

Background: Small ankyrin 1 (sAnk1) is required for stability of the network sarcoplasmic reticulum and shares transmembrane similarity with sarcolipin.
Results: sAnk1-SERCA interactions and their effects on SERCA were demonstrated by co-immunoprecipitation, FRET, blot overlay, and Ca2+-ATPase assays.
Conclusion: sAnk1 binds SERCA and reduces the affinity of SERCA for Ca2+.
Significance: sAnk1 may play a role in maintaining Ca2+ homeostasis in skeletal muscle.
Keywords: calcium ATPase, membrane protein, sarcoplasmic reticulum (SR), skeletal muscle, transmembrane domain, SERCA, small ankyrin 1, calcium regulation
Abstract
Small ankyrin 1 (sAnk1) is a 17-kDa transmembrane (TM) protein that binds to the cytoskeletal protein, obscurin, and stabilizes the network sarcoplasmic reticulum in skeletal muscle. We report that sAnk1 shares homology in its TM amino acid sequence with sarcolipin, a small protein inhibitor of the sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA). Here we investigate whether sAnk1 and SERCA1 interact. Our results indicate that sAnk1 interacts specifically with SERCA1 in sarcoplasmic reticulum vesicles isolated from rabbit skeletal muscle, and in COS7 cells transfected to express these proteins. This interaction was demonstrated by co-immunoprecipitation and an anisotropy-based FRET method. Binding was reduced ∼2-fold by the replacement of all of the TM amino acids of sAnk1 with leucines by mutagenesis. This suggests that, like sarcolipin, sAnk1 interacts with SERCA1 at least in part via its TM domain. Binding of the cytoplasmic domain of sAnk1 to SERCA1 was also detected in vitro. ATPase activity assays show that co-expression of sAnk1 with SERCA1 leads to a reduction of the apparent Ca2+ affinity of SERCA1 but that the effect of sAnk1 is less than that of sarcolipin. The sAnk1 TM mutant has no effect on SERCA1 activity. Our results suggest that sAnk1 interacts with SERCA1 through its TM and cytoplasmic domains to regulate SERCA1 activity and modulate sequestration of Ca2+ in the sarcoplasmic reticulum lumen. The identification of sAnk1 as a novel regulator of SERCA1 has significant implications for muscle physiology and the development of therapeutic approaches to treat heart failure and muscular dystrophies linked to Ca2+ misregulation.
Introduction
The mechanisms that regulate calcium homeostasis are critical to the function and viability of eukaryotic cells. In muscle, maintaining low resting intracellular Ca2+ ([Ca2+]i < 100 nm) compared with that found extracellularly (∼2 mm) or within the lumen of the sarcoplasmic reticulum (SR)3 (free, ∼0.4 mm; total, ∼2 mm) (1–4) is critical to excitation-contraction coupling (5). The sarco(endo)plasmic reticulum calcium ATPase (SERCA) is the enzyme that pumps Ca2+ from the cytoplasm into the lumen of the SR, leading to muscle relaxation following contraction. In mammals, there are three ATP2A genes that encode more than 10 different SERCA isoforms (6). The ubiquitous expression of one or more SERCA isoforms highlights its importance in the Ca2+ dynamics of muscle and non-muscle cells alike. Alterations in SERCA expression and activity are linked to several forms of muscular dystrophy and cardiomyopathies, including heart failure (5, 7–10). In addition, age-related alterations in SERCA levels have been observed in both animal models of aging and senescent human myocardium, suggesting that changes in SERCA activity may also be relevant to the aging process (6).
The small transmembrane (TM) proteins, phospholamban (PLN) and sarcolipin (SLN), are the two most well known regulators of SERCA activity. PLN is expressed at high levels in the ventricles of the heart and at lower levels in the atria and in slow-twitch skeletal muscle (11–13). SLN expression is more prominent in the atria and in fast-twitch skeletal muscle of larger mammals (10, 14–18). SLN and PLN share extensive homology in their TM sequences (14, 19, 20), which mediate their binding to several of the TM helices of SERCA (21–27). The TM sequences also mediate homo- and hetero-oligomerization of PLN and SLN (19, 28–35). SLN and PLN also interact with SERCA via their lumenal and cytoplasmic sequences, respectively (36–38).
The binding of PLN or SLN to SERCA is associated with a reduction in the apparent Ca2+ affinity of SERCA (39), and both proteins together have been reported to have a synergistic effect, leading to superinhibition of SERCA, presumably through forming a ternary complex (24, 28). Recent studies demonstrate that PLN and SLN can be co-expressed in both human and rodent skeletal muscle tissue, suggesting that superinhibition of SERCA activity may play a significant role in the regulation of intracellular Ca2+ (37, 40). Another small SR protein, myoregulin (MLN), which, like SLN, interacts with SERCA1 and inhibits its activity, has also been reported recently (41).
