autónoma de Madrid). 2C11 and H146 hybridomas were provided by E. Palmer (University Hospital Basel). Antibodies from eBioscience included anti-Thy1.2 (30-H12), anti-Thy1.1 (OX-7), anti-CD8β (53-5.8), anti-CD4 (GK1.5), anti-CD69 (H1.2F3), anti-PD1 (RMP1–30), anti-CD11a (2D7), anti-CD44 (IM7), anti-CD25 (3C7), anti-IRF4 (3E4), and anti–T-bet (ebio4B10). Polyclonal antibodies from Jackson ImmunoResearch included Ms IgG, Ms IgG Fab (control Ms IgG), anti-Ms IgG fluorescein isothiocyanate, and anti–Armenian hamster IgG horseradish peroxidase (HRP). Depletion antibodies against CD4 (GK1.5) and CD8 (2.43) and anti–CTLA-4 (9H10) were purchased from BioXcell. For cell and lysate incubation and stimulation, mAbs and Fabs were used at 10 μg/ml.
Mono-7D6-Fab generation and purification
Mono-7D6-Fab was generated and purified as previously described by Nelson et al. (21). Briefly, 7D6 mAb was cleaved with the cysteine protease papain (Sigma-Aldrich) for 24 hours at 37°C, and the digestion was quenched with 30 mM iodoacetamide (Sigma-Aldrich). The digestion was dialyzed with frequent buffer exchanges in phosphate-buffered saline (PBS) at 4°C. Next, Fc fragment–containing species were removed by incubating the digestion with Protein A Sepharose beads (GE Healthcare) at 4°C overnight. Fab preparations were sterile-filtered, and total protein was quantified using a Nanodrop spectrophotometer (Thermo Scientific). The resulting 7D6-Fab preparations were subjected to size exclusion chromatography (SEC) using two Superdex 200 10/300 GL columns (GE Healthcare) aligned in tandem on an AKTA FPLC system (GE Healthcare). SEC was equilibrated in PBS + 2 M l-proline (PBS Pro 2M) and operated in a chromatography refrigerator at 10° to 16°C. 7D6-Fab preparations were injected over the columns in a sample volume of 1 to 2 ml, and elution of protein was monitored by UV absorbance at 280 nm. The Mono-7D6-Fab species was identified and collected according to its peak volume of elution, as previously described by Nelson et al. (21). Absence of Fab aggregates in the resulting Mono-7D6-Fab samples was tested in a T cell stimulation assay monitoring the up-regulation of the activation marker CD69 and through analysis of Fab sensitivity to fragmentation by SDS–polyacrylamide gel electrophoresis (PAGE) and Western blot analysis, as previously described by Nelson et al. (21).
T cell activation assays
For the monitoring of T cell responses, cells from the pooled spleen and lymph nodes of individual mice of each specified genotype were stimulated for 24 hours (CD69, IRF4, and T-bet up-regulation, TCR/CD3 down-regulation) or 48 hours (CD25 up-regulation) with the indicated peptides (0.2 nM) and/or respective Fab (10 μg/ml) or mAbs (10 μg/ml). Next, we stained the samples on ice with fluorophore-conjugated mAbs to detect Thy1.2, CD8, TCR/CD3, and the respective activation marker, and analyzed them by flow cytometry. For intracellular staining of the transcription factors T-bet and IRF4, after the staining of OT-I T cell surface markers, samples were fixed and permeabilized with the FOXP3/Transcription Factor Staining Buffer Set (eBioscience) according to the manufacturer’s protocol. For the monitoring of T cell division, cells from the pooled spleen and lymph nodes of OT-I mice were labeled with carboxyfluorescein diacetate succinimidyl ester (CFSE) before being incubated with the indicated peptides (0.2 nM) and Mono-7D6-Fab or control Ms IgG Fab (10 μg/ml) in the presence of mouse interleukin-7 (25 U/ml; from the supernatant of interleukin-7–producing J558 cells provided by M. Daniels of University of Missouri) for 96 hours. Afterward, the cells were stained on ice with fluorophore-conjugated mAbs to detect Thy1.2 and CD8 at the cell surface, and with propidium iodide (Sigma-Aldrich) for dead-cell exclusion immediately before flow cytometry. Samples were collected on a BD Accuri C6 Flow Cytometer (BD Biosciences), and data were analyzed using CFlow Plus software.
