

Force parameters of human β-cardiac myosin with the hypertrophic cardiomyopathy mutation R403Q show loss of molecular motor function.
Keywords: High pressure, Crystallography, Metallic alloy, Chemical bonding, electron density, maximum entropy method
Abstract
Hypertrophic cardiomyopathy (HCM) is the most frequently occurring inherited cardiovascular disease. It is caused by mutations in genes encoding the force-generating machinery of the cardiac sarcomere, including human β-cardiac myosin. We present a detailed characterization of the most debated HCM-causing mutation in human β-cardiac myosin, R403Q. Despite numerous studies, most performed with nonhuman or noncardiac myosin, there is no consensus about the mechanism of action of this mutation on the function of the enzyme. We use recombinant human β-cardiac myosin and new methodologies to characterize in vitro contractility parameters of the R403Q myosin compared to wild type. We extend our studies beyond pure actin filaments to include the interaction of myosin with regulated actin filaments containing tropomyosin and troponin. We find that, with pure actin, the intrinsic force generated by R403Q is ~15% lower than that generated by wild type. The unloaded velocity is, however, ~10% higher for R403Q myosin, resulting in a load-dependent velocity curve that has the characteristics of lower contractility at higher external loads compared to wild type. With regulated actin filaments, there is no increase in the unloaded velocity and the contractility of the R403Q myosin is lower than that of wild type at all loads. Unlike that with pure actin, the actin-activated adenosine triphosphatase activity for R403Q myosin with Ca2+-regulated actin filaments is ~30% lower than that for wild type, predicting a lower unloaded duty ratio of the motor. Overall, the contractility parameters studied fit with a loss of human β-cardiac myosin contractility as a result of the R403Q mutation.
INTRODUCTION
Heart disease, in various forms, is the leading cause of death in the United States. Hypertrophic cardiomyopathy (HCM) is the most common cause of genetic heart disease (1). One in every 500 individuals suffers from HCM, and this number could be even greater (2). Clinically, HCM presents with ventricular wall thickening and myocyte enlargement. These hearts are often characterized as clinically hyperdynamic, on the basis of physical exam findings and echocardiographic findings with supranormal ejection fraction, whereas the earliest hallmark of the HCM heart is diastolic dysfunction (3). Pathologically, there is increased fibrosis of the muscle and myofibrillar disarray (4). Up to 60 to 70% of HCM cases are due to mutations in genes encoding proteins of the cardiac sarcomere (5). Between 30 and 50% of those mutations are found in the gene that encodes human β-cardiac myosin, the motor that powers ventricular contraction (6–8).
More than two decades ago, the first cardiomyopathy-causing mutation to be discovered, R403Q, was mapped to the human β-cardiac myosin gene, MYH7 (9). Since then, more than 400 different mutations in β-cardiac myosin have been suggested to be HCM-causing (10). These mutations are distributed throughout the molecule (10). Although a number of these mutations have been studied in the last decade [for review, see Moore et al. (11)], there is no clear agreement as to the molecular mechanism(s) by which these mutations give rise to the disease state [for review, see (12–16)].
Because of its early discovery and clinical severity, numerous studies have focused on the molecular effects of the R403Q mutation on myosin function. Therefore, there is more known about the biochemical and biophysical effects of this mutation than for any other myosin mutation. There has been considerable controversy in the literature regarding the effects of the R403Q mutation on myosin function. An early study on R403Q mutant human β-cardiac myosin isolated from biopsies of cardiac and soleus slow skeletal muscle, both of which express the β-cardiac myosin heavy chain, reported a decrease in actin sliding velocity by the mutant myosin (17), whereas a later study that used protein isolated from cardiac tissues showed an increase in actin velocity (18). This discrepancy may reflect the fact that the activity of myosin isolated from biopsy samples can be variable, depending on tissue handling. Furthermore, these biopsy samples yield an undefined mixture of wild-type and mutant myosins, the ratio of which can change from patient to patient (19). In the case of cardiac muscle, there is also a variable amount of α-cardiac myosin present. This complicates quantitative analysis of the effects of the mutant on myosin function. Later studies using mouse α-cardiac myosin containing the R403Q mutation consistently reported an increase in velocity and an increase in the adenosine triphosphatase (ATPase) activity (20). Although a general consensus about the actin motility and ATPase activity being higher was developing, the effect of R403Q on the force production was different in different studies. Using purified mouse α-cardiac myosin, Tyska et al. (20) found that the intrinsic force produced by R403Q did not show any change, although they reported a twofold increase in ensemble force. This increase in ensemble force for R403Q was also seen by Debold et al. (21) in the same mouse myosin backbone. Conversely, Palmiter et al. (18) reported no change in ensemble force for R403Q using a heterogeneous mixture of wild-type and mutant myosin from human cardiac muscle biopsy samples. The results of the skinned fiber and myofibril studies are also contradictory. The maximum tension developed in human soleus muscle skinned fiber biopsy samples was lower in one study (22), and this was also the case for cardiac myofibrils from R403Q patients in another study (23). However, Palmer et al. (24) showed no change in maximal isometric tension for R403Q mouse myofibrils in one study and increased maximal tension in a subsequent study (25). Blanchard et al. (26) found increased tension at submaximal Ca2+ but marginally lower tension at maximal Ca2+ concentrations. Similarly, experiments performed in totally different myosin backgrounds, such as chicken smooth muscle and Dictyostelium myosin, showed results consistent with some studies and not with others (27, 28). In short, these results from different laboratories have not yielded a consistent view of the effects of the R403Q mutation on the biochemical and biophysical properties of myosin, although the mouse α-cardiac R403Q myosin results generally suggested gain of function by the R403Q mutation.
Why are there such discrepancies? Evolutionarily, muscle myosin in different organisms is designed to work in a very particular manner, that is, to generate a certain level of force and to slide actin filaments at a particular velocity. Although the overall architecture of the actomyosin network is similar, there are differences in the sequences of myosin, which bring about changes in function. For example, in cardiac muscle, the faster and lower force-generating human α-cardiac myosin motor domain (1 to 843 amino acids) has 79 amino acid residues that are different from the β-isoform. For a mouse, this difference is 64 residues. Moreover, the mouse α-cardiac myosin, where most of the studies with R403Q have been performed, has 84 amino acid residues that are different from the human β-cardiac myosin. Therefore, studying the effects of single disease-causing mutations in nonhuman cardiac myosin is less than ideal. In seminal studies by Lowey et al. (29), the effects of the R403Q mutation in mouse cardiac myosin were shown to depend on the mouse isoform into which the mutation was introduced. In mouse α-cardiac myosin, they saw an increase in both ATPase activity and actin sliding velocity, whereas in the mouse β-cardiac myosin, there was no significant change in the actin velocity and actually a slight decrease in the ATPase activity (29). Similarly, the adenosine diphosphate (ADP) release rate for R403Q in mouse α-cardiac myosin was ~20% higher than that for wild type but was unaltered when studied in the mouse β-cardiac myosin background (30). It is therefore clear that the differences in myosin sequence in different isoforms and from different species do alter the changes in biomechanics due to the disease-causing mutation. Human cardiac biopsy samples could theoretically provide material to study, but these are difficult to obtain and furthermore yield poor amounts of a heterogeneous population of mutated and wild-type motor. This all speaks to the importance of studying the HCM mutations using reconstituted purified human β-cardiac myosin.
Until recently, expression of human cardiac muscle myosin was unsuccessful with common recombinant protein expression systems. This problem has now been addressed by a recently developed muscle cell expression system (31, 32) that provides a source of active recombinant human β-cardiac muscle myosin (33, 34) and can now be used to study cardiomyopathic mutations in human β-cardiac myosin (35–37).
Here, we focus on the HCM-causing R403Q mutation using purified human β-cardiac myosin. We compare the in vitro contractility parameters that relate to ensemble force (Fensemble) of the mutant and wild-type proteins. The ensemble force can be approximated as Fensemble = f(ts/tc)Nt, where f is the intrinsic force of an individual myosin motor; ts/tc is the duty ratio, defined as the total fraction of myosin heads in the force-producing state at any moment; and Nt is the total number of myosin motors that are functionally available to interact with actin filaments in the sarcomere (16). In this equation, ts is the duration that myosin remains attached to actin during the chemomechanical cycle (the strongly bound state time) and tc is the total time to complete one chemomechanical cycle. Changes in any of these parameters or a combination of these parameters will change Fensemble. Here, we experimentally measure f, ts/tc, and relative Fensemble for wild-type and R403Q human β-cardiac myosin motors. Our data show that all three of these parameters are lower for R403Q, as compared to wild type, consistent with the mutation being a loss of function for those myosin heads that are functionally available for interaction with actin in the heart.
RESULTS
Although HCM is a devastating disease, patients are often asymptomatic in the early years of the disease, and underlying biochemical and biophysical changes in the sarcomere contractile apparatus due to HCM mutations are likely to be small. Thus, many experiments are required to obtain statistically significant comparisons between the biomechanics of mutant and wild-type human β-cardiac myosin. All of the results presented here are therefore replicates of many experiments (“exp”) using multiple independent preparations of myosin (“preps”).
For all of our experiments, we used the purified short subfragment 1 (sS1) construct of human β-cardiac myosin containing a truncated human myosin heavy chain (residues 1 to 808) and the human ventricular essential light chain (ELC) (fig. S1). We refer to this construct as human β-cardiac sS1 or sS1. Many studies have shown that the catalytic domain of myosin is the myosin motor, and the light-chain binding region acts as a lever arm to amplify the movement of the converter region of the catalytic domain [for review, see (38–40)]. Thus, the fundamental contractility properties of the myosin are expressed by the sS1, and the fundamental changes in these parameters due to HCM mutations likely reflect the impact of these mutations on the full-length myosin. However, we recognize that the two-headed nature of the full-length myosin and the presence of the phosphorylatable regulatory light chain (RLC) may affect these parameters, and our future studies will compare the results described here with the more complex two-headed heavy meromyosin.
Here, we describe the effects of the R403Q mutation on human β-cardiac sS1 function at both single-molecule and ensemble levels with pure actin and with the more physiologically relevant regulated thin filaments (RTFs) containing actin, tropomyosin, and the troponin complex.
Biomechanics of R403Q human β-cardiac sS1 with pure actin
R403Q human β-cardiac sS1 generates higher actin velocities than does wild type in an unloaded in vitro motility assay
Figure 1A shows the position of R403Q (green spheres) in β-cardiac S1 with respect to actin and tropomyosin. The location of this mutation near the actin-tropomyosin binding site suggests that it might alter the actin-myosin cross-bridge kinetics. The maximum speed at which muscle motion can be attained is limited by these actin-myosin kinetics. We therefore characterized the motion-generating capability of the R403Q mutant myosin in an in vitro motility assay by measuring the movement of individual rhodamine-phalloidin–labeled actin filaments over a myosin-coated surface (41). Representative experimental traces of wild-type and R403Q sS1 unloaded motility are shown in fig. S2. Actin filament velocity movies were automatically and objectively analyzed as described elsewhere, Aksel et al. (37) to measure four velocity parameters (see the legend to Fig. 1). Briefly, these parameters are MVEL, MVEL20, TOP5%, and PLATEAU. MVEL is the mean velocity of all moving filaments. MVEL20 is the mean velocity of all moving filaments after applying a 20% tolerance filter. The tolerance filter removes all intermittently moving filaments whose deviation in velocity is 20% or more than its mean velocity. TOP5% is the mean of the top 5% of the velocity distribution across different actin filament lengths, and PLATEAU is the mean velocity obtained from fitting a single exponential function to the maximum velocities across different actin filaments.
Fig. 1. The position of R403 in the myosin molecule in the context of actin and tropomyosin and its effects on unloaded motility.
