

The first isolable gold(III)-CO complex reveals unexpected differences in CO bonding to gold and platinum catalysts.
Keywords: Gold, Catalysis, Carbonyl complex, mechanisms, water-gas shift, Organometallic, pincer ligand, DFT, CO bonding
Abstract
The water-gas shift (WGS) reaction is an important process for the generation of hydrogen. Heterogeneous gold catalysts exhibit good WGS activity, but the nature of the active site, the oxidation state, and competing reaction mechanisms are very much matters of debate. Homogeneous gold WGS systems that could shed light on the mechanism are conspicuous by their absence: gold(I)–CO is inactive and gold(III)–CO complexes were unknown. We report the synthesis of the first example of an isolable CO complex of Au(III). Its reactivity demonstrates fundamental differences between the CO adducts of the neighboring d8 ions Pt(II) and Au(III): whereas Pt(II)-CO is stable to moisture, Au(III)–CO compounds are extremely susceptible to nucleophilic attack and show WGS reactivity at low temperature. The key to understanding these dramatic differences is the donation/back-donation ratio of the M–CO bond: gold-CO shows substantially less back-bonding than Pt-CO, irrespective of closely similar ν(CO) frequencies. Key WGS intermediates include the gold-CO2 complex [(C^N^C)Au]2(μ-CO2), which reductively eliminates CO2. The species identified here are in accord with Au(III) as active species and a carboxylate WGS mechanism.
INTRODUCTION
The water-gas shift (WGS) reaction is an important process for the industrial generation of hydrogen, as well as for improving the purity of H2 for fuel cell applications by removing CO (1). Because the reaction is exothermic (CO + H2O → CO2 + H2, ΔHr = −41.2 kJ/mol), it is favored by lower reaction temperatures, which has encouraged the development of low-temperature catalysts. Heterogeneous catalysts including those based on platinum and gold supported on various metal oxides show good activity (2, 3), with gold exhibiting a significantly lower activation energy than platinum (4, 5). However, the mechanism of the WGS reaction is as yet poorly understood, with redox, formate, and carboxylate pathways being discussed (2, 3, 6–8). There is uncertainty in particular about the nature of the active species: its oxidation state, whether it is dispersed and mononuclear, or whether it is a metal nanoparticle or a metal ion. Song and Hensen (8) have summarized the mechanistic complexity and favor the involvement of gold nanoclusters, whereas Flytzani-Stephanopoulos and colleagues put forward cogent arguments for dispersed gold in high oxidation states, formulated as Au(O)x(OH)y(Na)z with gold in an octahedral coordination geometry (9, 10).
Homogeneous complexes offer obvious advantages for the study of reaction mechanisms. Studies on homogeneous WGS systems go back to Hieber’s work on iron carbonyls (11); this and later work on Ru and other metal carbonyls (12) suggested the predominance of the carboxylate mechanism (Fig. 1).
Fig. 1. Principles of the carboxylate mechanism, based on the classical work on iron and ruthenium carbonyls (11, 12).
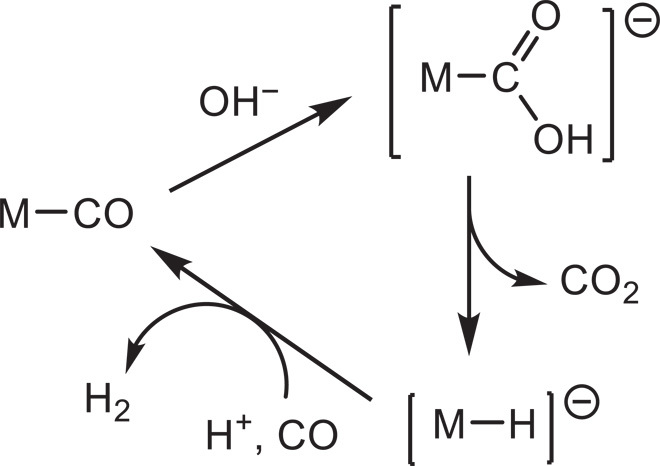
However, no such studies exist for gold catalysts. Until now, well-defined gold–CO complexes that could be used for mechanistic investigations were only known for gold(I), for example, AuCl(CO) (13, 14) and the cations [LAuCO]+ (L = phosphine or carbene ligand) (15–19). No WGS activity has been reported for any of these. By contrast, isolable CO complexes of Au(III) were unknown; hence, their reactivity could not be explored.
The lack of Au(III)–CO complexes is surprising. Au(III) and Pt(II) are isoelectronic d8 ions that typically give isostructural complexes. Carbonyl complexes of Pt(II) have been known since the 19th century; they were the first transition metal–CO complexes ever made (20). Evidently, the platinum-gold analogy, which generally serves well, does not extend to CO adducts. However, although Pt(II) carbonyls show characteristically high ν(CO) frequencies, which suggest electrophilic carbonyl C atoms (21, 22), they have been proven to be stable to the attack of water and do not show WGS-type reactivity (23, 24).
Given the strongly positive redox potential of Au3+ (E0 = 1.52 V) and the reducing power of CO, the absence of isolable Au(III) carbonyl derivatives seemed entirely plausible. However, we found that CO complexes of Au(III) are readily accessible through the appropriate choice of stabilizing ligands and report here the synthesis of [(C^N^C)Au–CO]+X− salts, where X is a noncoordinating anion. The cyclometalated C^N^C pincer ligand [(C^N^C) = 2,6-bis(4-ButC6H3)2pyridine dianion] had previously enabled the isolation of Au(III) hydride, alkene, and peroxide complexes (25–27). Moreover, these Au(III)–CO complexes show facile WGS-type reactivity, in stark contrast to their Pt(II) congeners.
RESULTS
Treatment of (C^N^C)AuOAcF (OAcF = trifluoroacetate) with B(C6F5)3 in dichloromethane at −30°C gave a single product in quantitative yield [by 1H NMR (nuclear magnetic resonance) spectroscopy], formulated as [(C^N^C)Au]+[(C6F5)3BOAcF] (1). This intermediate is sufficiently stable at −20°C to permit subsequent reactions with weak donor ligands. Thus, treatment with CO gas cleanly generated [(C^N^C)Au–CO]+[(C6F5)3BOAcF]− (2a), which can be precipitated with light petroleum and was isolated as a yellow microcrystalline solid (Fig. 2). The 13C-labeled analog [(C^N^C)Au–13CO]+ (2a-13C) was similarly obtained. The same product was obtained when the ethylene complex [(C^N^C)Au(C2H4)]+[(C6F5)3BOAcF]− (26) was exposed to CO at −20°C. The ligand exchange process is monitored by 1H NMR spectroscopy (fig. S6). Over a period of 30 min, the signal for bound ethylene at δ 6.25 disappeared, accompanied by spectral changes confirming the quantitative formation of the CO complex. The hexafluorophosphate salt 2b was prepared by reacting (C^N^C)AuOAcF with [Ph3C][PF6] and CO in CH2Cl2 at −30°C. The CO complexes 2a, b are temperature-sensitive and must be handled at temperatures lower than −10°C.
