

MED12 is methylated by CARM1, and methylated MED12 serves as a sensor for anticancer drug response in human breast cancer.
Keywords: CARM1, RNA polymerase II mediator complex, methylation, breast cancer, chemotherapy
Abstract
The RNA polymerase II mediator complex subunit 12 (MED12) is frequently mutated in human cancers, and loss of MED12 has been shown to induce drug resistance through activation of transforming growth factor–β receptor (TGF-βR) signaling. We identified MED12 as a substrate for coactivator-associated arginine methyltransferase 1 (CARM1). Not only are the expression levels of CARM1 and MED12 positively correlated, but their high expression also predicts better prognosis in human breast cancers after chemotherapy. MED12 was methylated at R1862 and R1912 by CARM1, and mutation of these sites in cell lines resulted in resistance to chemotherapy drugs. Furthermore, we showed that the methylation-dependent drug response mechanism is distinct from activation of TGF-βR signaling, because methylated MED12 potently suppresses p21/WAF1 transcription. Cells defective in MED12 methylation have up-regulated p21 protein, which correlates with poor prognosis in breast cancer patients treated with chemotherapy. Collectively, this study identifies MED12 methylation as a sensor for predicting response to commonly used chemotherapy drugs in human cancers.
INTRODUCTION
Coactivator-associated arginine methyltransferase 1 (CARM1), also known as PRMT4, is a type I protein arginine methyltransferase (PRMT) that asymmetrically dimethylates proteins on arginine residues (1). CARM1 is overexpressed in human breast cancers, and elevated levels of CARM1 correlate with poor prognosis (2). The roles of protein arginine methylation by CARM1 in carcinogenesis are poorly understood because of the lack of effective methods to identify nonhistone substrates. We recently generated CARM1 knockout cell lines and used a newly developed anti–dimethylated arginine antibody (α-ADMA) for substrate identification. This antibody detected both BAF155 and MED12 in CARM1 wild-type cells but not in CARM1 knockout cells (3), implying that like BAF155, MED12 could be a substrate for CARM1.
The MED12 gene is located at Xq13 and encodes a subunit in a ~2-MD complex known as Mediator (MED), which plays essential roles in transcriptional regulation (4). To date, MED subunits including MED1, MED28, MED12, CDK8 (cyclin-dependent kinase 8), and cyclin C have been associated with various types of human cancers (5). MED12 is frequently mutated in human cancers. MED12 germ-line missense mutations cause intellectual disability and dysmorphic features in FG syndrome (6). Somatic mutations in MED12 occur in 70% of uterine leiomyomas, and these mutations disrupt the interaction between MED12 and cyclin C–CDK8 (7). Recently, recurrent mutations on MED12 were identified through exome sequencing in prostate cancer (8) and breast fibroadenoma (9). In addition, MED12 has been linked to cancer drug resistance. A portion of MED12 existing in the cytosol physically binds to the immature form of transforming growth factor–β receptor 2 (TGF-βR2) during secretion and prevents its glycosylation and delivery to the cell surface (10). Consequently, loss of MED12 increases cell surface expression of TGF-βR2, leading to extracellular signal–regulated kinase (ERK) signaling activation, which accounts for multidrug resistance in colon and lung cancers (10). In another independent study, by using a genome-scale CRISPR-Cas9 knockout (GeCKO) library screening, MED12 was found as the top hit whose loss in melanoma cells led to resistance to vemurafenib, a therapeutic RAF inhibitor (11). These studies underscored the roles of MED12 in mediating drug response in human cancers, that is, loss of MED12 renders cells resistant to cancer drugs. However, MED12 mutation spectra vary in different cancer types, and whether single site mutation in MED12 is sufficient to mediate drug response is unknown.
RESULTS
The expression levels of CARM1 and its substrate MED12 are positively correlated
We have shown that MED12 protein was immunoprecipitated by the α-ADMA antibody in a CARM1-dependent manner (3), implying it as a putative CARM1 substrate. To directly test this possibility, we performed an in vitro methylation assay where recombinant CARM1 and MED12 purified from human embryonic kidney (HEK) 293T CARM1KO cells were incubated with [3H]SAM (S-adenosyl-l-methionine). As expected, hypomethylated MED12 was methylated by CARM1 in vitro (Fig. 1A). However, MED12 was not methylated by PRMT1 and PRMT6, two of the other major type I PRMTs (fig. S1, A and B). In keeping with this finding, MED12 precipitated from parental but not the corresponding CARM1KO cell lines was recognized by the α-ADMA antibody (Fig. 1B). We then investigated if the expression levels of CARM1 and MED12 were positively correlated in breast cancer cell lines or clinical specimens. Using a Breast Cancer Gene-Expression Miner v3.0 software, we observed a weak yet significant positive correlation between MED12 and CARM1 (Fig. 1C and fig. S1C), but not other PRMTs (fig. S1D), in human breast tumors (n = 5790). Moreover, MED12 and CARM1 were positively correlated at the protein level in a panel of breast cancer cell lines (Fig. 1D and fig. S1E) and primary tumors (Fig. 1E). Both antibodies stained tumors more strongly than the surrounding stroma in IHC (Fig. 1F). The significant correlation of CARM1 and MED12 expression levels implies that MED12 methylation by CARM1 may have a functional role in breast cancer.
Fig. 1. Positive correlation between the expression of CARM1 and MED12 in breast cancer cell lines and human breast tumors.
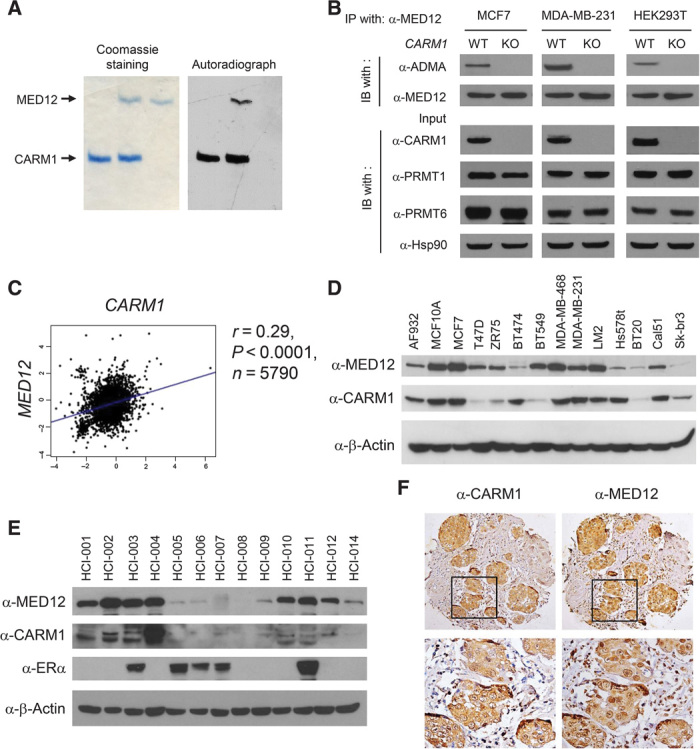
(A) Coomassie brilliant blue staining (left panel) and autoradiograph (right panel) of in vitro methylated MED12 by CARM1 in the presence of [3H]SAM. 3xFLAG-tagged MED12 protein was purified from HEK293T CARM1KO cells. (B) Western blot analyses of immunoprecipitated (IP) MED12 and input lysates from CARM1WT or CARM1KO cell lines using indicated antibodies. WT, wild type; IB, immunoblotting. (C) The Pearson correlation plot depicts the positive correlation between CARM1 and MED12 mRNA expression in 5790 human breast tumor cases collected in the bc-GenExMiner database. (D) Western blot analyses of MED12 and CARM1 proteins in human breast cancer and normal epithelial cell lines. β-Actin was used as an internal control. (E) Western blot analyses of MED12, CARM1, and ERα in patient-derived human breast tumor grafts (36). β-Actin was used as an internal control. (F) Immunohistochemical (IHC) staining of MED12 and CARM1 in human breast tumors.
