

A global metagenome assessment reveals a low risk of methylmercury production in humans and a high potential in Arctic permafrost.
Keywords: Mercury methylation, microbiology, mercury, methylmercury, hgcAB
Abstract
Mercury (Hg) methylation produces the neurotoxic, highly bioaccumulative methylmercury (MeHg). The highly conserved nature of the recently identified Hg methylation genes hgcAB provides a foundation for broadly evaluating spatial and niche-specific patterns of microbial Hg methylation potential in nature. We queried hgcAB diversity and distribution in >3500 publicly available microbial metagenomes, encompassing a broad range of environments and generating a new global view of Hg methylation potential. The hgcAB genes were found in nearly all anaerobic (but not aerobic) environments, including oxygenated layers of the open ocean. Critically, hgcAB was effectively absent in ~1500 human and mammalian microbiomes, suggesting a low risk of endogenous MeHg production. New potential methylation habitats were identified, including invertebrate digestive tracts, thawing permafrost soils, coastal “dead zones,” soils, sediments, and extreme environments, suggesting multiple routes for MeHg entry into food webs. Several new taxonomic groups capable of methylating Hg emerged, including lineages having no cultured representatives. Phylogenetic analysis points to an evolutionary relationship between hgcA and genes encoding corrinoid iron-sulfur proteins functioning in the ancient Wood-Ljungdahl carbon fixation pathway, suggesting that methanogenic Archaea may have been the first to perform these biotransformations.
INTRODUCTION
Mercury (Hg) is a pervasive global contaminant of concern that affects human and ecosystem health through the production and bioaccumulation of the neurotoxic methylmercury (MeHg) (1). Although concerted international efforts have attempted to curb Hg release into the environment (2), between 3 and 15% of women of child-bearing age in the United States and ~25% of women of child-bearing age in Korea and many other countries exceed the U.S. Environmental Protection Agency MeHg reference dose of 0.1 μg/kg body weight/day (1, 3, 4). Production of MeHg is a microbial process that is initially associated with sulfate-reducing bacteria and later associated with Fe(III)-reducing bacteria (5, 6). The identification of two genes (hgcAB) essential for Hg methylation (7) led to the discovery of Hg methylation capability in a much more diverse group of microorganisms, including syntrophic Deltaproteobacteria, Firmicutes, and Archaea (5). The predictability of Hg methylation based on the presence of hgcAB (5) has provided the foundation for studying the genetic, evolutionary, and biochemical aspects of Hg methylation. We now know that cultured microorganisms harboring hgcAB originate from a wide range of habitats and phylogenetic positions in the tree of life and that, consequently, Hg methylation is more phylogenetically diverse and prevalent than previously realized (5).
The gene hgcA encodes a homolog of corrinoid iron-sulfur proteins (CFeSP; PFam3599) involved in the reductive acetyl coenzyme A (acetyl-CoA) [Wood-Ljungdahl (WL)] carbon fixation pathway. It has a cobalamin binding domain and a transmembrane domain with no close similarity to other proteins. HgcB is almost always present next to hgcA and encodes a small iron-sulfur cluster-containing ferredoxin (PFam13237) that is distinct from those involved in the WL pathway. Although both genes are essential for methylation, neither is required for survival in Desulfovibrio desulfuricans ND132 or Geobacter sulfurreducens (7). The gene pair hgcAB appears to be rare but is widely dispersed among sequenced Bacteria and Archaea (7) as hgcAB orthologs have only been identified in ~100 microbial genomes among ~7000 sequenced species. Because they are not widely distributed among microorganisms (7) and no alternate physiological function has yet been identified, our understanding of these proteins is incomplete.
With the exception of a few syntrophic and fermentative Firmicutes (5), Hg methylators are heterotrophic bacteria that use sulfate, iron, and carbon dioxide (CO2) as terminal electron acceptors. Phylogenetic analysis of available HgcAB amino acid sequences grouped methylators into five clades: three in Deltaproteobacteria [sulfate-reducing Desulfovibrionales, Fe(III)-reducing Desulfuromonadales, and syntrophic Syntrophobacterales], a disparate group of Firmicutes, and a narrow group of Archaea, the methylotrophic Methanomicrobia (5). We have strong evidence that the gene pair endows microorganisms with the ability to produce MeHg. Every hgcAB+ microorganism tested is capable of methylating Hg in culture (5). Of about 100 cultured hgcAB+ microorganisms known as of this writing, about 50 have been explicitly tested for methylation ability in culture, including several Firmicutes, syntrophs, and methanogens (5, 8–11). We have also identified sporadic hgcAB in the genomes of a few species of Dehalococcoides (Chloroflexi), in Chrysiogenetes, and in members of the uncultured candidate phyla OP9 and ACD79, possibly resulting from horizontal gene transfer. However, these organisms have not been tested for their ability to methylate Hg.
Identification of hgcAB as a Hg methylation biomarker provides new opportunities to evaluate microbial Hg methylation distribution in nature and to identify the types of microorganisms responsible, without making rate measurements that are not feasible for many environments. In this study, we queried >3500 publicly available microbial metagenomes in GenBank and Integrated Microbial Genomes and Metagenomes (IMGM) (12, 13) that were part of ~200 metagenomic projects (table S1). These data encompassed ~823 million gene sequences and represented a wide range of environments on Earth, allowing us to evaluate hgcAB prevalence, global and taxonomic distribution, and relative abundance. Implicit in this study was an assessment of the potential to methylate Hg from a wide variety of environments in the context of what is known about MeHg levels and risks in specific niches, including the human body. We specifically evaluated the prevalence and distribution of newly discovered types of Hg methylators while addressing long-standing questions about the evolutionary history of Hg methylation and its relationship with known biological processes. Figure 1 provides an overview of the environmental and geographic breadth of available metagenomes investigated. Colored symbols in the figure show hgcA abundance in each metagenome overlaid with Hg emissions estimated by country (2) to highlight the intersection of Hg availability and the presence of the Hg methylation genes.
Fig. 1. Global frequency and abundance of HgcA based on the metagenomic projects evaluated.
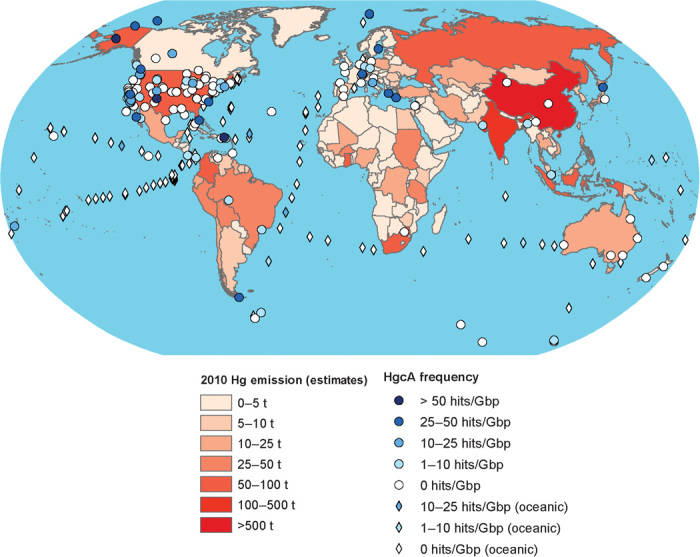
Overlay is the estimated continental emission of Hg (in tons) based on “UNEP Global Mercury Assessment 2013: Sources, Emissions, Releases and Environmental Transport” (2). Diamonds represent pelagic ocean water samples, whereas circles represent all other samples. The abundance of Hg emissions and hgcA is colored according to the accompanying legend.
RESULTS
The presence of HgcA was evaluated in ~3500 available environmental microbial genomes comprising about 8 × 108 genes and 8 × 1011 nucleotides (table S1). We primarily used the assembled and annotated data publicly available in the U.S. Department of Energy (DOE) Joint Genome Institute IMGM comparative analysis system, which consists of data generated under numerous metagenome sequencing projects, including the Human Microbiome Project (13, 14). To search for orthologs of HgcA and HgcB, we generated hidden Markov model (HMM) profiles for full-length proteins and for the HgcA cobalamin binding domain and transmembrane domain. Because of the highly fragmented nature of metagenomic data, we expected many, if not most, genes and encoded proteins (including HgcA and HgcB) in microbial metagenomes to be incomplete. Reliably identifying such proteins and protein fragments (including previously unknown variants) and distinguishing them from their relatives (other CFeSP and ferredoxins) in large data sets using simple sequence alignment–based algorithms [for example, Basic Local Alignment Search Tool (BLAST)] are difficult. We used profile HMM–based searches, which are more sensitive and accurate in identifying homologs compared with other similar methods (15).