Small ankyrin 1 (sAnk1, also known as Ank1.5), an alternatively spliced product of the ANK1 gene, is a 155-amino acid TM protein (42–44). The 82 C-terminal cytoplasmic residues share homology with the larger members of the ankyrin superfamily, whereas the 73 N-terminal residues are unique to sAnk1 and include a TM domain in its most N-terminal sequence (44–46). sAnk1 localizes to the network compartment of the SR (nSR) (44, 45, 47–52) and colocalizes with SERCA1 in the nSR surrounding Z-disks (43, 49). The C terminus of sAnk1 protrudes into the cytoplasm (45), where it can interact with the giant myofibrillar proteins, obscurin and titin (50, 51, 53). These interactions provide a potential connection between the nSR membrane and the underlying contractile apparatus and are thought help to organize the SR membrane around each sarcomere (50, 54, 55).
In a 2011 study, Ackermann et al. (49) examined the effects of reducing the expression of sAnk1 in mouse myofibers using siRNA targeted to the 5′-UTR of its mRNA. Decreases in sAnk1 mRNA and protein levels were accompanied by a reduction in both SERCA and SLN protein (but not mRNA) levels. Consistent with these results, Ca2+ uptake kinetics and lumenal SR Ca2+ stores were reduced in myofibers depleted of sAnk1 (49). Reintroducing sAnk1 by transfection rescued SERCA localization. Remarkably, the loss of sAnk1 significantly disrupted SERCA and SLN localization within the nSR but had much smaller effects on proteins of the triad junction and sarcomere (49). More recently, Giacomello et al. (56) showed that muscle cells lacking sAnk1 due to homologous recombination have a compartment that was both reduced in size and slower to take up Ca2+. These observations suggested that sAnk1 may play a broader role than initially believed in the organization and stabilization of the nSR. One possibility that we considered is that sAnk1 can interact directly with SERCA and that in its absence, the nSR membrane either does not form or is unstable if it does. Here we test this hypothesis in experiments to examine the interactions between sAnk1 and SERCA.
Experimental Procedures
Materials
The chemiluminescence kit used for immunoblotting was from Applied Biosystems (Foster City, CA). Thapsagargin, A23187, and ATP were from Sigma. Dynabeads coupled with sheep anti-mouse IgG or sheep anti-rabbit IgG and Lipofectamine were from Invitrogen. Pi ColorLock ALS reagents were from Novus Biologicals (Littleton, CO). Amylose resin was from New England Biolabs (Ipswich, MA). All buffers were supplemented with Complete Protease Inhibitor Mixture tablets (Roche Applied Science).
Antibodies
Primary antibodies against sAnk1 were made by injecting rabbits with the C-terminal sequence of sAnk1 (C-aminocaproic acid-VKRASLKRGKQ-OH) linked to BSA. Antibody generation and affinity purification were carried out by 21st Century Biochemicals, Inc. (Marlborough, MA). Other primary antibodies used include SERCA1 (IIH11 mAb), ryanodine receptor-1 (RyR1) (34C mAb), and triadin (GE 4.90 mAb) from Thermo Scientific (Waltham, MA); junctophilin-1 (JPH1) (ab57425 mAb) from Abcam (Cambridge, MA); JPH1 (40-5100 rabbit pAb) from Invitrogen; FLAG (M2 mAb and rabbit pAb, Sigma; mCherry (rabbit pAb, Biovision (Milpitas, CA)); maltose-binding protein (MBP) (E8302 mAb); mouse IgG1κ (MOPC-21, Sigma); rabbit IgG (Jackson ImmunoResearch (West Grove, PA)); and SLN (rabbit pAb, Proteintech Group (Chicago, IL)).
cDNA Construction
Rabbit cDNAs encoding SERCA1 and an N-terminal FLAG-tagged sarcolipin in the pMT2 vector were gifts from Dr. David MacLennan (University of Toronto). SERCA1 cDNA was extracted by digestion with EcoRI and inserted into the pCDNA3.1(−) vector. A C-terminal FLAG-tagged sAnk1 was generated by digesting sAnk1 cDNA from the pmCherry-N1 vector constructed previously (48) with EcoRI and BamHI and ligating it into p3xFLAG-CMV-14. Sense (5′-CCGATCATGGAGCGATCCACCCGGGAGCTGTGTCTCAACTTCACTGTTGTCCTTATTACAGTGATCCTTATTTGGCTCCTTGTGAGGTCCTACCAGTACTGAG-3′) and antisense (5′-AATTCTCAGTACTGGTAGGACCTCACAAGGAGCCAAATAAGGATCACTGTAATAAGGACAACAGTGAAGTTGAGACACAGCTCCCGGGTGGATCGCTCCATGATCGGAGCT-3′) oligomers of the rabbit SLN coding region were synthesized with SacI (5′) and EcoRI (3′) overhanging restriction sites (Integrated DNA Technologies, Coralville, IA) and hybridized. The hybridized SLN oligomer, as well as cDNAs encoding SERCA1 (5′ SacI, 3′ EcoRI), sAnk1 (5′ KpnI, 3′ EcoRI), and dysferlin (5′ KpnI, 3′ BclI) were all inserted into the pmCerulean3-C1 (CFP) and pmVenus-C1 (YFP) vectors. Construction of the mCerulean3:mVenus conjugate has been described (57, 58).