Fig. 2. Synthetic routes to Au(III)-CO complexes.
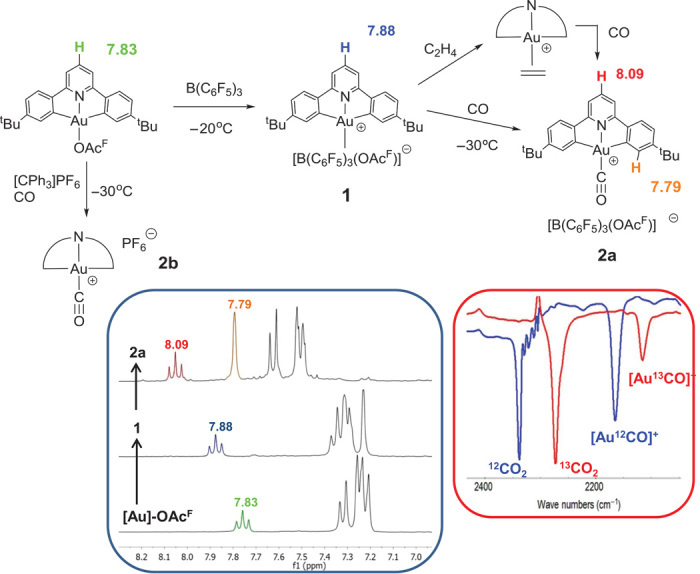
Left inset shows the diagnostic aromatic region of the 1H NMR spectra of (C^N^C)AuOAcF (bottom), intermediate [(C^N^C)Au]+ [(C6F5)3BOAcF] (1) (middle), and product [(C^N^C)Au-CO]+ [(C6F5)3BOAcF] (2a) (top) (300 MHz, CD2Cl2, −30°C), confirming quantitative generation of the CO complex. Right inset shows the CO stretching bands of 2a and of 2a-13C in CH2Cl2 solution at −20°C, accompanied by the corresponding bands for 12CO2 and 13CO2, respectively, resulting from the reaction of the Au(III)-CO complex with traces of moisture.
These reactions can be conveniently monitored by 1H NMR spectroscopy (Fig. 2). The signals in the aromatic region of the spectrum are highly diagnostic, in particular the triplet resonance of the H atom in the para position of the pyridine ring, and they confirm that, within detection limits, the reactions are clean and quantitative. The 13C NMR signal of coordinated CO in 2a, b is observed at δ 167.6 (cf. δ 184 for free CO).
The most sensitive tools for probing the nature of the Au(III)–CO bond in these compounds are vibrational spectroscopy and chemical reactivity. The νCO stretching frequency of 2a is observed at 2167 cm−1, compared to 2143 cm−1 of free 12CO. The νCO stretch of the 13C-labeled version 2a-13C is found at 2143 cm−1. The value found for 2a is close to that of CO bound to Au3+ centers in titania-supported heterogeneous gold–CO oxidation catalysts (2158 cm−1) (28), which suggests that the CO bonding in our complexes closely mirrors that found in heterogeneous catalysts (29).
On the other hand, the differences in infrared (IR) parameters between structurally related Pt(II) and Au(III) are more pronounced; for example, the Pt(II) pincer complex [(N^N^C)Pt–CO]+ shows a ν(CO) vibration at 2094 cm−1 (24), that is, substantially lower than that of 2 [(N^N^C) = 2-(C5H3N)-6-(C6H4)pyridine]. Evidently, the back-bonding contribution in Au(III)-CO is significantly weaker than that in its Pt(II) congeners. This is reflected in dramatic differences in chemical reactivity: whereas the [(N^N^C)Pt–CO]+ compound could be recrystallized from boiling methanol, the Au complexes 2a, b are highly sensitive to temperature and nucleophilic attack on CO (vide infra).
To explain the reactivity differences between the carbonyls of Pt(II) and Au(III), we probed the nature of the Au–CO interaction by density functional theory (DFT) calculations. Simulation of the [(C^N^C)Au(CO)]+ cation revealed that the highest occupied molecular orbital (HOMO) shows no electron density in the Au–C region (Fig. 3), and there is also no evidence for an Au–C π-bonding contribution in other high-energy occupied orbitals, that is, HOMO-1, HOMO-2, and HOMO-3 (fig. S20). The lowest unoccupied molecular orbital (LUMO) does show π-symmetry around the Au–C vector but is some 0.138 Ha higher in energy than the HOMO (for comparison, the HOMO-1 is only 0.021 Ha below the HOMO).
Fig. 3. Molecular orbitals involved in Au-CO bonding, showing, from left to right, HOMO-1, HOMO, and LUMO in [(C^N^C)Au(CO)]+ as simulated by DFT.
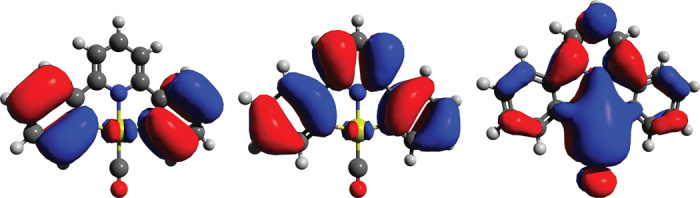
None of the lower-lying occupied orbitals shows any π-bonding interactions along the Au–CO vector.
Natural bond orbital analysis classifies the Au–C interaction as a single bond, with major contributions from gold derived from the 6s, 6py, and 6dx2y2 molecular orbitals. Support for formulating the Au–CO interaction as a single σ-bond was provided by further analysis of the DFT structure. The donation/back-donation (d/b) ratio, as estimated by charge decomposition analysis (CDA), proved to be particularly informative: [(C^N^C)Au(CO)]+ shows a d/b ratio of 2.26, compared to a value of only 1.54 [(N^N^C)Pt(CO)]+, in line with a relatively stronger back-bonding in Pt(II)–CO complexes.
This view is further reinforced by the bond analysis of cis-PtCl2(CO)2, a complex that shows νCO stretching frequencies of 2178 and 2137 cm−1 (νs and νas, respectively, in benzene solution) (22), closely comparable to the value of 2167 cm−1 found for 2. However, the d/b ratio of PtCl2(CO)2 is only 0.65; that is, despite the high stretching frequency, the back-donation from Pt(II) to CO is relatively much stronger than that in Au(III). Although high CO frequencies above those found for free CO (2143 cm−1) are generally taken as diagnostic for high electrophilicity, it is the d/b ratio, rather than the νCO value, that best explains the chemical reactivity.