For the development of HgcA and HgcB HMMs that would enable us to distinguish them from more distant homologs, profiles were built using the full range of known hgcAB sequences (that is, the genomes of about 40 species), including all known groups of methylators in Bacteria and Archaea (7). The effectiveness of identifying true HgcA and HgcB candidates and discriminating against their WL pathway–associated CFeSP and ferredoxin homologs in profile searches encompassing all microbial proteome databases (UniProt) was confirmed by protein sequence alignments and identification of conserved motifs and domains. This enabled metagenomic searches that successfully recovered fragmented gene sequences. This combined assessment of distinct scoring based on HMM profile searches and phylogenetic sequence fingerprints provided the resolving power needed to differentiate HgcA identified in metagenomic data sets, assign them to bacterial and archaeal lineages, and even predict the existence of novel yet uncultured taxa that can perform Hg methylation (fig. S1). In all but one case, hgcA and hgcB were colocalized, suggestive of coexpression and regulation (7).
With this approach, ~3100 hgcA genes were identified and taxonomically characterized, along with hgcB, when large scaffolds were present. These represent a subset of >33,000 sequences encoding PFam3599 superfamily proteins and range from fragments containing the conserved domains to full-length genes associated with hgcB. About two-thirds (140 of 203) of the metagenomic projects did not reveal any hgcA, although half of them contained corrinoid FeS protein genes, suggesting that the anaerobic WL pathway is not necessarily correlated with Hg methylation potential, as also indicated by genomic data (figs. S2 and S3). Furthermore, although the amount of hgcA per gigabase of sequence data was not proportional to the size of the metagenome, hgcA was generally absent in the smallest metagenomes. Of the >2200 individual metagenomes where hgcA was not present (among ~3500 evaluated), 1250 metagenomes were from the human microbiota, with only a single human sample revealing the presence of a potential Hg-methylating organism. When animal microbiomes and engineered consortia data sets (70 projects) were excluded, only a third of the metagenomic projects lacked hgcA, suggesting that these genes are rather broadly or globally distributed in many open environmental settings. The size of available metagenomes varied tremendously from several megabase pairs to more than 100 Gbp; therefore, we normalized hgcA abundance to each data set size (table S1). Relative abundance of hgcA was then compared between metagenomes based on general environment types. Because many of the data sets that revealed no hgcA were large, not only the presence but also the absence of hgcA is significant, suggesting a low potential for MeHg generation at those locations.
High abundance and broad diversity of hgcAB in sediments and wetland soils
To date, most Hg-methylating microorganisms have been isolated from aquatic sediments, wetlands, saturated soils, and low-oxygen subsurface environments (5)—niches thought to be most important in MeHg production (1). The available metagenomes were taken from several habitats of known methylation potential, including marine and freshwater sediments and marshes (1). High rates of Hg methylation have been measured specifically in Spartina marshes similar to the Sippewissett Marsh (16, 17) and in the Hg-contaminated San Francisco Bay Delta marshes and rice paddies that include the Twitchell Island (18). Saturated agricultural soils, such as rice paddies, are potential MeHg production sites that can lead to human exposure through rice consumption (19, 20). In our analyses, these environments all showed abundant hgcA per gigabase pair of sequence and spanned diverse geographic locations from the Arctic and the Baltic Sea to the Southern Hemisphere (Figs. 1 and 2). Most of these environments also contained a very broad hgcA phylogenetic diversity (Fig. 3), with representatives in every known microbial taxon linked to Hg methylation.
Fig. 2. Distribution and relative abundance of HgcA in metagenomic projects, by environment type.

Each metagenomic project includes a wide-ranging number of metagenome sequencing data sets. Each bar represents a metagenome project. Gray bars indicate metagenome size. Colored bars represent HgcA abundance normalized to 1 Gb of metagenomic sequence. Overall, about two-thirds of 3500 metagenomes and 140 of 203 metagenome projects did not reveal any hgcA. PCE, polychloroethylene.
Fig. 3. Maximum likelihood phylogeny of HgcA proteins in complete genomic and metagenomic sequences.

Open circles at major nodes denote bootstrap support values >50. Gray-shaded clades represent the fused HgcAB sequences. Sequences with colored circles and color naming indicate full-length HgcA from metagenomes, with the color corresponding to the metagenome classification in Fig. 2.
Critically, high hgcAB abundance was observed in thawing permafrost soils (Figs. 1 and 2; for example, Bonanza Creek, AK). Long-range atmospheric Hg transport to the Arctic has resulted in elevated risks in wildlife and humans (21). Thawing permafrost soils can potentially further increase MeHg risk in the Arctic through release of inorganic Hg (22, 23) and increased microbial activity (24). MeHg production in Arctic marine and freshwater sediments has been demonstrated (25–27) and higher MeHg production in Arctic soils has been inferred from runoff in northern rivers (28), but MeHg production in permafrost soils has not been explicitly demonstrated. However, a recent high-latitude study showed that microbial activity and growth occur at subzero temperatures (29), suggesting that microbial Hg methylation can occur in frozen permafrost and will likely increase once thawed. The hgcAB genes in permafrost metagenomes were taxonomically diverse but consisted mostly of methanogens (Figs. 3 and 4), along with several unclassified Firmicutes (Fig. 3). Methanomicrobia, the group of methanogens known to encode hgcAB, was dominant in thawing permafrost soils (30), supporting the potential for high MeHg production in thawing Arctic soils. Importantly, complementary metatranscriptome data sets were also available for thawing permafrost soils in Bonanza Creek, AK (Fig. 4). These data showed that hgcA was indeed expressed in many of the bacterial strains identified in Deltaproteobacteria and Firmicutes, and particularly in the methanogenic Archaea, thus indicating that the Hg-methylating genes are now active in those Arctic communities.
Fig. 4. Closest family and genera assignments for metagenomic HgcA genes/fragments for metagenomic data sets containing hgcA.
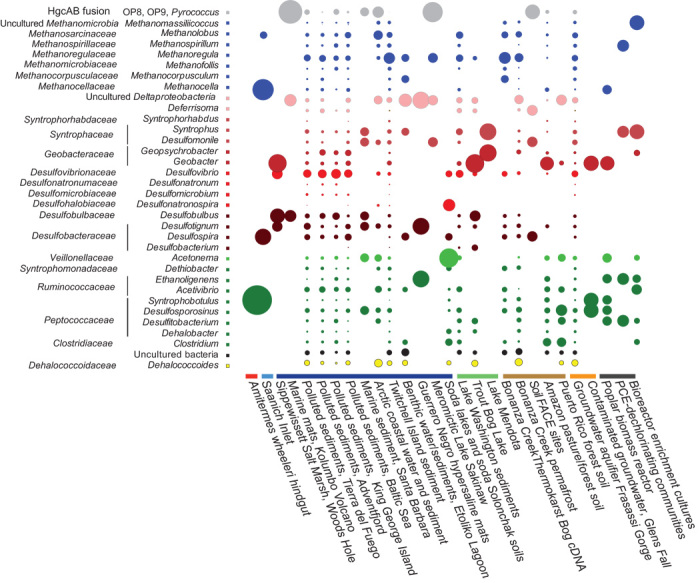
Assignments were made using MEGAN.5, a program that evaluates the taxonomic and functional content of short-read metagenomic data sets. Circle size represents the relative abundance (square root–transformed) of taxonomically assigned hgcA sequence types in each metagenome. The metagenomes are arranged by environment type, as classified in Fig. 2. PCE, polychloroethylene.
In more temperate regions, hgcAB was absent or rare in metagenomes from oxic forest soils, such as grassland and agricultural settings, but was readily detected in tropical forest soils (Puerto Rico and Brazil), including sites converted into pasture land (Figs. 2 and 4). Deforestation can increase moisture content and lower oxygen content in tropical soils (31). In combination with Hg contamination from the artisanal gold industry (2), this may lead to increased risk in humans.
Lake hypolimia
The anoxic bottom waters of lakes contained abundant hgcA (Fig. 2), but methylators were generally less diverse than in sediments and soils (Fig. 4). Phylotypes in metagenomes from the eutrophic stratified Lake Mendota consisted primarily of Geopsychrobacter and Syntrophobacter spp., whereas the bottom waters of the dystrophic Trout Bog, WI, were dominated by Geobacter spp. (Fig. 4). Anaerobic lake bottoms are well-known MeHg production sites that can transfer significant MeHg into freshwater food webs (32). Geobacter spp. are particularly efficient at MeHg generation, at least in the laboratory (5), although their relative importance to Hg methylation in nature remains poorly understood (33–37).