CD3-PD assay, immunoprecipitation, and Western blots
CD3-PD assay had been previously described as a means to detect CD3Δc (6–8, 10, 12, 13). Briefly, 30 × 106 to 50 × 106 cells from the pooled spleen and lymph nodes of B6 or Pmel-1 mice were lysed in an isotonic buffer containing 1% Digitonin (Sigma-Aldrich), and 1-ml postnuclear lysates were obtained. Samples were precleared by incubation with GST beads (for 1 hour at 4°C) in the presence of 10 μg of the indicated immunoglobulins before specific pull-down with GST-SH3.1 beads (for 4 to 12 hours at 4°C), followed by reducing SDS-PAGE (13% gel), nitrocelullose transfer, and Western blot analysis of CD3ζ (H146). Where indicated, TCR/CD3 was immunoprecipitated with Protein G Sepharose beads (GE Healthcare) bound to the mAb APA1/1 (Ms IgG, specific for the CD3ε cytoplasmic domain). Samples were subjected to reducing SDS-PAGE (12% gel), transferred to nitrocellulose, and subjected to Western blot analysis with the antiphosphotyrosine antibody 4G10 biotin (EMD Millipore) and SA-HRP (R&D Systems).
Calcium flux assay
Cells (5 × 106) from the pooled spleen and lymph nodes of B6 mice were resuspended in Hanks’ balanced salt solution (HBSS) + 10 mM Hepes (pH 7) and loaded with 0.05 mM Indo1/AM (Molecular Probes) at 37°C for 30 min. The cells were washed and resuspended in HBSS + 10 mM Hepes + 0.5% bovine serum albumin (pH 7.4). Next, the cells were stained with anti-Thy1.2 for 15 min at room temperature. Calcium flux was measured using a 405-nm/495-nm fluorescence ratio on a BD LSRII Flow Cytometer (BD Biosciences). Basal condition was first measured at 37°C for 2 min; this was followed by the addition of indicated stimuli and resulting calcium flux for 5 min. Data were analyzed using the kinetic analysis features of FlowJo software (Tree Star).
IFN-γ enzyme-linked immunosorbent assay
Mouse IFN-γ ELISA (enzyme-linked immunosorbent assay) MAX Deluxe Sets were purchased from BioLegend. Cells from the pooled spleen and lymph nodes of Pmel-1 mice were stimulated with the indicated doses of the peptide mgp100 in the presence of Ms IgG Fab or Mono-7D6-Fab (10 μg/ml). After 72 hours, supernatants were collected and ELISAs were performed in triplicate according to the manufacturer’s protocol. Samples were read on a Thermomax microplate reader (Molecular Devices).
Serum cytokine quantification
Blood serum was prepared using serum separation tubes (BD Biosciences). The concentration of cytokines in mouse serum was determined in duplicate samples, using a multiplex bead assay according to the manufacturer’s protocol and using their software (Bio-Plex Pro Mouse Cytokine, Chemokine, and Growth Factor Assays; Bio-Rad) and hardware (Bio-Plex Multiplex microsphere plate reader; Bio-Rad).
B16-F10 tumor experiments
Age-matched mice were intravenously injected with 0.5 × 106 B16-F10 melanoma cells in 200 μl of PBS through the tail vein. Subsequently, mice were retro-orbitally injected with 10 μg of Mono-7D6-Fab or Ms IgG Fab in PBS Pro 2M either on the same day of tumor injection (day 0) or 72 hours later (day 3). Between 21 and 28 days after tumor injection, mice were sacrificed and their lungs were harvested for tumor burden quantification. To deplete T cell subsets from mice during the course of some experiments, we administered 100 μg of anti-CD4 or anti-CD8 antibody intraperitoneally into tumor-bearing mice on days −1, 3, 6, 10, and 17. T cell depletion was periodically monitored in peripheral blood draws by flow cytometry. Efficient depletion was defined as a >90% reduction in the relevant T cell subset. To block CTLA-4 and PD1 receptors in mice, we injected them intraperitoneally with either 100 μg of anti–CTLA-4 mAb (9H10) and 50 μg of anti-PD1 mAb (G4; provided by H. Dong of Mayo Clinic) or control IgG antibodies on days 3, 5, 7, and 9.