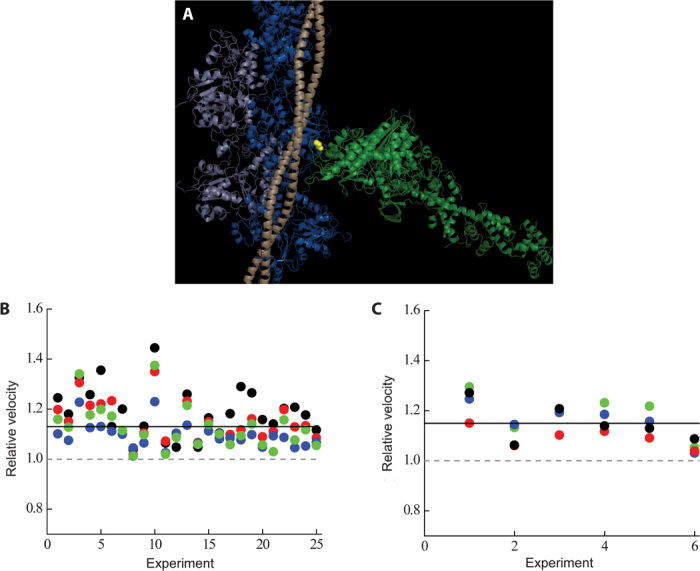
(A) The R403 residue (yellow spheres) resides in a loop at the actin-binding domain of the myosin molecule (light green) and recently has been postulated to interact with both actin (light and dark blue) and tropomyosin (light wheat) (45). The structure is a modification of the Protein Data Bank (PDB) file, 2TM1 [submitted by Behrmann et al. (45)] with a chimera of the human β-cardiac myosin motor domain (PDB: 4DB1; residues 1 to 777) and chicken lever arm (PDB: 2MYS; residues 780 to 843) along with the human ELC and RLC (the S1 domain of myosin, light green) docked in. (B) Normalized motility parameters for human β-cardiac R403Q with respect to wild type. Experimental outcomes of MVEL (black), MVEL20 (red), TOP5% (blue), and PLATEAU (green) for different experiments, performed at 23°C, are shown for R403Q with respect to wild type (the wild-type value is normalized to 1 and is denoted as a gray dashed line). The solid line denotes ~15% increase in all the motility parameters for human β-cardiac sS1 R403Q (P < 0.01). (C) Same as (B), except that the experiments were performed at 30°C. A ~15% increase for all velocity parameters of R403Q sS1 was also observed at this temperature (P < 0.01). MVEL is the mean of the velocity distribution of moving actin filaments and excludes any filaments that are stuck. The percentage of stuck filaments for the human β-cardiac sS1, after dead-head removal (see Unloaded and loaded in vitro motility under Materials and Methods), is typically 5 to 10% for a given myosin preparation. MVEL20 is a 20% tolerance-filtered mean velocity. This parameter is calculated by eliminating intermittently moving filaments with velocities fluctuating more than 20% of the mean velocity. TOP5% is the mean of the top 5% of the velocities of all moving filaments. PLATEAU is the mean velocity obtained from fitting a single exponential function to the maximum velocities across different actin lengths. See fig. S2 for a representative example of these parameters. A complete description of this methodology can be found in (37), and the FAST software used for analysis is available for downloading on our Web site (http://spudlab.stanford.edu/FAST.html).
The velocity parameter that relates most closely to the average velocity usually reported in the field is the mean velocity of reasonably smooth-moving filaments (here, the mean velocity with a 20% filter to eliminate filaments that are not moving relatively smoothly, MVEL20). We also report the mean velocity of moving filaments that includes filaments that move intermittently (MVEL). This parameter gives us the mean velocity without filtering for smoothly moving filaments. Intermittently moving filaments result from filaments encountering a load, such as a rigor myosin head, while they move. Although very low in number, such myosin “dead heads” lead to stuck filaments. Often the number of stuck filaments on the surface is not reported in motility reports, but this is an important parameter because it is a reflection of the quality of the myosin preparation (see fig. S2, A and B, upper right panels). Typically, in all our experiments, the percentage of stuck filaments varied from 2 to 8%. Furthermore, in loaded motility assays, increasing numbers of filaments become immobile as more load molecules are added to the surface, and the fraction of stuck filaments becomes an important reportable parameter. We therefore also report the percentage of stuck filaments (%STUCK; see fig. S2, A and B, upper right panels). This particular parameter is discussed in detail below (see The ensemble force metric of R403Q human β-cardiac sS1 is significantly lower than that of wild type). Finally, it is also useful to know the mean velocity of the TOP5% fastest-moving filaments, as well as the PLATEAU mean velocity value where the velocity no longer depends on actin filament length. Reporting all of these values provides the reader with a much clearer understanding of the quality and meaning of the motility data.
The mean values from many actin velocity measurements using wild-type human β-cardiac sS1 at 23°C were as follows: MVEL, 470 ± 10 nm/s; MVEL20, 550 ± 20 nm/s; TOP5%, 780 ± 20 nm/s; and PLATEAU, 610 ± 20 nm/s (n = 25 exp, 4 preps). The mean values from many actin velocity measurements using R403Q human β-cardiac sS1 were as follows: MVEL, 590 ± 20 nm/s; MVEL20, 630 ± 20 nm/s; TOP5%, 860 ± 20 nm/s; and PLATEAU, 690 ± 20 nm/s (n = 25 exp, 4 preps), all consistently higher than the wild-type values. Figure 1B shows the velocities of R403Q normalized to the corresponding wild-type controls (set to 1.0, dashed line). The different colors represent the four different velocity parameters. Regardless of which parameter is used, R403Q human β-cardiac sS1 showed a significant ~15% (P < 0.01) increase in actin filament velocities. Motility experiments performed at a higher (30°C) temperature yielded similar results (~15% increase, P < 0.01, Fig. 1C).
Unloaded velocity is related to the displacement generated by the myosin power stroke (the step size, d) and the duration that myosin remains attached to actin during the chemomechanical cycle (the strongly bound state time ts), so that velocity is ∝ d/ts. Thus, the increase in the velocity for R403Q human β-cardiac sS1 could be due to either an increase in d or a decrease in ts or both. We therefore measured the step size of single sS1 molecules using the dual-beam laser trap assay and the ADP release rate, which reflects ts, from the actin-sS1-ADP complex using stopped-flow kinetics. Note that both of these parameters may be somewhat different in the context of the motility assay, which may not be truly unloaded even in the absence of added load molecules (see below).
The step size and unloaded ADP release rate for R403Q human β-cardiac sS1 are similar to those for wild type
The magnitude of the displacement of an optically trapped bead at low trap stiffness (~0.06 to 0.1 pN/nm) is a measure of the step size (d) of the myosin produced during the power stroke. There was no statistical difference in the step size of R403Q human β-cardiac sS1 (6.0 ± 1 nm, n = 4 exp, 3 preps) as compared to wild type (5.8 ± 1 nm, n = 14 exp, 10 preps) (fig. S3). Note, however, that it is very difficult to obtain step size data that would robustly distinguish a difference of less than 10%. This result is similar to that found by Palmiter et al. (18) using myosin purified from human cardiac biopsy samples.
ADP release is the rate-limiting step for the strongly bound (to actin) state time (ts) of myosin in the ATPase cycle. The inverse of the ADP release rate constant is ts. We measured the ADP release rate constant for R403Q in solution under two different conditions and found no significant difference between R403Q human β-cardiac sS1 and wild type (table S1). Thus, all we can conclude is that the values for d and ts are very similar for R403Q human β-cardiac sS1 and wild type under true unloaded conditions, and we cannot be sure whether one or the other or both are contributing to the 15% increase in velocity by the R403Q human β-cardiac sS1. It is also possible that one or both of these parameters change under low-load conditions, which may reflect the “unloaded” motility assay. To further explore the effects of the R403Q mutation, we measured tc, the total cycle time of the myosin ATPase, and f, the intrinsic force of the single molecule, both of which can affect the Fensemble of the contractile complex.
R403Q human β-cardiac sS1 has a lower tc than wild type
To determine tc, we measured the maximal rate of ATP hydrolysis (kcat) by the actin-myosin complex (tc = 1/kcat). An increase of 30 to 125% in kcat was previously reported for this mutation in the mouse α-cardiac myosin background in three independent studies (20, 24, 29). When studied in the mouse β-cardiac myosin background, however, there was no difference in kcat between the R403Q mutant enzyme and wild type (29). For the human β-cardiac sS1, we found the kcat for R403Q sS1 (7.6 ± 0.5 s−1; n = 9 exp, 4 preps) to be ~25% higher (P < 0.05) than that for the wild-type motor (6.0 ± 0.5 s−1; n = 8 exp, 4 preps) (Fig. 2A). Thus, the calculated tc was ~170 ± 2 ms for wild-type human β-cardiac sS1 compared to 130 ± 2 ms for the R403Q mutant form. This statistically significant difference in values of tc points to a fundamental alteration in the cross-bridge kinetics between R403Q and wild-type human β-cardiac sS1. Specifically, this ~25% decrease in tc for the R403Q myosin would contribute toward a higher duty ratio (ts/tc). The Km for actin from the actin-activated myosin ATPase assay was not statistically different between wild-type (40 ± 6 μM; n = 8 exp, 4 preps) and R403Q human β-cardiac sS1 (50 ± 7 μM; n = 9 exp, 4 preps).
Fig. 2. Enzymatic and mechanical properties of wild-type and R403Q human β-cardiac sS1.
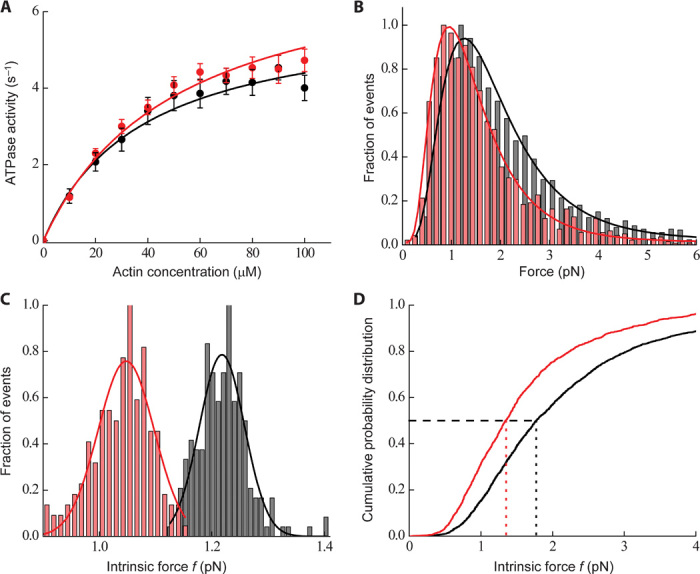
(A) Actin-activated ATPase activities (circles) of human wild-type (black) and R403Q mutant (red) β-cardiac sS1 at 23°C. The data points shown are the average of multiple experiments (n = 4 exp, 3 preps). The data are fit (solid lines, black for wild-type and red for R403Q) to the Michaelis-Menten equation to obtain the kcat and Km values. Errors reported are SEM. (B) Histograms of all single-molecule force events for wild-type (gray bars) (~2500 events, n = 23 exp, 10 preps) and R403Q human β-cardiac sS1 (red bars) (~1800 events, n = 10 exp, 4 preps). We used a Gaussian distribution function with a cutoff to take into account the missing small force events. The force distributions showed a long tail in the higher force regimes. These high-force events are a small fraction, but they can easily alter the fitting results and hence the interpretation of the data. To extract the mean force, we instead used a double Gaussian distribution (black line for wild type and red line for R403Q) and the fit results using the first Gaussian peak were used to bootstrap the data (see Optical trap setup and single-molecule measurements under Materials and Methods). For R403Q, the force distribution is shifted toward lower force values as compared to wild-type human β-cardiac sS1. (C) Distribution of mean forces obtained from bootstrapping of 300 single events from the total number of force events [as shown in (B), ~2500 events from n = 23 exp, 10 preps for wild type and ~1800 events from n = 10 exp, 4 preps for R403Q]. A Gaussian function was fit to the bootstrapped mean histogram (solid line, black for wild type and red for R403Q) to obtain a mean force value. The mean force of wild type and R403Q is significantly different from each other within 95% confidence interval. (D) Cumulative frequency distribution (solid line, black for wild type and red for R403Q) calculated from all single-molecule force events (~2500 events from n = 23 exp, 10 preps for wild type and ~1800 events from n = 10 exp, 4 preps for R403Q). Vertical dotted lines (black for wild type and red for R403Q) show a half-probability (shown as a black horizontal dashed line) of the force distribution, which is smaller for R403Q as compared to wild type.
A single R403Q human β-cardiac sS1 molecule produces a lower intrinsic force than does wild type
We used the dual-beam optical trap to measure the intrinsic force f for the R403Q and wild-type human β-cardiac sS1. The motions of the trapped actin-bound beads were followed with high spatial and temporal resolution under high trap stiffness, achieved by a feedback system. This high-frequency feedback signal moves the trap position in response to a single myosin stroke, and this can be used to quantify f generated by a single myosin molecule (42). Many such single-molecule events from several molecules obtained from different preparations were combined to produce a force histogram (Fig. 2B).
This force histogram was processed to obtain an average intrinsic force (Fig. 2C) (see Materials and Methods). For R403Q, the average intrinsic force produced by a single sS1 molecule was 1.05 ± 0.05 pN (n = 10 exp, 4 preps), which is significantly lower by ~15% (P < 0.01) than the force generated by the wild-type sS1 under the same conditions (1.25 ± 0.05 pN, n = 23 exp, 10 preps). Additionally, we used all the individual single-molecule force events to create a cumulative frequency distribution. This measures the probability of occurrence of any particular force event. For R403Q, a probability of 0.5 (black horizontal dashed line in Fig. 2D) occurred at a force value lower than that for wild type (compare red and black vertical dotted lines in Fig. 2D). This suggests that a single molecule of R403Q is more probable to generate a lower intrinsic force compared to a wild-type molecule.