Extreme environments
Metagenomes from extreme environments, including soda lakes, hypersaline and hypersulfidic waters (Sakinaw Lake and Etoliko), saltern microbial mats, and thermal sites, were positive for various hgcAB phylotypes (Fig. 2), many from unclassified or uncultured organisms. Soda lakes appear to have a high diversity of novel Firmicutes-type HgcA, which are distinct from those from any cultured species (Figs. 3 and 4). The hypersaline/sulfidic bottom waters of the permanently stratified Sakinaw Lake, an isolated former fjord, were particularly rich in hgcAB fusion genes corresponding to representatives of the uncultured divisions OP8 and OP9 (Figs. 3 and 4). Sakinaw Lake also contains methylating methanogens and syntrophic Deltaproteobacteria, in agreement with metabolic inferences and based on community and genomic data (38).
Pelagic marine water columns
Marine fish are the main route of MeHg exposure in many human populations (39), but the source of MeHg in marine water columns remains unknown. Coastal sediments and terrestrial runoff (1) supply MeHg to coastal fisheries, and hgcAB is common to metagenomes from several coastal and estuarine sediments in the Atlantic and Pacific Basins. However, several lines of evidence support the idea that open-ocean fish acquire their burden from MeHg produced de novo in pelagic water columns, including mass balance models (40, 41) and the Hg-stable isotope composition of marine fish (42).
Depth profiles from recent efforts to map MeHg in the open ocean consistently showed MeHg accumulation in the subthermocline oxygen minimum zone (OMZ) (43), which is commonly attributed to in situ MeHg production associated with microbial remineralization of sinking organic particles (41). However, the OMZ is generally not fully anoxic and may not support the strict anaerobes understood to produce MeHg, unless they are present in anaerobic niches in decaying particles. Methylation rates have rarely been measured explicitly in blue waters, but recent incubations (44, 45) have demonstrated in situ MeHg production in oxic surface waters.
Nevertheless, although hgcAB appeared to be abundant in marine sediments (Figs. 2 to 4), it was rarely found in pelagic marine water column metagenomes, and the sequences found were distinct from hgcAB in confirmed methylating organisms, as judged by an identity matrix of sequence similarities (table S2). Available metagenomic data included several profiles through the OMZ (Bermuda Atlantic Time-Series Study, ALOHA, Palmer time-series stations, and Deepwater Horizon event) (Figs. 1 and 2, and table S1). Overall, of 138 samples analyzed, only 7 showed any evidence of hgcAB. Several oxic and suboxic coastal waters also lacked hgcAB, including large metagenomes representing the expanding OMZ in the Eastern North Pacific Ocean (46) and ammonia-oxidizing waters in Monterey Bay, CA.
Intriguingly, rare and distinct hgcA-like sequences were found at depth in several metagenomes from the mesopelagic equatorial Pacific Ocean and the Southern Atlantic Ocean (Global Malaspina, mesopelagic equatorial Pacific, and Southern Atlantic Ocean metagenomes; Figs. 2 and 3). Even if the characteristic cap helix domain is present in those proteins, some distinct amino acid substitutions are present, although none is predicted to abolish Hg methylation based on recent mutagenesis studies in a model Hg-methylating bacterium (47). Because no MeHg-generating organisms in culture encode such variants of HgcA, their methylation potential remains unknown. In addition, those marine sequences included the hgcA transmembrane domain but lacked hgcB in the few contigs that extended beyond hgcA.
Either hgcAB+ organisms were rare in the open ocean, their abundance was too low to be captured in these metagenomes (many were relatively shallow), or the depth at which methylation occurred was narrow and was not captured in these profiles. Alternatively, hgcAB may not be responsible for MeHg production in the blue ocean, leaving open the possibility of noncellular methylation by agents such as methyliodide, dimethylsulfide, or humic matter (48–50) or methylation by another unidentified metabolic pathway. Deeper sequencing of targeted marine samples, with concurrent measurements of inorganic and MeHg, will be required to better understand the distribution of hgcAB in marine water columns and to identify any organisms encoding these genes. Identification, isolation, and testing of organisms encoding such distinct hgcA sequences could provide key information on the origin of MeHg in the open ocean.
However, our metagenomic analysis hinted at MeHg production potential in coastal “dead zone” waters. We found abundant Hg methylation genes of limited diversity in the fully anoxic bottom waters of a stratified fjord, Saanich Inlet, Canada (46) (Figs. 2 to 4). This is significant because anoxic coastal waters are expanding globally as a result of global warming and increased terrestrial nutrient fluxes (51). MeHg production in these areas represents another potential hazard to human and ecosystem health, especially considering humans’ large consumption of fish derived from coastal waters (39).
Potential for MeHg production in engineered systems and contaminated sites
Microbial systems engineered for biomass fermentation or chlorinated compound degradation, including engineered subsurface environments, often contained abundant hgcAB (Fig. 2). However, diversity was often limited, especially in bioreactors, reflecting restricted species richness (Figs. 3 and 4). The MeHg production potential of dechlorinating systems is consistent with the dechlorination capacity of some Hg-methylating Deltaproteobacteria and Clostridia (5) but may also be linked to the presence of hgcAB in some Dehalococcoides. In contrast to more engineered systems, a subsurface sulfidic aquifer in Frasassi, Italy, displayed relatively high hgcAB abundance and phylogenetic diversity (Figs. 1 and 4). Similarly, untreated contaminated sites in several locations across the United States and Canada resulting from human mining or disposal of pollutants revealed abundant hgcA (Figs. 1 and 2). Hence, our data justify the inclusion of MeHg production monitoring in the risk management of water and wastewater reclamation systems and in the remediation of legacy contaminants.
One of our goals in identifying the genes for Hg methylation and in evaluating their presence in available microbial metagenomic data was to look for environments where MeHg production may have gone unrecognized. On the basis of this study, wastewater, bioreactors, and contaminated groundwater are some environments that might bear more attention to MeHg production, although there are limited examples of MeHg in contaminated groundwater (52–56), wastewater (57–59), and bioreactors (60, 61).
Absence of hgcAB from human and vertebrate microbiomes
Because humans are exposed to inorganic Hg (from dental amalgams, food, and industrial pollutants), potential Hg methylation by the human gut microbiome would have tremendous health implications. The complexity of Hg transformation routes inside the body has continued to make risk assessment difficult; to date, the capacity of the gut microbiome for Hg methylation remains unknown. To evaluate hgcAB distribution in human populations, we searched a vast number of metagenomes generated under the Human Microbiome Project (14) and other human microbiome studies worldwide (table S1). The data sets included >850 assembled metagenomes representing 297 healthy individuals from North America, Europe, and Asia, with samples collected from a variety of body sites (including oral and fecal). No hgcAB was found in any of these metagenomes encompassing >100 million predicted microbial genes (Fig. 2). We routinely identified abundant carbon monoxide dehydrogenase/acetyl-CoA synthase genes (indicative of the WL pathway) (table S1) and a few methyl coenzyme M reductase genes (mcrA, signatures of the methanogenic Archaea) in fecal (but not in oral) microbiomes. This demonstrates that relatives of Hg methylators (but not the Hg methylators themselves) were present and represented in metagenomic data sets, confirming the findings of cultured and genomic sequencing studies.
So far, no bacterial genomes from mammalian microbiota, including oral and intestinal Deltaproteobacteria, have encoded HgcAB. Among the typical mammalian intestinal methanogens, the sequenced species of Methanosphaera and Methanobrevibacter (both Methanomicrobiales) also lack hgcAB. However, a recently isolated methanogen from human feces, Methanomassiliicoccus luminyensis (62), does have hgcAB, and we show for the first time that this organism methylates Hg in culture. In duplicate experiments (each with triplicates in the presence of 20 μM sulfide), M. luminyensis methylated 25 ± 3% and 37 ± 9% of the 1 nM 201Hg provided (figs. S4 and S5). Me201Hg production in live cultures was significantly higher than those in spent medium and sterile medium controls, with a detection limit of 0.2 pM. MeHg production by M. luminyensis was about 5 to 8 pmol of MeHg per microgram of protein produced. Only a few other measurements of methylation rate by methanogens have been found. Normalized to protein production, M. luminyensis methylated at a rate similar to that of Methanomethylovorans hollandica and well above that of Methanolobus tindarius (5).