This difference in d/b ratios between the two d8 systems Pt(II) and Au(III) has consequences. The low-temperature solution IR spectra of 2a and 2a-13C in CH2Cl2 were always accompanied by bands at 2338 cm−1 for 12CO2 and 2273 cm−1 for 13CO2 (Fig. 2). Because the CO2 must have originated from the CO complex, this observation pointed to a gold-mediated WGS reaction due to the presence of traces of moisture condensation under the recording conditions (−20°C), an indication of the facile nucleophilic attack by water on the cationic gold–CO complex, in contrast to Pt(II) carbonyls. To demonstrate the reaction pathways of the Au(III)–CO system, we decided to use (C^N^C)AuOH (30) as a surrogate for water, because this would allow precise stoichiometry control and facilitate the reaction monitoring by NMR spectroscopy. If WGS reactions were observed, the process would of course lead to the formation of (C^N^C)Au–H instead of H2, and again, this gold hydride gives a unique NMR signature (25). The expected reactions are summarized in Fig. 4. Reactions A, B, and C are part of a WGS cycle according to the carboxylate mechanism.
Fig. 4. Gold(III)-mediated WGS reactions showing reaction steps A to C, the formation of the Au(III)-CO2 complex 3 via pathway D, and the reductive elimination of CO2 from 3 (step E).

The colors indicate the H atoms that are diagnostic for monitoring these processes by 1H NMR spectroscopy, with associated chemical shifts. In the molecular structure of [(C^N^C)Au]2(μ-κC:κO-CO2)·∙C6H6 (3·∙C6H6), thermal ellipsoids are set at 50% probability level and hydrogen atoms and the solvent were omitted for clarity. The CO2 group is 50% disordered between two positions; the box shows the central core of the other position. Selected bond distances (in angstoms) and angles (in degrees) are as follows: Au(1)-C(26) 2.11(1); C(26)-O(1a) 1.18(1); C(26)-O(2′) 1.29(2); O(2′)-Au(1′) 2.036(9); Au(1)-N(1) 1.999(6); Au(1)-C(1) 2.064(8); Au(1)-C(17) 2.068(8); N(1)-Au(1)-C(26) 169.5(5); Au(1)-C(26)-O(1a) 129(1); Au(1)-C(26)-O(2′) 104(1); O(1a)-C(26)-O(2) 126(1); Au(1′)-O(2′)-C(26) 114(1); N(1′)-Au(1′)-O(2′) 160.5(3).
Bubbling CO through a solution of (C^N^C)AuOH for 30 s at room temperature, followed by the replacement of excess CO by N2, does indeed generate the hydride (C^N^C)AuH, in agreement with reaction steps A and B. This sequence implies the formation of an unstable carboxylate intermediate (C^N^C)Au–COOH, which readily decomposes by β-H elimination, to give CO2 and (C^N^C)AuH (Fig. 4).
Hydrolysis of the gold(III) hydride, with liberation of H2 and regeneration of (C^N^C)AuOH (reaction C), would close the cycle. However, this step cannot proceed under the neutral reaction conditions used for the NMR experiments because the Au–H bond in (C^N^C)AuH is highly covalent; the complex is stable to water and mild acids. DFT calculations confirm the observed reactivity, with enthalpy values of −141 and −28 kJ mol−1 for reaction steps A and B, respectively, whereas C is endothermic (+95 kJ mol−1).
A different outcome was observed when a benzene solution of (C^N^C)AuOH was exposed to CO and left to crystallize in the dark for 20 hours. The yellow crystalline product was identified as the CO2 complex (C^N^C)Au(μ-κC:κO-CO2)Au(C^N^C)·C6H6 (3·C6H6). The formation can be explained by the reaction of (C^N^C)Au-COOH with (C^N^C)AuOH with elimination of water, reaction D (Fig. 4), with a calculated reaction enthalpy of −31 kJ mol−1. The same product was obtained by treating the gold(III) oxide {(C^N^C)Au}2(μ-O) with CO; this reaction proceeds even with the crystalline oxide in the solid state. Such reactivity is central to CO oxidation with heterogeneous gold catalysts (28). Complex 3 is the first CO2 complex of gold in any oxidation state.
The crystal structure of 3·C6H6 confirmed the presence of a bridging CO2 ligand that adopts a μ-κC:κ2O position between two Au(III) centers (Fig. 4). There is disorder because CO2 gives two linkage isomers, each with 50% occupancy, similar to that described before for complex [2,6-C6H3(CH2PiPr2)2Pd]2(μ-CO2) (31). The Au–C bond to CO2, 2.11(1) Å, is long compared to conventional Au–C(sp3) and Au–C(sp2) bonds [for comparison, the bonds to ethyl and aryl ligands in (C^N^C)AuEt and (C^N)Au(C6H4F)(OAcF) are 2.042(8) and 2.029(7) Å, respectively (32)]. The short C═O (1.18 Å) and long C–O (1.29 Å) distances, together with the interatomic angles, point to a low degree of charge delocalization in the CO2 ligand.
Complex 3 is stable under ambient conditions in the solid state. However, heating the solid to 80° to 120°C under vacuum leads to the reductive elimination of CO2 and formation of the known (25, 27, 33) thermally stable Au(II) dimer, [Au(C^N^C)]2 (Fig. 4). The CO2 elimination also proceeds easily in solution, and even though it forms a gold(II) product, it is calculated to be exothermic, ΔH = −96 kJ mol−1.
Computational models of the WGS process on gold nanoclusters tend to assume that the CO2 product is weakly C-bonded to a gold atom before desorption (6, 8). The binuclear CO2 bonding in 3 would seem to suggest the possibility of alternative bonding modes and the involvement of gold species in higher oxidation states during this process.
The evidence for the Au–COOH intermediate mainly relies on the observation of the follow-on products, 3 and (C^N^C)AuH. This pathway gains support, however, by the analogous reaction of CO with the alkoxide (C^N^C)AuOMe (30), which gives the methyl carboxylate (C^N^C)Au-COOMe (4) as a white crystalline solid. Compound 4 is thermally stable, and there is no reaction of 4 with excess (C^N^C)AuOH to produce 3 and MeOH.
As an alternative to the carboxylate pathway in the WGS reaction, a formate pathway has been suggested, where CO formally inserts into the O–H bond of Au–OH to give Au–OC(O)H (7, 8). Alternatively, formic acid may be generated by hydrogenolysis of a metal–COOH species. The search for this reaction pathway was, however, unsuccessful for our system: Complex 4 is stable under 4 bar of H2 up to 80°C without any sign for the presence of methyl formate. The attempted hydrogenolysis of 3 (4 bar of H2) exclusively led to the reductive elimination of CO2. In any case, in the present system, the hydrogenolysis of Au–COOH to Au–H + HCOOH is effectively thermoneutral (by DFT, ΔH = −4 kJ mol−1).