A lower intrinsic force for R403Q human β-cardiac sS1 means that less work can be obtained from a single sS1 molecule per ATPase cycle. This could be reflected in a lower ΔG, the maximum amount of energy that can be “freed” from the system to perform useful work, upon rebinding of myosin to actin into its strongly bound rigor state after the hydrolysis of ATP (43, 44). Because ΔG is proportional to the natural logarithm of the equilibrium association constant for binding, the R403Q human β-cardiac sS1 is expected to have a weaker affinity for the strongly bound rigor state of actin and sS1 compared to wild type. Alternatively, a more compliant myosin molecule could also lead to less work performed by the motor. To tease apart these possibilities, we compared the affinities of the strongly bound actin-myosin rigor state of R403Q human β-cardiac sS1 and wild type using a stopped-flow approach.
The binding affinity of R403Q human β-cardiac sS1 to actin is twofold lower than that of wild type
We measured the equilibrium dissociation constant (KA, table S1) for the strongly bound actin-sS1 (actin·sS1) state (Fig. 3, A and B). The R403Q human β-cardiac sS1 showed a twofold reduction (P < 0.05) in actin affinity (dissociation constant of 19.7 ± 6.2 nM at 25 mM ionic strength) of the actin·sS1 strongly bound state compared to wild type (dissociation constant of 10.0 ± 1.8 nM). Increasing the ionic strength to 100 mM to weaken the affinity and make the measurement more precise showed a similar twofold reduction (P < 0.05) in affinity (R403Q, 63.2 ± 16.7 nM and wild type, 23.4 ± 2.5 nM at 100 mM ionic strength) (table S1; see KA under “sS1-affinity for actin”).
Fig. 3. Rigor binding actin affinity of wild type and R403Q human β-cardiac sS1.
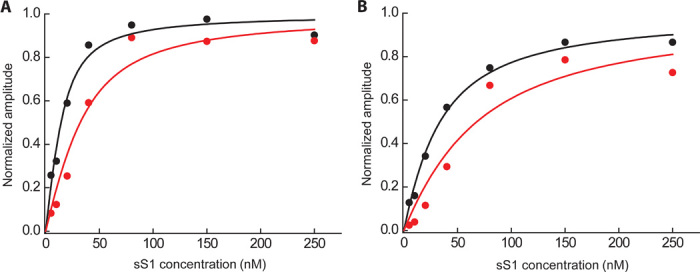
(A and B) Phalloidin-stabilized actin was incubated with various amounts of wild-type (black) and R403Q (red) human β-cardiac sS1 before mixing with 20 μM ATP at either (A) 25 mM KCl or (B) 100 mM KCl. The binding experiment at 25 mM ionic strength (A) was repeated at 100 mM ionic strength (B) to ensure that the changes observed were not due to nonspecific interactions. The pyrene fluorescence of the free actin is quenched upon binding of the myosin. Hence, the amount of actin that is tightly bound to myosin at equilibrium can be determined by observing the amplitude change of increase in fluorescence after rapidly mixing the equilibrium actin-sS1 mixture with ATP, which dissociates the actin·sS1 complex. This amplitude change as a function of myosin concentration, starting with a fixed concentration of actin, generates a curve that reflects the equilibrium dissociation constant of the binding (KA; see table S1). A single exponential function was fit to the raw fluorescence trace, and the amplitude from the fit was plotted as a function of different sS1 concentrations. One representative experimental curve is shown in (A) and (B). The best fit of the amplitude dependence on sS1 concentration was determined using the quadratic equation describing the binding isotherm as before (34, 36).
The twofold reduction in affinity (increase in KA; see table S1) is the largest change in all the rate and equilibrium constants we measured. We found no significant differences in the binding of either ATP or ADP to sS1 (table S1). The ADP binding to actin·sS1 and the actin binding to ADP·sS1 were also unaltered (table S1). For R403Q sS1, a significant increase (P < 0.05) was observed in the equilibrium association constant of ATP binding to actin·sS1 (K1′; see 1/K1′ in table S1), and a decrease was observed in the first-order rate constant of strong-to-weak binding transition of actin·sS1 (k+2′, P < 0.0001; see table S1), such that the effects nullified the overall second-order ATP-induced actin·sS1 dissociation rate constant (K1′k+2′; see table S1). Note, however, that as for the d and ts data presented above, it is very difficult to distinguish between these measured parameters with such small differences. Thus, all we can say is that there are no other major changes in the rate constants we measured aside from the significant change that we observed in the affinity of the strongly bound actin·sS1 rigor complex.
The results described so far suggest that the R403Q human β-cardiac sS1 has a small but significant increase in velocity compared to wild type and a small but significant decrease in intrinsic force. The unloaded velocity measurements (zero load regime) and the isometric intrinsic force measurements (zero velocity regime) do not inform about the power output of the system because the relevant power output of the heart is the product of load and velocity under the loads experienced during systolic contraction. We therefore used a loaded in vitro motility assay to obtain more information about the load-dependent velocity relationships involving these proteins and to assess an Fensemble metric.
The ensemble force metric of R403Q human β-cardiac sS1 is significantly lower than that of wild type
In the context of the heart, cardiac myosin generates power that propels blood throughout the body. Every time a heart beats, this process takes place under a variable external load created by the blood pressure in the circulatory system. It is necessary to mimic the conditions of the intact muscle and recognize myosin function under opposing load to understand collective myosin behavior. We used a loaded in vitro motility assay to study such ensemble-loaded behaviors (37). Briefly, various ratios of human β-cardiac sS1 and utrophin, an actin-binding protein that puts a load on the actin, were attached to the motility surface using a common SNAP-PDZ attachment system (see Unloaded and loaded in vitro motility under Materials and Methods) and the velocities of the gliding actin filaments were measured at different concentrations of utrophin [for details of the methodology, see Aksel et al. (37)]. At zero utrophin concentration, the velocity (which we consider the unloaded velocity) was the highest, which then progressively decayed as more utrophin was introduced into the motility chamber, finally slowing down the filaments to the point that we could no longer observe movement. Qualitatively, this decay in the velocity profile can be attributed to the increasing number of intermittently moving filaments at higher utrophin concentrations. Intuitively, this decay rate is related to the ensemble force generated by the ensemble of myosin molecules on the surface—the more force the myosins produce, the more utrophin is needed to slow down the actin filaments.
Figure 4A shows a representative curve of a loaded motility experiment. The mean velocity (MVEL, including intermittently moving filaments) profile of R403Q (red circles) started with a higher unloaded velocity but decayed more sharply than did that of wild type (black circles). This type of behavior leads to a crossover point, suggesting a lower relative Fensemble generated by the R403Q mutant, contributed to by the lower intrinsic force we measured with the laser trap.
Fig. 4. Loaded motility parameters for wild type and R403Q human β-cardiac sS1 with pure actin.
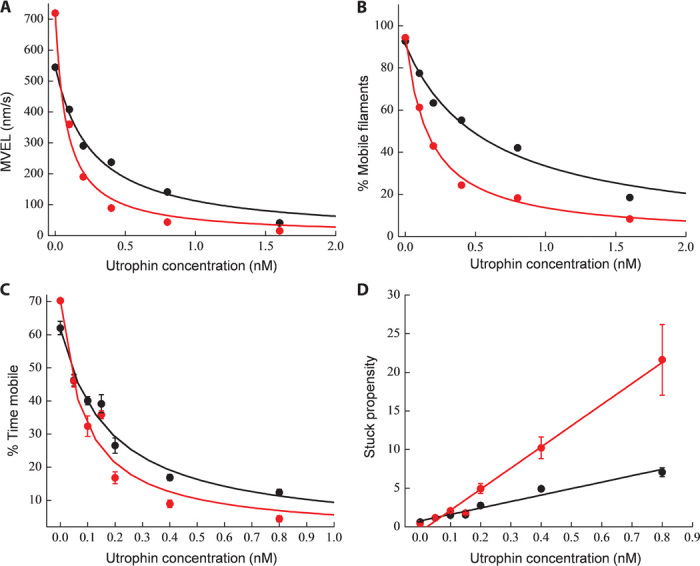
(A and B) Representative MVEL (A) and percentage of mobile filaments (B) as a function of utrophin concentration for wild-type (black circles) and R403Q (red circles) human β-cardiac sS1. Solid lines are exponential fits as a guide to the eye (black for wild type and red for R403Q). (C) Percent time mobile data for wild type (black circles) (n = 4 exp, 4 preps) and R403Q (red circles) (n = 4 exp, 4 preps). Mathematically, percent time mobile is defined as the product of percentage of mobile filaments (A) and MVEL (B) normalized by unloaded TOP5% velocity as derived by Aksel et al. (37). The solid line fits (black for wild type and red for R403Q) are according to the Stop model described by Aksel et al. (37). Errors reported are SEM. (D) Alternative representation of percent time mobile data as stuck propensity, which is equivalent to (100 − percent time mobile) divided by percent time mobile (black circles for wild type and red circles for R403Q), which, when modeled, gives a linear relationship with utrophin concentration. Errors reported are SEM. In such a formulism, a line with a higher slope corresponds to a lower Ks value and therefore an indicator of lower Fensemble. Unloaded stuck propensity is subtracted from each point in (D). The solid lines (black for wild type and red for R403Q) show the best linear equation fits to the data points. The steeper slope for R403Q as compared to wild type suggests it to be a lower-force generator.
The fraction of moving actin filaments at any given utrophin concentration is another strong indicator of the ensemble force—the more force the myosins produce, the more utrophin is needed to completely block movement of the actin filaments. Filaments with a mean velocity of less than one pixel length per second (80 nm/s) are classified as stuck. The time-weighted fraction of such stuck filaments is represented as %STUCK (see fig. S2, A and B, upper right panels). The percentage of mobile filaments is defined as (100 − %STUCK). Analysis of the mobile filament fraction showed that under unloaded conditions, ~95% of the filaments glide smoothly, but the number of gliding filaments decreases with increasing utrophin concentration. At nearly all utrophin concentrations, R403Q β-cardiac sS1 had a smaller percentage of mobile filaments (red circles, Fig. 4B) compared to wild type (black circles, Fig. 4B). As observed with the mean velocity profile, the percentage of mobile filaments also decreased faster as a function of utrophin concentration for R403Q human β-cardiac sS1 than for wild type. That is, R403Q needs less utrophin than wild type to stop the same number of filaments. This result also suggests that R403Q generates a lower Fensemble than does wild type.
Although the profiles of decreasing MVEL and decreasing percentage of mobile filaments as a function of utrophin concentration intuitively relate to the Fensemble, they do not capture the entire effect of utrophin for two reasons: (i) the effective density of utrophin on the surface is reduced by some small number of permanently stuck actin filaments that reduces available utrophin on the surface, leading to overestimation of MVEL, and (ii) mobile actin filaments do not always continuously move without interruption. They have periods of uninterrupted mobility and periods of “being stuck” when they encounter an utrophin molecule, leading to an overestimation of percentage of mobile filaments (37). To correct this, we used two parameters named “percent time mobile” (which is the percentage of mobile filaments corrected for the times spent as immobile) and “stuck propensity” (see the legend to Fig. 4), as derived by Aksel et al. (37). Briefly, percent time mobile is defined as the product of “percentage of mobile filaments” and “MVEL” normalized by unloaded “TOP5%” velocity. This correction gives a true estimate of the mobile time fraction of all filaments. A quick mathematical linearization provides an alternative representation of percent time mobile data as stuck propensity, which is equivalent to (100 − percent time mobile) divided by percent time mobile. Aksel et al. (37) modeled both percent time mobile and stuck propensity as a function of utrophin concentration and derived an expression that we interpret to be a force-dependent parameter, Ks. This parameter is inversely proportional to the actin-utrophin binding affinity and is related to Fensemble—the greater the force generated by the myosin, the lesser is the time of an actin-utrophin interaction and hence a higher Ks (37). Conversely, a weaker ensemble of myosin would not be able to disrupt the actin-utrophin interaction and would thereby lead to a lower Ks.
Combining data from four different experiments from four different sS1 preparations, we show in Fig. 4C a plot of percent time mobile for wild type β-cardiac sS1 (black circles) and R403Q (red circles) that recapitulates a very similar behavior as observed with the MVEL and percentage of mobile filaments. R403Q β-cardiac sS1 had a sharper decay profile than wild type. Fitting the Stop model described by Aksel et al. (37) to the experiments (solid lines, black for wild type and red for R403Q) gave a Ks of 0.10 ± 0.02 nM for wild type and 0.05 ± 0.02 nM for R403Q, consistent with a significant decrease (P < 0.01) in the Fensemble for R403Q human β-cardiac sS1 as compared to wild type. We want to emphasize that although we consider Ks to be a metric of ensemble force, given the complexity of the loaded assay, the percentage of change in Ks observed should not be construed as a similar percentage of change in ensemble force. Rather, we consider the data to indicate that this contractility parameter points to hypocontractility of R403Q sS1.