Adoptive transfer of CTLs
T cells from the pooled spleen and lymph nodes of OT-I or Pmel-1 mice were activated using plate-bound anti-CD3 (2C11; 10 μg/ml) for 72 hours in the presence of mouse IL-2 (25 U/ml; from the supernatant of IL-2–producing p3x63AG8 cells provided by E. Palmer). Samples were removed from the plate-bound stimulus and cultured for an additional 48 hours in the presence of IL-2 before being washed and adoptively transferred (2 × 106 cells per mouse) by retro-orbital injection into recipient mice. After the adoptive transfer, Fab were injected intravenously as previously described. Where indicated, mice were injected intraperitoneally with 120,000 U of human recombinant IL-2 (Proleukin; Prometheus Laboratories Inc.), and were injected with an additional 60,000 U injected 24 and 48 hours later, as described previously, Overwijk et al. (38).
Melanoma quantification from digital lung images
A code for objective quantification of melanoma visible in lungs was written using MATLAB software (MathWorks). Images of lung ventral sides were compared in each data set, where a single set of image settings was applied to all images in a specific experiment that had been acquired under identical illumination conditions. Because organ size was not equal between specimens, the proportion of dark pixels in the lungs was used to compare relative melanoma occupation. First, image transformation was performed using contrast-limited adaptive histogram equalization (http://mathworks.es/es/help/images/ref/adapthisteq.html), with appropriate parameters (including exponential histogram shape) applied. A threshold gray level (set to 35) was applied to the image’s contrast to quantify dark pixels. Finally, the total sum of each dark pixel count was divided by the total image-viewable pixels and multiplied by 100 to provide an arbitrary unit (AU) scale representing an objective relative quantification of melanoma visible in each lung.
Statistical analysis
Statistical analysis was performed using Student’s t test, where data were either paired or unpaired. Survival analysis was performed using Mantel-Cox log-rank test (GraphPad Prism).
Acknowledgments
We thank R. Stiles and T. Davis for excellent technical support. Funding: This work was supported by the Mayo Foundation (D.G. and A.G.S.) and the NIH [grant R01AI097187 (to D.G.), grant T32 AI07425-16 (supporting investigator, M.M.H.; principal investigator, L.R.P.), and grant R25GM55252 (supporting investigator, M.M.H.)]. Author contributions: M.M.H., A.D.N., C.A.P., E.E.R., M.J.H., and G.R. performed the experiments. C.M.-M. designed and programmed the digital pixelometry for lung melanoma analysis. M.M.H., L.R.P., G.R., A.G.S., and D.G. designed the experiments, interpreted the data, and/or wrote the manuscript. Competing interests: A.G.S. and D.G. report a pending patent on monovalent anti-CD3 adjuvants. Data and materials availability: All data needed to evaluate the findings in the paper are present in the paper and Supplementary Materials.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/1/9/e1500415/DC1
Fig. S1. Mono-7D6-Fab bound to OT-I T cells does not alter antigen binding to the OT-I TCR.
Fig. S2. Mono-7D6-Fab increases T cell responses to weak antigens in vitro.
Fig. S3. Mono-7D6-Fab increases T cell IRF4 expression in response to weak antigens in vitro.
Fig. S4. CD4+ and CD8+ T cell subsets play a role in the anti-melanoma effects of Mono-7D6-Fab.
Fig. S5. Mono-7D6-Fab anti-melanoma metastasis effect requires T cells of relevant antigenic specificity or clone identity.
Fig. S6. Mono-7D6-Fab shows efficacy against pre-established metastases.
Fig. S7. Mono-7D6-Fab can increase Pmel-1 T cell response to mgp100 in vitro.
Fig. S8. TCR co-potentiation model.