This work has shown that, given suitable supporting ligands, CO complexes of Au(III) can indeed be isolated, nearly 150 years after the preparation of the isoelectronic Pt(II) analogs. This has allowed a detailed comparison of their reactivity. The susceptibility to nucleophilic attack and DFT modeling suggest a minimal contribution by back-donation to the Au–CO bond, which contrasts with structurally related Pt(II)–CO complexes. The d/b ratio, obtained by way of CDA, proved to be a more reliable indicator of chemical reactivity than the νCO frequencies and explains subtle but important differences in metal–CO bonding between the neighboring elements platinum and gold. The result is WGS-type reactivity in the gold system at low temperature and the absence of such reactions for platinum(II) carbonyls. These studies have enabled the use of a homogeneous gold system to probe the viability of the carboxylic mechanism proposed for the WGS reaction catalyzed on gold surfaces and provide support for gold(III) ions in this process.
MATERIALS AND METHODS
General
Unless otherwise stated, all manipulations were performed using standard Schlenk techniques under dry nitrogen or using Saffron Scientific or MBRAUN glove boxes. Nitrogen was purified by passing through columns of supported P2O5 with moisture indicator and activated 4 Å molecular sieves. Anhydrous solvents were freshly distilled from appropriate drying agents. (C^N^C)AuOH (30), (C^N^C)Au(OAcF) (30), [(C^N^C)Au]2O (27), (C^N^C)AuH (25), [(C^N^C)Au]2 (25), [(C^N^C)Au(η2-C2H4)][B(C6F5)3(OAcF)] (26), and B(C6F5)3 (34) were prepared using literature methods. [Ph3C][PF6] (Sigma) was used as purchased. Natural abundance CO (BOC) and 13CO (Euriso-Top) were used as purchased or dried before use passing through columns with activated 4 Å molecular sieves.
1H, 13C{1H}, and 19F spectra were recorded using a Bruker Avance DPX-300 or a Bruker Avance DPX-500 spectrometer. Deuterated solvents were dried over CaH2, degassed by three freeze-pump-thaw cycles, and stored on 4 Å molecular sieves before use. 1H NMR spectra (300.13 MHz) were referenced to the residual protons of the deuterated solvent used. 13C{1H} NMR spectra (75.47 MHz) were referenced internally to the D-coupled 13C resonances of the NMR solvent. IR spectra were recorded using a Perkin Elmer Spectrum 65 FT-IR spectrometer with a diamond attenuated total reflectance attachment or using liquid cells with KBr plates.
Synthesis of [(C^N^C)Au][B(C6F5)3OAcF] (1)
(C^N^C)AuOAcF (15 mg, 23.6 μmol) and B(C6F5)3 (12 mg, 23.6 μmol) were charged into a J. Young NMR tube and cooled to −78°C. To this, we added precooled (−78°C) CD2Cl2 (0.6 ml), generating a yellow solution. The sample was inserted into an NMR spectrometer probe head precooled to −40°C. Obvious signs of decomposition were observed above −20°C by 1H NMR spectroscopy, which coincided with a darkening of the solution from yellow to brown. The numbering system used for assigning the 1H and 13C NMR signals of the C^N^C ligand is as follows:
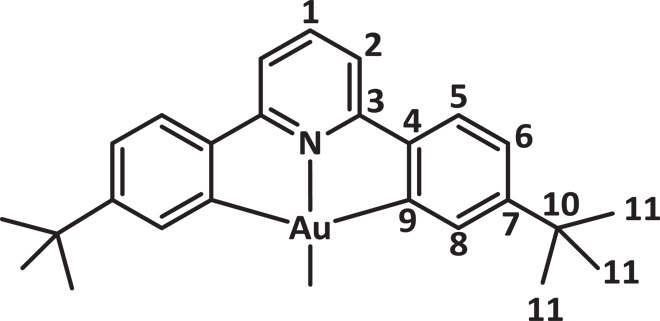
1H NMR (fig. S1, 300 MHz, CD2Cl2, −25°C) δ 7.88 (t, J = 8.0 Hz, 1H, H1), 7.41 to 7.33 (m, 5H), 7.23 (d, J = 1.0 Hz, 2H, H8), 1.22 (s, 18H, H11). 13C{1H} NMR (75 MHz, CD2Cl2, −25°C) δ 163.92 (C3), 157.65 (C7), 144.65 (C4), 144.42 (C1), 127.14 (C8), 126.57 (C5), 118.05 (C2), 35.69 (C10), 30.54 (C11). Resonances for C9, C2, and the B(C6F5)3OAcF anion were not observed because of the short acquisition time. 19F NMR (282 MHz, C6D6) δ −76.96 (s, 3F, OAcF), −135.08 (d, 6F, J = 22.5 Hz, o-C6F5), −161.47 (t, 3F, J = 20.3 Hz, p-C6F5), −166.38 to −166.56 (m, 6F, m-C6F5).
Synthesis of [(C^N^C)AuCO][B(C6F5)3OAcF] (2a)
Method 1
(C^N^C)AuOAcF (100 mg, 0.16 mmol) and B(C6F5)3 (80.5 mg, 0.16 mmol) were charged into a Schlenk flask and cooled to −78°C. To this, we added CH2Cl2 (30 ml) precooled to −78°C. The mixture was warmed to −30°C and CO gas was bubbled through the mixture for a few seconds. The mixture was kept at −30°C for 1 hour. While keeping the mixture below −10°C, the solvent was evaporated, yielding 2a as a bright yellow powder.
Sample preparation for IR spectroscopy: an aliquot of a CH2Cl2 solution of 2a was injected into a liquid IR cell that was precooled with dry ice. The IR spectrum was recorded immediately (fig. S2). The compound proved thermally too labile for elemental analysis.
Method 2
In a J. Young NMR tube, [(C^N^C)Au][B(C6F5)3OAcF] (1) was generated as described above by mixing (C^N^C)AuOAcF (15 mg, 23.6 μmol) with B(C6F5)3 (12 mg, 23.6 μmol) at −50°C. The 1H NMR spectrum of 1 was then recorded at −40°C. 12CO or 13CO was bubbled through the solution for a few seconds, and the sample was injected into the NMR spectrometer precooled to −40°C. The conversion from 1 to 2a could be monitored over the course of 2 hours at −20°C (figs. S3 to S5).
Method 3
In a J. Young NMR tube, [(C^N^C)Au][B(C6F5)3OAcF] (1) was generated as described above by mixing (C^N^C)AuOAcF (15 mg, 23.6 μmol) with B(C6F5)3 (12 mg, 23.6 μmol) at −50°C. The 1H NMR spectrum of 1 was recorded at −40°C. Ethylene was then added to generate [(C^N^C)Au(C2H4)][B(C6F5)3OAcF], and the mixture was treated with CO gas for a few seconds at −40°C. The solution was transferred into the NMR spectrometer probe precooled to −20°C, and the conversion of [(C^N^C)Au(C2H4)]+ to [(C^N^C)Au(CO)]+ was monitored (fig. S6).