Transfection
COS7 cells (American Type Culture Collection, Manassas, VA) were cultured in DMEM supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin in an atmosphere of 10% CO2, 90% air and transiently transfected with cDNA at a concentration of 1 μg/ml and either Lipofectamine 2000 (Invitrogen) or LipoD293 (SignaGen, Gaithersburg, MD), according to the manufacturer's protocols. When two cDNAs were transfected together, we used a ratio of 1:2 (SERCA1 cDNA/X, where X was sAnk1 or SLN cDNA). Cells were seeded to achieve ∼70–80% confluence at the time of transfection and incubated for 48 h to allow protein expression. Cells seeded on 35-cm tissue culture plates with glass bottoms (MatTek, Ashland, MA) were washed with Hanks' balanced salt solution + 0.1% bovine serum albumin and used for anisotropy-based fluorescence resonance energy transfer (AFRET). Cells in 10-cm tissue culture dishes were washed twice with PBS, detached with a cell scraper, homogenized, and either subjected to differential centrifugation (59) to obtain a microsomal fraction for ATPase measurements or subjected to centrifugation at 12,000 × g and solubilized in a solution containing 0.5% Tween 20 (see below) for co-immunoprecipitation studies.
Co-immunoprecipitation
Co-IP experiments were performed as described (60), with preparations of SR vesicles isolated by the method of Eletr and Inesi (61, 62) from rabbit skeletal muscle from the back and hind limb (Pel-Freez, Rogers, AR) or from crude membrane extracts of COS7 cells prepared as described (59, 63). For the latter, COS7 cells were harvested in PBS at 48 h following transfection, collected by centrifugation, frozen in liquid N2, and stored at −80 °C until needed. Briefly, pellets were homogenized with 30 strokes of a glass Dounce homogenizer in resuspension buffer (0.25 m sucrose, 10 mm Tris-HCl, pH 7.5, 20 mm CaCl2, 3 mm 2-mercaptoethanol, 150 mm KCl) and solubilized with an equal volume of lysis buffer (40 mm HEPES-NaOH, pH 7.5, 300 mm NaCl, 2 mm EDTA, 1% Tween 20). SR vesicles were prepared for co-IP by the same methods for resuspension and solubilization. The muscle and cell extracts were precleared with 50 μl of uncoated Dynabeads. The precleared extracts were mixed with antibody-coated Dynabeads (5 μg of antibody, 50 μl of beads) and incubated at 4 °C for 4 h. Following ≥5 washes in washing buffer (20 mm HEPES, pH 7.5, 150 mm NaCl, 1 mm EDTA, 0.5% Tween 20), protein was eluted by boiling the beads in 70 μl of SDS-PAGE sample loading buffer. Proteins in the samples were separated by SDS-PAGE and analyzed via immunoblot as described (64).
Microscopy
Cultures of COS7 cells transfected as described above were fixed in PBS containing 4% paraformaldehyde and 4% sucrose for 15 min at room temperature and mounted in Vectashield (Vector Laboratories, Burlingame, CA). Confocal microscopy used a Zeiss 510 META system (Carl Zeiss) equipped with a ×63, 1.4 numerical aperture objective lens. Images were collected by exciting the samples at 458 nm (CFP) and 514 nm (YFP), provided by an argon laser, and collected through 480–520-nm (CFP) and 530–600-nm (YFP) band pass filters. Colocalization was assessed by Pearson's coefficients with ImageJ software (National Institutes of Health), and the Just Another Co-localization Plugin (65).
To study the interaction between SERCA1 and sAnk1 by AFRET, transfected COS7 cells on coverslips were washed with Hanks' balanced salt solution + 0.1% BSA without phenol red and examined with a specially equipped Zeiss AxioObserver microscope (66) with a ×20, 0.75 numerical aperture dry objective lens. Details of the filters used to collect emissions and equations used to analyze the data have been described (66, 67). Briefly, fields were illuminated with vertically polarized light, and the intensity of emitted light was measured in planes parallel (V) and perpendicular (H) to that used for illumination. Emissions were collected for donor, acceptor, and FRET (excite donor, collect acceptor) fluorescence in both planes. ImageJ software was used to stack and align the images (16-bit) and to collect mean intensity values from a minimum of 3 regions of interest/cell. Regions of interest were excluded if their mean pixel intensity value was <2000 above background. Microsoft Excel was used to calculate anisotropies and FRET, indicated by a non-zero difference between the anisotropy measured for the FRET channel and the anisotropy measured for the donor (Δr = rCFP − rFRET). The many advantages of AFRET over other FRET methods have been discussed (57, 68).