Both MED12 and CARM1 protein levels predict chemotherapy response in breast cancer patients
Loss of MED12 was recently shown to confer resistance to receptor tyrosine kinase (RTK) inhibitors in non–small cell lung cancer and colon cancer (10). To investigate if MED12 level predicts drug response in breast cancer patients, we performed Kaplan-Meier (KM) analyses of relapse-free survival (RFS) for three cohorts of breast cancer patients: untreated (Fig. 2A, left), treated with endocrine therapy (middle), and treated with chemotherapy (right). High MED12 mRNA level was significantly associated with better RFS of patients treated with chemotherapy agents (n = 274) (Fig. 2A, right), but not with those untreated (n = 1000) (Fig. 2A, left) or with those who underwent endocrine therapy (n = 849) (Fig. 2A, middle). Because MED12 is methylated by CARM1 and they share a positive correlation in breast cancers (Fig. 1), we investigated if CARM1 is linked to chemosensitivity using tissue microarrays (TMAs) containing 254 breast tumors from doxorubicin- or fluorouracil-treated patients with 100-month clinical follow-up. The CARM1 antibody staining in specimens was rank-ordered by intensity and divided into CARM1low (bottom 50% intensity) and CARM1high (top 50% intensity) groups. The KM analysis showed that the CARM1high patients have a better overall survival (OS) and disease-free survival (DFS) than CARM1low patients who underwent chemotherapy (Fig. 2B). Given the positive correlation of the expression levels of CARM1 and MED12 (Fig. 1) and their respective connection to the response to chemotherapy, we stratified patients into CARM1highMED12high and CARM1lowMED12low groups. The CARM1highMED12high patients exhibited a more marked OS (P < 0.005) and DFS (P < 0.001) (Fig. 2C) as compared with CARM1lowMED12low patients. These data suggest that CARM1 and MED12 proteins might converge in regulating drug sensitivity in human breast cancers. To test this hypothesis, we measured cell survival in paired CARM1WT and CARM1KO cell lines (MCF7, MDA-MB-231, and CAL51) after treatment with three commonly used chemotherapy drugs (Fig. 2D). Regardless of cell line, CARM1WT cells are more prone to death than CARM1KO cells after drug treatment. To further investigate whether MED12 is involved in CARM1-dependent chemosensitivity, MED12 was knocked down in CARM1WT and CARM1KO MDA-MB-231 cells that were subjected to fluorouracil or doxorubicin treatment (Fig. 2E). In Fig. 2F, although either knockdown of MED12 or knockout of CARM1 renders drug resistance, knockdown of MED12 in CARM1KO cells failed to elicit additional effects. Similar results were obtained in CARM1WT and CARM1KO MCF7 cells (Fig. 2, G and H). These data indicate that the role of CARM1 in sensitizing chemotherapy drugs could be due to its methylation of MED12.
Fig. 2. Higher levels of MED12 and CARM1 correlate with better response to chemotherapy drugs in cell line models and clinical cohorts.
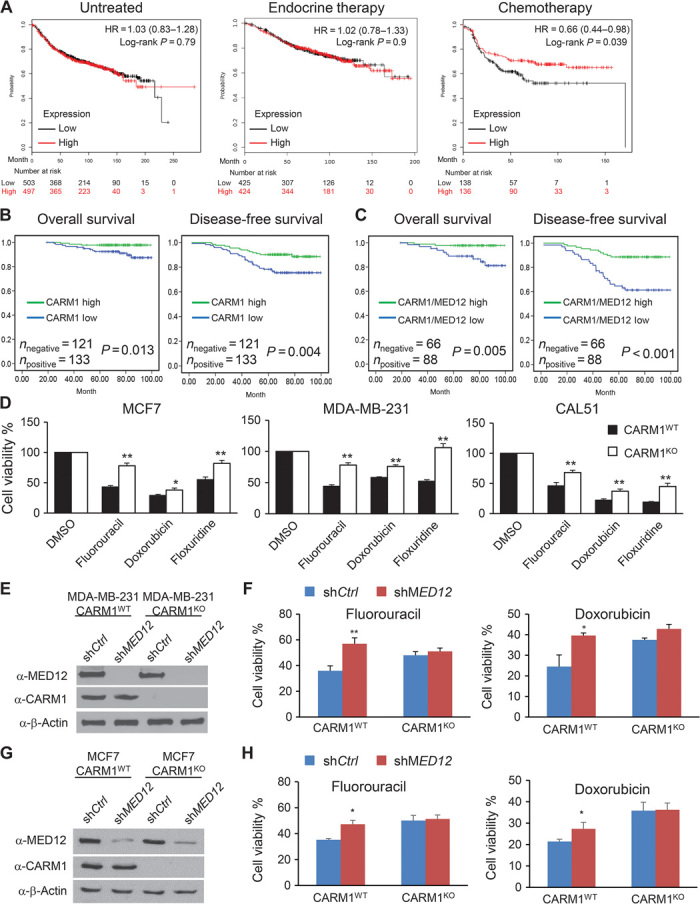
(A) KM curves stratified by MED12 mRNA levels depicting the probability of RFS in untreated, endocrine therapy–treated, or chemotherapy-treated breast cancer patients. Patient samples were divided into MED12high and MED12low groups based on the median of the expression level of MED12. Affymetrix gene ID 214275_at was used to plot the survival curves of MED12 using data sets from Gene Expression Omnibus (GEO) (Affymetrix HG-U133A and HGU-133+2 microarrays), European Genome-Phenome Archive (EGA), and The Cancer Genome Atlas (TCGA). HR, hazard ratio. (B and C) High levels of CARM1 and MED12 proteins correlate with better survival after 5-fluorouracil (5-FU) or doxorubicin treatment in breast cancer patients with 100-month follow-up (n = 254). KM estimates of DFS and OS of human patients according to the expression levels of CARM1 (n = 254) or CARM1/MED12 (n = 154). Comparison was made between groups with high or low levels of CARM1 alone or high or low levels of CARM1 and MED12. P value refers to two-sided log-rank tests. (D) Cell viability analyses of three paired CARM1WT and CARM1KO breast cancer cell lines after incubating with 1 μM 5-FU, doxorubicin, or floxuridine for 72 hours. Cell viability was determined by MTT assays. Quantitative data are presented as averages ± SD. Student’s t test was used for statistical analysis. *P < 0.05; **P < 0.01. (E) Western blotting shows the knockdown of MED12 in both CARM1WT and CARM1KO MDA-MB-231 cells. (F) Cell viability analyses of MDA-MB-231 CARM1WT shCtrl, MDA-MB-231 CARM1WT shMED12, MDA-MB-231 CARM1KO shCtrl, and MDA-MB-231 CARM1KO shMED12 cells after treatment with 5-FU or doxorubicin for 72 hours. Quantitative data are presented as averages ± SD. Student’s t test was used for statistical analysis. *P < 0.05; **P < 0.01. (G) Western blotting shows the knockdown of MED12 in both CARM1WT and CARM1KO MCF7 cells. (H) Cell viability analyses of MCF7 CARM1WT shCtrl, MCF7 CARM1WT shMED12,MCF7 CARM1KO shCtrl, and MCF7 CARM1KO shMED12 cells after treatment with 5-FU or doxorubicin for 72 hours. Quantitative data are presented as averages ± SD. Student’s t test was used for statistical analysis. *P < 0.05.
MED12 is methylated by CARM1 at R1862 and R1912
The PMeS software predicted 16 putative arginine methylation sites on MED12. On the basis of the distribution of these sites, we constructed six MED12 truncation proteins (Fig. 3A). The plasmids encoding FLAG-tagged truncated proteins were transiently transfected into HEK293T cells. The resulting proteins were immunoprecipitated using α-FLAG antibody and detected with the α-ADMA antibody in Western blot (Fig. 3B). Notably, the two MED12 fragments containing the proline-glutamine-leucine–rich (PQL) domain were recognized by the α-ADMA antibody, indicating that they harbor methylation sites. To map the MED12 region(s) that directly binds to CARM1, we linked each of the MED12 fragments to a T7 promoter and a FLAG tag, which are suitable for in vitro transcription/translation assays. In Fig. 3C, both full-length MED12 and the PQL domain alone were able to bind to bacterially expressed glutathione S-transferase (GST)–CARM1. These results narrowed down the MED12 methylation site(s) to the PQL domain, which consists of six arginine residues. These arginine residues were individually mutated to lysine in a FLAG-tagged, PQL domain–expressing plasmid. After transient transfection of individual plasmids to HEK293T and immunoprecipitation using α-FLAG antibody, only FLAG-MED12R1862K and FLAG-MED12R1912K mutant proteins showed substantially reduced methylation detected by the α-ADMA antibody (Fig. 3D). We subsequently mutated both residues to lysine and found that the double mutant cannot be detected by the α-ADMA antibody (Fig. 3E). Consistently, although recombinant MED12 proteins carrying single site mutations showed reduced methylation as compared with the wild-type MED12 in the in vitro methylation assay, the methylation was undetectable with the double mutant (MED12R1862K/R1912K) (Fig. 3F). Thus, MED12 is methylated by CARM1 at R1862 and R1912. To further distinguish the major methylation site, we fused two DNA sequences encoding 21–amino acid peptides containing R1862 or R1912 sites to GST in a bacterial expression vector (fig. S2A). The two GST-peptide fusion proteins were purified and used for in vitro methylation assays. In Fig. 3G, mutation at R1862 had a stronger effect on methylation than that of R1912, indicating that R1862 is the major methylation site. Both sites are highly conserved among higher eukaryotes (fig. S2B). To determine whether endogenous MED12 is dimethylated in breast cancer cells, we synthesized a peptide encompassing asymmetric dimethylated R1862 (fig. S2C) and used it as antigen to generate a rabbit polyclonal antibody specific for dimethylated MED12 (me-MED12). Western blotting showed that the me-MED12 antibody recognized MED12WT but not MED12R1862K protein transiently expressed in HEK293T cells (fig. S2D). Having demonstrated the antibody specificity, we next used this antibody to detect endogenous, methylated MED12 protein. Cell lysates from MDA-MB-231 cells either with CARM1KO or expressing MED12 short hairpin RNA (shRNA) were used as negative controls. Methylated MED12 can be detected in parental MDA-MB-231 cells but not in CARM1KO- or in MED12 shRNA–expressing cells (Fig. 3H). This result further affirms the antibody specificity and shows that the endogenous MED12 is methylated by CARM1.