1H NMR (300 MHz, CD2Cl2, −20°C) δ 8.05 (t, J = 8 Hz, 1H, H1), 7.79 (d, J = 2 Hz, 2H, H8), 7.62 (d, J = 8 Hz, 2H, H5), 7.52 to 7. 48 (dd, J = 8, 2Hz, H2 overlapping with d, J = 8 Hz, H6, 4H), 1.34 (s, 18H). 13C{1H} NMR (75 MHz, CD2Cl2, −20°C) δ 168.22 (C9), 167.35 (AuCO), 166.67 (C3), 159.25 (C7), 146.22 (C4), 144.78 (C1), 133.91 (C8), 127.17 (C5), 126.99 (C6), 118.74 (C2), 35.76 (C10), 30.55 (C11). Resonances for the [B(C6F5)3OAcF]− anion were not observed because of the short acquisition time. 19F NMR (282 MHz, CD2Cl2, −40°C) δ −76.68 (s, 3F, OAcF), −133.56 (br s, 3F, o-C6F5), −159.36 (br s, 3F, p-C6F5), −164.86 (br s, 6F, m-C6F5). IR (CH2Cl2 solution): ν(12CO) 2167 cm−1.
Synthesis of [(C^N^C)Au(13CO)][B(C6F5)3OAcF] (2a-13C)
The same conditions used for the synthesis of 2a were applied, but 13CO was used instead of 12CO. The complex shows the same spectroscopic pattern to 2a except for a clear higher intensity of the signal at 167.35 parts per million (ppm) in the 13C NMR and a shift of the ν(CO) band in the IR spectrum. IR (CH2Cl2 solution): ν(13CO) 2143 cm−1 (fig. S2).
Synthesis of [(C^N^C)AuCO][PF6] (2b)
(C^N^C)AuOAcF (100 mg, 0.16 mmol) and [Ph3C][PF6] (62 mg, 0.16 mmol) were charged into a Schlenk flask and cooled to −78°C. To this, we added CH2Cl2 (30 ml) precooled to −78°C. The mixture was warmed to −30°C and CO gas was bubbled through the mixture for a few seconds. The mixture was kept at −30°C for 1 hour. While keeping the mixture below −20°C, the CH2Cl2 solution was layered with light petroleum (1:1 v/v) and was stored at −20°C. The spectroscopic parameters were identical to those of 2a, but the compound proved thermally too unstable to allow isolation.
Synthesis of [(C^N^C)Au]2(μ-κC:κO-CO2) (3)
Method 1
CO gas was bubbled for 30 s through a solution of (C^N^C)AuOH (100 mg, 0.18 mmol) in benzene (30 ml). The solution was then left to stand in the dark for 20 hours, yielding 3 as dark-yellow crystals (41 mg, 40%). The crystals were suitable for x-ray diffraction.
1H NMR (500 MHz, CD2Cl2, 293 K; fig. S7): δ 8.18 (d, J = 2.1 Hz, 2H, H8 or 8′), 7.83 (t, J = 8.2 Hz, 1H, H1 or 1′), 7.80 (d, J ~ 2 Hz, 2H, H8 or 8′) overlapped with (t, J ~ 8 Hz, 1H, H1 or 1′), 7.53 (d, J = 8.2 Hz, 2H, H5 or 5′), 7.42 (d, J = 8.0 Hz, 2H, H2 or 2′), 7.36 (d, J = 8.2 Hz, 2H, H5 or 5′), 7.28 (d, J = 8.0 Hz, 2H, H2 or 2′), 7.23 (dd, J = 8.2, 2.1 Hz, 2H, H6 or 6′), 7.19 (dd, J = 8.2, 2.1 Hz, 2H, H6 or 6′), 1.20 (s, 18H, H11 or 11′), 1.18 (s, 18H, H11 or 11′). 13C{1H} NMR (126 MHz, CD2Cl2, 293 K) δ 170.69 (-CO2-), 168.06 (C9/C9′), 164.94 (C3/C3′), 163.50 (C3/C3′), 154.99 (C7/C7′), 154.39 (C7/C7′), 146.83 (C4/C4′), 144.65 (C4/C4′), 143.26 (C1/C1′), 141.79 (C1/C1′), 134.41 (C8/C8′), 131.65 (C8/C8′), 124.68 (C5/C5′), 124.65 (C5/C5′) 124.14 (C6/C6′), 123.45 (C6/C6′), 116.19 (C2/C2′), 115.81 (C2/C2′), 35.17 (C10/C10′), 35.04 (C10/C10′), 31.08 (C11/C11′), 30.93 (C11/C11′). Anal. calcd. (found) for C51H50N2Au2O2 (1116.89): C 54.84 (54.70); 4.51 (4.68); 2.51 (2.47).
Method 2
CO gas was bubbled for 30 s through a solution of [(C^N^C)Au]2O (20 mg, 0.018 mmol) in CH2Cl2 (5 ml). This solution was layered with light petroleum (boiling point, 40° to 60°C) and allowed to stand at −30°C for 48 hours, yielding 3 as a polycrystalline solid (10 mg, 49%) with identical spectroscopic properties to the sample obtained by method 1.
Synthesis of [(C^N^C)Au]2(μ-κC:κO-13CO2) (3-13C)
The same conditions used for the synthesis of 3 were applied, but 13CO was used instead of 12CO. The 13C NMR spectrum of the mixture was recorded (fig. S8). The main difference is the higher intensity of the signal at 170.69 ppm, which corresponds to the bridging CO2 ligand.
Thermolysis of [(C^N^C)Au]2(μ-κC:κO-CO2) (3 and 3-13C)
Method 1
A Schlenk flask was charged with 3 (20 mg, 0.017 mmol). The dry solid was heated at 120°C under vacuum for 16 hours. The resulting residue was dissolved in CD2Cl2. The quantitative conversion of 3 to [(C^N^C)Au]2 was confirmed by 1H NMR spectroscopy.
Method 2
13CO gas was bubbled for 30 s through a solution of [(C^N^C)Au]2O (5 mg, 0.005 mmol) in CD2Cl2 (5 ml) and stored under 13CO for an additional 5 min. After this time, the reaction was subjected to three freeze-pump-thaw cycles and stored under N2 at 60°C. Monitoring by 1H and 13C NMR spectroscopy at 25°C confirmed the conversion of [(C^N^C)Au]2(μ-κC:κO-13CO2) 3-13C to [(C^N^C)Au]2 and 13CO2 (fig. S9).
Reactivity of (C^N^C)AuOH with CO
Fast reaction conditions: CO gas bubbled for 30 s through a solution of (C^N^C)AuOH (5 mg, 9 μmol) in CD2Cl2 (2 ml). Then, the reaction is subjected to three freeze-pump-thaw cycles, stored under N2, and monitored by 1H NMR spectroscopy at 25°C. The spectrum showed additional signals that corresponded to the formation of (C^N^C)AuH (figs. S10 and S11).