Blot Overlay Assay
The blot overlay assay was performed as described (50, 69) with minor modifications. Aliquots containing 10 μg of SR vesicle protein, purified as described above, were separated on 4–12% SDS-polyacrylamide gels and transferred to PVDF membranes. Blots were placed in overlay buffer (50 mm Tris, pH 7.5, 120 mm NaCl, 3% BSA, 2 mm dithiothreitol, 0.5% Nonidet P-40, 0.1% Tween 20) for 4 h at 25 °C and then incubated with 3 μg/ml MBP or sAnk1(29–155)-MBP in overlay buffer for 12 h at 4 °C. The generation and purification of these fusion proteins were as described (51). A separate section of the membrane was reserved to identify SERCA1. Blots were washed extensively with overlay buffer and once with TBS, pH 7.4, plus 0.5% Tween 20 (TBST). The blots were blocked in TBST + 4% nonfat dry milk for 4 h at 25 °C and probed with antibodies to MBP (overlay portion) and SERCA1 (non-overlay portion). Immunoblots were completed as described above.
MBP Pull-down Assay
MBP and sAnk1 (29–155)-MBP fusion protein (20 μg) were bound to 50 μl of amylose resin in MBP column buffer (20 mm Tris-HCl, 200 mm NaCl, 1 mm EDTA). Resin was washed extensively with column buffer and once with IP lysis buffer (see above) and then mixed with 250 μg of protein from COS7 cell lysates transfected to express SERCA1 (see above) and incubated for 12 h at 4 °C. After the amylose resin was washed extensively in IP buffer, bound proteins were eluted by boiling in 50 μl of SDS-PAGE sample loading buffer and analyzed via SDS-PAGE and immunoblotting methods (64).
Assay of Ca2+-ATPase Activity
ATPase activity was measured in microsomes prepared as described (59) from transfected COS7 cells with a colorimetric assay to detect inorganic phosphate (Pi) released during ATP hydrolysis. Microsomal vesicles (20 μg/ml) were incubated in assay buffer (20 mm MOPS, pH 7.0, 100 mm KCl, 5 mm NaN3, 5 mm MgCl2, 1 mm EGTA, 2.5 mm ATP, 2 μm ionophore A23187). Reactions were initiated by the addition of different amounts of CaCl2 to achieve desired [Ca2+]free, as calculated by Maxchelator software. Following a 30-min incubation at 37 °C, reactions were loaded in triplicate onto 96-well plates and terminated by the addition of Pi ColorLock ALS reagent to detect Pi. Absorbance was read 30 min later at 635 nm on a Tecan Infinite M1000 Pro spectrophotometer. Immunoblots were used to measure levels of SERCA1 in each experimental group. Densitometric analysis with ImageJ quantified the relative expression levels of SERCA1 in the different cell preparations to normalize the measured ATPase activity. Activity values are reported as percentage of the maximum level of activity when SERCA1 was expressed alone.
Transmembrane Modeling and Protein Docking
The TM regions of sAnk1 and SLN were modeled with I-TASSER (70) and the sequences of the human proteins. ClusPro version 2.0 was used for automated protein docking simulation (71–74). The sAnk1 TM model obtained from I-TASSER was docked to the crystal structure of SERCA1 (Protein Data Bank entry 4H1W). Models were visualized and annotated with DeepView, the Swiss-PdbViewer version 4.10 (75).
Statistics
Values are reported as mean ± S.E. Each data point for AFRET experiments represents the average Δr value from all valid regions of interest (see “Microscopy” for criteria). t tests compared these values with a theoretical mean of zero to determine whether the mean AFRET value was statistically significant. Student's t tests were used to compare sAnk1 (WT) and sAnk1 mutant, sAnk1 (all-L), for co-IP and AFRET experiments with p < 0.05 being considered significant. Results of assays of ATPase activity were fit to the equation for an allosteric sigmoidal model, Y = Vmax × [S]h/(K′ + [S]h (76, 77), from data acquired in four experiments conducted on microsomes from three independent transfections. KCa2+ ([Ca2+]free resulting in half-maximal activation) was calculated using nonlinear regression analysis. All graphs were produced with GraphPad Prism version 5 software (La Jolla, CA). Statistics were evaluated with one-way analysis of variance with p < 0.05 being considered significant.
Protein Accession Numbers
The NCBI accession numbers for the proteins studied here are as follows: SERCA1 (rabbit, NP_001082787), sAnk1 (human, NP_065211.2; mouse, NP_001264213; rat, not available), SLN (human, NP_003054; mouse, NP_079816; rat, NP_001013265), PLN (mouse, NP_001135399), MLN (mouse, NP_001291668).
Results
The TM Domain of sAnk1 Shares Sequence Similarity with SLN
The observation that sAnk1 colocalizes with SERCA1 and is required for the structural integrity of the nSR suggested a possible interaction between sAnk1 and SERCA1. SLN is the major regulator of SERCA1 activity in skeletal muscle, and it is known to associate with SERCA through TM interactions (22). For these reasons, we compared the TM amino acid sequences of sAnk1 and SLN. Because sAnk1 and SLN are oriented in opposite directions within the SR membrane (Fig. 1A), we aligned the two sequences accordingly (Fig. 1B). The TM domains of these proteins were 29% identical and 53% conserved, for an overall similarity of 82% (Fig. 1C). We also found that sAnk1 shared considerable sequence similarity with PLN (76%) and, to a lesser extent, with MLN (47%; Fig. 1D).