Fig. 3. CARM1 directly interacts with MED12 and methylates MED12 at two sites R1862 and R1912.
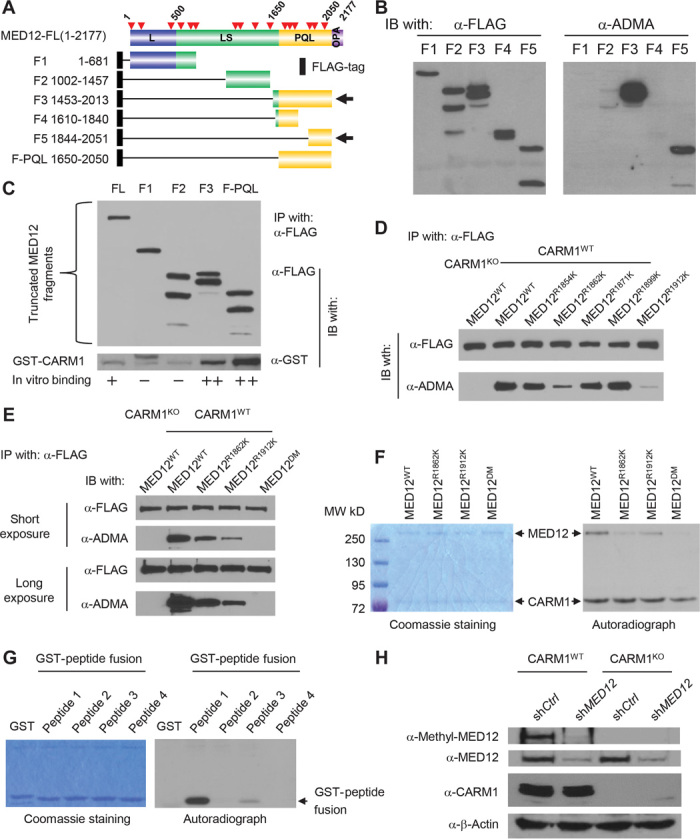
(A) Schematic diagram of full-length MED12 domain structure and the truncation constructs, which are all fused to a FLAG tag cassette via the N terminus. Red arrows depict the putative arginine methylation sites predicted by the PMeS program. (B) Western blot analysis of α-FLAG immunoprecipitated MED12 fragments detected by the α-FLAG antibody (left panel) or the α-ADMA antibody (right panel) from HEK293 cell lysates transiently transfected with the corresponding MED12-expressing plasmids. (C) Mapping of CARM1-interacting domain to the PQL domain of MED12 using GST pull-down assay. FLAG-tagged MED12 fragment proteins were incubated with GST-CARM1 and then immunoprecipitated and detected with α-FLAG antibody in a Western blot (upper panel). The presence of GST-CARM1 in immunoprecipitates was detected by the anti-GST antibody in a Western blot (lower panel). (D) Western blot analysis of immunoprecipitated FLAG-tagged WT or mutant MED12 proteins using the α-FLAG or α-ADMA antibodies. (E) Western blot analysis of immunoprecipitated FLAG-tagged WT or mutant MED12 proteins using the α-FLAG or α-ADMA antibodies. (F) Coomassie brilliant blue staining (left panel) and autoradiograph (right panel) of in vitro methylated recombinant MED12WT, MED12R1862K, MED12R1912K, and MED12DM proteins by CARM1 in the presence of [3H]SAM. (G) Coomassie brilliant blue staining (left panel) and autoradiograph (right panel) of in vitro methylated GST-peptide fusion proteins by CARM1 in the presence of [3H]SAM. (H) Western blot analysis of total MED12, me-MED12, and CARM1 in control shRNA– or MED12 shRNA–expressing MDA-MB-231 CARM1WT and MDA-MB-231 CARM1KO cells. β-Actin was used as an internal control.
MED12 methylation sensitizes cells to 5-fluorouracil but not to RTK inhibitors
Next, we determined whether MED12 methylation is required for sensitizing cells to chemotherapy drugs. To substitute endogenous MED12 with MED12R1862K/R1912K (MED12DM) methyl-defective mutant, we infected MDA-MB-231 cells with retrovirus expressing shRNA-resistant MED12WT (12), MED12DM, or green fluorescent protein (GFP), selected for survival clones by neomycin for 4 weeks, and then lentivirally expressed MED12 shRNA to knock down the endogenous MED12 (Fig. 4A). Figure 4A shows that endogenous MED12 was silenced in MDA-MB-231–shMED12–GFP cells and that MDA-MB-231–shMED12–MED12WT and MDA-MB-231–shMED12–MED12DM cells express reconstituted MED12WT or MED12DM protein to similar levels as the endogenous MED12 protein, respectively. Knockdown of MED12 decreased growth of MDA-MB-231 cells (fig. S3A) as had been reported in other cancer types (10), whereas MED12 methylation status had no effect on cell proliferation (fig. S3B). These cell lines were subjected to chemosensitivity studies using a panel of 97 U.S. Food and Drug Administration (FDA)–approved anticancer drugs. In Fig. 4 (B and C), a large number of drugs conferred differential sensitivity in response to knockdown of MED12 (as in table S1A), whereas fewer drugs (table S1B) including 5-FU and floxuridine, an analog of 5-FU, differed in sensitivity between MED12WT- and MED12DM-expressing cells. Dose-dependent cell viability assays validated that MED12WT-expressing MDA-MB-231–shMED12 cells are more prone to die after treatment with 5-FU or floxuridine than MED12DM-expressing cells (Fig. 4D). The differential response to drugs was significant and highly reproducible, and was also apparent in colony formation assays (Fig. 4E). MED12 was recently shown to control response to cancer drugs through inhibiting TGF-β receptor signaling (10). Loss of MED12 resulted in activation of TGF-β receptor signaling and phosphorylation of ERK, resulting in resistance to RTK inhibitors in cancer cells (10). To discern if resistance mechanisms to 5-FU and RTK inhibitors are both dependent on MED12 methylation, parental, MED12 knockdown, MED12WT- and MED12DM-reconstituted MDA-MB-231 cells were treated with RTK inhibitors selumentinib and crizotinib, and compared with 5-FU. As expected, knockdown of MED12 resulted in resistance to all drugs (Fig. 4F). Restoring either MED12WT or MED12DM could restore RTK inhibitor sensitivity in MED12 knockdown cells; however, restoring only MED12WT but not MED12DM restored 5-FU sensitivity (Fig. 4F). These results suggest that different mechanisms are involved in mediating response to RTK inhibitors and 5-FU, and the response to 5-FU but not to RTK inhibitors is MED12 methylation–dependent. It has been shown that MED12 loss induces an epithelial-to-mesenchymal transition (EMT)–like phenotype that is associated with chemotherapy resistance in colon and lung cancer cells (10). To determine whether MED12 loss triggers EMT in breast cancer, we silenced MED12 in luminal MCF7 cells with two independent shRNAs and found that the expression levels of TGF-βR2 downstream genes and EMT markers were not affected by loss of MED12 (fig. S3C); this is in contrast to findings in other cancer types (10). Consistent with the gene expression data, cell morphology was not altered by the loss of MED12 or MED12 methylation status (fig. S3D). Nonetheless, loss of MED12 triggered ERK activation as shown in other cancer types (10), and ERK activation was abrogated by reexpression of either MED12WT or MED12DM (fig. S3E). These data suggest that induction of EMT by loss of MED12 could be cell type–dependent. Although MED12 methylation status does not influence ERK activation, nor does it attribute to resistance to RTK inhibitors, the response to chemotherapy agents such as 5-FU and doxorubicin is MED12 methylation–dependent. To confirm this finding in vivo, MED12WT- or MED12DM-expressing MDA-MB-231–shMED12 cells were xenografted in nude mice and subjected to 5-FU treatment. Two weeks after transplantation, the mice were treated with either phosphate-buffered saline (PBS) or 5-FU, and tumor volumes were measured every 4 days. The tumor growth curve (Fig. 4G) as well as tumor size measurement (Fig. 4H) showed that MDA-MB-231–shMED12–MED12WT grafts were sensitive to 5-FU treatment. In contrast, MED12DM-expressing tumors are insensitive to 5-FU (Fig. 4, G and H).