However, M. luminyensis was not detected in any of the assembled human microbiomes. This organism has been argued to be predominantly found in the intestinal microbiota of the elderly (63), which were not well represented in the Human Microbiome Project data sets. To explore the potential age difference in the distribution of Methanomassiliicoccus spp., we searched for methanogen-type hgcAB in unassembled metagenomic sequences (>500 billion base pairs) from the gut microbiota of 145 elderly Swedish women (64) and MetaHIT project data sets (65), including 418 fecal metagenomes covering a wide age range in several European countries. Frequently finding mcrA with Mega BLAST confirmed that archaeal DNA was recovered and sequenced from these samples. Only one single-sequence read (out of >30 billion combined reads from the two project data sets), retrieved from the search using hgcA query, represented an hgcA-like sequence (Fig. 2). That read, identified from the study of Karlsson et al. (64), represents a methanogenic HgcA that is not identical to M. luminyensis HgcA and likely belongs to a yet uncultured human gut methanogen. The inferred protein sequence based on that read, in alignment with that of M. luminyensis HgcA, is shown later. A highly conserved and catalytically critical cysteine residue (47) (shaded in box below) appears to have been replaced by a glycine in the human microbiome sequence read. If that substitution does not represent a sequence error, the resulting protein should be incapable of Hg methylation. These findings suggest that M. luminyensis (and other hgcA-encoding organisms) are extremely rare in human populations across multiple continents.
| gnl|SRA|ERR260153.9799443.1 | 1 TKGINVWGAAGKGTFGTDELVGRISETGLSTKG+NVW AAGKGTFGT+ELV RI +TGL+ |
| M. luminyensis B10 | 77 TKGVNVWCAAGKGTFGTEELVRRIEDTGLA 106 |
Another closely related species that was subsequently isolated from the human gut, Methanomassiliicoccus intestinalis (66), does not harbor hgcAB. Therefore, it appears that the risk of Hg methylation in the human body by the endogenous microbiota is quite low but perhaps not zero.
HgcAB was also absent in other vertebrate microbiomes, including mice, pandas, ruminants, and birds, but was detectable in invertebrate microbiomes, including termites and beetles (Fig. 2). This is not unexpected considering that several hgcAB+ bacteria (for example, Acetonema longum and Clostridium termitidis) were isolated from termite guts and MeHg formation has been documented in such invertebrates (67). A marine gutless oligochaete (Olavius algarvensis) also harbored an uncultured Deltaproteobacteria endosymbiont (68) that encodes hgcAB (Figs. 3 and 4). Clearly, such invertebrates can contribute MeHg, further complicating MeHg source tracking of, and entry into, terrestrial and aquatic food webs.
Novel Hg-methylating organisms and hgcAB evolution
An unexpected finding of our in-depth exploration of global metagenomes was the discovery of a family of rare hgcA-like proteins that are fused with hgcB (fig. S6). This protein family has a limited distribution (Fig. 3), and the only cultured organism encoding this fusion protein is the hyperthermophilic archaeon Pyrococcus furiosus. As in Sakinaw Lake, several aquatic metagenomes revealed deep-branching uncultured bacterial lineages (OP8 and OP9) encoding these fusions (Figs. 3 and 4).
No Me201Hg production higher than that of controls was detected with P. furiosus grown in the laboratory at near-boiling temperatures, with well-growing cells on media with and without S0 in either medium (fig. S7). It is critical to assess Me201Hg levels in various controls because of low MeHg production levels in many media. Me201Hg concentrations in cultures were generally within the detection limit or just higher than the detection limit (average, 0.2 pM) and were not significantly higher than the Me201Hg concentrations in spent and fresh medium controls with matched chemistry. Because Hg methylation depends on intracellular Hg transport and other yet unknown biochemical transformations, the absence of MeHg generation in P. furiosus does not necessarily reflect the inability of the fused HgcAB to catalyze that reaction. The origin of the fused HgcAB is unclear, but P. furiosus likely acquired it from a distant lineage because other Pyrococcus spp. and nonmethanogenic Archaea lack the fused HgcAB (Fig. 3).
Horizontal gene transfer appears to play a significant role in hgcAB distribution and WL pathway utilization across Bacteria and Archaea and is most evident in environments supporting syntrophic interactions. hgcAB distribution and WL pathway utilization are restricted to specific lineages of Deltaproteobacteria, of Firmicutes, and of the methanogenic Archaea, which include many syntrophs. Occasionally, species of Dehalococcoides, Clostridia, Spirochaetes, and others that typically do not harbor hgcAB or the WL pathway appear to acquire them from syntrophic free-living or invertebrate gut communities. When the taxonomic distribution of HgcA across all metagenomes was analyzed for co-occurrence with MEGAN (69), we observed coassociations between sulfate reducers, fermenters, and methanogens that have been shown to be syntrophic (fig. S8). A recent study also found abundant hgcAB+ syntrophs in Florida everglades (70).
An obvious question is, “Where in the microbial world did hgcA appear, and how is the evolution of hgcAB linked to that of CFeSP functioning in the WL pathway?” Answering this question may also provide clues to the physiological function of HgcAB. Although we could not unequivocally resolve the origin of hgcAB, phylogenetic analysis of the cobalamin binding domain suggests a deep divergence for WL pathway–associated CFeSP and HgcA, which is possibly linked to associations with other protein domains (the FeS centers in carbon monoxide dehydrogenase/acetyl-CoA synthase and the transmembrane domain in HgcA; Fig. 5). Ancestral CFeSP and HgcAB appear in methanogens, although unambiguously assigning a specific origin lineage is difficult. The paralogous relationship between WL pathway–associated CFeSP and HgcA also enabled inquiries about which type of WL pathway appeared first and in which microbial lineage. Our analysis is consistent with previously proposed scenarios for the early metabolism and evolution of the WL pathway (71) in that hydrogenotrophic methanogens may have first encoded this enzymatic complex, followed by secondary branching of aceticlastic and methylotrophic taxa.
Fig. 5. Maximum likelihood phylogeny of the cobalamin binding domain of HgcA and the WL pathway carbon monoxide dehydrogenase/acetyl-CoA synthase γ subunit.

HgcA and HgcAB clades were collapsed. Bootstrap support at nodes is indicated by black circles (>75) or white circles (50 to 75). Some archaeal and bacterial taxa were collapsed and color-coded. Blue and yellow branches indicate archaeal and bacterial lines of divergence that would suggest a potential archaeal origin for bacterial CFeSP. Cand, candidate phylum.
DISCUSSION
This broad analysis of hgcAB offers the first global look at the distribution and diversity of Hg-methylating microorganisms. To our knowledge, this is the first comprehensive analysis of the presence and distribution of specific genes that are directly linked to a specific biogeochemical transformation across many environments on Earth. The gene pair hgcAB was found in nearly every anaerobic environment, except in vertebrate gut microbiomes, but was not regularly found in any aerobic settings, including the open ocean. Several new potential methylation habitats were uncovered, and suspected habitats were confirmed to have MeHg production potential, including thawing permafrost soils, coastal dead zones, bioreactors for contaminant degradation, saturated agricultural soils, and extreme anaerobic environments.
Taxonomic characterization of hgcAB+ organisms confirmed the role of sulfate-reducing and Fe(III)-reducing Deltaproteobacteria in many habitats known for Hg methylation but also demonstrated the potential role of syntrophic Deltaproteobacteria, methanogens, and Firmicutes in MeHg production. Where found, hgcAB phylogenetic diversity in deeply sequenced metagenomes usually encompassed all anaerobic metabolic processes and taxa associated with Hg methylation, including primary and secondary fermentors, sulfate-reducing and Fe(III)-reducing bacteria, and methanogens, suggesting that there is potential for widespread Hg methylation by organisms using a wide variety of carbon sources and electron acceptors in most anaerobic environments in nature. This may be most significant in thawing northern latitudes caused by climate change. As the permafrost of today becomes the active layer of tomorrow, microbial metabolism of copious carbon resources, combined with enhanced Hg deposition in the Arctic, is likely to increase MeHg risk in Arctic ecosystems.
On the basis of the presence of hgcAB and its abundance, we have generated a new picture of where Hg methylation is likely to occur, with important implications for environmental and human health. hgcAB is effectively absent in the microbiota of the anaerobic digestive system of vertebrates. The human microbiome commonly contains microorganisms from all major clades and metabolic guilds that harbor hgcAB+ organisms (34). However, the native function of hgcAB remains unknown, and its elucidation is crucial for a more predictive understanding of what dictates the presence of Hg methylators, and therefore methylation potentials, in open and host-associated environments.
MATERIALS AND METHODS
Because of the highly fragmented nature of metagenomic data, we expected many, if not most, genes and encoded proteins (including HgcA and HgcB) in microbial metagenomes to be incomplete. Reliably identifying such proteins and protein fragments (including previously unknown variants) and distinguishing them from their relatives (other CFeSP and ferredoxins) in large data sets using simple sequence alignment–based algorithms (for example, BLAST) are difficult. We therefore used profile HMM–based searches, which are more sensitive and accurate in identifying homologs compared with BLAST or other similar methods (15).
For the development of profile HMMs for HgcA and HgcB that would enable us to distinguish them from more distant homologs, HgcA and HgcB encoded in Hg-methylating bacterial and archaeal genomes were aligned, and the presence of conserved motifs and protein domains was confirmed as described by Gilmour et al. (5) and Parks et al. (7). With the exception of Desulfovibrio inopinatus, hgcB is present immediately downstream of hgcA (or is separated by one gene in the genome of Desulfovibrio africanus). This suggests a strong coregulation between expression and coevolution. The two genes appear to be distant in the draft genome of D. inopinatus (on different contigs); however, because of the unfinished state of that genome, this separation may be subject to change.