REFERENCES AND NOTES
- 1.Alarcón B., Gil D., Delgado P., Schamel W. W., Initiation of TCR signaling: Regulation within CD3 dimers. Immunol. Rev. 191, 38–46 (2003). [DOI] [PubMed] [Google Scholar]
- 2.van der Merwe P. A., Dushek O., Mechanisms for T cell receptor triggering. Nat. Rev. Immunol. 11, 47–55 (2011). [DOI] [PubMed] [Google Scholar]
- 3.Beddoe T., Chen Z., Clements C. S., Ely L. K., Bushell S. R., Vivian J. P., Kjer-Nielsen L., Pang S. S., Dunstone M. A., Liu Y. C., Macdonald W. A., Perugini M. A., Wilce M. C. J., Burrows S. R., Purcell A. W., Tiganis T., Bottomley S. P., McCluskey J., Rossjohn J., Antigen ligation triggers a conformational change within the constant domain of the αβ T cell receptor. Immunity 30, 777–788 (2009). [DOI] [PubMed] [Google Scholar]
- 4.Janeway C. A., Jr, Ligands for the T-cell receptor: Hard times for avidity models. Immunol. Today 16, 223–225 (1995). [DOI] [PubMed] [Google Scholar]
- 5.Janeway C. A. Jr, Dianzani U., Portoles P., Rath S., Reich E.-P., Rojo J., Yagi J., Murphy D. B., Cross-linking and conformational change in T-cell receptors: Role in activation and in repertoire selection. Cold Spring Harb. Symp. Quant. Biol. 54, 657–666 (1989). [DOI] [PubMed] [Google Scholar]
- 6.Kjer-Nielsen L., Clements C. S., Brooks A. G., Purcell A. W., McCluskey J., Rossjohn J., The 1.5 Å crystal structure of a highly selected antiviral T cell receptor provides evidence for a structural basis of immunodominance. Structure 10, 1521–1532 (2002). [DOI] [PubMed] [Google Scholar]
- 7.Rojo J. M., Janeway C. A. Jr, The biologic activity of anti–T cell receptor V region monoclonal antibodies is determined by the epitope recognized. J. Immunol. 140, 1081–1088 (1988). [PubMed] [Google Scholar]
- 8.Rojo J. M., Saizawa K., Janeway C. A. Jr, Physical association of CD4 and the T-cell receptor can be induced by anti–T-cell receptor antibodies. Proc. Natl. Acad. Sci. U.S.A. 86, 3311–3315 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gil D., Schamel W. W. A., Montoya M., Sánchez-Madrid F., Alarcón B., Recruitment of Nck by CD3ε reveals a ligand-induced conformational change essential for T cell receptor signaling and synapse formation. Cell 109, 901–912 (2002). [DOI] [PubMed] [Google Scholar]
- 10.Minguet S., Swamy M., Alarcón B., Luescher I. F., Schamel W. W. A., Full activation of the T cell receptor requires both clustering and conformational changes at CD3. Immunity 26, 43–54 (2007). [DOI] [PubMed] [Google Scholar]
- 11.Risueno R. M., Schamel W. W., Alarcon B., T cell receptor engagement triggers its CD3ε and CD3ζ subunits to adopt a compact, locked conformation. PLOS One 3, e1747 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu C., Gagnon E., Call M. E., Schnell J. R., Schwieters C. D., Carman C. V., Chou J. J., Wucherpfennig K. W., Regulation of T cell receptor activation by dynamic membrane binding of the CD3ε cytoplasmic tyrosine-based motif. Cell 135, 702–713 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sun Z.-Y. J., Kim K. S., Wagner G., Reinherz E. L., Mechanisms contributing to T cell receptor signaling and assembly revealed by the solution structure of an ectodomain fragment of the CD3εγ heterodimer. Cell 105, 913–923 (2001). [DOI] [PubMed] [Google Scholar]
- 14.Kuhns M. S., Davis M. M., Garcia K. C., Deconstructing the form and function of the TCR/CD3 complex. Immunity 24, 133–139 (2006). [DOI] [PubMed] [Google Scholar]
- 15.Kim S. T., Takeuchi K., Sun Z.-Y. J., Touma M., Castro C. E., Fahmy A., Lang M. J., Wagner G., Reinherz E. L., The αβ T cell receptor is an anisotropic mechanosensor. J. Biol. Chem. 284, 31028–31037 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim S. T., Touma M., Takeuchi K., Sun Z.-Y. J., Dave V. P., Kappes D. J., Wagner G., Reinherz E. L., Distinctive CD3 heterodimeric ectodomain topologies maximize antigen-triggered activation of αβ T cell receptors. J. Immunol. 185, 2951–2959 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Das D. K., Feng Y., Mallis R. J., Li X., Keskin D. B., Hussey R. E., Brady S. K., Wang J.-H., Wagner G., Reinherz E. L., Lang M. J., Force-dependent transition in the T-cell receptor β-subunit allosterically regulates peptide discrimination and pMHC bond lifetime. Proc. Natl. Acad. Sci. U.S.A. 112, 1517–1522 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krogsgaard M., Prado N., Adams E. J., He X.-L., Chow D.-C., Wilson D. B., Garcia K. C., Davis M. M., Evidence that structural rearrangements and/or flexibility during TCR binding can contribute to T cell activation. Mol. Cell 12, 1367–1378 (2003). [DOI] [PubMed] [Google Scholar]