FIGURE 1.

Sequence comparison in transmembrane regions of sAnk1 and SLN. A, relative orientation of sAnk1 and SLN within the SR membrane. B, residues that are identical or conserved in sAnk1 and SLN are highlighted in dark and light gray, respectively. C, percent similarity between sAnk1 and SLN. D, mouse amino acid sequence comparison of the transmembrane regions of sAnk1, SLN, PLN, and MLN. Similar residues are highlighted in gray.
To determine the significance of the sequence similarity between sAnk1 and SLN, we searched a database comprising of 13,607 TM sequences for the identical residues shared between sAnk1 and SLN (78). We found that 0.75% of sequences in this database contain the sequence TVLL or its reverse, LLVT, suggesting that the sequence similarity is significant. We also used the values of Senes et al. (78) for the frequency at which each amino acid occurs at a particular position in TM sequences, to calculate the chances of the 17FXXXXXTVLL8 sequence occurring randomly. This probability was <0.00002. Consistent with these observations, NCBI BLAST alignment of the TM domains of sAnk1 and SLN gave an E-value of 7E−4. These results indicate that the similarity between these sequences is highly significant (79).
Interestingly, two of the residues that sAnk1 shares with SLN, Val-19 and Leu-21 (Val-10 and Leu-8 of sAnk1), mediate the ability of SLN to reduce the affinity of SERCA1 for Ca2+ (20). Therefore, we hypothesized that, like SLN, sAnk1 interacts with SERCA1 and that this interaction occurs at least in part via its TM domain.
Interaction of sAnk1 and SERCA1
We first studied the interaction between SERCA1 and sAnk1 by co-IP, performed with SR vesicles prepared from rabbit skeletal muscle (see “Experimental Procedures”). Immunoblots of the proteins in the precipitate indicated that IP of SERCA1 resulted in specific co-IP of sAnk1 (Fig. 2A, left). Similarly, blots of immunoprecipitates generated with antibodies specific to sAnk1 showed co-IP of SERCA1 (Fig. 2A, right). RyR1 is the calcium release channel located in the SR membrane of skeletal muscle and is known to interact with several proteins at the triad junction, including the transmembrane proteins triadin and JPH1 (80, 81). To confirm the specific interaction between SERCA1 and sAnk1, antibodies specific for JPH1 were compared with antibodies to sAnk1 in their ability to co-immunoprecipitate SERCA from SR vesicles. Again, Western blot analysis of the eluates confirmed that SERCA1 coimmunoprecipitated with sAnk1 but that triadin and RyR1 did not. Antibodies to JPH1 resulted in co-IP of both RyR1 and triadin but not SERCA1 or sAnk1 (Fig. 2B). These results suggest that SERCA1 and sAnk1 interact specifically to form a distinct complex within the membranes of isolated SR vesicles.
FIGURE 2.

Co-IP of SERCA and sAnk1 from rabbit skeletal muscle. A, SR vesicles were solubilized and subjected to IP with antibodies to SERCA1 or sAnk1. B, specificity was demonstrated with an antibody to JPH1, which co-immunoprecipitated RyR1 and TRDN but not SERCA1 or sAnk1. Non-immune rabbit or mouse IgG were used as controls and did not immunoprecipitate either SERCA1 or sAnk1. The results show that SERCA1 and sAnk1 co-immunoprecipitate specifically from solubilized SR vesicles.
Interactions between SERCA and other proteins are commonly studied with exogenous expression systems to facilitate manipulation of experimental conditions. One such method involves the preparation of microsomes from transfected eukaryotic cells (63), which we have adopted here. We co-transfected COS7 cells with SERCA1-pcDNA3.1 and with either sAnk1-mCherry or empty mCherryN1 vector. We isolated a crude microsomal fraction from the cells ∼48 h later, dissolved them in detergent solution, and subjected them to IP with antibodies against SERCA1 or mCherry. Results showed that IP of either SERCA1 or sAnk1-mCherry led to co-IP of the other protein but that co-IP did not occur from homogenates containing mCherry and SERCA1 (Fig. 3A). In all cases, the level of specific IP and co-IP was severalfold greater than that of the nonspecific signals observed after IP with non-immune IgG. IP of SERCA1 or a FLAG-tagged sAnk1 variant (sAnk1-FLAG) was also able to co-immunoprecipitate the other protein when co-expressed in COS7 cells (Fig. 7A), suggesting that the association of these proteins in COS7 cells was not impacted significantly by the epitope tag. These results further support the idea that sAnk1 and SERCA1 interact specifically.
FIGURE 3.