Alignments were edited manually in Geneious (version R8), taking into account conserved sequence domains and secondary structure domains. High-variability regions that could not be confidently aligned were excluded. Several profile HMMs were generated. For an HMM of the entire HgcA sequence (HgcA trim), which includes the cobalamin binding domain and the distinct transmembrane domains, the alignment contained 245 amino acids from 41 representative HgcA sequences representing Hg-methylating Bacteria and Archaea. A profile for the characteristic cap helix domain of HgcA (HgcACap) was built based on a region (14 amino acids long) spanning that domain. A separate profile was also generated for the predicted membrane-anchored C-terminal domain of HgcA (HgcATrans), represented by an alignment region 68 amino acids long. For the HgcB profile, we used an alignment (72 amino acids) representing 48 sequences. To generate profile HMMs, we used the program hmmbuild of the HMMER v3.0 package (http://hmmer.org/) with default parameters. The sequence alignments and resulting profile HMMs are provided in the Supplementary Materials.
To test the sensitivity and specificity of HMMs in identifying HgcA and HgcB sequences in protein databases, we performed local searches using an hmmsearch (HMMER v3.0) of superfamily data sets (PFam3599 for HgcA and PFam13237 for HgcB) extracted from PFam and IMG (13) and of the comprehensive protein database UniProtKB (release 2014 04). Hit scores were retrieved, and corresponding sequences were aligned and grouped based on the presence of conserved domains and their affiliation with HgcAB or other superfamily types. With the HgcA trim profile search, the retrieved hits partitioned into a distinct cluster that exclusively contained HgcAs, a small group of sequences of moderate scoring strength that consisted of the fused HgcAB proteins (from P. furiosus and uncultured OP8 and OP9 bacteria), and a much weaker scoring cluster that included other CFeSP relatives (fig. S1). The profile HMMs constructed for HgcACap and the transmembrane domains exclusively retrieved HgcAs from both PFam3599 and UniProt. These searches not only resulted in the identification of all previously known HgcAs and HgcBs but also revealed a number of novel ones from the recently sequenced genomes of cultured Bacteria/Archaea and from assembled single cells and metagenome projects (OP9, OP8, and ACD78). Furthermore, all of the identified HgcAs from UniProt belong to PFam3599, confirming that HgcA is a subset of the PFam CFeSP superfamily profile HMM. The distinct scoring values, especially for HgcA trim, allowed us to unambiguously distinguish HgcA from the fused HgcAB and from WL pathway–associated CFeSP. Moreover, the HgcACap and HgcATrans profiles excluded non-HgcA proteins and, because they targeted only a fraction of the proteins, were complementary in the search for fragmented metagenome data, in conjunction with the HgcA trim profile. The profile for full-length HgcB also retrieved those proteins as a distinct group but with less separation from other ferredoxins (fig. S1). All of the identified HgcB had paired neighbor HgcAs that were identified by multiple HgcA profile HMMs. Because HgcB proteins are very small and lack highly conserved domains that distinguish them from other ferredoxins, we chose not to use their profile HMM in the search for metagenomes.
Identification of HgcA in microbial metagenomes
To search for the presence of HgcA in environmental microbial genomes, we primarily used the assembled and annotated data publicly available in the DOE Joint Genome Institute IMGM comparative analysis system, which combines data generated under numerous metagenome sequencing projects, including the Human Microbiome Project (13, 14). The primary search for HgcA was performed using the August 2013 release of IMGM, but additional subsequent searches were performed on updated data sets and additional releases through December 2014. A summary of the analyzed projects and their classifications based on the types of environments and samples they represent (based on available metadata) is provided (table S1).
As the first step in HgcA identification, we searched for and retrieved all genes encoding CFeSP proteins classified under PFam3599 from each metagenomic data set. Because some metagenomes contained a large amount of unassembled sequences, we searched both assembled and unassembled data, if available. The number of PFam3599 hits for each metagenomic project and environmental location is provided in table S1. The retrieved protein sequences were subjected to local searches with several HgcA profile HMMs. The distribution of hit scores was compared to that obtained from test searches of UniProt and was found to span the same overall distribution. Protein sequence alignments were generated for batches of retrieved metagenomic hits, and redundant sequences (retrieved by multiple HgcA profile HMMs) were consolidated. Metagenomic HgcAs (fragments and full length) were confirmed by identification of conserved sequence domains (cap helix and transmembrane regions). When hits were part of contigs greater than 1 kb, we also individually inspected them and confirmed the presence of downstream hgcB. The size of metagenomes varies by several orders of magnitude; therefore, to compare HgcA abundance between samples, projects, and environments, we normalized them to the size of the metagenome.
The geographical coordinates for environmental samples linked to individual projects were used to place them on a global map overlaid with data on Hg emissions, by country. Mapping was performed in ArcGIS (Esri) using 2010 data on Hg emissions, by country. Data on emissions were taken from the Technical Background Report for the Global Mercury Assessment 2013 (72). The geographical location of each metagenome was added using latitudinal/longitudinal georeferences obtained from the metadata of each sample in the JGI Gold Database and colored according to the number of hgcA sequences found for every 1 Gbp. The relative abundance of HgcA in those samples was also projected (Fig. 1).
In two large human gut microbiome projects (64, 65), protein PFam classifications were not available and a large fraction of data were not assembled. To search for the potential presence of M. luminyensis HgcA in those data sets, we performed a Mega BLAST analysis of raw Illumina sequences deposited in the National Center for Biotechnology Information small reads archive. As control, we also searched for the presence of a diagnostic methanogenic gene (mcrA) that is present in human gut Archaea (Methanomassiliicoccus and Methanobrevibacter spp.).
Phylogenetic analyses
HgcA and HgcB protein sequences were aligned using Muscle in Geneious v6. Concatenated alignments (HgcA followed by HgcB) were also generated and used to analyze the naturally fused HgcAB, which was used as an outgroup. Full-length sequences identified in metagenomic data were also added to the alignment for phylogenetic reconstruction. A test for the best model of evolution that fits the data was performed in Mega BLAST 5.2 (12) and determined to be WAG with γ-distributed rates. A search for the best tree was performed under maximum likelihood using WAG+G and 100 independent inferences. Searches under the same model and bootstrap analysis (100 replicates) were also performed using RAxML v7.4.2 (11).
We also investigated the phylogenetic relationship between HgcA and the CFeSP of the γ subunit of the acetyl-CoA synthase complex involved in the WL pathway. In that case, the alignment was limited to the homologous cobalamin binding domain in those families of proteins, which corresponds to the profile HMM for PFam3599. Maximum likelihood phylogenetic reconstruction was conducted under the WAG+G model in RAxML v7.4.2, using 100 independent inferences and bootstrap analysis (100 replicates). The best tree was rooted between HgcA-HgcAB and the γ subunit of WL pathway–associated CFeSP.
To explore the taxonomic diversity of fragmented metagenomic HgcA sequences, we used the software MEGAN5.2 (69). Taxonomic rank assignments based on the lowest common ancestor algorithm were performed against a database constructed from all HgcA sequences identified in all bacterial and archaeal genomes. Scaled inferred abundances were calculated for each individual metagenomic project at the genus level. Co-occurrence networks were generated using combined HgcA data from all metagenomes at various probability thresholds.
Methylation assays for M. luminyensis B10 (DSM-25720)
The culture was purchased from the Deutsche Sammlung von Mikroorganismen und Zellkulturen. Methylation assays were conducted during batch growth on a medium adapted from Dridi et al. (62) and on DSM-119. The carbon sources were 20 mM sodium acetate, trypticase peptone (1 g/liter), and yeast extract (1 g/liter). The salts were 4 mM KH2PO4, 170 mM NaCl, 2 mM MgSO4, 0.3 mM CaCl2, and 20 mM NH4Cl. Trace elements and vitamins from DSM-141, plus selenite and tungstate (up to a final concentration of 25 nM), were used. The medium was buffered with 10 mM Mops (pH 7.6), and resazurin was used as a redox indicator. After degassing and sterilization, the following components were added up to the given final concentrations: 20 mM bicarbonate, 20 mM Hepes, 10 μM Na2S, 500 mM cysteine, 50% MeOH (5 ml/liter), rumen fluid (100 ml/liter), fatty acid mixture (20 ml/liter), and 50 μM. Headspace was overpressurized to 20 psi with H2/CO2 (20%:80%).
Assays were conducted in triplicate bottles including cultures, uninoculated sterile medium controls, and spent medium controls. Spent medium was obtained by filtration (0.45 μm) of cultures in stationary phase in a glove box under 5% H2 in N2 atmosphere. Optical density and pH were followed daily to monitor growth. Other parameters were measured initially and again at the same time that the growing cultures reached stationary phase.