The same data presented in Fig. 4C are plotted in Fig. 4D using the stuck propensity parameter. The linear relationship (solid lines, black for wild type and red for R403Q) with utrophin concentration for R403Q β-cardiac sS1 showed a steeper dependence than did that for wild type, implying lower Fensemble generated by an ensemble of R403Q as compared to wild-type β-cardiac sS1.
In summary, with the pure actin system, R403Q human β-cardiac sS1 has a small but consistent increase in unloaded velocity and a small increase in ATPase kcat. The latter change would be consistent with an increase in the unloaded duty ratio, which would contribute to an increase in ensemble force production. However, this change is observed under unloaded conditions and may not reflect the situation in cardiac muscle, which is always under load. On the other hand, the lower intrinsic force measurements for the R403Q β-cardiac sS1 would contribute to a decrease in ensemble force production, and the loaded in vitro motility results are consistent with an overall loss of function as a result of the mutation.
Biomechanics of R403Q human β-cardiac sS1 with RTFs
In cardiac muscle, the regulated actin filaments (thin filaments) consist of actin with the Ca2+-regulatory components tropomyosin and the troponin complex [one copy each of troponin T (TnT), troponin I (TnI), and troponin C (TnC)] bound in a 7:1:1 ratio (the RTF). Myocardial force generation is the result of cyclic interactions between myosins and the RTFs in the sarcomere that are initiated upon the release of Ca2+ from the sarcoplasmic reticulum and binding of Ca2+ to TnC.
The loop of myosin that houses the HCM R403Q mutation is at the actin-binding interface of the myosin molecule (Fig. 1A), and a recent high-resolution electron microscopy study using Dictyostelium myosin and RTFs suggests that the R403 position of the human cardiac myosin may interact with tropomyosin and actin simultaneously (45). We therefore extended our study to include RTFs and found two interesting major differences from the results using pure actin alone.
In contrast to pure actin, the RTF-activated ATPase of R403Q human β-cardiac sS1 is lower than that of wild type, although the unloaded ADP release rate is similar
We measured the Ca2+ concentration dependence of the RTF-activated human β-cardiac sS1 ATPase activity of both R403Q mutant protein and wild type. In both cases, at pCa 8, there was less than 10% of the maximum activity observed at pCa 5 (P < 0.01), showing that nearly complete functional reconstitution was achieved (Fig. 5A). ATPase activity at different Ca2+ concentrations is plotted in Fig. 5A (black circles for wild type and red circles for R403Q). The pCa50 (pCa at half the maximal activity) for R403Q human β-cardiac sS1 (6.28 ± 0.05, n = 4 exp, 4 preps) was the same as that for wild type (6.25 ± 0.05, n = 4 exp, 4 preps), suggesting no change in the Ca2+ sensitivity. The maximal activity of R403Q human β-cardiac myosin was only ~50% (P < 0.01) of that for wild type. These pCa curves are carried out at subsaturating RTF concentrations (3 μM), and therefore, the decreased maximal ATPase activity for R403Q, seen in Fig. 5A, could be due to a change in either Km or kcat or both. Under similar pure actin conditions (~3 μM, in Fig. 2A), there was no difference in the activity of R403Q.
Fig. 5. Ca2+ sensitivity and RTF-activated ATPase activity of wild-type and R403Q human β-cardiac sS1.
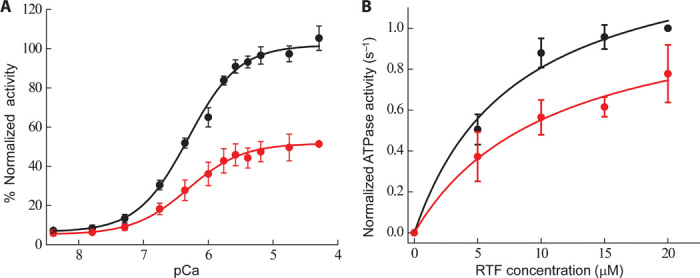
(A) Ca2+ sensitivity of RTF-activated ATPase activity of human β-cardiac sS1 wild type (black circles) and R403Q (red circles) at 30°C (n = 4 exp, 4 preps). For each sS1 preparation, average maximal wild-type activity was considered 100%, and all activities of the mutant R403Q sS1 were normalized to this value. The Hill equation was fit to the data. (B) RTF-activated ATPase activity of wild type (black circles) and R403Q (red circles) human β-cardiac sS1 at pCa 4 and 23°C. The average maximal wild-type activity was considered 1.0, and all activities of the mutant R403Q sS1 were normalized to this value. The data are fit (solid lines, black for wild type and red for R403Q) to the Michaelis-Menten equation to obtain the kcat and Km values. Errors reported are SEM.
To understand this difference in behavior, we compared the RTF-activated ATPase activity of R403Q β-cardiac sS1 with wild type. Generating such an ATPase curve is technically challenging, because of the viscosity and coaggregation of all the thin filament proteins at high concentration. We avoided this problem by performing the assay in mid-high concentrations of the thin filament proteins (up to 20 μM actin) at 30 mM KCl (instead of 5 mM KCl used for the actin-activated ATPase assay as in Fig. 2A). Under these conditions, consistent with our pCa data, R403Q sS1 showed a 30% decrease in the maximal ATPase activity, kcat (which corresponds to an increase in tc) (8.0 ± 0.5 s−1, n = 3 exp, 2 preps for wild type; 5.6 ± 0.5 s−1, n = 3 exp, 2 preps for R403Q) (Fig. 5B, black circles for wild type and red circles for R403Q). This result is opposite to what we found with pure actin, where R403Q sS1 showed a ~25% increase in the kcat.
The Km from the RTF-activated ATPase measurements was unaltered (8 ± 3 μM for wild type and 10 ± 4 μM for R403Q). As with pure actin, we also measured the ADP release rate constant with RTFs and found no significant difference between R403Q and wild type human β-cardiac sS1 (wild type, 67 ± 10 s−1; R403Q, 72 ± 10 s−1).
R403Q human β-cardiac sS1 and wild type generate similar RTF velocities in an unloaded in vitro motility assay
To assess the mechanical properties of the six-component regulated system, we first performed the unloaded in vitro motility experiments with the RTFs. These experiments were performed at 2.5 mM KCl, rather than at the 25 mM KCl condition used in the pure actin experiments above, because of diminished affinity of RTFs on an R403Q-attached surface at 25 mM KCl. Because of this change in ionic strength, for each experiment with the RTFs, a replicate experiment with pure actin at 2.5 mM KCl was performed as a control.
For the pure actin controls at 2.5 mM KCl, R403Q unloaded motility values were as follows: MVEL, 720 ± 40 nm/s; MVEL20, 800 ± 40 nm/s; TOP5%, 1100 ± 50 nm/s; and PLATEAU, 900 ± 50 nm/s (n = 15 exp, 4 preps). For wild-type human β-cardiac myosin, the values were as follows: MVEL, 540 ± 30 nm/s; MVEL20, 630 ± 30 nm/s; TOP5%, 960 ± 50 nm/s; and PLATEAU, 750 ± 30 nm/s (n = 15 exp, 4 preps). Thus, at this lower salt concentration than used in the experiments described in Fig. 1, we still observed a significant increase (~25% under these conditions, P < 0.01) in unloaded velocity using pure actin for the R403Q human β-cardiac sS1 compared to wild type (Fig. 6A). Strikingly, however, this increase was not seen when we used RTFs (Fig. 6B). With the RTFs, R403Q human β-cardiac sS1 unloaded motility values were as follows: MVEL, 1010 ± 30 nm/s; MVEL20, 1100 ± 30 nm/s; TOP5%, 1530 ± 4 nm/s; and PLATEAU, 1230 ± 30 nm/s (n = 15 exp, 4 preps). For wild type, the values were as follows: MVEL, 990 ± 40 nm/s; MVEL20, 1080 ± 40 nm/s; TOP5%, 1600 ± 50 nm/s; and PLATEAU, 1300 ± 30 nm/s (n = 15 exp, 4 preps).
Fig. 6. Unloaded motility of human β-cardiac sS1 with actin and RTFs.
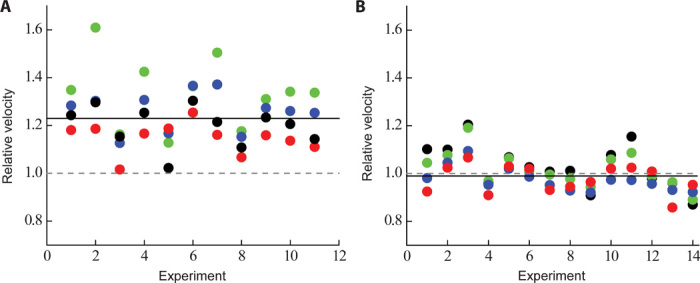
(A) Normalized motility parameters for human β-cardiac R403Q with respect to wild type. Experimental outcomes of MVEL (black), MVEL20 (red), TOP5% (blue), and PLATEAU (green) for different experiments with actin, performed at 23°C and 2.5 mM KCl, are shown for R403Q (n = 15 exp, 4 preps) with respect to wild type (n = 15 exp, 4 preps) (the wild type value is normalized to 1 and is denoted as a gray dashed line). The solid line is the overall mean of all the different normalized motility parameters and, in this case, denotes ~25% increase for R403Q human β-cardiac sS1. (B) Same as (A), except that the experiments were performed with RTFs. With RTFs, all the normalized motility parameters for R403Q (n = 15 exp, 4 preps) now show no difference as compared to wild type human β-cardiac sS1 (n = 15 exp, 4 preps) (compare the gray dashed line with the black solid line).
As before, we normalized the velocities of R403Q with the corresponding wild-type controls. Figure 6A shows that pure actin, at 2.5 mM KCl, had a ~25% higher velocity (predicting a decrease in ts) with R403Q human β-cardiac sS1 than with wild type, which is somewhat higher than what we observed for pure actin at 25 mM KCl (Fig. 1B). However, with RTFs, this difference was no longer seen (no change in ts) (Fig. 6B). This difference between pure actin and RTFs may result from the fact that the arginine in the 403 position of cardiac myosin probably interacts with the actin-tropomyosin interface differently from pure actin alone.
A ~30% increase in tc (see above) along with no change in ts for the R403Q myosin would contribute to a lower duty ratio (ts/tc), unlike what was observed with pure actin. Given these results, we used the loaded motility assay to reexamine the load-velocity relationship of wild-type and R403Q human β-cardiac sS1 now using RTFs.
With RTFs, the ensemble force metric of R403Q human β-cardiac sS1 is significantly lower than that of wild type
Figure 7A shows representative velocity profiles of R403Q (red circles) and wild-type sS1 (black circles) with increasing utrophin concentration. Both R403Q and wild type started with similar unloaded velocities, unlike with pure actin, which showed a higher velocity for R403Q sS1. Similar to the pure actin results, however, with RTFs, the R403Q human β-cardiac sS1 load-dependent velocity curve decayed faster than did wild type. This suggests that the ensemble force generated with RTFs by R403Q human β-cardiac sS1 is weaker than that by wild type. That is, R403Q sS1 is not as effective as wild type in overcoming the external load. Because the unloaded velocities of wild type and R403Q were nearly identical (Fig. 6B), in contrast to what we found for pure actin alone (Fig. 6A), there is no obvious crossover point in the case with RTFs (compare Figs. 4A and 7A). Similarly, the fraction of mobile filaments (Fig. 7B) decayed more rapidly for R403Q human β-cardiac sS1 (red circles) than for wild type (black circles), again suggesting that the R403Q human β-cardiac myosin is a weaker motor than wild type. Combining data from four different experiments using four different sS1 preparations, we show in Fig. 7C a plot of percent time mobile for wild type (black circles) and R403Q (red circles) that reiterates a very similar behavior as observed with the MVEL and percentage of mobile filaments. R403Q had a sharper decay profile than wild type. Fitting the model described by Aksel et al. (37) to the experiments gave a Ks of 1.0 ± 0.02 nM for wild-type human β-cardiac sS1 and 0.7 ± 0.07 nM for R403Q, consistent with a significant decrease in the ensemble force (Fensemble) for R403Q human β-cardiac sS1 as compared to wild type. The same data presented in Fig. 7C are plotted in Fig. 7D using the stuck propensity parameter. The linear relationship with utrophin concentration for R403Q showed a steeper dependence than wild type, implying lower Fensemble generated by an ensemble of R403Q as compared to wild-type β-cardiac sS1.