Co-IP of SERCA and sAnk1 from COS7 cells. A, extracts of COS7 cells transfected as indicated below were subjected to IP with antibodies to SERCA1, sAnk1, or mCherry. Non-immune rabbit or mouse IgG was used as controls. The results show that SERCA1 and WT sAnk1 co-IP specifically in COS7 cells, consistent with the results from muscle tissue. B, compared with sAnk1 (WT), expression of sAnk1 (all-L) with SERCA1 led to a significant reduction in co-IP between the two proteins. C and D, quantitative comparison showed that co-IP was reduced by ∼50% for the mutant sAnk1 when normalized to co-IP of SERCA1 and sAnk1 (WT). Reduction in co-IP between SERCA1 and sAnk1 (all-L) ranged between 34 and 61%. This result was statistically significant by t test (C, p = 0.0024; D, p = 0.0033). Note that the bands observed in the IP lanes above mCherry protein (A) and above and below sAnk1(WT)-mCh are nonspecific bands due to the presence of the subunits of the antibodies used for IP. Error bars, S.E.
FIGURE 7.

The cytoplasmic domain of sAnk1 directly interacts with SERCA1 in vitro. A, top, extracts of COS7 cells transfected to express SERCA1 and WT sAnk1-FLAG were subjected to IP with antibodies to SERCA1 or FLAG. Non-immune mouse or rabbit IgG were used as controls. The results show that SERCA1 and sAnk1-FLAG specifically interact in COS7 cells and that the epitope tag on sAnk1 does not alter this interaction. Bottom, COS7 extracts were co-transfected to express SERCA1 and FLAG-tagged versions of full-length sAnk1 or the cytoplasmic domain, sAnk1(29–155). A FLAG empty vector was used as control. The results indicate that, like FLAG-tagged full-length sAnk1 (sAnk1 WT), a FLAG-tagged form of the cytoplasmic domain of sAnk1 (sAnk1(29–155)) also co-immunoprecipitates with SERCA1. B, amylose resin bound to bacterially expressed sAnk1(29–155)-MBP fusion protein was incubated with extracts of COS7 cells transfected to express SERCA1. MBP protein alone was used as a control. Densitometric analysis of the eluates after SDS-PAGE and blotting with antibodies to SERCA1 revealed a 9-fold increase in the amount of SERCA1 pulled down by sAnk1(29–155) relative to the MBP control (bottom). Ponceau staining was used as a loading control (top). C, SR vesicle preparations were used for blot overlay assays. Blots were first overlaid with sAnk1(29–155)-MBP or MBP protein alone, followed by incubation with antibodies to MBP. The labeled band at ∼110 kDa (left) indicates that sAnk1(29–155)-MBP, but not MBP protein alone, can bind to SERCA1 directly. Ponceau staining was used as a loading control (right).
We next assessed the interaction of SERCA and sAnk1 in COS7 cells co-transfected with SERCA1-CFP and sAnk1-YFP. Both proteins distributed in a reticular pattern, typical of localization to the endoplasmic reticulum (ER; Fig. 4), although some may reside in other membrane compartments, including the Golgi complex. Co-transfections with DS-Red-KDEL (a gift of Dr. S. Feng, University of Maryland, Baltimore, MD) confirmed the identity of this membrane compartment as the ER (not shown).
FIGURE 4.

Colocalization of fluorescent fusion proteins in COS7 cells. A–F, COS7 cells were transfected with cDNA encoding the indicated fluorescent fusion proteins. Significant colocalization was observed when SERCA1-CFP was co-expressed with SLN-YFP, sAnk1-YFP, and sAnk1 (all-L)-YFP, and when SERCA1-CFP or sAnk1-CFP was co-expressed with dysferlin-YFP (see merged panels A3–F3, respectively), as measured by Pearson's correlation coefficient. The values of Pearson's coefficient are included at the bottom of the merged image for each pair of proteins studied (>0.7 for each). Scale bar, 10 μm.
Analysis with Pearson's correlation coefficient showed significant colocalization of SERCA1-CFP and sAnk1-YFP (0.74 ± 0.03; Fig. 4A). The level of codistribution was comparable when the CFP and YFP moieties were switched (0.83 ± 0.02; Fig. 4B). Colocalization studies of SERCA1-CFP and SLN-YFP (0.74 ± 0.031; Fig. 4C) and SERCA1-CFP and sAnk1 (all-L)-YFP (0.72 ± 0.01; Fig. 4D) gave similar results.
We studied the co-transfected COS7 cells further using an AFRET assay to determine whether SERCA1 and sAnk1 reside within molecular distances (≤10 nm) of each other in living cells. The method is based on the principle that a fluorescently labeled protein excited with polarized light will emit highly polarized photons, leading to large anisotropy values (57, 68). A nearby acceptor fluorescent protein is not fully constrained to the original polarization plane, resulting in reduced anisotropy if FRET occurs between the donor and acceptor. By setting a minimum intensity cut-off required for inclusion of any region of interest, we ensured that AFRET was examined only in areas of the cell that co-expressed donor and acceptor.