All bottles were amended at T0 with 1 nM enriched 201HgCl2 (obtained from Oak Ridge National Laboratory at 98.1% enrichment). Aqueous 201Hg concentrations were assessed at the end of the assay to evaluate losses to bottle walls and to more accurately assess the fraction of Me201Hg production. All measurements were made in an unfiltered medium.
Methylation assays for P. furiosus (DSM-3638)
The type strain of P. furiosus was obtained from the German Collection of Microorganisms and Cell Cultures. Methylation assays were conducted during batch growth on a medium adapted from DSM-337. The carbon sources were 15 mM maltose and yeast extract (1 g/liter). The salts were 0.5 M NaCl, 15 mM MgSO4, 14 mM MgCl2, 10 mM KCl, 5 mM NH4Cl, and 1 mM CaCl2. The medium was buffered with 10 mM Mops (pH 7.0), and resazurin was used as a redox indicator. After degassing and sterilization, the following components were added up to the given final concentrations: 1 mM K2HPO4, 2 mM bicarbonate, 100 μM Na2S, and 500 mM cysteine. Trace elements and vitamins from DSM-141, plus tungstate (up to a final concentration of 100 nM), were used.
Methylation assays were conducted in media with and without elemental sulfur (added to 150 μM). The medium with S0 was also amended with trypticase peptone (5 g/liter). The headspace for the medium with S0 was overpressurized to 20 psi with H2/CO2 (20%:80%). The headspace for the medium without S0 was N2. All cultures were grown in bottles with a high headspace-to-liquid ratio, and pressure was monitored and adjusted during growth.
Assays were conducted in triplicate bottles, including cultures, uninoculated sterile medium controls, and spent medium controls. Spent medium was obtained by filtration (0.45 μm) of cultures in stationary phase in a glove box under 5% H2 in N2 atmosphere. Optical density and pH were followed daily to monitor growth. Other parameters were measured initially and again at the same time that the growing cultures reached stationary phase. Cultures were incubated at 95°C.
P. furiosus assays were amended at T0 with 10 nM enriched 201HgCl2 (obtained from Oak Ridge National Laboratory at 98.1% enrichment). Aqueous 201Hg concentrations were assessed at the end of the assay to evaluate losses to bottle walls and to more accurately assess the fraction of Me201Hg production. All measurements were made in an unfiltered medium.
Hg and MeHg analysis
Total Hg was quantified in digested samples with isotope dilution, using stannous chloride reduction coupled online to inductively coupled plasma mass spectrometry (ICP-MS). Total Hg in medium (unfiltered samples) was digested in hot HNO3/H2SO4 (7:4, v/v) before analysis. In the text, the use of the term “201Hg” or “Me201Hg” implies an excess concentration of the enriched isotope above any contamination from natural Hg abundance. MeHg was determined by isotope dilution gas chromatography ICP-MS following aqueous-phase distillation and ethylation. Methods are described in detail by Gilmour et al. (5). Detection limits are specific for each analysis and are given in Results. Standards and certified reference materials purchased from multiple sources and MeHg standards synthesized in-house were used to ensure adequate recovery and precise and accurate analyses.
Funding
This research was supported by the DOE, Office of Science, Office of Biological and Environmental Research, through the Mercury Scientific Focus Area at Oak Ridge National Laboratory. M.P. was also supported by NIH grants R01HG004857 and R01DE024463. Oak Ridge National Laboratory is managed by UT-Battelle LLC for the U.S. Department of Energy under contract DE-AC05-00OR22725. Author contributions: M.P., S.D.B., C.C.B., A.V.P., and D.A.E. designed the study. M.P., A.C.S., and C.C.B. performed the comparative metagenomic analyses. M.P., A.S., and C.C.G. performed the experiments. M.P., S.D.B., B.R.C., and C.C.B. performed the analyses. M.P., S.D.B., C.C.G., C.C.B., and D.A.E. wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All raw data can be obtained from the JGI Gold Database (http://gold.jgi.doe.gov/) and NCBI Sequence Read Archive (www.ncbi.nlm.nih.gov/sra). Individual sequencing project identifications are listed in column B of table S1.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/1/9/e1500675/DC1
Table S1. List of metagenomic projects with hgcA counts.
Table S2. Identity matrix of sequence similarities.
Fig. S1. Whisker plot distribution of genomic and metagenomic PFam3599 protein hits to various hidden Markov profiles.
Fig. S2. Distribution of HgcA and WL pathway–associated CFeSP encoding genes in methanogenic Archaea.
Fig. S3. Distribution of HgcA, HgcB, and WL pathway–associated CFeSP in genomes of Deltaproteobacteria and Firmicutes.
Fig. S4. Hg methylation assays for M. luminyensis B10.
Fig. S5. Repeat methylation assay for M. luminyensis B10.
Fig. S6. Schematic representation of the domain architectures of HgcA, HgcB, and the fusion HgcAB.
Fig. S7. Hg methylation assays for P. furiosus.
Fig. S8. MEGAN-based co-occurrence profiles of the closest genera-assigned HgcAs based on all metagenomes.
REFERENCES AND NOTES
- 1.Driscoll C. T., Mason R. P., Chan H. M., Jacob D. J., Pirrone N., Mercury as a global pollutant: Sources, pathways, and effects. Environ. Sci. Technol. 47, 4967–4983 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.UNEP, UNEP Global Mercury Assessment 2013: Sources, Emissions, Releases and Environmental Transport (United Nations Environment Programme Chemicals Branch, Geneva, Switzerland, 2013). [Google Scholar]
- 3.Kim N. S., Lee B. K., Blood total mercury and fish consumption in the Korean general population in KNHANES III. Sci. Total Environ. 408, 4841− 4847 (2005). [DOI] [PubMed] [Google Scholar]
- 4.Mahaffey K. R., Clickner R. P., Jeffries R. A., Adult women’s blood mercury concentrations vary regionally in the United States: Association with patterns of fish consumption (NHANES 1999–2004). Environ. Health Perspect. 117, 47–53 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gilmour C. C., Podar M., Bullock A. L., Graham A. M., Brown S. D., Somenahally A. C., Johs A., Hurt R. A. Jr, Bailey K. L., Elias D. A., Mercury methylation by novel microorganisms from new environments. Environ. Sci. Technol. 47, 11810–11820 (2013). [DOI] [PubMed] [Google Scholar]
- 6.Hsu-Kim H., Kucharzyk K. H., Zhang T., Deshusses M. A., Mechanisms regulating mercury bioavailability for methylating microorganisms in the aquatic environment: A critical review. Environ. Sci. Technol. 47, 2441–2456 (2013). [DOI] [PubMed] [Google Scholar]
- 7.Parks J. M., Johs A., Podar M., Bridou R., Hurt R. A. Jr, Smith S. D., Tomanicek S. J., Qian Y., Brown S. D., Brandt C. C., Palumbo A. V., Smith J. C., Wall J. D., Elias D. A., Liang L., The genetic basis for bacterial mercury methylation. Science 339, 1332–1335 (2013). [DOI] [PubMed] [Google Scholar]
- 8.Brown S. D., Gilmour C. C., Kucken A. M., Wall J. D., Elias D. A., Brandt C. C., Podar M., Chertkov O., Held B., Bruce D. C., Detter J. C., Tapia R., Han C. S., Goodwin L. A., Cheng J.-F., Pitluck S., Woyke T., Mikhailova N., Ivanova N. N., Han J., Lucas S., Lapidus A. L., Land M. L., Hauser L. J., Palumbo A. V., Genome sequence of the mercury-methylating strain Desulfovibrio desulfuricans ND132. J. Bacteriol. 193, 2078–2079 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gilmour C. C., Elias D. A., Kucken A. M., Brown S. D., Palumbo A. V., Schadt C. W., Wall J. D., Sulfate-reducing bacterium Desulfovibrio desulfuricans ND132 as a model for understanding bacterial mercury methylation. Appl. Environ. Microbiol. 77, 3938–3951 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Graham A. M., Bullock A. L., Maizel A. C., Elias D. A., Gilmour C. C., Detailed assessment of the kinetics of Hg-cell association, Hg methylation, and methylmercury degradation in several Desulfovibrio species. Appl. Environ. Microbiol. 78, 7337–7346 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu R.-Q., Reinfelder J. R., Hines M. E., Barkay T., Mercury methylation by the methanogen Methanospirillum hungatei. Appl. Environ. Microbiol. 79, 6325–6330 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Benson D. A., Karsch-Mizrachi I., Clark K., Lipman D. J., Ostell J., Sayers E. W., GenBank. Nucleic Acids Res. 40, D48–D53 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Markowitz V. M., Chen I.-M. A., Palaniappan K., Chu K., Szeto E., Grechkin Y., Ratner A., Jacob B., Huang J., Williams P., Huntemann M., Anderson I., Mavromatis K., Ivanova N. N., Kyrpides N. C., IMG: The Integrated Microbial Genomes database and comparative analysis system. Nucleic Acids Res. 40, D115–D122 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.The Human Microbiome Project Consortium , Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eddy S. R., A new generation of homology search tools based on probabilistic inference. Genome Inform. 23, 205–211 (2009). [PubMed] [Google Scholar]