Fig. 7. Loaded motility parameters for wild-type and R403Q human β-cardiac sS1 with RTFs.
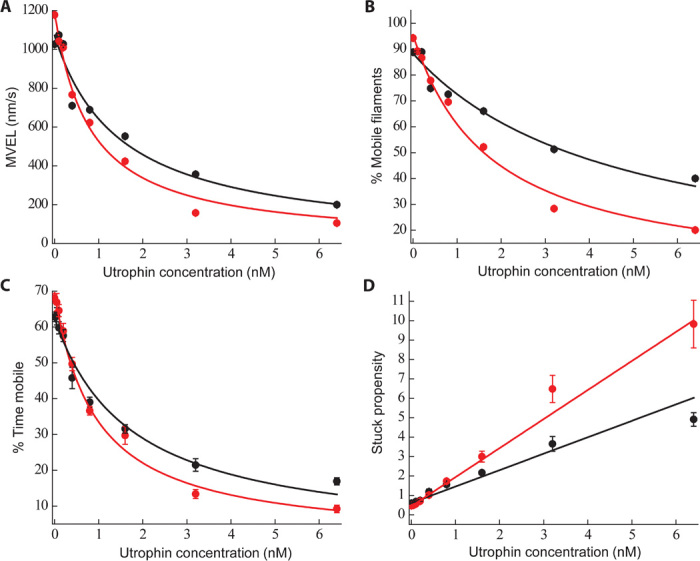
(A and B) Representative MVEL (A) and percentage of mobile filaments (B) as a function of utrophin concentration for wild type (black circles) and R403Q (red circles). Solid lines are exponential fits as a guide to the eye (black for wild type and red for R403Q). (C) Percent time mobile data for wild type (black circles) (n = 4 exp, 4 preps) and R403Q (red circles) (n = 4 exp, 4 preps). Percent time mobile is defined as the product of percentage of mobile filaments (A) and MVEL (B) normalized by unloaded TOP5% velocity. Errors reported are SEM. Fits are according to the Stop model described by Aksel et al. (37). (D) Alternative representation of percent time mobile data as stuck propensity, which is equivalent to (100 − percent time mobile) divided by percent time mobile (black circles for wild type and red circles for R403Q). Errors reported are SEM. Unloaded stuck propensity is subtracted from each point. The solid line (black for wild type and red for R403Q) shows the best linear equation fit to the data points. The steeper slope for R403Q as compared to wild type suggests it to be a lower-force generator.
Summarizing all the experiments with RTFs, we found that (i) the unloaded velocity of R403Q human β-cardiac sS1 is the same as wild type (no change in ts), and (ii) R403Q shows a small decrease in ATPase kcat (increase in tc), thereby predicting a lower unloaded duty ratio (ts/tc). These results suggest loss of function by the R403Q mutation, which is corroborated by a lower Fensemble, as predicted by the loaded in vitro motility assay.
DISCUSSION
The ensemble force produced by ventricular cardiac myosin determines the force of contraction of the heart. In the equation for ensemble force, Fensemble = f(ts/tc)Nt, the intrinsic force produced by each myosin head, f, is an intrinsic motor property. The intrinsic force of a motor is generally considered as f = kd, where d is the displacement due to the stroke and k is the overall spring constant of the motor. The overall spring constant is the summation of all individual atomic interactions in the molecule, each of which acts as a tiny spring. A ~15% decrease in intrinsic force of the human β-cardiac sS1 as a result of the R403Q mutation is significant and suggests that this mutation causes a slight local weakening of the spring.
Further contributing to a lower Fensemble for R403Q human β-cardiac sS1 is the apparently lower duty ratio (ts/tc), as judged by measurements of ts (from unloaded velocity) and tc (from ATPase) with the RTFs. Although the measurements of unloaded velocity (and hence ts) were performed under unloaded conditions, the load-dependent velocity curves are consistent with a conclusion of lower Fensemble for R403Q sS1. Another possible explanation for the increased sensitivity of the velocity to load in the loaded motility assay is that the R403Q sS1 ADP release rate is more sensitive to load than wild type. Thus, a faster decrease in the ADP release rate as a function of load (faster increase in ts) will lead to a steeper decrease in velocity as a function of load. Both of these parameters would manifest as a change in Ks—a proxy for contractility. For both mechanisms, a depressed load-dependent velocity curve, as in the case for R403Q, leads to lower velocity at higher loads, consistent with lower overall contractility. The conclusion from the results presented here is that with RTFs, the R403Q mutation in human β-cardiac sS1 results in changes in fundamental contractility parameters consistent with loss of molecular motor function.
In general, the results presented here on the effects of the HCM mutation R403Q on the f and ts/tc by the cardiac contractile system differ significantly from those of previous studies. As described in the Introduction, many earlier studies used the mouse ventricular α-cardiac myosin and one force parameter or the other was measured. Although not uniform in their conclusions, results most often suggested gain of function due to the R403Q mutation (20, 21, 24, 26, 29). The primary difference between our study and earlier ones is our use of purified, homogeneous human ventricular β-cardiac myosin. The differences we see compared to earlier studies are probably not surprising given, for example, the difference of more than 80 residues between the human β-cardiac myosin head domain and that of the mouse α-cardiac myosin and the differences in activity seen by Lowey et al. (29, 30) when studying this mutation in mouse α-cardiac versus β-cardiac myosin. Previous studies of human biopsy material have resulted in conflicting results (17, 18), which may reflect differences in quality and the relative ratios of wild type to mutant motor that are unavoidable with biopsy specimens. Furthermore, these studies were limited to unloaded in vitro motility and step size studies, because the amount of motor obtainable from biopsy material precludes doing many of the solution kinetic and ensemble force assays we performed with homogeneous recombinant human sS1. This emphasizes the importance of using the human reconstituted system for such characterizations. Finally, although a previous study of myofibrils from R403Q patients also showed decreased maximal force production (23), these samples came from patients with late-stage disease and likely contained numerous secondary changes in sarcomeric protein expression and posttranslational modifications (46) that would confound analysis as to the cause of the observed decrease in force production.
Clinical observations suggest that, even in the earliest stages of human HCM, the mutant heart manifests diastolic dysfunction by echocardiographic assessment, and the presence of supranormal ejection fraction together with diastolic dysfunction is highly predictive of the development of hypertrophy in preclinical patients with mutations in β-cardiac myosin (47–49). The force parameters f and ts/tc for the HCM mutant R453C measured in the human β-cardiac myosin are consistent with a gain of function for this mutation (35), in keeping with clinical observations in HCM patients (47–49). Hence, it is unexpected to find using the human system that these force parameters measured in R403Q myosin gave rise to loss of function. An important point is that patients with R403Q mutations are hypercontractile in the setting of hypertrophied hearts. The increased wall thickness results in decreased wall stress and smaller end systolic volumes and is often accompanied by mitral regurgitation, which enhances preload and decreases afterload. Thus, the enhanced systolic function in these patients may not reflect significant intrinsic myocardial hypercontractility. The key will be to study individuals with R403Q mutations but no hypertrophy (identified by screening family members of affected individuals) to see if they are hypercontractile in the absence of hypertrophy. This has been done for some β-cardiac myosin HCM mutations (48) but not for R403Q as far as we know.
These studies represent a first step in a long series of studies needed to understand how the typically heterozygous cardiomyopathy missense mutations contribute quantitatively to the sarcomere dysfunction and why the phenotype develops only with time, despite the presence of a congenital mechanical defect. Our thesis is that it is important to first lay the foundation of the effects of these mutations on the contractile machinery itself. Only then can one begin to add the necessary complexity to begin to unravel the consequences of these mutations. We believe that this study is the most comprehensive to date in beginning to lay the needed foundation.
Notably, for R403Q human β-cardiac sS1, the changes in the parameters f and ts/tc are relatively small, and another parameter in the Fensemble equation that could change is the total number of myosin motors that are functionally available to interact with actin filaments in the sarcomere (Nt). Changes in Nt could tip the balance between hypercontractility and hypocontractility in HCM mutations, as was recently suggested (50). This possibility needs to be addressed in future studies. Additionally, further studies of R403Q in the context of a double-headed myosin subfragment containing a phosphorylatable RLC are required to fully appreciate the effects of this mutation on the force- and motion-generating capability of the myosin molecule. Such studies will allow us to determine whether the loss of function in the R403Q sS1 translates when studied with an HMM (heavy meromyosin) construct that includes the subfragment 2 region of the myosin molecule. Such efforts are currently under way in our laboratory.
MATERIALS AND METHODS
Materials
Myosin constructs and protein expression
Human β-cardiac sS1: Wild-type and R403Q human β-cardiac sS1 were constructed and produced using a modified AdEasy Vector System (Qbiogene Inc.). The cloning, expression, and purification methodologies are described in detail elsewhere (35). Briefly, complementary DNA (cDNA) for MYH7 (human β-cardiac myosin) and MYL3 (human ventricular ELC) were purchased from Open Biosystems (Thermo). A truncated version of MYH7 (residues 1 to 808), corresponding to an sS1, followed by a flexible GSG (Gly-Ser-Gly) linker was made with either (i) a C-terminal enhanced green fluorescent protein (eGFP) linker (for ATPase and single-molecule optical trap measurements) or (ii) a C-terminal eight-residue (RGSIDTWV) PDZ binding peptide (for motility and ADP release experiments) (fig. S1A). Human ventricular ELC with an N-terminal FLAG tag (DYKDDDDK) and tobacco etch virus (TEV) protease site was coexpressed as previously described (fig. S1A), Sommese et al. (35). The purity of the expressed proteins is shown in fig. S1B. Purified fractions were stored in column buffer [10 mM imidazole, 4 mM MgCl2, 1 mM dithiothreitol (DTT), and ~200 mM NaCl] containing 10% sucrose and were flash-frozen before storage at −80°C. Frozen proteins exhibited similar ATPase and motility properties as compared to their fresh counterparts. Before any experiment, the myosin constructs were exchanged into the appropriate buffer conditions using Amicon centrifugal filter units (Millipore) followed by centrifugation at 350,000g for 10 min to remove any aggregated protein. For all other kinetic measurements, sS1 with a C-terminal 6× His-purification tag was used. This was prepared essentially as per the above protocol and was described by Deacon et al. and Bloemink et al. (34, 36).
Additional protein purification
Actin: Chicken and rabbit skeletal actin (which have identical amino acid sequences) and bovine α-cardiac actin were prepared from muscle acetone powders, using slight modifications to previously described protocols (51). In addition, we used purified bovine G-actin gifted to us by MyoKardia Inc. After preparation, actin was stored in its F form in 2 mM tris (pH 8), 50 mM KCl, 0.2 mM CaCl2, 2 mM ATP, 2 mM MgCl2, 1 mM DTT, and 0.02% sodium azide. Actin was cycled from G- to F-actin freshly for each assay and used only for up to a week before being recycled again. For optical trap measurements, biotin-labeled rabbit skeletal G-actin was purchased from Cytoskeleton. F-actin was stored at 4°C, and G-actin was frozen at −80°C for future use.
For kinetic experiments, rabbit skeletal muscle actin was labeled with pyrene similarly to previously published methods (52). Briefly, F-actin was dialyzed into 50 mM Hepes (pH 7.5), 100 mM KCl, and 0.2 mM CaCl2 at 4°C. F-actin was labeled with fivefold excess pyrene-maleimide overnight at 23°C in the dark. The reaction was quenched with 10-fold excess DTT, and undissolved pyrene was sedimented in a tabletop centrifuge at 11,000g for 15 min. F-actin was then cycled into G-actin at 4°C by dialysis in G buffer [2 mM tris (pH 8), 0.2 mM ATP, 0.2 mM CaCl2, and 1 mM DTT] for ~12 hours at 4°C. Any remaining F-actin was pelleted at 38,000g for 1 hour at 4°C. Pyrene-labeled G-actin was dialyzed into 20 mM Hepes (pH 7.5), 10 mM KCl, 2 mM MgCl2, and 1 mM DTT and used within a week before recycling the actin again from F to G. The concentration and percentage of labeling of G-actin were determined by measuring the Bradford assay for protein concentration and the extinction coefficient of pyrene at 344 nm (22,000 M−1 cm−1). Except for the ATPase experiments (which used chicken skeletal actin) and the transient kinetic experiments (which used rabbit skeletal actin), all other mechanochemical assays were performed with bovine cardiac actin.
Gelsolin: Full-length human gelsolin was expressed and purified on the basis of previous methods (53) as described elsewhere (35). Final fractions of gelsolin were dialyzed into buffer D (53), flash-frozen in liquid nitrogen, and stored at −80°C.