Reaction of (C^N^C)AuOH and [(C^N^C)Au]2O with CO in the solid state
A Schlenk flask was charged with (C^N^C)AuOH (5 mg, 9 μmol) or [(C^N^C)Au]2O (5 mg, 4 μmol) and pressurized with 2 bar of CO. After stirring for 8 hours, the flask was subjected to three cycles of evacuation followed by N2 addition. One portion of the solid was used to record the IR spectrum; the rest was dissolved in CD2Cl2. The 1H NMR spectrum confirmed the presence of a mixture of the corresponding starting material together with [(C^N^C)Au]2(μ-κC:κO-CO2) 3 and [(C^N^C)Au]2 (fig. S12). The IR spectrum showed the emergence of a shoulder at 1624 cm−1 for the ν(C═O) band (fig. S13).
Attempted hydrogenolysis of [(C^N^C)Au]2(μ-κC:κO-CO2) 3.
A high-pressure NMR tube was charged with [(C^N^C)Au]2(μ-κC:κO-CO2) 3 (5 mg, 4 μmol) in 5 ml of CD2Cl2, pressurized with 4 bar of H2, and warmed to 60°C. After 8 hours, full conversion into [(C^N^C)Au]2 was observed, without any detectable amounts of formic acid.
Synthesis of (C^N^C)AuCO2Me 4.
CO gas was bubbled through a solution of (C^N^C)AuOMe (10 mg, 18 μmol) in CH2Cl2 for 5 min, in the presence of 4 Å molecular sieves. After stirring under a CO atmosphere for an additional 5 min, filtration through celite, evaporation of the filtrate to dryness, and washing with light petroleum afforded 4 as a white solid that was dried in vacuo (figs. S14 to S16). 1H NMR (300 MHz, CD2Cl2, 20°C): δ 7.83 (t, J = 8.0 Hz, 1H, H1), 7.73 (d, J = 2.0 Hz, 2H, H8), 7.55 (d, J = 8.2 Hz, 2H, H5), 7.45 (d, J = 8.0 Hz, 2H, H2), 7.30 (dd, J = 8.2, 2.0 Hz, H6), 3.93 (s, 3H, Me, CO2Me), 1.34 (s, 18H). 13C{1H} NMR (75 MHz, CD2Cl2, 20°C): δ 175.55 (COOMe), 156.03 (C8), 154.99 (C9), 147.64 (C7), 143.16 (C4), 134.84 (C1), 125.95 (C5), 125.59 (C3), 124.73 (C6), 117.10 (C2), 52.69 (COOCH3), 36.04 (C10), 31.88 (C11). Anal. calcd. (found) for C28H30N1Au1O2 (609.51): C 55.18 (55.67); 4.96 (4.75); 2.30 (2.60). IR: ν(12C═O) 1675 cm−1.
Reaction of (C^N^C)AuOMe with 13CO
13CO gas was bubbled through a solution of (C^N^C)AuOMe (5 mg, 9 μmol) in CD2Cl2 (2 ml) for 10 min at −30°C (fig. S17). Further, CD2Cl2 was added to restore the evaporation losses. The mixture was subjected to three freeze-pump-thaw cycles, stored under N2, and monitored by 1H and 13C NMR spectroscopy. The 1H NMR spectrum shows a pattern identical to 4, but the signal at 3.93 ppm appeared as a 13C-coupled doublet. In addition, because of the difficulty of drying 13CO as thoroughly as 12CO, the complex appeared mixed with 3-13C and [(C^N^C)Au]2, because of the hydrolysis of (C^N^C)AuOMe, which generates CH3OH and (C^N^C)AuOH and opens the path to 4 and reductive CO2 elimination (figs. S17 and S18).
Attempt of hydrogenolysis of [(C^N^C)AuCO2Me] 4.
A high-pressure NMR tube was charged with (C^N^C)AuCO2Me 4 (5 mg, 9 μmol) in CD2Cl2 (5 ml), pressurized with 4 bar of H2, and warmed to 60°C. No formation of methyl formate was detectable after 1 week.
Funding
This work was supported by the European Research Council (ERC), the Leverhulme Trust, and Johnson Matthey PLC. M.B. is an ERC Advanced Investigator Award holder (grant no. 338944-GOCAT). D.-A.R. thanks the University of East Anglia for a studentship. Author contributions: M.B. conceived and designed the research; D.-A.R., J.F.-C., and J.M. performed the experiments; J.A.W. provided the DFT analysis; M.B., D.-A.R., and J.F.-C. wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: Crystallographic data for this paper can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif, quoting CCDC no. 1410466.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/1/9/e1500761/DC1
Text
Fig. S1. 1H NMR (CD2Cl2, −25°C) spectrum of 1.
Fig. S2. Superposition of the IR spectra of [(C^N^C)Au12CO][B(C6F5)3OAcF] 2a and [(C^N^C)Au13CO][B(C6F5)3OAcF] 2a-13C.
Fig. S3. 1H NMR spectrum of 2a (CD2Cl2, −20°C).
Fig. S4. Stacked plot of the aromatic region of the 1H NMR spectra (CD2Cl2, −20°C) of (C^N^C)AuOAcF, [(C^N^C)Au(CH2Cl2)]+ 1, and [(C^N^C)Au(CO)]+ 2a.
Fig. S5. 13C NMR (CD2Cl2, −20°C) spectrum of 2a13.
Fig. S6. Monitoring by 1H NMR (CD2Cl2, −20°C) of the conversion of [(C^N^C)Au(η2-C2H4)]+ to [(C^N^C)Au(13CO)]+ (2a).
Fig. S7. 1H NMR spectrum of 3 (CD2Cl2, 25°C). The inset shows the t-butyl resonances.
Fig. S8. 13C NMR spectrum of complex 3-13C (CD2Cl2, 25°C).
Fig. S9. Monitoring by 1H and 13C NMR spectroscopy of the thermolysis of complex 3-13C in CD2Cl2.
Fig. S10. 1H NMR spectra of a solution of (C^N^C)AuOH in CD2Cl2 under 2 bar of CO at room temperature at different reaction times.
Fig. S11. Aromatic and hydride regions of the 1H NMR spectra of a solution of (C^N^C)AuOH in CD2Cl2 and after CO addition for 30 s.
Fig. S12. 1H NMR spectra in CD2Cl2 at room temperature of the aromatic region of [(C^N^C)Au]2O before and after its exposure to 2 bar of CO in the solid state.
Fig. S13. Superposition of the IR spectra of [(C^N^C)Au]2O in the solid state and after exposure to 2 bar of CO for 8 hours.
Fig. S14. 1H NMR spectrum of 4 (CD2Cl2, 25°C).
Fig. S15. Superposition of the IR spectra of (C^N^C)AuCO2Me 4 (red) and (C^N^C)AuOMe (blue) in the solid state.