- 16.Merritt K. A., Amirbahman A., Mercury methylation dynamics in estuarine and coastal marine environments—A critical review. Earth Sci. Rev. 96, 54–66 (2009). [Google Scholar]
- 17.Mitchell C. P. J., Gilmour C. C., Methylmercury production in a Chesapeake Bay salt marsh. J. Geophys. Res. 113, G00C04 (2008). [Google Scholar]
- 18.Marvin-DiPasquale M., Windham-Myers L., Agee J. L., Kakouros E., Kieu L. H., Fleck J. A., Alpers C. N., Stricker C. A., Methylmercury production in sediment from agricultural and non-agricultural wetlands in the Yolo Bypass, California, USA. Sci. Total Environ. 484, 288–299 (2014). [DOI] [PubMed] [Google Scholar]
- 19.Zhang H., Feng X., Larssen T., Qiu G., Vogt R. D., In inland China, rice, rather than fish, is the major pathway for methylmercury exposure. Environ. Health Perspect. 118, 1183–1188 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang H., Feng X., Larssen T., Shang L., Li P., Bioaccumulation of methylmercury versus inorganic mercury in rice (Oryza sativa L.) grain. Environ. Sci. Technol. 44, 4499–4504 (2010). [DOI] [PubMed] [Google Scholar]
- 21.AMAP, AMAP Assessment 2011: Mercury in the Arctic (Arctic Monitoring and Assessment Programme, Oslo, Norway, 2011). [Google Scholar]
- 22.Macdonald R. W., Harner T., Fyfe J., Recent climate change in the Arctic and its impact on contaminant pathways and interpretation of temporal trend data. Sci. Total Environ. 342, 5–86 (2005). [DOI] [PubMed] [Google Scholar]
- 23.Schuster P. F., Striegl R. G., Aiken G. R., Krabbenhoft D. P., Dewild J. F., Butler K., Kamark B., Dornblaser M., Mercury export from the Yukon River Basin and potential response to a changing climate. Environ. Sci. Technol. 45, 9262–9267 (2011). [DOI] [PubMed] [Google Scholar]
- 24.Jansson J. K., Taş N., The microbial ecology of permafrost. Nat. Rev. Microbiol. 12, 414–425 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Lehnherr I., St. Louis V. L., Kirk J. L., Methylmercury cycling in High Arctic wetland ponds: Controls on sedimentary production. Environ. Sci. Technol. 46, 10523–10531 (2012). [DOI] [PubMed] [Google Scholar]
- 26.Lehnherr I., St. Louis V. L., Emmerton C. A., Barker J. D., Kirk J. L., Methylmercury cycling in high Arctic wetland ponds: Sources and sinks. Environ. Sci. Technol. 46, 10514–10522 (2012). [DOI] [PubMed] [Google Scholar]
- 27.St. Pierre K. A., Chétélat J., Yumvihoze E., Poulain A. J., Temperature and the sulfur cycle control monomethylmercury cycling in high Arctic coastal marine sediments from Allen Bay, Nunavut, Canada. Environ. Sci. Technol. 48, 2680–2687 (2014). [DOI] [PubMed] [Google Scholar]
- 28.Kirk J. L., St. Louis V. L., Multiyear total and methyl mercury exports from two major sub-Arctic rivers draining into Hudson Bay, Canada. Environ. Sci. Technol. 43, 2254–2261 (2009). [DOI] [PubMed] [Google Scholar]
- 29.Tuorto S. J., Darias P., McGuinness L. R., Panikov N., Zhang T., Häggblom M. M., Kerkhof L. J., Bacterial genome replication at subzero temperatures in permafrost. ISME J. 8, 139–149 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mackelprang R., Waldrop M. P., DeAngelis K. M., David M. M., Chavarria K. L., Blazewicz S. J., Rubin E. M., Jansson J. K., Metagenomic analysis of a permafrost microbial community reveals a rapid response to thaw. Nature 480, 368–371 (2011). [DOI] [PubMed] [Google Scholar]
- 31.Paula F. S., Rodrigues J. L., Zhou J., Wu L., Mueller R. C., Mirza B. S., Bohannan B. J. M., Nüsslein K., Deng Y., Tiedje J. M., Pellizari V. H., Land use change alters functional gene diversity, composition and abundance in Amazon forest soil microbial communities. Mol. Ecol. 23, 2988–2999 (2014). [DOI] [PubMed] [Google Scholar]
- 32.Harris R. C., Rudd J. W. M., Amyot M., Babiarz C. L., Beaty K. G., Blanchfield P. J., Bodaly R. A., Branfireun B. A., Gilmour C. C., Graydon J. A., Heyes A., Hintelmann H., Hurley J. P., Kelly C. A., Krabbenhoft D. P., Lindberg S. E., Mason R. P., Paterson M. J., Podemski C. L., Robinson A., Sandilands K. A., Southworth G. R., St. Louis V. L., Tatem M. T., Whole-ecosystem study shows rapid fish-mercury response to changes in mercury deposition. Proc. Natl. Acad. Sci. U.S.A. 104, 16586–16591 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mehrotra A. S., Sedlak D. L., Decrease in net mercury methylation rates following iron amendment to anoxic wetland sediment slurries. Environ. Sci. Technol. 39, 2564–2570 (2005). [DOI] [PubMed] [Google Scholar]
- 34.Warner K. A., Roden E. E., Bonzongo J.-C., Microbial mercury transformation in anoxic freshwater sediments under iron-reducing and other electron-accepting conditions. Environ. Sci. Technol. 37, 2159–2165 (2003). [DOI] [PubMed] [Google Scholar]
- 35.Han S., Obraztsova A., Pretto P., Choe K.-Y., Gieskes J., Deheyn D. D., Tebo B. M., Biogeochemical factors affecting mercury methylation in sediments of the Venice Lagoon, Italy. Environ. Toxicol. Chem. 26, 655–663 (2007). [DOI] [PubMed] [Google Scholar]
- 36.Ulrich P. D., Sedlak D. L., Impact of iron amendment on net methylmercury export from tidal wetland microcosms. Environ. Sci. Technol. 44, 7659–7665 (2010). [DOI] [PubMed] [Google Scholar]
- 37.Warner K. A., Bonzongo J.-C. J., Roden E. E., Ward G. M., Green A. C., Chaubey I., Lyons W. B., Arrington D. A., Effect of watershed parameters on mercury distribution in different environmental compartments in the Mobile Alabama River Basin, USA. Sci. Total Environ. 347, 187–207 (2005). [DOI] [PubMed] [Google Scholar]
- 38.Gies E. A., Konwar K. M., Beatty J. T., Hallam S. J., Illuminating microbial dark matter in meromictic Sakinaw Lake. Appl. Environ. Microbiol. 80, 6807–6818 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sunderland E. M., Mercury exposure from domestic and imported estuarine and marine fish in the U.S. seafood market. Environ. Health Perspect. 115, 235–242 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mason R. P., Choi A. L., Fitzgerald W. F., Hammerschmidt C. R., Lamborg C. H., Soerensen A. L., Sunderland E. M., Mercury biogeochemical cycling in the ocean and policy implications. Environ. Res. 119, 101–117 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sunderland E. M., Krabbenhoft D. P., Moreau J. W., Strode S. A., Landing W. M., Mercury sources, distribution, and bioavailability in the North Pacific Ocean: Insights from data and models. Global Biogeochem. Cycles 23, GB2010 (2009). [Google Scholar]
- 42.Blum J. D., Popp B. N., Drazen J. C., Choy C. A., Johnson M. W., Methylmercury production below the mixed layer in the North Pacific Ocean. Nat. Geosci. 6, 879–884 (2013). [Google Scholar]
- 43.Lamborg C., Bowman K., Hammerschmidt C., Gilmour C., Munson K., Selin N., Tseng C.-M., Mercury in the anthropocene ocean. Oceanography 27, 76–87 (2014). [Google Scholar]
- 44.Lehnherr I., St. Louis V. L., Kirk J. L., Methylation of inorganic mercury in polar marine waters. Nat. Geosci. 4, 298–302 (2011). [Google Scholar]