Tropomyosin: Tropomyosin was purified from bovine cardiac tissue according to the protocol of Smillie (54) with a few modifications as described by Sommese et al. (55). Purified tropomysin was dialyzed in 20 mM imidazole (pH 7.5), 300 mM KCl, and 1 mM DTT before flash-freezing and storing at −80°C.
Troponin: Human adult cardiac troponin subunit (TNNT2, TNNI3, and TNNC2) expression and purification were based on previously published methods (56, 57). For details, see Sommese et al. (55). TnT, TnI, and TnC were purified and stored in storage buffer [20 mM imidazole (pH 7.5), 1 M KCl, 1 mM MgCl2, and 1 mM DTT]. These proteins were then flash-frozen and stored at −80°C for future use. Troponin complexes were formed according to Szczesna et al. (58) with slight modifications. Components were mixed at a molar ratio of 1.3:1.3:1 (TnI/TnT/TnC) for 1 hour on ice. Complexes were then dialyzed at 4°C in six sequential steps into complex buffer [20 mM imidazole (pH 7.5), 2 mM MgCl2, and 1 mM DTT] containing 0.7, 0.5, 0.3, 0.1, and 0.01 M KCl twice, for 6 to 12 hours each. Complexes were flash-frozen in complex buffer before storing them at −80°C. The cDNAs for human adult cardiac TnI, TnC, and TnT in carbenicillin-selective pET-3d plasmids were obtained from J. Potter (University of Miami).
RTF complex formation: For all experiments involving the RTFs, RTFs were formed by mixing excess tropomyosin and troponin complex to actin on ice and then incubated overnight. The final molar ratio was 7:2:2 of actin/tropomyosin/troponin for all experiments except the pCa curves, which used a ratio of 7:2:4.
Utrophin: Mouse utrophin with an eight-residue (RGSIDTWV) PDZ binding peptide was expressed in bacterial cells as previously published, Aksel et al. (37). The purified protein was concentrated and dialyzed overnight against 150 mM NaCl, 25 mM tris, and 1 mM DTT (pH 8.0) at 4°C before flash-freezing in liquid nitrogen and storing at −80°C.
PDZ18: The SNAP-PDZ18 fusion construct was expressed in bacterial cells as described elsewhere, Aksel et al. (37). Eluted protein was concentrated and the buffer was exchanged to 150 mM NaCl and 25 mM tris (pH 8.0). The purified protein was flash-frozen in liquid nitrogen and stored at −80°C.
Methods
Kinetic measurements
ADP release rates at 23°C were measured in a stopped-flow instrument (HiTech SF-61DX2, TgK Scientific Ltd.) in 25 mM Hepes (pH 7.5), 25 mM KCl, 4 mM MgCl2, 0.2 mM CaCl2, and 1 mM DTT containing 50 μM ADP. At least six different traces were collected and fit individually. Pyrene-labeled actin was mixed with human β-cardiac sS1-ADP to a final concentration of 2 μM pyrene-actin, 2 μM sS1-ADP, and 50 μM ADP. After an incubation of at least 15 min, this mixture was rapidly mixed with buffer containing 2 mM ATP. ADP release rate measurements with RTFs were performed as above except pyrene-actin was replaced by pyrene-labeled RTFs, which were made by mixing pyrene-labeled bovine actin/bovine tropomyosin/human troponin complex at a 7:2:2 ratio (that is, 3.5 μM actin, 1 μM tropomyosin, and 1 μM troponin, respectively) and incubating overnight on ice.
All kinetic experiments at 20°C were performed as previously described, Bloemink et al. (36), apart from the applied buffer conditions. Measurements were performed in 25 mM KCl buffered at pH 7.0 with 20 mM Mops, 5 mM MgCl2, and 1 mM DTT, unless stated otherwise. From the kinetic measurements involving ATP binding to actin·sS1 (see table S1), we were able to estimate K1′k+2′ from the slope of the rate constant versus ATP concentration plot at low ATP concentrations (34, 36). At higher ATP concentrations, there was evidence of saturation of the observed rate constant, but the maximum value was too fast to measure reliably. We therefore repeated the measurement at a lower temperature (10°C) where the hyperbolic dependence of rate constants on ATP concentration was well defined, allowing both K1′ and k+2′ to be defined. For Fig. 3, the fluorescence amplitudes are plotted as described by Kurzawa and Geeves (59). Briefly, the amplitude of the fluorescence transient is defined as the difference between the signal at the start and end of the transient. The amplitude is then normalized by the maximum amplitude, which is observed at the highest concentration of sS1 used.
Actin-activated ATPase assay
For actin-activated ATPase, gelsolin was added to actin at a ratio of 1:1000. Gelsolin at this concentration was used to decrease the viscosity of the actin and thereby decrease pipetting error. Actin-activated ATPase assays were then performed as previously described using a colorimetric readout (60). Briefly, sS1 was diluted to a final concentration of 0.03 to 0.1 μM (with three to five times as much for the basal myosin ATPase control in the absence of actin to amplify the signal) with 2 mM ATP and actin at concentrations ranging from 0 to 100 μM. The final buffer conditions were 25 mM imidazole (pH 7.5), 5 mM KCl, 3 mM MgCl2, and 1 mM DTT. The reaction was performed at 23°C with shaking in a Thermo Scientific Multiskan GO, and four to five time points were taken for each concentration. The sS1 activity was linear over the time period of the assay, and hence, an ATP-regenerating system was not necessary. Basal activity (<0.2 s−1) was subtracted to get actin-activated ATPase activity. The Michaelis-Menten equation was fit to the data to determine the maximal activity (kcat) and the associated actin constant for myosin (Km) using OriginLab.
RTF-activated ATPase assay
To determine the steady-state Ca2+-dependent RTF ATPase rate for the human β-cardiac sS1 under different Ca2+ concentrations, we used an NADH (reduced form of nicotinamide adenine dinucleotide) coupled assay (61). The assay was performed with freshly prepared sS1 and RTFs at 30°C. The concentrations used were 3.5 μM actin, 1 μM tropomyosin, 2 μM troponin complex, and 0.3 to 0.5 μM sS1. The final buffer conditions were 25 mM imidazole (pH 7.5), 10 mM KCl, 3 mM MgCl2, 1 mM DTT, 2 mM ATP, the Ca2+ buffers [2 mM EGTA, 4 mM nitrilotriacetic acid (NTA), and varying concentrations of CaCl2], and the NADH-coupling system (61). The Ca2+ buffers with EGTA and NTA were calculated using the pCa calculator developed by Dweck et al. (62). All buffers were carefully adjusted to be pH 7.5 at 30°C in the final reactions, and the Hill equation was fit to the data. For each sS1 preparation, average maximal wild-type activity was considered 100%, and all activities of the mutant R403Q sS1 were normalized to this value.
A very similar procedure was adapted to measure the RTF-activated ATPase rate at pCa 4 for the human β-cardiac sS1 in a Thermo Scientific Multiskan GO plate reader. Briefly, sS1 was used at a final concentration of 0.1 to 0.3 μM, and RTFs were used at final concentrations ranging from 0 to 30 μM actin. The final buffer conditions were 25 mM imidazole (pH 7.5), 30 mM KCl, 3 mM MgCl2, 1 mM DTT, and 2 mM ATP. The reaction was performed at 23°C. The average maximal wild-type activity was considered to be 1, and all activities of the mutant R403Q sS1 were normalized to this value. The Michaelis-Menten equation was fit to the data to determine the maximal activity (kcat) and the associated actin apparent affinity constant (Km) for myosin using OriginLab.
Unloaded and loaded in vitro motility
The basic method followed our previously described motility assay, Kron et al. (41) with some modifications. Coverslips (VWR micro cover glass) were coated with a mixture of 0.2% nitrocellulose (Ernest Fullam Inc.) and 0.2% collodion (Electron Microscopy Sciences) dissolved in amyl acetate (Sigma) and air-dried for a few hours before use. A permanent double-sided tape (Scotch) was used to construct four channels in each slide (Gold Seal) in which four different experiments were performed.
Tenfold molar excess of F-actin was added in the presence of 4 mM ATP, incubated for 10 min, and sedimented at 350,000g for 20 min (dead-heading) to reduce the number of partially inactivated myosin heads in sS1 preparations. MgCl2 was added to 50 mM (to form F-actin para-crystals) and incubated for 10 min and the mixture was resedimented at 350,000g for 40 min to eliminate residual actin in the supernatant. The supernatant was collected and its concentration was measured using the Bradford reagent (Bio-Rad). A mock clean-up procedure containing actin and MgCl2 without sS1 was also performed simultaneously and was used as the blank for the concentration determination. The quality of the sS1 clean-up was assessed by the percentage of stuck filaments under unloaded conditions. We repeated the sS1 clean-up procedure until the percentage of stuck filaments dropped below 10%. Before any experiments, dead-headed sS1 was diluted in ABBSA {assay buffer [AB; 25 mM imidazole (pH 7.5), 25 mM KCl, 4 mM MgCl2, 1 mM EGTA, and 1 mM DTT] with bovine serum albumin (BSA; 1 mg/ml) diluted in AB}, unless otherwise stated.
For motility experiments, reagents were sequentially flown into the channels in the following order: (i) 10 μl of 4 μM SNAP-PDZ18 diluted in AB and incubated for 2 min; (ii) 20 μl of ABBSA to block the surface from nonspecific attachments and incubated for 2 min; (iii) 10 μl of a mixture of eight-residue (RGSIDTWV)–tagged human cardiac sS1 (~0.05 to 0.1 mg/ml for actin motility and 0.2 to 0.3 mg/ml for RTF motility) and utrophin at desired concentrations and incubated for 5 min (before mixing sS1 and utrophin, sS1 and utrophin dilutions were prepared in ABBSA; for unloaded motility, utrophin was skipped in this step); (iv) 20 μl of ABBSA to wash any unattached proteins; and (v) finally, 10 μl of the GO solution {1 to 5 nM tetramethylrhodamine (TMR)-phalloidin (Invitrogen)–labeled bovine actin, 2 mM ATP (Calbiochem), an oxygen-scavenging system [0.2% glucose, glucose oxidase (0.11 mg/ml; Calbiochem), and catalase (0.018 mg/ml; Calbiochem)], and an ATP regeneration system [1 mM phosphocreatine (Calbiochem) and creatine phosphokinase (0.1 mg/ml; Calbiochem)]} in ABBSA.
For RTF motility experiments, all the above procedures were kept the same except for the following modifications in the GO solution: (i) the final GO solution was made in ABBSA containing 2.5 mM KCl instead of the conventional 25 mM KCl used for actin-alone experiments; (ii) along with all its components, the GO solution had 100 nM excess tropomyosin and troponin complex; and (iii) 1 to 5 nM TMR-phalloidin–labeled bovine actin was replaced by 1 to 5 nM TMR-phalloidin–labeled RTF (made overnight by mixing a 7:2:2 ratio of TMR-phalloidin–labeled bovine actin/bovine tropomyosin/human troponin complex, that is, 3.5 μM actin, 1 μM tropomyosin, and 1 μM troponin in AB containing 100 mM KCl instead of 25 mM KCl).
For all experiments, movies were obtained at 23°C unless otherwise mentioned, at a frame rate of 1 Hz using a Nikon Ti-E inverted microscope with Andor iXon+EMCCD camera model DU885. All experiments were repeated with at least four different fresh protein preparations. At each condition, at least three different movies with a duration of 30 s were recorded. Experiments with wild-type and R403Q human β-cardiac sS1 were carried out in parallel for direct comparison, therefore minimizing any differences in different preparations. Filament tracking and analysis of movies, both under unloaded and loaded conditions, were performed by a recently published method, FAST (Fast Automated Spud Trekker), Aksel et al. (37).
Optical trap setup and single-molecule measurements
The dual-beam laser trap techniques used in our experiments are described in detail elsewhere, Sung et al. (42). Briefly, a 1064-nm laser light from a diode-pumped Nd:YAG laser (YLR-10-1064-LP), with a high numerical aperture (1.45) oil immersion objective lens (TIRF, CFI Plan Apo, 60×, Nikon), was used to produce a dual-beam optical trap. A back-focal plane interferometry method using an 845-nm laser beam (LU0845, Lumics) along with an infrared-enhanced quadrant photodiode detector (QPD; QP45-Q-HVSD, Pacific Silicon Sensor) was used to detect bead displacement (63).
All trap experiments were performed in a manner similar to those described for the in vitro motility assay. A nitrocellulose-coated sample chamber, coated with 1.5-μm-diameter silica beads as platforms, was prepared. Anti–green fluorescent protein antibody (~1 to 10 μg/ml) (Abcam) was flowed through the chamber, followed by blocking the surface with ABBSA. Dead-head cleaned human β-cardiac sS1 was then anchored to the antibody on the surface through its C-terminal eGFP tag. A typical concentration of ~10 to 100 pM was used to ensure single-molecule binding events. The chamber was washed with ABBSA. Finally, ABBSA containing ATP, TMR-phalloidin–labeled biotin-actin filaments, streptavidin-coated polystyrene beads (Polysciences), 1 mM phalloidin (Invitrogen), and the oxygen-scavenging and ATP regeneration systems described above was flowed through the chamber. The chamber was sealed with vacuum grease, and each such slide was used for up to 1 hour. All measurements were performed at 23°C.