Fig. 4. Methylation of MED12 renders cells sensitive to chemotherapy drugs in vitro and in vivo.
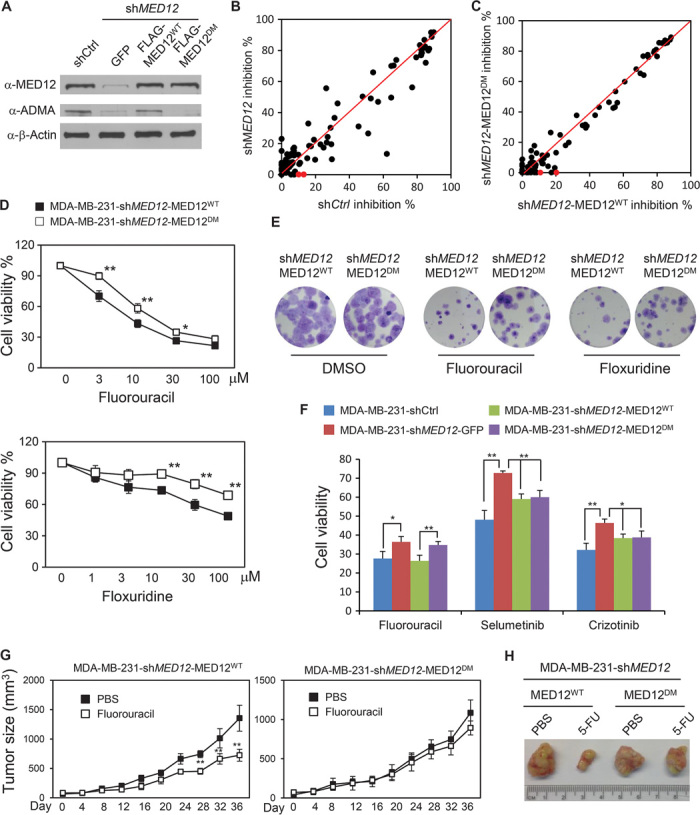
(A) Western blotting analysis of MED12 in MDA-MB-231 cells and shMED12-expressing MDA-MB-231 cells restored with GFP, MED12WT, or MED12DM using anti-MED12 antibody against whole-cell lysate or the α-ADMA antibody against immunoprecipitated MED12. (B) Cell survival inhibition rate plot of MDA-MB-231 shCtrl cells (x axis) or MDA-MB-231 shMED12 cells (y axis) after a 72-hour treatment with 97 FDA-approved cancer drugs (1 μM). Red dots denote 5-FU and its analog floxuridine. The inhibition rate was calculated as the difference of MTTDMSO and MTTdrug normalized to MTTDMSO. (C) Cell survival inhibition rate plot of MDA-MB-231–shMED12–MED12WT cells (x axis) or MDA-MB-231–shMED12–MED12DM cells (y axis) after a 72-hour treatment with 97 FDA-approved cancer drugs (1 μM). Red dots denote 5-FU and its analog floxuridine. (D) Cell viability curves for MDA-MB-231–shMED12–MED12WT and MDA-MB-231–shMED12–MED12DM cells after treatment with various concentrations of fluorouracil or floxuridine for 72 hours. (E) Colony yields after 2 weeks of treatment with 0.3 μM fluorouracil or floxuridine in MDA-MB-231–shMED12–MED12WT and MDA-MB-231–shMED12–MED12DM cells. (F) Cell viability assays for the indicated cell lines after 72 hours of treatment with 10 μM 5-FU, selumetinib, or crizotinib. (G) Tumor growth curve of MDA-MB-231–shMED12–MED12WT and MDA-MB-231–shMED12–MED12DM cell grafts in nude mice (n = 5). Student’s t test was used for statistical analysis. **P < 0.01; *P < 0.05. (H) Representative photographs of tumors derived from MDA-MB-231–shMED12–MED12WT and MDA-MB-231–shMED12–MED12DM cell grafts treated with PBS or 5-FU.
Suppression of p21 expression by methylated MED12 is involved in conferring drug sensitivity
Given that loss of MED12 results in multidrug resistance in diversified cell types and the methylation status of MED12 also affects response to some chemotherapy drugs, we took a genomic approach to identify methylated MED12-responsive genes to explain the mechanism of resistance. We reasoned that the genes regulated by methylated MED12 protein should be sensitive to loss of either MED12 or CARM1; thus, MED12 was knocked down in CARM1WT and CARM1KO MDA-MB-231 cells for Affymetrix microarray analyses. More than 800 genes whose mRNA levels were affected by loss of CARM1 and MED12 (fold > 1.5, P < 0.05) were identified, among which 444 and 410 genes were up-regulated or down-regulated, respectively (fig. S4A). Among the complete list of differentially expressed genes (table S2), a panel of genes including KRT14, KIF5A, PRSS2, and CD74 previously shown to mediate chemosensitivity were selected for validation (13–17). Real-time quantitative polymerase chain reaction (qPCR) analyses showed that the mRNA levels of these genes were indeed affected by loss of either MED12 or CARM1 (Fig. 5A and fig. S4B), indicating that they are co-regulated by CARM1 and MED12. Next, we examined their expression levels in MDA-MB-231 cells reconstituted with either MED12WT or MED12DM. In Fig. 5B, not only loss of MED12, but also lack of MED12 methylation affected the mRNA levels of KRT14, KIF5A, PRSS2, and CD74 genes, suggesting that the expression of these four genes is sensitive to MED12 methylation status. p21/WAF1 was among the top up-regulated genes upon loss of MED12 or CARM1 (fig. S4B). Thus, MED12 appears to suppress the p21 gene as either loss of MED12 or lack of MED12 methylation led to the up-regulation of p21/WAF1. The p21 protein is involved in the DNA damage response, and up-regulation of p21 has been shown to mediate drug response (18). Among the five putative downstream effectors of methylated MED12, we examined the effects of CD74, PRSS2, and p21 on mediating drug response. Because CD74 and PRSS2 are activated and p21 is suppressed by methylated MED12, we stably knocked down CD74, PRSS2, or overexpressed p21 in MDA-MB-231 cells and performed dose-responsive 5-FU sensitivity assays. Neither CD74 nor PRSS2 knockdown affected 5-FU response as measured by cell survival (fig. S4, C to F), whereas overexpressing p21 rendered cells more resistant to 5-FU (fig. S4, G and H). Consistent with the identification of p21 as a MED12-suppressed gene, knockdown of MED12 significantly increased p21 protein in MDA-MB-231 cells (Fig. 5C). Further, silencing of CARM1 alone increased the p21 protein level, and knockdown of MED12 in CARM1KO cell lines did not elicit additional effect on the p21 protein level. These results suggest that p21 is suppressed by methyl-MED12. Using KM analyses, we discovered that higher p21 expression level was strongly associated with worse RFS in breast patients treated with chemotherapy (n = 274) (fig. S4I, right) but not with endocrine therapy (fig. S4I, middle). These data implied that p21 protein, which is normally suppressed by methyl-MED12, could be a major effector of methyl-MED12 in regulating drug response. To delineate the contribution of MED12 methylation at R1862 and R1912 to p21 inhibition, the p21 levels were determined in MED12WT-, MED12R1862K-, or MED12R1912K-overexpressing MDA-MB-231 cells (Fig. 5D). Although p21 protein was similarly suppressed in MED12WT- and MED12R1912K-expressing cells, the suppression was abolished by mutation of MED12 at R1862 (Fig. 5D). These data suggest that methylation of MED12 at R1862 largely contributes to p21 inhibition, which reconciles with the identification of R1862 of MED12 as the major CARM1 methylation site (Fig. 3G).
Fig. 5. MED12 methylation enhances its association to p21 gene locus and suppresses p21 transcription.