Our results with COS7 cells transfected with cDNAs encoding SERCA1-CFP and sAnk1-YFP are shown in Fig. 5A. A construct encoding the CFP and YFP proteins tethered to one another was used as an intramolecular control to ensure effective transfection and detection of AFRET (Δrmean = 0.125 ± 0.004; data not shown). The results indicate that the FRET donor-acceptor pair, SERCA1-CFP and sAnk1-YFP, exhibited energy transfer (Δrmean = 0.026 ± 0.004). This value was significantly different from zero (p < 0.0001) but less than that obtained in similar studies of SERCA1-CFP and SLN-YFP (Δrmean = 0.064 ± 0.006; p < .0001). Dysferlin, a protein of the transverse tubules of skeletal muscle (82), was used as a negative control. In COS7 cells, YFP-tagged dysferlin localizes primarily to the ER with SERCA1 and sAnk1 (Pearson's coefficient = 0.47 ± 0.04 and 0.56 ± 0.12, respectively; Fig. 4, E and F), with small amounts in the plasma membrane and, perhaps, the Golgi complex. When YFP-dysferlin was used as the acceptor, FRET measured in the intracellular membrane compartment was not observed when SERCA1-CFP or sAnk1-CFP was co-expressed as the donor (Δrmean = −0.035 ± 0.006 and −0.028 ± 0.006, respectively). Additionally, there was no AFRET observed in the absence of acceptor (SERCA alone; Δrmean = −0.033 ± 0.003). These data indicate that sAnk1 and SERCA1, co-expressed as fluorescent fusion proteins in COS7 cells, reside within molecular distances of one another.
FIGURE 5.

AFRET of sAnk1 and SERCA and SLN and SERCA in COS7 cells. A, COS7 cells were transfected with SERCA-CFP together with either sAnk1-YFP, sAnk1 (all-L)-YFP, SLN-YFP, or dysferlin-YFP. SERCA-CFP alone was used as a control. One day post-transfection, AFRET was measured and expressed as Δr (rCFP-rFRET). Each point represents the average AFRET for a single cell. t tests for each sample set were performed against a theoretical mean of zero. *, mean is statistically greater than zero (p < 0.0001). Results show energy transfer between sAnk1 and SERCA1 and between SLN and SERCA1 but not between SERCA1 and dysferlin, between sAnk1 and dysferlin, or in SERCA1 alone. B, when compared with sAnk1 via t test (WT), sAnk1 (all-L) showed a significant reduction in Δr, suggesting reduced binding due to the mutated TM domain (***, p = 0.0008). Error bars, S.E.
sAnk1 Interacts with SERCA1 through Its Transmembrane Domain
The TM domains of SLN and PLN play a pivotal role in the ability of each to interact with SERCA (24). Due to the sequence similarity between sAnk1 and SLN, we hypothesized that the TM domain of sAnk1 would play a role in its association with SERCA1. To test this, we constructed a sAnk1 mutant that had TM amino acid residues 4–20 mutated to leucine, sAnk1 (all-L). Importantly, sAnk1 (all-L) is expressed and targeted in a manner indistinguishable from WT sAnk1 (Figs. 3 and 4). Analysis of its colocalization with SERCA1 gave Pearson's coefficients that were identical to wild type (0.72 ± 0.01), indicating that it too concentrates with SERCA1 in the ER. Experiments in COS7 cell extracts showed that antibodies to sAnk1 coimmunoprecipitated only 49 ± 6% of SERCA1 from microsomes with sAnk1 (all-L) compared with WT (Fig. 3, B (right) and D). Similarly, antibodies to SERCA1 coimmunoprecipitated 49 ± 5% of sAnk1 (all-L) compared with WT sAnk1 (Fig. 3, B (left) and C). These results suggest that the TM domain mediates binding of sAnk1 to SERCA1 in the microsomes of COS7 cells.
We next utilized AFRET to determine whether the sAnk1 (all-L) mutant interacts with SERCA1 in living cells. As expected, sAnk1 (all-L) showed a reduced AFRET signal (Δrmean = 0.017 ± 0.005) compared with that of WT sAnk1 (Δrmean = 0.043 ± 0.005; Fig. 5B; p = 0.0008). As above, SERCA-CFP and SLN-YFP were used as a positive control (Δrmean = 0.054 ± 0.006). These results support the idea that the TM domain of sAnk1 plays a role in binding to SERCA1 in living cells.
Effect of sAnk1 on SERCA1 Ca2+-ATPase Activity
We used a colorimetric assay of Pi release to determine whether sAnk1 alters SERCA1 ATPase activity. We prepared microsomes from COS7 cells that were transfected to express SERCA1 alone or SERCA1 in combination with FLAG-SLN or sAnk1-FLAG and assayed them for Ca2+-dependent, thapsigargin-sensitive ATPase activity. The microsomes containing only SERCA1 showed the highest apparent affinity for Ca2+, with a KCa2+ ([Ca2+]free resulting in half-maximal activation) of 468 nm, equivalent to a pCa2+ = 6.33; Table 1). Expression of a C-terminal FLAG-tagged sAnk1 variant with SERCA1 significantly inhibited SERCA1 by reducing its apparent Ca2+ affinity by 0.18 pCa2+ units in this experiment (p < 0.05; Fig. 6, A and B; the reduction was 0.20 for all studies; Table 1). This was significantly less than the inhibition obtained following co-expression of SERCA1 with SLN carrying an N-terminal FLAG tag (FLAG-SLN), which shifted the apparent Ca2+ affinity of SERCA1 by −0.38 pCa2+ units (p < 0.05). These results suggest that sAnk1 inhibits SERCA1 by reducing its affinity for Ca2+ but that it is a less potent inhibitor than SLN.