Fig. S16. 1H NMR monitoring of the conversion of (C^N^C)AuOMe into (C^N^C)AuCO2Me 4 under 2 bar of 12CO at 25°C.
Fig. S17. Reactivity of (C^N^C)AuOMe and 13CO in the presence of moisture.
Fig. S18. Reaction of (C^N^C)AuOMe with CO.
Fig. S19. HOMO-1, HOMO-2, and HOMO-3 for [(C^N^C)Au(CO)]+.
Fig. S20. Enthalpy and Gibbs free energy values for reaction steps as calculated by DFT (T = 298.15 K).
Table S1. Selected crystal data and structure refinement details for 3·C6H6.
Table S2. CDA and d/b ratios of 2, [(C^N^N)Pt(CO)]+, and Pt(CO)2Cl2.
DFT coordinates
REFERENCES AND NOTES
- 1.LeValley T. L., Richard A. R., Fan M., The progress in water gas shift and steam reforming hydrogen production technologies—A review. Int. J. Hydrogen Energy 30, 16983–17000 (2014). [Google Scholar]
- 2.Gonzalez-Castaño M., Reina T. R., Ivanova S., Centeno M. A., Odriozola J. A., Pt vs. Au in water–gas shift reaction. J. Catal. 314, 1–9 (2014). [Google Scholar]
- 3.Rodriguez J. A., Senanayake S. D., Stacchiola D., Liu P., Hrbek J., The activation of gold and the water-gas shift reaction: Insights from studies with model catalysts. Acc. Chem. Res. 47, 773–782 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Fu Q., Saltsburg H., Flytzani-Stephanopoulos M., Active nonmetallic Au and Pt species on ceria-based water-gas shift catalysts. Science 301, 935–938 (2003). [DOI] [PubMed] [Google Scholar]
- 5.Shekhar M., Wang J., Lee W.-S., Williams W. D., Kim S. M., Stach E. A., Miller J. T., Delgass W. N., Ribeiro F. H., Size and support effects for the water-gas shift catalysis over gold nanoparticles supported on model Al2O3 and TiO2. J. Am. Chem. Soc. 134, 4700− 4708 (2012). [DOI] [PubMed] [Google Scholar]
- 6.Hussain A., Gracia J., Nieuwenhuys B. E., Niemantsverdriet J. W. H., Explicit role of Au and TiO2 in a bifunctional Au/TiO2 catalyst for the water-gas shift reaction: A DFT study. ChemCatChem 5, 2479–2488 (2013). [Google Scholar]
- 7.Bond G., Mechanisms of the gold-catalysed water-gas shift. Gold Bull. 42, 337–342 (2009). [Google Scholar]
- 8.Song W., Hensen E. J. M., Mechanistic aspects of the water-gas shift reaction on isolated and clustered Au atoms on CeO2(110): A density functional theory study. ACS Catal. 4, 1885–1892 (2014). [Google Scholar]
- 9.Yang M., Li S., Wang Y., Herron J. A., Xu Y., Allard L. F., Lee S., Huang J., Mavrikakis M., Flytzani-Stephanopoulos M., Catalytically active Au-O(OH)x- species stabilized by alkali ions on zeolites and mesoporous oxides. Science 346, 1498–1501 (2014). [DOI] [PubMed] [Google Scholar]
- 10.Flytzani-Stephanopoulos M., Gold atoms stabilized on various supports catalyze the water-gas shift reaction. Acc. Chem. Res. 47, 783–792 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Hieber W., Leutert F., Über metallcarbonyle. XII. Die Basenreaktion des Eisenpentacarbonyls und die Bildung des Eisencarbonylwasserstoffs. Z. Anorg. Allg. Chem. 204, 145–164 (1932). [Google Scholar]
- 12.Ford P. C., The water-gas shift reaction: Homogeneous catalysis by ruthenium and other metal carbonyls. Acc. Chem. Res. 14, 31–37 (1981). [Google Scholar]
- 13.Manchot W., Gall H., Über eine Kohlenoxyd-Verbindung des Goldes. Ber. Dtsch. Chem. Ges. 58, 2175–2178 (1925). [Google Scholar]
- 14.Belli Dell’Amico D., Calderazzo F., Murray H. H., Fackler J. P., Carbonylchlorogold(I). Inorg. Synth. 24, 236–238 (1986). [Google Scholar]
- 15.Adelhelm M., Bacher W., Höhn E. G., Jacob E., Dicarbonylgold(I) hexafluorouranate(VI), Au(CO)2UF6. Chem. Ber. 124, 1559− 1561 (1991). [Google Scholar]
- 16.Willner H., J. Schaebs, Hwang G., Mistry F., Jones R., Trotter J., Aubke F., Bis(carbonyl)gold(I) undecafluorodiantimonate(V), [Au(CO)2][Sb2F11]: Synthesis, vibrational, and carbon-13 NMR study and the molecular structure of bis(acetonitrile)gold(I) hexafluoroantimonate(V), [Au(NCCH3)2][SbF6]. J. Am. Chem. Soc. 114, 8972–8980 (1992). [Google Scholar]
- 17.Dash C., Kroll P., Yousufuddin M., Dias H. V. R., Isolable, gold carbonyl complexes supported by N-heterocyclic carbenes. Chem. Commun. 47, 4478− 4480 (2011). [DOI] [PubMed] [Google Scholar]
- 18.Dias H. V. R., Dash C., Yousufuddin M., Celik M. A., Frenking G., Cationic gold carbonyl complex on a phosphine support. Inorg. Chem. 50, 4253− 4255 (2011). [DOI] [PubMed] [Google Scholar]
- 19.Celik M. A., Dash C., Adiraju V. A. K., Dias A., Yousufuddin M., Frenking G., Dias H. V. R., End-on and side-on π-acid ligand adducts of gold(I): Carbonyl, cyanide, isocyanide, and cyclooctyne gold(I) complexes supported by N-heterocyclic carbenes and phosphines. Inorg. Chem. 52, 729–742 (2013). [DOI] [PubMed] [Google Scholar]
- 20.Schützenberger P., Sur quelques réactions donnant lieu à la formation de l’oxychlorure de carbone et sur un nouveau composé volatil de platine. Ann. Chim. Phys. 15, 100–106 (1868). [Google Scholar]
- 21.von Ahsen B., Wartchow R., Willner H., Jonas V., Aubke F., Bis(carbonyl)platinum(II) derivatives: Molecular structure of cis-Pt(CO)2(SO3F)2, complete vibrational analysis of cis-Pt(CO)2Cl2, and attempted synthesis of cis-Pt(CO)2F2. Inorg. Chem. 39, 4424–4432 (2000). [Google Scholar]