- 45.Monperrus M., Tessier E., Amouroux D., Leynaert A., Huonnic P., Donard O. F. X., Mercury methylation, demethylation and reduction rates in coastal and marine surface waters of the Mediterranean Sea. Mar. Chem. 107, 49–63 (2007). [Google Scholar]
- 46.Hawley A. K., Brewer H. M., Norbeck A. D., Paša-Tolić L., Hallam S. J., Metaproteomics reveals differential modes of metabolic coupling among ubiquitous oxygen minimum zone microbes. Proc. Natl. Acad. Sci. U.S.A. 111, 11395–11400 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Smith S. D., Bridou R., Johs A., Parks J. M., Elias D. A., Hurt R. A. Jr, Brown S. D., Podar M., Wall J. D., Site-directed mutagenesis of HgcA and HgcB reveals amino acid residues important for mercury methylation. Appl. Environ. Microbiol. 81, 3205–3217 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Larose C., Dommergue A., De Angelis M., Cossa D., Averty B., Marusczak N., Soumis N., Schneider D., Ferrari C., Springtime changes in snow chemistry lead to new insights into mercury methylation in the Arctic. Geochim. Cosmochim. Acta 74, 6263–6275 (2010). [Google Scholar]
- 49.Weber J. H., Review of possible paths for abiotic methylation of mercury(II) in the aquatic environment. Chemosphere 26, 2063–2077 (1993). [Google Scholar]
- 50.Yin Y., Li Y., Tai C., Cai Y., Jiang G., Fumigant methyl iodide can methylate inorganic mercury species in natural waters. Nat. Commun. 5, 4633 (2014). [DOI] [PubMed] [Google Scholar]
- 51.Diaz R. J., Rosenberg R., Spreading dead zones and consequences for marine ecosystems. Science 321, 926–929 (2008). [DOI] [PubMed] [Google Scholar]
- 52.Mosher J. J., Vishnivetskaya T. A., Elias D. A., Podar M., Brooks S. C., Brown S. D., Brandt C. C., Palumbo A. V., Characterization of the Deltaproteobacteria in contaminated and uncontaminated stream sediments and identification of potential mercury methylators. Aquat. Microb. Ecol. 66, 271–282 (2012). [Google Scholar]
- 53.Vishnivetskaya T. A., Mosher J. J., Palumbo A. V., Yang Z. K., Podar M., Brown S. D., Brooks S. C., Gu B., Southworth G. R., Drake M. M., Brandt C. C., Elias D. A., Mercury and other heavy metals influence bacterial community structure in contaminated Tennessee streams. Appl. Environ. Microbiol. 77, 302–311 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brooks S. C., Southworth G. R., History of mercury use and environmental contamination at the Oak Ridge Y-12 plant. Environ. Pollut. 159, 219–228 (2011). [DOI] [PubMed] [Google Scholar]
- 55.Porat I., Vishnivetskaya T. A., Mosher J. J., Brandt C. C., Yang Z. K., Brooks S. C., Liang L., Drake M. M., Podar M., Brown S. D., Palumbo A. V., Characterization of archaeal community in contaminated and uncontaminated surface stream sediments. Microb. Ecol. 60, 784–795 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dong W., Liang L., Brooks S., Southworth G., Gu B., Roles of dissolved organic matter in the speciation of mercury and methylmercury in a contaminated ecosystem in Oak Ridge, Tennessee. Environ. Chem. 7, 94–102 (2010). [Google Scholar]
- 57.Balogh S. J., Nollet Y. H., Methylmercury input to the Mississippi River from a large metropolitan wastewater treatment plant. Sci. Total Environ. 406, 145–153 (2008). [DOI] [PubMed] [Google Scholar]
- 58.Bodaly R. A., Rudd W. M., Flett R. J., Effect of urban sewage treatment on total and methyl mercury concentrations in effluents. Biogeochemistry 40, 279–291 (1998). [Google Scholar]
- 59.Gilmour C. C., Bloom N. S., A case study of mercury and methylmercury dynamics in a Hg-contaminated municipal waste-water treatment-plant. Water Air Soil Pollut. 80, 799–803 (1995). [Google Scholar]
- 60.Shih R., Robertson W. D., Schiff S. L., Rudolph D. L., Nitrate controls methyl mercury production in a streambed bioreactor. J. Environ. Qual. 40, 1586–1592 (2011). [DOI] [PubMed] [Google Scholar]
- 61.Zhao X., Rockne K. J., Drummond J. L., Aeration prevents methyl mercury production in dental wastewater. J. Environ. Sci. Health 47, 598–604 (2012). [DOI] [PubMed] [Google Scholar]
- 62.Dridi B., Fardeau M.-L., Ollivier B., Raoult D., Drancourt M., Methanomassiliicoccus luminyensis gen. nov., sp nov., a methanogenic archaeon isolated from human faeces. Int. J. Syst. Evol. Microbiol. 62, 1902–1907 (2012). [DOI] [PubMed] [Google Scholar]
- 63.Dridi B., Henry M., Richet H., Raoult D., Drancourt M., Age-related prevalence of Methanomassiliicoccus luminyensis in the human gut microbiome. APMIS 120, 773–777 (2012). [DOI] [PubMed] [Google Scholar]
- 64.Karlsson F. H., Tremaroli V., Nookaew I., Bergström G., Behre C. J., Fagerberg B., Nielsen J., Bäckhed F., Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature 498, 99–103 (2013). [DOI] [PubMed] [Google Scholar]
- 65.Nielsen H. B., Almeida M., Juncker A. S., Rasmussen S., Li J., Sunagawa S., Plichta D. R., Gautier L., Pedersen A. G., Le Chatelier E., Pelletier E., Bonde I., Nielsen T., Manichanh C., Arumugam M., Batto J.-M., Quintanilha Dos Santos M. B., Blom N., Borruel N., Burgdorf K. S., Boumezbeur F., Casellas F., Doré J., Dworzynski P., Guarner F., Hansen T., Hildebrand F., Kaas R. S., Kennedy S., Kristiansen K., Kultima J. R., Léonard P., Levenez F., Lund O., Moumen B., Le Paslier D., Pons N., Pedersen O., Prifti E., Qin J., Raes J., Sørensen S., Tap J., Tims S., Ussery D. W., Yamada T.; MetaHIT Consortium, Renault P., Sicheritz-Ponten T., Bork P., Wang J., Brunak S., Ehrlich S. D.; MetaHIT Consortium , Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes. Nat. Biotechnol. 32, 822–828 (2014). [DOI] [PubMed] [Google Scholar]
- 66.Borrel G., Harris H. M., Parisot N., Gaci N., Tottey W., Mihajlovski A., Deane J., Gribaldo S., Bardot O., Peyretaillade E., Peyret P., O’Toole P. W., Brugère J.-F., Genome sequence of “Candidatus Methanomassiliicoccus intestinalis” Issoire-Mx1, a third Thermoplasmatales-related methanogenic archaeon from human feces. Genome Announc. 1, e00453-13 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Limper U., Knopf B., König H., Production of methyl mercury in the gut of the Australian termite Mastotermes darwiniensis. J. Appl. Entomol. 132, 168–176 (2008). [Google Scholar]
- 68.Woyke T., Teeling H., Ivanova N. N., Huntemann M., Richter M., Gloeckner F. O., Boffelli D., Anderson I. J., Barry K. W., Shapiro H. J., Symbiosis insights through metagenomic analysis of a microbial consortium. Nature 443, 950–955 (2006). [DOI] [PubMed] [Google Scholar]
- 69.Huson D. H., Mitra S., Ruscheweyh H.-J., Weber N., Schuster S. C., Integrative analysis of environmental sequences using MEGAN4. Genome Res. 21, 1552–1560 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bae H.-S., Dierberg F. E., Ogram A., Syntrophs dominate sequences associated with the mercury methylation–related gene hgcA in the water conservation areas of the Florida everglades. Appl. Environ. Microbiol. 80, 6517–6526 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lindahl P. A., Chang B., The evolution of acetyl-CoA synthase. Orig. Life Evol. Biosph. 31, 403–434 (2001). [DOI] [PubMed] [Google Scholar]
- 72.AMAP/UNEP, Technical Background Report for the Global Mercury Assessment 2013 (UNEP Chemicals Branch, Geneva, Switzerland, 2013). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/1/9/e1500675/DC1
Table S1. List of metagenomic projects with hgcA counts.
Table S2. Identity matrix of sequence similarities.
Fig. S1. Whisker plot distribution of genomic and metagenomic PFam3599 protein hits to various hidden Markov profiles.
Fig. S2. Distribution of HgcA and WL pathway–associated CFeSP encoding genes in methanogenic Archaea.
Fig. S3. Distribution of HgcA, HgcB, and WL pathway–associated CFeSP in genomes of Deltaproteobacteria and Firmicutes.
Fig. S4. Hg methylation assays for M. luminyensis B10.
Fig. S5. Repeat methylation assay for M. luminyensis B10.
Fig. S6. Schematic representation of the domain architectures of HgcA, HgcB, and the fusion HgcAB.
Fig. S7. Hg methylation assays for P. furiosus.
Fig. S8. MEGAN-based co-occurrence profiles of the closest genera-assigned HgcAs based on all metagenomes.