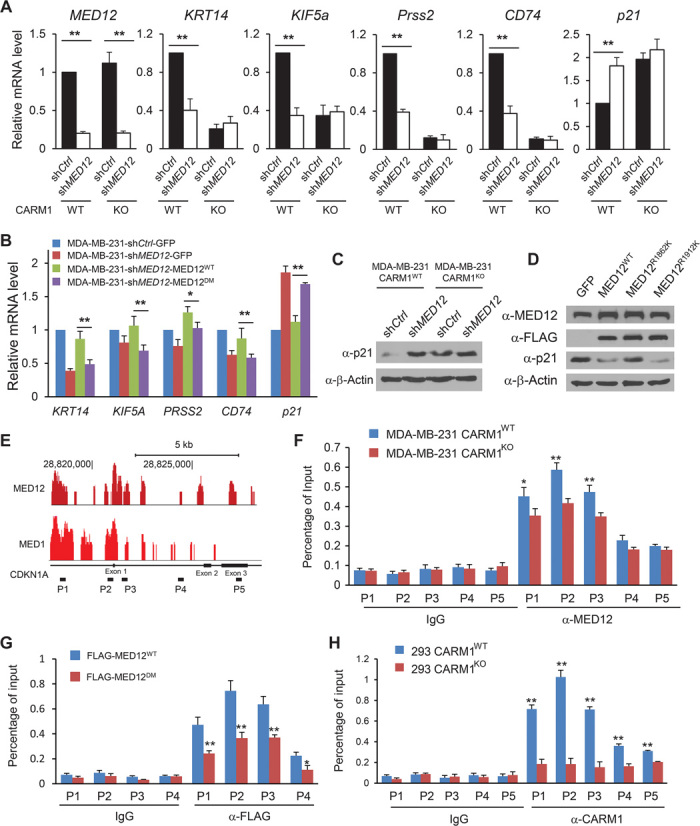
(A) Relative mRNA levels of MED12, KRT14, KIF5a, PRSS2, CD74, and p21 in control shRNA– or shMED12-expressing MDA-MB-231 CARM1WT or MDA-MB-231 CARM1KO cells were determined by real-time qPCR. β-Actin was used as an internal control. (B) Real-time qPCR analyses of KRT14, KIF5A, PRSS2, CD74, and p21 mRNA levels in MDA-MB-231–shCtrl-GFP, MDA-MB-231–shMED12-GFP, MDA-MB-231–shMED12-MED12WT, and MDA-MB-231–shMED12-MED12DM cells. β-Actin was used as an internal control. (C) Western blot analysis of p21 protein in control shRNA– or shMED12-expressing MDA-MB-231 CARM1WT and MDA-MB-231 CARM1KO cells. (D) Western blot analysis of p21 protein in MDA-MB-231 cells overexpressing GFP or FLAG-tagged MED12WT, MED12R1862K, and MED12R1912K. (E) The raw reads of ChIP-seq tracing of MED1 (GSM560353) and MED12 (GSM560354) enrichment peaks on the mouse p21 gene locus retrieved from the GEO database (19). (F) ChIP-qPCR analysis of MED12 binding to the five genomic regions of the p21 gene in MDA-MB-231 CARM1WT or MDA-MB-231 CARM1KO cells. Normal rabbit immunoglobulin G (IgG) was used as antibody control. (G) ChIP-qPCR analysis of FLAG-tagged MED12 binding to four genomic regions of the p21 gene in HEK293 cells transiently expressing FLAG-MED12WT or FLAG-MED12DM. (H) ChIP-qPCR analysis of CARM1 binding to five genomic regions of the p21 gene in HEK293 CARM1WT or HEK293 CARM1KO cells. **P < 0.01; *P < 0.05.
Methylation of MED12 suppresses p21 transcription by enhanced genomic association
The mediator complex was reported to be associated with the p21 promoter region as well as the gene body in a MED12 chromatin immunoprecipitation (ChIP)–seq experiment using mouse embryonic fibroblasts (19). As illustrated in Fig. 5E, both MED12 and MED1 peaks were found adjacent to the p21 promoter and gene body based on the ChIP-seq data (19). To affirm that MED12 methylation does not affect the integrity of MED, we immunoprecipitated MED12 from MED12WT or MED12DM transiently transfected HEK293T cells and measured the levels of MED12 interacting proteins such as G9a, CDK8, and MED30 in a Western blot (fig. S5). The result showed that MED12DM interacts with all proteins in a manner similar to MED12WT, indicating that MED12 methylation does not affect MED assembly. We next examined whether methylation of MED12 enhanced the chromatin association of the mediator complex to the p21 gene, thus suppressing its expression. Five pairs of qPCR primers were designed on the p21 gene locus based on published MED12 ChIP-seq data (19). ChIP-qPCR results showed that MED12 association to the p21 gene locus, in particular in the promoter and exon 1 regions, was sensitive to CARM1 level, that is, stronger association of MED12 to the p21 promoter, and exon 1 was detected in parental MDA-MB-231 than CARM1KO cells (Fig. 5F). This result implies that methylation of MED12 by CARM1 may promote the association of MED12 to the p21 locus, thus suppressing p21 transcription. To confirm this finding in a different cell line, we transiently overexpressed MED12WT or MED12R1862K in HEK293 cells and measured the p21 mRNA levels. Expressing MED12WT but not MED12R1862K inhibited the p21 mRNA levels (fig. S6A). Correspondingly, overexpressing MED12WT but not MED12R1862K in HEK293 cells reduced the p21 protein level (fig. S6B). These results confirmed that suppression of p21 by methylated MED12 is not specific to MDA-MB-231. Because methylation of MED12 appears to suppress p21 in HEK293 cells, we transiently transfected HEK293 cells with Flag-tagged MED12WT or MED12DM and performed MED12 ChIP-qPCR on the p21 gene locus using α-FLAG antibody. In Fig. 5G, mutation of MED12 weakened the association of MED12 to the p21 gene locus. Because CARM1 directly interacts with the PQL domain of MED12 protein, we also measured the association of CARM1 to the p21 gene locus by ChIP-qPCR. Figure 5H showed that CARM1 could also be detected in the same p21 gene locus as MED12. Together, the results suggest that CARM1 and MED12 might form a repressive complex at the p21 gene locus and suppress p21 transcription.
DISCUSSION
Here, we reported that MED12, a key component in the CDK submodule of MED, is a novel substrate of CARM1 and identified R1862 and R1912 as methylation sites on MED12. The most striking finding of this study lies in the identification of an unexpected arginine methylation–dependent mechanism for drug resistance. MED12 has previously been linked to multidrug resistance (10). In conformity with the previous study, we also showed that knockdown of MED12 in MDA-MB-231 breast cancer cell line resulted in multidrug resistance (Fig. 4B). Reconstituting with MED12-carrying mutations at two methylation sites in MDA-MB-231 cells is sufficient to induce resistance to several DNA-damaging agents (Fig. 4C) including 5-FU, doxorubicin, and floxuridine, which are commonly used drugs for chemotherapy. Unlike knockdown of MED12, mutation at MED12 methylation sites does not induce TGF-βR activation and phosphorylation of ERK, nor does it induce an EMT phenotype (fig. S3), suggesting that the MED12 methylation–dependent drug resistance uses a different mechanism from that of TGF-βR. Because CARM1 is the only enzyme that mediates MED12 methylation at R1862 and R1912, we also evaluated the role of CARM1 in chemoresistance by comparing drug response in CARM1 knockout cells and the paired parental cells. Indeed, knockout of CARM1 also induced resistance to the same set of drugs in multiple breast cancer cell lines (Fig. 2D). Although CARM1 has been implicated in DNA damage response pathways (20), to our knowledge, this is the first report on loss of CARM1-induced drug resistance. In alignment with the in vitro findings, breast cancer patients expressing high levels of CARM1 and MED12 exhibited better long-term survival after adjuvant chemotherapy (Fig. 2C). Thus, high expression of CARM1 and its substrate MED12 may serve as independent prognostic biomarkers for predicting response to chemotherapy.
In contrast to other MED subunits whose expression levels are deregulated in cancers, MED12 is frequently mutated in human cancers. More than 700 mutations on MED12 could be identified in the COSMIC (Catalogue of Somatic Mutations in Cancer) cancer mutation database. The mutation sites are dispersedly distributed on MED12 proteins. Little is known about how these mutations disrupt normal functions of MED12 and cause human diseases. Because MED12 methylation occurs on R1862 and R1912, we searched the COSMIC database (http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/) for mutations at these sites and found that R1862 is mutated in a lung carcinoma, and a somatic, homozygous mutation at R1912 was found in a melanoma patient who was resistant to a combined RAF/MEK (mitogen-activated protein kinase kinase) inhibitor treatment (21). In the TCGA database, 27 of 1093 breast tumors carry mutation(s) in MED12; however, no mutation could be found at R1912 and R1862. Given that the TCGA sequenced tumors are primary tumors, we speculate that the low MED12 mutation frequency could reflect the low mutation rate in primary tumors, and chemotherapy treatment might induce mutations at R methylation sites of MED12. This hypothesis has to be tested by deep sequencing of chemotherapy-resistant breast tumors. Because our results show that mutations on R1862 and R1912 of MED12 abrogate sensitivity to commonly used chemotherapy drugs, if mutations of these sites are indeed found in recurrent tumors after chemotherapy, periodically sequencing MED12 in tumors in search of these mutations could help predict the patients’ response to chemotherapy.