Each of the two beams trapped a streptavidin-coated polystyrene bead (1 μm diameter) that was attached to the ends of a fluorescently labeled biotin-actin filament. The bead-actin-bead system, called an actin dumbbell, was stretched to remove compliance and was brought into close proximity to the surface of a silica bead pedestal. A typical trap stiffness of ~0.05 to 0.09 pN/nm was used for the step size measurements estimated from the equipartition theorem or the power spectral density analysis (64). Stroke size measurements were collected at low ATP concentration, typically 0.25 to 0.5 μM, to ensure long binding times. Gaussian distribution with a least-squares fitting was used to extract the mean stroke size from the individual molecules.
To measure the intrinsic force produced by a single myosin molecule, we used a stiffer trap of 0.2 to 0.3 pN/nm. This was produced by using a position feedback control in which the QPD output was fed into the driver of the acousto-optic deflector (IntraAction). A detailed isometric force measurement procedure is described elsewhere (65–67). Force measurements were carried out under similar conditions as stroke size measurements. Individual force events were collected from several single molecules. The number of total force events from individual molecules on average was 50 to 200. Such small binding events were not always sufficient to obtain good statistics. We therefore combined the force events of each preparation (typically two or three molecules in each preparation) to generate a force histogram. We then used a Gaussian distribution function with a cutoff to take into account the missing low force events. Additionally, we have always observed that for all of our sS1 constructs, the force distribution is accompanied with a long tail. The exact reason for this is unknown, but this phenomenon is commonly reported in the myosin single-molecule force literature (65, 68, 69). A possible explanation could be that each time a single myosin head attaches to the actin filament, it takes up a different orientation of attachment and hence changes the compliance of the system, giving rise to a long tail. Another possibility is that there are two different conformations of the myosin molecule, that is, open and closed nucleotide pocket, which can give two different force distributions. A third but very remote possibility is that two sS1 heads work simultaneously to give rise to higher forces, although this seems to be very unlikely given the working concentration of sS1 at which these experiments are performed. Nevertheless, these high-force events are a small fraction of total events, but they can easily alter the fitting results and hence the interpretation of the data. Instead, we chose a double Gaussian function to fit the force histogram data and have reported the first peak as the average force generated by the myosin molecule. This force histogram was further subjected for bootstrapping for estimating a variance of the mean force (see below).
Briefly, we used the simplest bootstrapping method, which involves taking the original single-molecule force data set (>1000 events), for each sample type, and resampling it to a size of 300 events (about one-third of the original events). The bootstrapped sample was taken from the original sample set using the “sampling with replacement” method. This process was repeated 1000 times, and for each of these bootstrap samples, we computed its mean (each of these are called bootstrap estimates) by fitting a double Gaussian function to the bootstrapped distribution (to account for the long tail in the force distributions in the higher force regimes) and taking the first Gaussian peak (see Fig. 2B). This gives a histogram of bootstrap estimates. A Gaussian function was then fit to this distribution to obtain a mean and variance of the bootstrapped means, and we report this mean as the intrinsic force generated by a single myosin molecule.
Acknowledgments
We thank S. Miller (University of California, San Francisco) for letting S.N. use her stopped-flow instrument. We thank A. Borrayo (Spudich laboratory) for his technical help of maintaining virus and cell stocks. We thank C. Vera for help with one of the sS1 constructs. Funding: This project was funded by NIH grants GM33289 and HL117138 to J.A.S., NIH grants GM29090 and HL117138 to L.A.L., and grants PG30200 from the British Heart Foundation and PG085309 from the Wellcome Trust to M.A.G. S.N. is a recipient of a Stanford Dean’s Postdoctoral Fellowship. Author contributions: Steady-state ATPase with actin and pCa ATPase were performed by R.F.S. with help from S.S. and K.M.R. Steady-state ATPase with RTFs, some of the kinetic experiments involving ADP release, unloaded and loaded motility experiments with pure actin and RTFs, and single-molecule laser trap experiments were performed by S.N. with help from S.S. and K.M.R. The bulk of the stopped-flow kinetic measurements were performed by Z.U. with help from A.C. and S.L. S.N. and J.A.S. wrote the manuscript. S.N., R.F.S., Z.U., L.A.L., M.A.G., K.M.R., and J.A.S. contributed to data analysis/interpretation and editing of the final manuscript. A.C., S.L., and S.S. contributed to new reagents. Competing interests: J.A.S. is a founder of and owns shares in Cytokinetics Inc. and MyoKardia Inc., biotech companies that are developing therapeutics that target the sarcomere. L.A.L. is a founder of and owns shares in MyoKardia Inc., a biotech company that is developing therapeutics that target the sarcomere. Data and materials availability: A complete version of the FAST program (37) for motility analysis is available for downloading on our Web site (http://spudlab.stanford.edu/FAST.html).
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/1/9/e1500511/DC1
Fig. S1. Schematic of the different constructs of the human β-cardiac sS1 used in our experiments and relative purity of the wild type and R403Q protein preparations.
Fig. S2. Representative experimental traces of wild-type and R403Q sS1 unloaded motility.
Fig. S3. Single-molecule stroke size measurements of wild type and R403Q human β-cardiac sS1.
Table S1. Rate constants and equilibrium constants for the various kinetic steps of wild type and R403Q human β-cardiac sS1.
Reaction schemes
Scheme 1. Kinetic schemes for interaction of myosin only with nucleotide.
Scheme 2. Kinetic schemes for interaction of myosin with actin and nucleotide.
Scheme 3. A combined equilibrium scheme for myosin, actin, and ADP binding to each other.
Reference (70)
REFERENCES AND NOTES
- 1.Maron B. J., Gardin J. M., Flack J. M., Gidding S. S., Kurosaki T. T., Bild D. E., Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA Study. Coronary Artery Risk Development in (Young) Adults. Circulation 92, 785–789 (1995). [DOI] [PubMed] [Google Scholar]
- 2.Semsarian C., Ingles J., Maron M. S., Maron B. J., New perspectives on the prevalence of hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 65, 1249–1254 (2015). [DOI] [PubMed] [Google Scholar]
- 3.C. E. Seidman, J. G. Seidman, in The Metabolic and Molecular Bases of Inherited Disease, C. R. Scriver, A. L. Beaudet, D. Valle, W. S. Sly, B. Childs, K. W. Kinzler, B. Vogelstein, Eds. (McGraw-Hill, New York, 2000), pp. 5433–5452. [Google Scholar]
- 4.Harvey P. A., Leinwand L. A., The cell biology of disease: Cellular mechanisms of cardiomyopathy. J. Cell Biol. 194, 355–365 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Watkins H., Ashrafian H., McKenna W. J., The genetics of hypertrophic cardiomyopathy: Teare redux. Heart 94, 1264–1268 (2008). [DOI] [PubMed] [Google Scholar]
- 6.Watkins H., Thierfelder L., Hwang D. S., McKenna W., Seidman J. G., Seidman C. E., Sporadic hypertrophic cardiomyopathy due to de novo myosin mutations. J. Clin. Invest. 90, 1666–1671 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alfares A. A., Kelly M. A., McDermott G., Funke B. H., Lebo M. S., Baxter S. B., Shen J., McLaughlin H. M., Clark E. H., Babb L. J., Cox S. W., DePalma S. R., Ho C. Y., Seidman J. G., Seidman C. E., Rehm H. L., Results of clinical genetic testing of 2,912 probands with hypertrophic cardiomyopathy: Expanded panels offer limited additional sensitivity. Genet. Med. 10.1038/gim.2014.205 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Richard P., Charron P., Carrier L., Ledeuil C., Cheav T., Pichereau C., Benaiche A., Isnard R., Dubourg O., Burban M., Gueffet J.-P., Millaire A., Desnos M., Schwartz K., Hainque B., Komajda M.; EUROGENE Heart Failure Project , Hypertrophic cardiomyopathy: Distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation 107, 2227–2232 (2003). [DOI] [PubMed] [Google Scholar]
- 9.Geisterfer-Lowrance A. A. T., Kass S., Tanigawa G., Vosberg H.-P., McKenna W., Seidman C. E., Seidman J. G., A molecular basis for familial hypertrophic cardiomyopathy: A β cardiac myosin heavy chain gene missense mutation. Cell 62, 999–1006 (1990). [DOI] [PubMed] [Google Scholar]
- 10.Colegrave M., Peckham M., Structural implications of β-cardiac myosin heavy chain mutations in human disease. Anat. Rec. 297, 1670–1680 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Moore J. R., Leinwand L., Warshaw D. M., Understanding cardiomyopathy phenotypes based on the functional impact of mutations in the myosin motor. Circ. Res. 111, 375–385 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lowey S., Functional consequences of mutations in the myosin heavy chain at sites implicated in familial hypertrophic cardiomyopathy. Trends Cardiovasc. Med. 12, 348–354 (2002). [DOI] [PubMed] [Google Scholar]
- 13.Tardiff J. C., Sarcomeric proteins and familial hypertrophic cardiomyopathy: Linking mutations in structural proteins to complex cardiovascular phenotypes. Heart Fail. Rev. 10, 237–248 (2005). [DOI] [PubMed] [Google Scholar]
- 14.Frey N., Luedde M., Katus H. A., Mechanisms of disease: Hypertrophic cardiomyopathy. Nat. Rev. Cardiol. 9, 91–100 (2012). [DOI] [PubMed] [Google Scholar]
- 15.Maron B. J., Ommen S. R., Semsarian C., Spirito P., Olivotto I., Maron M. S., Hypertrophic cardiomyopathy: Present and future, with translation into contemporary cardiovascular medicine. J. Am. Coll. Cardiol. 64, 83–99 (2014). [DOI] [PubMed] [Google Scholar]
- 16.Spudich J. A., Hypertrophic and dilated cardiomyopathy: Four decades of basic research on muscle lead to potential therapeutic approaches to these devastating genetic diseases. Biophys. J. 106, 1236–1249 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cuda G., Fananapazir L., Epstein N. D., Sellers J. R., The in vitro motility activity of β-cardiac myosin depends on the nature of the β-myosin heavy chain gene mutation in hypertrophic cardiomyopathy. J. Muscle Res. Cell Motil. 18, 275–283 (1997). [DOI] [PubMed] [Google Scholar]
- 18.Palmiter K. A., Tyska M. J., Haeberle J. R., Alpert N. R., Fananapazir L., Warshaw D. M., R403Q and L908V mutant β-cardiac myosin from patients with familial hypertrophic cardiomyopathy exhibit enhanced mechanical performance at the single molecule level. J. Muscle Res. Cell Motil. 21, 609–620 (2000). [DOI] [PubMed] [Google Scholar]
- 19.Helms A. S., Davis F. M., Coleman D., Bartolone S. N., Glazier A. A., Pagani F., Yob J. M., Sadayappan S., Pedersen E., Lyons R., Westfall M. V., Jones R., Russell M. W., Day S. M., Sarcomere mutation-specific expression patterns in human hypertrophic cardiomyopathy. Circ. Cardiovasc. Genet. 7, 434–443 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tyska M. J., Hayes E., Giewat M., Seidman C. E., Seidman J. G., Warshaw D. M., Single-molecule mechanics of R403Q cardiac myosin isolated from the mouse model of familial hypertrophic cardiomyopathy. Circ. Res. 86, 737–744 (2000). [DOI] [PubMed] [Google Scholar]
- 21.Debold E. P., Schmitt J. P., Patlak J. B., Beck S. E., Moore J. R., Seidman J. G., Seidman C., Warshaw D. M., Hypertrophic and dilated cardiomyopathy mutations differentially affect the molecular force generation of mouse α-cardiac myosin in the laser trap assay. Am. J. Physiol. Heart Circ. Physiol. 293, H284–H291 (2007). [DOI] [PubMed] [Google Scholar]