- 19.Wang Y., Becker D., Vass T., White J., Marrack P., Kappler J. W., A conserved CXXC motif in CD3ε is critical for T cell development and TCR signaling. PLOS Biol. 7, e1000253 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hawse W. F., Champion M. M., Joyce M. V., Hellman L. M., Hossain M., Ryan V., Pierce B. G., Weng Z., Baker B. M., Cutting edge: Evidence for a dynamically driven T cell signaling mechanism. J. Immunol. 188, 5819–5823 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nelson A. D., Hoffmann M. M., Parks C. A., Dasari S., Schrum A. G., Gil D., IgG Fab fragments forming bivalent complexes by a conformational mechanism that is reversible by osmolytes. J. Biol. Chem. 287, 42936–42950 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de la Cruz J., Kruger T., Parks C. A., Silge R. L., van Oers N. S. C., Luescher I. F., Schrum A. G., Gil D., Basal and antigen-induced exposure of the proline-rich sequence in CD3ε. J. Immunol. 186, 2282–2290 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gil D., Schrum A. G., Alarcón B., Palmer E., T cell receptor engagement by peptide–MHC ligands induces a conformational change in the CD3 complex of thymocytes. J. Exp. Med. 201, 517–522 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Risueño R. M., Gil D., Fernández E., Sánchez-Madrid F., Alarcón B., Ligand-induced conformational change in the T-cell receptor associated with productive immune synapses. Blood 106, 601–608 (2005). [DOI] [PubMed] [Google Scholar]
- 25.Tailor P., Tsai S., Shameli A., Serra P., Wang J., Robbins S., Nagata M., Szymczak-Workman A. L., Vignali D. A. A., Santamaria P., The proline-rich sequence of CD3ε as an amplifier of low-avidity TCR signaling. J. Immunol. 181, 243–255 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gil D., Schrum A. G., Daniels M. A., Palmer E., A role for CD8 in the developmental tuning of antigen recognition and CD3 conformational change. J. Immunol. 180, 3900–3909 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hodi F. S., O’Day S. J., McDermott D. F., Weber R. W., Sosman J. A., Haanen J. B., Gonzalez R., Robert C., Schadendorf D., Hassel J. C., Akerley W., van den Eertwegh A. J. M., Lutzky J., Lorigan P., Vaubel J. M., Linette G. P., Hogg D., Ottensmeier C. H., Lebbé C., Peschel C., Quirt I., Clark J. I., Wolchok J. D., Weber J. S., Tian J., Yellin M. J., Nichol G. M., Hoos A., Urba W. J., Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 363, 711–723 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Coulie P. G., Uyttenhove C., Wauters P., Manolios N., Klausner R. D., Samelson L. E., Van Snick J., Identification of a murine monoclonal antibody specific for an allotypic determinant on mouse CD3. Eur. J. Immunol. 21, 1703–1709 (1991). [DOI] [PubMed] [Google Scholar]
- 29.Salmond R. J., Brownlie R. J., Morrison V. L., Zamoyska R., The tyrosine phosphatase PTPN22 discriminates weak self peptides from strong agonist TCR signals. Nat. Immunol. 15, 875–883 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alegre M., Vandenabeele P., Flamand V., Moser M., Leo O., Abramowicz D., Urbain J., Fiers W., Goldman M., Hypothermia and hypoglycemia induced by anti-CD3 monoclonal antibody in mice: Role of tumor necrosis factor. Eur. J. Immunol. 20, 707–710 (1990). [DOI] [PubMed] [Google Scholar]
- 31.Chowdhary V. R., Tilahun A. Y., Clark C. R., Grande J. P., Rajagopalan G., Chronic exposure to staphylococcal superantigen elicits a systemic inflammatory disease mimicking lupus. J. Immunol. 189, 2054–2062 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fidler I. J., Gersten D. M., Budmen M. B., Characterization in vivo and in vitro of tumor cells selected for resistance to syngeneic lymphocyte-mediated cytotoxicity. Cancer Res. 36, 3160–3165 (1976). [PubMed] [Google Scholar]