MED12 resides in the CDK8 submodule that both positively and negatively regulates transcription (22, 23). Previous studies have shown that both MED12 and MED13 are engaged in submodule-dependent repression (24); however, only MED12 can activate CDK8 kinase (25). Given the essential role of MED12 in mediating transcriptional repression, it is not surprising that MED12 strongly represses the p21 gene as evident by the association of both MED12 and MED1 to the p21 gene locus in ChIP-seq (19). MED12 association with p21 is methylation-sensitive because the methylation-defective mutant exhibited reduced binding to the p21 gene locus. The mechanism by which the methylated MED12 tethers more strongly to the p21 promoter is not clear. MED12 is known to interact directly with a number of transcription factors. In particular, the PQL domain where methylation sites reside mediates direct interaction with SOX9, Gli3, β-catenin, and AICD (amyloid precursor protein intracellular domain) transcription factors (26–29). Although we did not detect differential interaction between MED12WT and MED12DM with G9a (fig. S5), it is possible that methylated MED12 promotes interaction with other proteins that enhance recruitment of MED12 to the p21 gene locus.
Induction of p21, an inhibitor of cyclin E/Cdk2, negatively regulates cell growth in various in vitro and in vivo models. However, overexpression of p21 protein in colon cancer cells increased resistance to apoptosis-inducing agents and abrogated endogenous p21-mediated differentiation (30). Consistent with the in vitro data, expression of p21, especially in combination with p53 mutation in colorectal cancers, predicts resistance to the combination chemotherapy with gefitinib (31). Furthermore, high levels of constitutive p21 expression were associated with chemoresistance in acute myelogenous leukemia (18). Here, we revealed that overexpressing p21 in MDA-MB-231 cells is sufficient to induce 5-FU resistance, an effect mimicking the loss of MED12 methylation. We found that high p21 expression in patients treated with chemotherapy strongly correlates with poor RFS. Although up-regulation of the p21 level was observed in MDA-MB-231–shMED12–MED12DM cells as compared to MDA-MB-231–shMED12–MED12WT cells, the differential p21 levels only affected drug sensitivity, without altering the cell growth rate. Similar growth rate–independent function of p21 in modulating anticancer drug sensitivity was also observed in some other tumor models (32, 33). For example, Oshimori and colleagues found that knockdown of p21 did not affect the cell proliferation of basal cells from HRasG12V-derived squamous cell carcinomas of mice transduced with lentivirus harboring scramble of p21 shRNAs. However, the cells with p21 knockdown are more sensitive to anticancer therapeutics, such as cisplatin (32). Although we could not exclude other mechanisms by which MED12 methylation modulates drug sensitivity, induction of p21 in response to the suppression of MED12 or loss of MED12 methylation clearly places p21 as a major downstream effector that mediates MED12 methylation–specific drug response.
MATERIALS and METHODS
Cell culture and cell lines
Generation of CARM1 knockout cell lines had been described previously (3). To knock down gene expression in cells, lentivirus-mediated RNA interference (RNAi) was performed with the pGIPZ shRNA system (Open Biosystems). After 6 hours of transfection, the medium was changed. The virus particles were harvested from medium between 24 and 48 hours and filtered through 0.45-μm syringe filter (Thermo Scientific). To infect the cells, specific retroviruses or lentiviruses were mixed with an equal volume of fresh media supplemented with 10% fetal bovine serum. Polybrene (5 μg/ml; Sigma) was added to increase the infection efficiency. Medium was changed after 6 hours of infection. Cells were selected with puromycin (2 μg/ml; RPI) for knocking down MED12, CD74, or PRSS2 for a week to generate stable cell lines. pGIPZ plasmids encoding shRNA target MED12, CD74, and PRSS2 are as follows: shMED12-1 antisense: TTGGTAAGCGCACAGGACG; shMED12-2 antisense: TGGACTGCATGTCGACAGT; shMED12-RNAi resistance antisense: TCACTCATCTCATGTTATA; shCD74 antisense: CATTGTTGGAGATAAGGTC; shprss2 antisense: TGATGAACTGTTCATTCCC. To overexpress MED12 or p21 in cells, retrovirus-mediated overexpression was performed with the pLNCX system. Full-length human p21 cDNA (complementary DNA) was cloned from HEK293 cells with the following primers: forward: 3′-GCGAAGCTTATGTCAGAACCGGCTGGGGATGT-5′; reverse: 3′-GCGGTTAACTTAGGGCTTCCTCTTGGAGAAG-5′. Full-length human MED12 cDNA was subcloned from p3xFLAG-CMV-10 plasmid (12) into pLNCX plasmid. Cells were selected with G418 (800 μg/ml; RPI) for 2 weeks to generate stable cell lines.
Immunoprecipitation
Cells were lysed in Triton lysis buffer [50 mM tris (pH 8.0), 150 mM NaCl, 0.5% Triton X-100, 10% glycerol, 1 mM dithiothreitol, protease inhibitors, and benzonase]. After centrifugation at 13,000g for 15 min, the supernatants were collected and incubated with primary antibody at 4°C for 2 hours with rotation. After incubation with immobilized protein A (Replicen), samples were washed with lysis buffer four times, and proteins were resuspended in SDS sample loading buffer and subjected to SDS–polyacrylamide gel electrophoresis.
MDA-MB-231 xenograft and 5-FU treatment in nude mice
All animal work was performed in accordance with protocols approved by the Research Animal Resource Center of the University of Wisconsin–Madison. Athymic nude mice at 5 to 6 weeks old were used for xenograft experiments. MDA-MB-231–shMED12–MED12WT (1 × 106) or MDA-MB-231–shMED12–MED12DM cells were resuspended in 0.1 ml of PBS, and were injected into the right fat pad of each nude mouse. Two weeks after transplantation, the mice inoculated with each cell line were divided into two groups (n = 5) and further treated with either PBS or fluorouracil (10 mg/kg) every other day. The tumor size was measured every 4 days. At the end of the experiment, the mice were sacrificed and representative tumor tissues were isolated and photographed.
Human primary tumor samples
There were a total of 282 formalin-fixed and paraffin-embedded tumor samples from 2006 to 2008 in our study. The clinical pathological information for all tumors was obtained. All subjects received a radical mastectomy or modified radical mastectomy. The axillary lymph nodes were routinely dissected, and lymph node metastasis was determined on the basis of histological examination. Tumor size was defined as the maximum tumor diameter measured on the tumor samples at the time of surgery. Histological types of the total 282 samples were defined according to the World Health Organization (WHO) classification criteria (2007). Clinical stage was also defined according to the WHO classification criteria (2007). The tumor samples were obtained from a tissue bank maintained at the Institute of Pathology and Southwest Cancer Center, Southwest Hospital, Third Military Medical University, Chongqing, China. After surgery, most of these subjects received adjuvant chemotherapy alone (cyclophosphamide, methotrexate, and fluorouracil or an anthracycline-based regimen) or combined chemotherapy and endocrine therapy, with or without radiotherapy.
In vitro methylation assay
In vitro methylation assays were performed as previously described (34).
ChIP assay
ChIP assays were performed as described previously (35), using the anti-MED12 (Abcam ab70842) and anti-CARM1 antibodies. The sequences of the primers used for qPCR of p21 genomic regions are as follows: P1 forward: 3′-CTGTCCTCCCCGAGGTCA-5′, P1 reverse: 3′-ACATCTCAGGCTGCTCAGAGTCT-5′; P2 forward: 3′-TATATCAGGGCCGCGCTG-5′, P2 reverse: 3′-GGCTCCACAAGGAACTGACTTC-5′; P3 forward: 3′-CCAGGAAGGGCGAGGAAA-5′, P3 reverse: 3′-GGGACCGATCCTAGACGAACTT-5′; P4 forward: 3′-AGTCACTCAGCCCTGGAGTCAA-5′, P4 reverse: 3′-GGAGAGTGAGTTTGCCCATGA-5′; P5 forward: 3′-CCTCCCACAATGCTGAATATACAG-5′, P5 reverse: 3′-AGTCACTAAGAATCATTTATTGAGCACC-5′.