- 22.Browning J., Goggin P. L., Goodfellow R. J., Norton M. G., Rattray A. J. M., Taylor B. F., Mink J., Vibrational and nuclear magnetic resonance spectroscopic studies on some carbonyl complexes of gold, palladium, platinum, rhodium, and iridium. J. Chem. Soc. Dalton Trans. 2061–2067 (1977). [Google Scholar]
- 23.Pt(II) carbonyl complexes can be formed by hydrolysis of Pt-CF3 and are stable to refluxing methanol: Appleton T. G., Berry R. D., Hall J. R., Neale D. W., Displacement of norbornadiene (NBD) from Pt(CF3)2(NBD) by weak donor ligands L, and reactions of cis-Pt(CF3)2L2 with water and acids. J. Organomet. Chem. 364, 249–273 (1989). [Google Scholar]
- 24.Lai S.-W., Lam H.-W., Lu W., Cheung K.-K., Che C.-M., Observation of low-energy metal–metal-to-ligand charge transfer absorption and emission: Electronic spectroscopy of cyclometalated platinum(II) complexes with isocyanide ligands. Organometallics 21, 226–234 (2002). [Google Scholar]
- 25.Roşca D.-A., Smith D. A., Hughes D. L., Bochmann M., A thermally stable gold(III) hydride: Synthesis, reactivity, and reductive condensation as a route to gold(II) complexes. Angew. Chem. Int. Ed. 51, 10643–10646 (2012). [DOI] [PubMed] [Google Scholar]
- 26.Savjani N., Roşca D.-A., Schormann M., Bochmann M., Gold(III) olefin complexes. Angew. Chem. Int. Ed. 52, 874–877 (2013). [DOI] [PubMed] [Google Scholar]
- 27.Roşca D.-A., Wright J. A., Hughes D. L., Bochmann M., Gold peroxide complexes and the conversion of hydroperoxides into gold hydrides by successive oxygen-transfer reactions. Nat. Commun. 4, 2167 (2013). [DOI] [PubMed] [Google Scholar]
- 28.Grünert W., Großmann D., Noei H., Pohl M.-M., Sinev I., De Toni A., Wang Y., Muhler M., Low-temperature oxidation of carbon monoxide with gold(III) ions supported on titanium oxide. Angew. Chem. Int. Ed. 53, 3245–3249 (2014). [DOI] [PubMed] [Google Scholar]
- 29.The 13C NMR signal of coordinated CO in 2a, b is observed at δ 167.6 (cf. δ 184 for free CO); this value falls within the range observed, for example, for [Au(CO)]+ and [Au(CO)2]+ in superacidic solution (δ 158 to 172) and is minimally influenced by the d8 versus d10 electron configuration of the metal center; see (16).
- 30.Roşca D.-A., Smith D. A., Bochmann M., Cyclometallated gold(III) hydroxides as versatile synthons for Au–N, Au–C complexes and luminescent compounds. Chem. Commun. 48, 7247–7249 (2012). [DOI] [PubMed] [Google Scholar]
- 31.Cámpora J., Palma P., del Río D., Álvarez E., CO insertion reactions into the M−OH bonds of monomeric nickel and palladium hydroxides. Reversible decarbonylation of a hydroxycarbonyl palladium complex. Organometallics 23, 1652–1655 (2004). [Google Scholar]
- 32.Smith D. A., Roşca D.-A., Bochmann M., Selective Au–C cleavage in (C^N^C)Au(III) aryl and alkyl pincer complexes. Organometallics 31, 5998–6000 (2012). [Google Scholar]
- 33.Dann T., Roşca D.-A., Wildgoose G. G., Wright J. A., Bochmann M., Electrochemistry of AuII and AuIII pincer complexes: Determination of the AuII–AuII bond energy. Chem. Commun. 49, 10169–10171 (2013). [DOI] [PubMed] [Google Scholar]
- 34.S. J. Lancaster, http://cssp.chemspider.com/Article.aspx?id=215.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/1/9/e1500761/DC1
Text
Fig. S1. 1H NMR (CD2Cl2, −25°C) spectrum of 1.
Fig. S2. Superposition of the IR spectra of [(C^N^C)Au12CO][B(C6F5)3OAcF] 2a and [(C^N^C)Au13CO][B(C6F5)3OAcF] 2a-13C.
Fig. S3. 1H NMR spectrum of 2a (CD2Cl2, −20°C).
Fig. S4. Stacked plot of the aromatic region of the 1H NMR spectra (CD2Cl2, −20°C) of (C^N^C)AuOAcF, [(C^N^C)Au(CH2Cl2)]+ 1, and [(C^N^C)Au(CO)]+ 2a.
Fig. S5. 13C NMR (CD2Cl2, −20°C) spectrum of 2a13.
Fig. S6. Monitoring by 1H NMR (CD2Cl2, −20°C) of the conversion of [(C^N^C)Au(η2-C2H4)]+ to [(C^N^C)Au(13CO)]+ (2a).
Fig. S7. 1H NMR spectrum of 3 (CD2Cl2, 25°C). The inset shows the t-butyl resonances.
Fig. S8. 13C NMR spectrum of complex 3-13C (CD2Cl2, 25°C).
Fig. S9. Monitoring by 1H and 13C NMR spectroscopy of the thermolysis of complex 3-13C in CD2Cl2.
Fig. S10. 1H NMR spectra of a solution of (C^N^C)AuOH in CD2Cl2 under 2 bar of CO at room temperature at different reaction times.
Fig. S11. Aromatic and hydride regions of the 1H NMR spectra of a solution of (C^N^C)AuOH in CD2Cl2 and after CO addition for 30 s.
Fig. S12. 1H NMR spectra in CD2Cl2 at room temperature of the aromatic region of [(C^N^C)Au]2O before and after its exposure to 2 bar of CO in the solid state.
Fig. S13. Superposition of the IR spectra of [(C^N^C)Au]2O in the solid state and after exposure to 2 bar of CO for 8 hours.
Fig. S14. 1H NMR spectrum of 4 (CD2Cl2, 25°C).
Fig. S15. Superposition of the IR spectra of (C^N^C)AuCO2Me 4 (red) and (C^N^C)AuOMe (blue) in the solid state.
Fig. S16. 1H NMR monitoring of the conversion of (C^N^C)AuOMe into (C^N^C)AuCO2Me 4 under 2 bar of 12CO at 25°C.
Fig. S17. Reactivity of (C^N^C)AuOMe and 13CO in the presence of moisture.
Fig. S18. Reaction of (C^N^C)AuOMe with CO.
Fig. S19. HOMO-1, HOMO-2, and HOMO-3 for [(C^N^C)Au(CO)]+.
Fig. S20. Enthalpy and Gibbs free energy values for reaction steps as calculated by DFT (T = 298.15 K).
Table S1. Selected crystal data and structure refinement details for 3·C6H6.
Table S2. CDA and d/b ratios of 2, [(C^N^N)Pt(CO)]+, and Pt(CO)2Cl2.
DFT coordinates