- 33.Liu X., Gibbons R. M., Harrington S. M., Krco C. J., Markovic S. N., Kwon E. D., Dong H., Endogenous tumor-reactive CD8+ T cells are differentiated effector cells expressing high levels of CD11a and PD-1 but are unable to control tumor growth. Oncoimmunology 2, e23972 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang N., Wang B., Salio M., Allen D., She J., Terhorst C., Expression of a CD3 epsilon transgene in CD3 epsilon(null) mice does not restore CD3 gamma and delta expression but efficiently rescues T cell development from a subpopulation of prothymocytes. Int. Immunol. 10, 1777–1788 (1998). [DOI] [PubMed] [Google Scholar]
- 35.Ferrer A., Schrum A. G., Gil D., A PCR-based method to genotype mice knocked out for all four CD3 subunits, the standard recipient strain for retrogenic TCR/CD3 bone marrow reconstitution technology. Biores. Open Access 2, 222–226 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Overwijk W. W., Theoret M. R., Finkelstein S. E., Surman D. R., de Jong L. A., Vyth-Dreese F. A., Dellemijn T. A., Antony P. A., Spiess P. J., Palmer D. C., Heimann D. M., Klebanoff C. A., Yu Z., Hwang L. N., Feigenbaum L., Kruisbeek A. M., Rosenberg S. A., Restifo N. P., Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J. Exp. Med. 198, 569–580 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Overwijk W. W., Tsung A., Irvine K. R., Parkhurst M. R., Goletz T. J., Tsung K., Carroll M. W., Liu C., Moss B., Rosenberg S. A., Restifo N. P., gp100/pmel 17 is a murine tumor rejection antigen: Induction of “self”-reactive, tumoricidal T cells using high-affinity, altered peptide ligand. J. Exp. Med. 188, 277–286 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Overwijk W. W., Restifo N. P., B16 as a mouse model for human melanoma. Curr. Protoc. Immunol. Chapter 20, Unit 20 1 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Das R., Verma R., Sznol M., Boddupalli C. S., Gettinger S. N., Kluger H., Callahan M., Wolchok J. D., Halaban R., Dhodapkar M. V., Dhodapkar K. M., Combination therapy with anti–CTLA-4 and anti–PD-1 leads to distinct immunologic changes in vivo. J. Immunol. 194, 950–959 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yao S., Zhu Y., Chen L., Advances in targeting cell surface signalling molecules for immune modulation. Nat. Rev. Drug Discov. 12, 130–146 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boniface J. J., Rabinowitz J. D., Wülfing C., Hampl J., Reich Z., Altman J. D., Kantor R. M., Beeson C., McConnell H. M., Davis M. M., Initiation of signal transduction through the T cell receptor requires the multivalent engagement of peptide/MHC ligands. Immunity 9, 459–466 (1998). [DOI] [PubMed] [Google Scholar]
- 42.Stone J. D., Stern L. J., CD8 T cells, like CD4 T cells, are triggered by multivalent engagement of TCRs by MHC-peptide ligands but not by monovalent engagement. J. Immunol. 176, 1498–1505 (2006). [DOI] [PubMed] [Google Scholar]
- 43.González-Martín A., Gómez L., Lustgarten J., Mira E., Mañes S., Maximal T cell–mediated antitumor responses rely upon CCR5 expression in both CD4+ and CD8+ T cells. Cancer Res. 71, 5455–5466 (2011). [DOI] [PubMed] [Google Scholar]
- 44.Johnson A. J., Njenga M. K., Hansen M. J., Kuhns S. T., Chen L., Rodriguez M., Pease L. R., Prevalent class I–restricted T-cell response to the Theiler’s virus epitope Db:VP2121–130 in the absence of endogenous CD4 help, tumor necrosis factor alpha, gamma interferon, perforin, or costimulation through CD28. J. Virol. 73, 3702–3708 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/1/9/e1500415/DC1
Fig. S1. Mono-7D6-Fab bound to OT-I T cells does not alter antigen binding to the OT-I TCR.
Fig. S2. Mono-7D6-Fab increases T cell responses to weak antigens in vitro.
Fig. S3. Mono-7D6-Fab increases T cell IRF4 expression in response to weak antigens in vitro.
Fig. S4. CD4+ and CD8+ T cell subsets play a role in the anti-melanoma effects of Mono-7D6-Fab.
Fig. S5. Mono-7D6-Fab anti-melanoma metastasis effect requires T cells of relevant antigenic specificity or clone identity.
Fig. S6. Mono-7D6-Fab shows efficacy against pre-established metastases.
Fig. S7. Mono-7D6-Fab can increase Pmel-1 T cell response to mgp100 in vitro.
Fig. S8. TCR co-potentiation model.