- 22.Lankford E. B., Epstein N. D., Fananapazir L., Sweeney H. L., Abnormal contractile properties of muscle fibers expressing β-myosin heavy chain gene mutations in patients with hypertrophic cardiomyopathy. J. Clin. Invest. 95, 1409–1414 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Belus A., Piroddi N., Scellini B., Tesi C., D’Amati G., Girolami F., Yacoub M., Cecchi F., Olivotto I., Poggesi C., The familial hypertrophic cardiomyopathy-associated myosin mutation R403Q accelerates tension generation and relaxation of human cardiac myofibrils. J. Physiol. 586, 3639–3644 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Palmer B. M., Fishbaugher D. E., Schmitt J. P., Wang Y., Alpert N. R., Seidman C. E., Seidman J. G., VanBuren P., Maughan D. W., Differential cross-bridge kinetics of FHC myosin mutations R403Q and R453C in heterozygous mouse myocardium. Am. J. Physiol. Heart Circ. Physiol. 287, H91–H99 (2004). [DOI] [PubMed] [Google Scholar]
- 25.Palmer B. M., Wang Y., Teekakirikul P., Hinson J. T., Fatkin D., Strouse S., VanBuren P., Seidman C. E., Seidman J. G., Maughan D. W., Myofilament mechanical performance is enhanced by R403Q myosin in mouse myocardium independent of sex. Am. J. Physiol. Heart Circ. Physiol. 294, H1939–H1947 (2008). [DOI] [PubMed] [Google Scholar]
- 26.Blanchard E., Seidman C., Seidman J. G., LeWinter M., Maughan D., Altered crossbridge kinetics in the αMHC403/+ mouse model of familial hypertrophic cardiomyopathy. Circ. Res. 84, 475–483 (1999). [DOI] [PubMed] [Google Scholar]
- 27.Yamashita H., Tyska M. J., Warshaw D. M., Lowey S., Trybus K. M., Functional consequences of mutations in the smooth muscle myosin heavy chain at sites implicated in familial hypertrophic cardiomyopathy. J. Biol. Chem. 275, 28045–28052 (2000). [DOI] [PubMed] [Google Scholar]
- 28.Fujita H., Sugiura S., Momomura S., Omata M., Sugi H., Sutoh K., Characterization of mutant myosins of Dictyostelium discoideum equivalent to human familial hypertrophic cardiomyopathy mutants. Molecular force level of mutant myosins may have a prognostic implication. J. Clin. Invest. 99, 1010–1015 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lowey S., Lesko L. M., Rovner A. S., Hodges A. R., White S. L., Low R. B., Rincon M., Gulick J., Robbins J., Functional effects of the hypertrophic cardiomyopathy R403Q mutation are different in an α- or β-myosin heavy chain backbone. J. Biol. Chem. 283, 20579–20589 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lowey S., Bretton V., Gulick J., Robbins J., Trybus K. M., Transgenic mouse α- and β-cardiac myosins containing the R403Q mutation show isoform-dependent transient kinetic differences. J. Biol. Chem. 288, 14780–14787 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu L., Srikakulam R., Winkelmann D. A., Unc45 activates Hsp90-dependent folding of the myosin motor domain. J. Biol. Chem. 283, 13185–13193 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Srikakulam R., Winkelmann D. A., Chaperone-mediated folding and assembly of myosin in striated muscle. J. Cell Sci. 117, 641–652 (2004). [DOI] [PubMed] [Google Scholar]
- 33.Resnicow D. I., Deacon J. C., Warrick H. M., Spudich J. A., Leinwand L. A., Functional diversity among a family of human skeletal muscle myosin motors. Proc. Natl. Acad. Sci. U.S.A. 107, 1053–1058 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deacon J. C., Bloemink M. J., Rezavandi H., Geeves M. A., Leinwand L. A., Erratum to: Identification of functional differences between recombinant human α and β cardiac myosin motors. Cell. Mol. Life Sci. 69, 4239–4255 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sommese R. F., Sung J., Nag S., Sutton S., Deacon J. C., Choe E., Leinwand L. A., Ruppel K., Spudich J. A., Molecular consequences of the R453C hypertrophic cardiomyopathy mutation on human β-cardiac myosin motor function. Proc. Natl. Acad. Sci. U.S.A. 110, 12607–12612 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bloemink M., Deacon J., Langer S., Vera C., Combs A., Leinwand L., Geeves M. A., The hypertrophic cardiomyopathy myosin mutation R453C alters ATP binding and hydrolysis of human cardiac β-myosin. J. Biol. Chem. 289, 5158–5167 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aksel T., Choe Yu E., Sutton S., Ruppel K. M., Spudich J. A., Ensemble force changes that result from human cardiac myosin mutations and a small-molecule effector. Cell Rep. 11, 910–920 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Geeves M. A., Holmes K. C., Structural mechanism of muscle contraction. Annu. Rev. Biochem. 68, 687–728 (1999). [DOI] [PubMed] [Google Scholar]
- 39.Geeves M. A., Holmes K. C., The molecular mechanism of muscle contraction. Adv. Protein Chem. 71, 161–193 (2005). [DOI] [PubMed] [Google Scholar]
- 40.Holmes K. C., Geeves M. A., The structural basis of muscle contraction. Philos. Trans. R. Soc. Lond. B Biol. Sci. 355, 419–431 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kron S. J., Toyoshima Y. Y., Uyeda T. Q. P., Spudich J. A., Assays for actin sliding movement over myosin-coated surfaces. Methods Enzymol. 196, 399–416 (1991). [DOI] [PubMed] [Google Scholar]
- 42.Sung J., Sivaramakrishnan S., Dunn A. R., Spudich J. A., Single-molecule dual-beam optical trap analysis of protein structure and function. Methods Enzymol. 475, 321–375 (2010). [DOI] [PubMed] [Google Scholar]
- 43.Cooke R., Actomyosin interaction in striated muscle. Physiol. Rev. 77, 671–697 (1997). [DOI] [PubMed] [Google Scholar]
- 44.Karatzaferi C., Chinn M. K., Cooke R., The force exerted by a muscle cross-bridge depends directly on the strength of the actomyosin bond. Biophys. J. 87, 2532–2544 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Behrmann E., Müller M., Penczek P. A., Mannherz H. G., Manstein D. J., Raunser S., Structure of the rigor actin-tropomyosin-myosin complex. Cell 150, 327–338 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sequeira V., Wijnker P. J. M., Nijenkamp L. L. A. M., Kuster D. W. D., Najafi A., Witjas-Paalberends E. R., Regan J. A., Boontje N., ten Cate F. J., Germans T., Carrier L., Sadayappan S., van Slegtenhorst M. A., Zaremba R., Foster D. B., Murphy A. M., Poggesi C., dos Remedios C., Stienen G. J. M., Ho C. Y., Michels M., van der Velden J., Perturbed length-dependent activation in human hypertrophic cardiomyopathy with missense sarcomeric gene mutations. Circ. Res. 112, 1491–1505 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.J. Wynne, E. Braunwald, in Braunwald’s Heart Disease: A Textbook of Cardiovascular Medicine (W.B. Saunders, Philadelphia, PA, 1992), pp. 1394–1450. [Google Scholar]
- 48.Ho C. Y., Sweitzer N. K., McDonough B., Maron B. J., Casey S. A., Seidman J. G., Seidman C. E., Solomon S. D., Assessment of diastolic function with Doppler tissue imaging to predict genotype in preclinical hypertrophic cardiomyopathy. Circulation 105, 2992–2997 (2002). [DOI] [PubMed] [Google Scholar]
- 49.Olivotto I., Maron B. J., Appelbaum E., Harrigan C. J., Salton C., Gibson C. M., Udelson J. E., O’Donnell C., Lesser J. R., Manning W. J., Maron M. S., Spectrum and clinical significance of systolic function and myocardial fibrosis assessed by cardiovascular magnetic resonance in hypertrophic cardiomyopathy. Am. J. Cardiol. 106, 261–267 (2010). [DOI] [PubMed] [Google Scholar]
- 50.Spudich J. A., The myosin mesa and a possible unifying hypothesis for the molecular basis of human hypertrophic cardiomyopathy. Biochem. Soc. Trans. 43, 64–72 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pardee J. D., Spudich J. A., Purification of muscle actin. Methods Enzymol. 85, 164–181 (1982). [DOI] [PubMed] [Google Scholar]
- 52.Kouyama T., Mihashi K., Fluorimetry study of N-(1-pyrenyl)iodoacetamide-labelled F-actin: Local structural change of actin protomer both on polymerization and on binding of heavy meromyosin. Eur. J. Biochem. 114, 33–38 (1981). [PubMed] [Google Scholar]
- 53.Way M., Gooch J., Pope B., Weeds A. G., Expression of human plasma gelsolin in Escherichia coli and dissection of actin binding sites by segmental deletion mutagenesis. J. Cell Biol. 109, 593–605 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smillie L. B., Preparation and identification of α- and β-tropomyosins. Methods Enzymol. 85, 234–241 (1982). [DOI] [PubMed] [Google Scholar]
- 55.Sommese R. F., Nag S., Sutton S., Miller S. M., Spudich J. A., Ruppel K. M., Effects of troponin T cardiomyopathy mutations on the calcium sensitivity of the regulated thin filament and the actomyosin cross-bridge kinetics of human β-cardiac myosin. PLOS One 8, e83403 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sheng Z., Pan B. S., Miller T. E., Potter J. D., Isolation, expression, and mutation of a rabbit skeletal muscle cDNA clone for troponin I. The role of the NH2 terminus of fast skeletal muscle troponin I in its biological activity. J. Biol. Chem. 267, 25407–25413 (1992). [PubMed] [Google Scholar]
- 57.Pan B. S., Potter J. D., Two genetically expressed troponin T fragments representing α and β isoforms exhibit functional differences. J. Biol. Chem. 267, 23052–23056 (1992). [PubMed] [Google Scholar]
- 58.Szczesna D., Guzman G., Miller T., Zhao J., Farokhi K., Ellemberger H., Potter J. D., The role of the four Ca2+ binding sites of troponin C in the regulation of skeletal muscle contraction. J. Biol. Chem. 271, 8381–8386 (1996). [DOI] [PubMed] [Google Scholar]
- 59.Kurzawa S. E., Geeves M. A., A novel stopped-flow method for measuring the affinity of actin for myosin head fragments using microgram quantities of protein. J. Muscle Res. Cell Motil. 17, 669–676 (1996). [DOI] [PubMed] [Google Scholar]
- 60.Trybus K. M., Biochemical studies of myosin. Methods 22, 327–335 (2000). [DOI] [PubMed] [Google Scholar]
- 61.De La Cruz E. M., Ostap E. M., Kinetic and equilibrium analysis of the myosin ATPase. Methods Enzymol. 455, 157–192 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dweck D., Reyes-Alfonso A. Jr, Potter J. D., Expanding the range of free calcium regulation in biological solutions. Anal. Biochem. 347, 303–315 (2005). [DOI] [PubMed] [Google Scholar]
- 63.Allersma M. W., Gittes F., deCastro M. J., Stewart R. J., Schmidt C. F., Two-dimensional tracking of ncd motility by back focal plane interferometry. Biophys. J. 74, 1074–1085 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Berg-Sørensen K., Flyvbjerg H., Power spectrum analysis for optical tweezers. Rev. Sci. Instrum. 75, 594–612 (2004). [Google Scholar]
- 65.Finer J. T., Simmons R. M., Spudich J. A., Single myosin molecule mechanics: Piconewton forces and nanometer steps. Nature 368, 113–119 (1994). [DOI] [PubMed] [Google Scholar]
- 66.Simmons R. M., Finer J. T., Chu S., Spudich J. A., Quantitative measurements of force and displacement using an optical trap. Biophys. J. 70, 1813–1822 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Takagi Y., Homsher E. E., Goldman Y. E., Shuman H., Force generation in single conventional actomyosin complexes under high dynamic load. Biophys. J. 90, 1295–1307 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Guilford W. H., Dupuis D. E., Kennedy G., Wu J., Patlak J. B., Warshaw D. M., Smooth muscle and skeletal muscle myosins produce similar unitary forces and displacements in the laser trap. Biophys. J. 72, 1006–1021 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Molloy J. E., Burns J. E., Kendrick-Jones J., Tregear R. T., White D. C. S., Movement and force produced by a single myosin head. Nature 378, 209–212 (1995). [DOI] [PubMed] [Google Scholar]
- 70.Huang J., Nagy S. S., Koide A., Rock R. S., Koide S., A peptide tag system for facile purification and single-molecule immobilization. Biochemistry 48, 11834–11836 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/1/9/e1500511/DC1
Fig. S1. Schematic of the different constructs of the human β-cardiac sS1 used in our experiments and relative purity of the wild type and R403Q protein preparations.
Fig. S2. Representative experimental traces of wild-type and R403Q sS1 unloaded motility.
Fig. S3. Single-molecule stroke size measurements of wild type and R403Q human β-cardiac sS1.
Table S1. Rate constants and equilibrium constants for the various kinetic steps of wild type and R403Q human β-cardiac sS1.
Reaction schemes
Scheme 1. Kinetic schemes for interaction of myosin only with nucleotide.
Scheme 2. Kinetic schemes for interaction of myosin with actin and nucleotide.
Scheme 3. A combined equilibrium scheme for myosin, actin, and ADP binding to each other.
Reference (70)