Quantitative real-time PCR
Total cellular RNA was extracted using the HP Total RNA Kit (VWR Scientific) according to the manufacturer’s instructions. RNA (1 μg) was reversely transcribed using SuperScript II RT (Invitrogen), and quantitative PCR was performed with SYBR Green dye (Roche Scientific) and a CFX96 instrument (Bio-Rad). Primer sequences (IDT) used in this study are as follows: CDKN1A forward: 3′-TGTCCGTCAGAACCCATGC-5′, CDKN1A reverse: 3′-AAAGTCGAAGTTCCATCGCTC-5′; CD74 forward: 3′-GGAAGATCAGAAGCCAGTCATG-5′, reverse: 3′-AGGATGGAAAAGCCTGTGTAC-5′; PRSS2 forward: 3′-TGAAGCCTCCTACCCTGGAA-5′, reverse: 3′-GTTGGCAGCTATGGTGTCCT-5′; KIF5A forward: 3′-AAAACGAATTGCTGAGGTGC-5′, reverse: 3′-TTTTGCTGATGTAGAGTCGGG-5′; KRT14 forward: 3′-GAAGTGAAGATCCGTGACTGG-5′, reverse: 3′-GCAGAAGGACATTGGCATTG-5′; MED12 forward: 3′-GTGGACCCATACCGTCCTGT-5′, reverse: 3′-AAGGCGCTGTCCTTGGGGCA-5′.
Generation of methylated MED12 (me-MED12)–specific antibody
Me-MED12–specific anti-peptide antibody was generated by Genemed Synthesis Inc. The KLH (keyhole limpet hemocyanin)–conjugated MED12 peptide DPYRPVR(me2)LPMQKLPTRC, with R1862 asymmetrically dimethylated, was synthesized. The peptide corresponding to the human MED12 amino acids 1856 to 1872 was used to immunize rabbits. To purify the dimethylated MED12–specific antibody, 10 mg of dimethyl peptide (column A) and 10 mg of control peptide (nonmethyl) (column B) were coupled separately to cyanogen bromide–activated agarose beads. Antisera (100 ml) were then incubated with the peptide-agarose column A. The unbound antiserum was washed with 1× PBS buffer. After several washes, the antibody was eluted with 0.1 M glycine, pH 2.5, and neutralized with 1 M tris, pH 8.0. The antibody was stabilized with 0.1% bovine serum albumin (elution A). Elution A was then incubated with column B, and then the same procedure was followed for elution. The flow through from column B was the dimethyl-specific antibody (me-MED12). The antisera and purified antibody were then tested by enzyme-linked immunosorbent assay.
Gene expression microarray analyses
Total RNAs of MDA-MB-231–CARM1WT–shCtrl, MDA-MB-231–CARM1WT–shMED12, MDA-MB-231–CARM1KO–shCtrl, and MDA-MB-231–CARM1KO–shMED12 cells were used for microarray analysis. By using the BIOARRAY HIGHYIELD RNA labeling kit (Enzo Life Sciences), the second-round cDNA was used to synthesize biotinylated antisense RNA, which was hybridized to Affymetrix HG-U133 Plus 2.0 microarrays containing 54,675 probesets for >47,000 transcripts and variants, including 38,500 human genes. A typical probeset contains eleven 25-mer oligonucleotide pairs (a perfect match and a mismatch control). Some genes are measured by multiple probesets. To identify genes co-regulated by CARM1 and MED12, we used an empirical Bayes hierarchical modeling approach called EBarrays. The method is also available in Bioconductor. The approach uses information across genes and arrays to fit a hierarchical Bayesian model, and provides posterior probabilities that quantify evidence that a gene is differentially expressed. Posterior probability thresholds can be set to control the false discovery rate at a desired level. In particular, an expression level heat map for genes whose posterior probability of differential expression is more than 99% is given.
Statistical analyses
Statistical analyses were performed with unpaired Student’s t tests, and P values <0.05 were considered statistically significant. Error bars in figures represent SD. Pearson correlations were calculated in GraphPad Prism 5 software.
Acknowledgments
We thank A. Welm for providing human breast tumors, A. Brinkman for editorial assistance, and G. Davis for helpful discussion. Funding: This project is supported by Department of Defense (DOD) Era of Hope Award (W81XWYH-11-1-0237) to W.X. Author contributions: L.W., H.Z., and W.X. conceived and designed the experiments, and drafted and revised the article. L.W., H.Z., and Z.Z. performed all the biochemical and cell line–related experiments and analyzed the data. L.W. performed the xenograft experiment. Q.W. and X.B. performed the TMA experiment and analyzed the data. T.G.B., X.B., and W.X. interpreted the data and drafted the article. Competing interests: The authors declare that they have no competing interests. Data and materials availability: Data will be made available upon request by emailing wxu@oncology.wisc.edu.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/1/9/e1500463/DC1
Fig. S1. In vitro methylation assays of MED12 by PRMT1/PRMT6 and correlation analyses of indicated genes in breast cancer specimens and cell lines.
Fig. S2. Western blotting analysis of endogenous dimethylated MED12R1862 using a methyl-specific MED12 rabbit polyclonal antibody.
Fig. S3. Mutation of MED12 methylation sites does not affect cell growth or EMT-associated gene expression.
Fig. S4. The mRNA level of CDKN1A/p21, a MED12 and CARM1 co-regulated gene, correlates with 5-FU response in vitro and predicts the probability of recurrence-free survival in breast cancer patients.
Fig. S5. Mutation of MED12 methylation sites does not affect the interaction of MED12 with other known interacting proteins.
Fig. S6. Suppression of p21 mRNA and protein levels is retained in MED12WT- but not MED12DM-expressing HEK293 cells.
Table S1. Primary hits from the FDA-approved oncology drug screening.
Table S2. Differentially expressed genes regulated by CARM1 and MED12.
REFERENCES AND NOTES
- 1.Bedford M. T., Clarke S. G., Protein arginine methylation in mammals: Who, what, and why. Mol. Cell 33, 1–13 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cheng H., Qin Y., Fan H., Su P., Zhang X., Zhang H., Zhou G., Overexpression of CARM1 in breast cancer is correlated with poorly characterized clinicopathologic parameters and molecular subtypes. Diagn. Pathol. 8, 129 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang L., Zhao Z., Meyer M. B., Saha S., Yu M., Guo A., Wisinski K. B., Huang W., Cai W., Pike J. W., Yuan M., Ahlquist P., Xu W., CARM1 methylates chromatin remodeling factor BAF155 to enhance tumor progression and metastasis. Cancer Cell 25, 21–36 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lewis B. A., Reinberg D., The mediator coactivator complex: Functional and physical roles in transcriptional regulation. J. Cell Sci. 116, 3667–3675 (2003). [DOI] [PubMed] [Google Scholar]
- 5.Schiano C., Casamassimi A., Rienzo M., de Nigris F., Sommese I., Napoli C., Involvement of mediator complex in malignancy. Biochim. Biophys. Acta 1845, 66–83 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Graham J. M. Jr, Schwartz C. E., MED12 related disorders. Am. J. Med. Genet. A 161, 2734–2740 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Turunen M., Spaeth J. M., Keskitalo S., Park M. J., Kivioja T., Clark A. D., Mäkinen N., Gao F., Palin K., Nurkkala H., Vähärautio A., Aavikko M., Kämpjärvi K., Vahteriso P., Kim C. A., Aaltonen L. A., Varjosalo M., Taipale J., Boyer T. G., Uterine leiomyoma-linked MED12 mutations disrupt mediator-associated CDK activity. Cell Rep. 7, 654–660 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barbieri C. E., Baca S. C., Lawrence M. S., Demichelis F., Blattner M., Theurillat J.-P., White T. A., Stojanov P., Vsn Allen E., Stansky N., Nickerson E., Chae S.-S., Boysen G., Auclair D., Onofrio R. C., Park K., Kitabayashi N., MacDonald T. Y., Sheikh K., Vuong T., Guiducci C., Cibulskis K., Sivachenko A., Carter S. L., Saksena G., Voet D., Hussain W. M., Ramos A. H., Winckler W., Redman M. C., Ardlie K., Tewari A. K., Mosquera J. M., Rupp N., Wild P. J., Moch H., Morrissey C., Nelson P. S., Kantoff P. W., Gabriel S. B., Golub T. R., Meyerson M., Lander E. S., Getz G., Rubin M. A., Garraway L. A., Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat. Genet. 44, 685–689 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lim W. K., Ong C. K., Tan J., Thike A. A., Ng C. C. Y., Rajasegaran V., Myint S. S., Nagarajan S., Nasir N. D. M., McPherson J. R., Cutcutache I., Poore G., Tay S. T., Ooi W. S., Tan V. K. M., Hartman M., Ong K. W., Tan B. K. T., Rozen S. G., Tan P. H., Tan P., Teh B. T., Exome sequencing identifies highly recurrent MED12 somatic mutations in breast fibroadenoma. Nat. Genet. 46, 877–880 (2014). [DOI] [PubMed] [Google Scholar]
- 10.Huang S., Hölzel M., Knijnenburg T., Schlicker A., Poepman P., McDermott U., Garnett M., Grernrum W., Sun C., Prahallad A., Groenendijik F. H., Mittempergher l., Nijkamp W., Neefjes J., Salazar R., Dijke P. t., Uramoto H., Tanaka F., Beijersbergen R. L., Wessels L. F. A., Bernards R., MED12 controls the response to multiple cancer drugs through regulation of TGF-β receptor signaling. Cell 151, 937–950 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shalem O., Sanjana N. E., Hartenian E., Shi X., Scott D. A., Mikkelsen T. S., Heckl D., Ebert B. L., Root D. E., Doench J. G., Zhang F., Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ding N., Zhou H., Esteve P.-O., Chin H. G., Kim S., Xu X., Joseph S. M., Friez M. J., Schwartz C. E., Pradhan S., Boyer T. G., Mediator links epigenetic silencing of neuronal gene expression with x-linked mental retardation. Mol. Cell 31, 347–359 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De S., Cipriano R., Jackson M. W., Stark G. R., Overexpression of kinesins mediates docetaxel resistance in breast cancer cells. Cancer Res. 69, 8035–8042 (2009). [DOI] [PubMed] [Google Scholar]
- 14.Tan M. H., De S., Bebek G., Orloff M. S., Wesolowski R., Downs-Kelly E., Budd G. T., Stark G. R., Eng C., Specific kinesin expression profiles associated with taxane resistance in basal-like breast cancer. Breast Cancer Res. Treat. 131, 849–858 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sherman-Baust C. A., Becker K. G., Wood W. H. III, Zhang Y., Morin P. J., Gene expression and pathway analysis of ovarian cancer cells selected for resistance to cisplatin, paclitaxel, or doxorubicin. J. Ovarian Res. 4, 21 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kitange G. J., Carlson B. L., Schroeder M. A., Decker P. A., Morlan B. W., Wu W., Ballman K. V., Giannini C., Sarkaria J. N., Expression of CD74 in high grade gliomas: A potential role in temozolomide resistance. J. Neuro-Oncol. 100, 177–186 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Törma H., Regulation of keratin expression by retinoids. Dermatoendocrinol. 3, 136–140 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang W., Kornblau S. M., Kobayashi T., Gambel A., Claxton D., Deisseroth A. B., High levels of constitutive WAF1/Cip1 protein are associated with chemoresistance in acute myelogenous leukemia. Clin. Cancer Res. 1, 1051–1057 (1995). [PubMed] [Google Scholar]
- 19.Kagey M. H., Newman J. J., Bilodeau S., Zhan Y., Orlando D. A., van Berkum N. L., Ebmeier C. C., Goossens J., Rahl P. B., Levine S. S., Taatjes D. J., Dekker J., Young R. A., Mediator and cohesin connect gene expression and chromatin architecture. Nature 472, 247 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee Y.-H., Stallcup M. R., Roles of protein arginine methylation in DNA damage signaling pathways. Is CARM1 a life-or-death decision point? Cell Cycle 10, 1343–1344 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wagle N., Van Allen E. M., Treacy D. J., Frederick D. T., Cooper Z. A., Taylor-Weiner A., Rosenberg M., Goetz E. M., Sullivan R. J., Farlow D. N., Friedrich D. C., Anderka K., Perrin D., Johannessen C. M., McKenna A., Cibulskis K., Kryukov G., Hodis E., Lawrence D. P., Fisher S., Getz G., Gabriel S. B., Carter S. L., Flaherty K. T., Wargo J. A., Garraway L. A., MAP kinase pathway alterations in BRAF-mutant melanoma patients with acquired resistance to combined RAF/MEK inhibition. Cancer Discov. 4, 61–68 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Firestein R., Bass A. J., Kim S. Y., Dunn I. F., Silver S. J., Guney I., Freed E., Ligon A. H., Vena N., Ogino S., Chheda M. G., Tamayo P., Finn S., Shrestha Y., Boehm J. S., Jain S., Bojarski E., Mermel C., Barretina J., Chan J. A., Baselga J., Tabernero J., Root D. E., Fuchs C. S., Loda M., Shivdasani R. A., Meyerson M., Hahn W. C., CDK8 is a colorectal cancer oncogene that regulates β-catenin activity. Nature 455, 547–551 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Galbraith M. D., Allen M. A., Bensard C. L., Wang X., Schwinn M. K., Qin B., Long H. W., Daniels D. L., Hahn W. C., Dowell R. D., Espinosa J. M., HIF1A employs CDK8-mediator to stimulate RNAPII elongation in response to hypoxia. Cell 153, 1327–1339 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Knuesel M. T., Meyer K. D., Bernecky C., Taatjes D. J., The human CDK8 subcomplex is a molecular switch that controls Mediator coactivator function. Genes Dev. 23, 439–451 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Knuesel M. T., Meyer K. D., Donner A. J., Espinosa J. M., Taatjes D. J., The human CDK8 subcomplex is a histone kinase that requires Med12 for activity and can function independently of mediator. Mol. Cell. Biol. 29, 650–661 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rau M. J., Fischer S., Neumann C. J., Zebrafish Trap230/Med12 is required as a coactivator for Sox9-dependent neural crest, cartilage and ear development. Dev. Biol. 296, 83–93 (2006). [DOI] [PubMed] [Google Scholar]
- 27.Kim S., Xu X., Hecht A., Boyer T. G., Mediator is a transducer of Wnt/β-catenin signaling. J. Biol. Chem. 281, 14066–14075 (2006). [DOI] [PubMed] [Google Scholar]
- 28.Zhou H., Kim S., Ishii S., Boyer T. G., Mediator modulates Gli3-dependent sonic hedgehog signaling. Mol. Cell. Biol. 26, 8667–8682 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu X., Zhou H., Boyer T. G., Mediator is a transducer of amyloid-precursor-protein-dependent nuclear signalling. EMBO Rep. 12, 216–222 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Izawa H., Yamamoto H., Damdinsuren B., Ikeda K., Tsujie M., Suzuki R., Kitani K., Seki Y., Hayashi T., Takemasa I., Ikeda M., Ohue M., Sekimoto M., Monden T., Monden M., Effects of p21cip1/waf1 overexpression on growth, apoptosis and differentiation in human colon carcinoma cells. Int. J. Oncol. 27, 69–76 (2005). [PubMed] [Google Scholar]
- 31.Ogino S., Meyerhardt J. A., Cantor M., Brahmandam M., Clark J. W., Namgyal C., Kawasaki T., Kinsella K., Michelini A. L., Enzinger P. C., Kulke M. H., Ryan D. P., Loda M., Fuchs C. S., Molecular alterations in tumors and response to combination chemotherapy with gefitinib for advanced colorectal cancer. Clin. Cancer Res. 11, 6650–6656 (2005). [DOI] [PubMed] [Google Scholar]
- 32.Oshimori N., Oristian D., Fuchs E., TGF-β promotes heterogeneity and drug resistance in squamous cell carcinoma. Cell 160, 963–976 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang Y., Geng L., Talmon G., Wang J., MicroRNA-520g confers drug resistance by regulating p21 expression in colorectal cancer. J. Biol. Chem. 290, 6215–6225 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang L., Charoensuksai P., Watson N. J., Wang X., Zhao Z., Coriano C. G., Kerr L. R., Xu W., CARM1 automethylation is controlled at the level of alternative splicing. Nucleic Acids Res. 41, 6870–6880 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu J., Xu W., Histone H3R17me2a mark recruits human RNA polymerase-associated factor 1 complex to activate transcription. Proc. Natl. Acad. Sci. U.S.A. 109, 5675–5680 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.DeRose Y. S., Wang G., Lin Y.-C., Bernard P. S., Buys S. S., Ebbert M. T. W., Factor R., Matsen C., Milash B. A., Nelson E., Neumayer L., Randall R. L., Stijleman I. J., Welm B. E., Welm A. L., Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat. Med. 17, 1514–1520 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/1/9/e1500463/DC1
Fig. S1. In vitro methylation assays of MED12 by PRMT1/PRMT6 and correlation analyses of indicated genes in breast cancer specimens and cell lines.
Fig. S2. Western blotting analysis of endogenous dimethylated MED12R1862 using a methyl-specific MED12 rabbit polyclonal antibody.
Fig. S3. Mutation of MED12 methylation sites does not affect cell growth or EMT-associated gene expression.
Fig. S4. The mRNA level of CDKN1A/p21, a MED12 and CARM1 co-regulated gene, correlates with 5-FU response in vitro and predicts the probability of recurrence-free survival in breast cancer patients.
Fig. S5. Mutation of MED12 methylation sites does not affect the interaction of MED12 with other known interacting proteins.
Fig. S6. Suppression of p21 mRNA and protein levels is retained in MED12WT- but not MED12DM-expressing HEK293 cells.
Table S1. Primary hits from the FDA-approved oncology drug screening.
Table S2. Differentially expressed genes regulated by CARM1 and MED12.