TABLE 1.
ATPase assays
Shown is a summary of the compiled results of all ATPase assays presented in Fig. 6. The KCa2+ ([Ca2+] required for half-maximal activation) is given in pCa2+ units (right-hand column) and nanomolar concentration (left-hand column). The change in KCa2+ (ΔKCa2+) relative to control (SERCA1 alone) is given in pCa2+ units. Results are mean values ± S.E.
|
KCa2+ |
ΔKCa2+ | n | ||
|---|---|---|---|---|
| Concentration | pCa2+ | |||
| nm | ||||
| SERCA1 alone | 479 ± 17 | 6.32 ± 0.015 | NAa | 8 |
| SERCA1 + sAnk1 | 759 ± 83 | 6.12 ± 0.046b,c,d | −0.20 | 8 |
| SERCA1 + SLN | 1122 ± 242 | 5.95 ± 0.081b,d | −0.37 | 4 |
| SERCA1 + sAnk1 (all-L) | 501 ± 64 | 6.3 ± 0.055e | −0.02 | 4 |
aNot applicable.
bSignificant difference (p < 0.05) in mean pCa2+ as measured by one-way analysis of variance compared with SERCA1 alone.
cSignificant difference (p < 0.05) in mean pCa2+ as measured by one-way analysis of variance compared with SERCA1 + SLN.
dSignificant difference (p < 0.05) in mean pCa2+ as measured by one-way analysis of variance compared with SERCA1 + sAnk1 (all-L).
eNot significantly different from SERCA1 alone.
FIGURE 6.

Ca2+-ATPase assay in COS7 microsomes. A, COS7 cells were transfected with the indicated cDNA construct(s). ATPase activity was determined at each [Ca2+]free compared with the Vmax measured for SERCA1 alone, following normalization of the levels of SERCA1 expression as determined by immunoblotting (see “Experimental Procedures”). Data were fitted to the equation for a general cooperative model for substrate binding. Results show that co-expression of sAnk1 with SERCA leads to a reduction of the apparent affinity of SERCA for Ca2+ but that the effect of sAnk1 is less than that of SLN. B, the KCa2+ ([Ca2+]free required for half-maximal activation) values were determined from each curve. Mean KCa2+ values were as follows: SERCA1, pCa = 6.33 (468 nm); SERCA1 + sAnk1, pCa = 6.15 (708 nm); SERCA1 + SLN, pCa = 5.95 (1122 nm). C, unlike sAnk1 (WT), sAnk1 (all-L) did not significantly alter the affinity of SERCA1 for Ca2+. D, mean KCa2+ values were as follows: SERCA1, pCa = 6.31 (488 nm); SERCA1 + sAnk1, pCa = 6.10 (794 nm); SERCA1 + sAnk1 (all-L), pCa = 6.30 (502 nm). Statistics used one-way analysis of variance. *, p < 0.05 versus SERCA1; **, p < 0.01 versus SERCA1; #, p < 0.05 versus SERCA1 + sAnk1 (all-L). Error bars, S.E.
To assess the role of the TM domain in sAnk1 inhibition of SERCA1 activity, we measured ATPase activity in microsomes containing sAnk1 (all-L) and SERCA1. Expression of SERCA1 alone gave a value for pCa2+ of 6.31 ± 0.028. Unlike WT sAnk1, which again significantly reduced the apparent Ca2+ affinity of SERCA1 (ΔKCa2+ = −0.22 pCa2+ units), the mutant sAnk1 failed to inhibit SERCA1 activity significantly (ΔKCa2+ = −0.01 pCa2+ units; Fig. 6, C and D). The compiled results and statistical analysis for all ATPase assays performed for this study are summarized in Table 1. These findings suggest that, like SLN, the contacts made between the TM domain of sAnk1 and SERCA1 are essential to the ability of sAnk1 to regulate SERCA1 activity.
sAnk1 Cytoplasmic Domain Interacts with SERCA1 in Vitro
In addition to TM contacts, SLN and PLN are known to make luminal and cytoplasmic contacts with SERCA1, respectively (36, 83, 84). Because sAnk1 also has a long cytoplasmic extension and because our co-IP studies suggested that mutation of the TM domain alone was not sufficient to eliminate sAnk1-SERCA1 association, we investigated the possible interaction of the sAnk1 cytoplasmic domain with SERCA1 in vitro.
We first performed co-IP studies on extracts of COS7 cells transfected to express SERCA1 and WT sAnk1 or the cytoplasmic domain of sAnk1 (sAnk1(29–155)), each with a FLAG epitope at its C terminus. Empty FLAG vector was used as a control. Immunoblots of the IP generated with antibodies to SERCA1 probed with anti-FLAG antibodies showed co-IP of not only the WT sAnk1, as we reported above, but also of sAnk1(29–155) (Fig. 7A, bottom). The intensity of the band in the co-IP was less for the cytoplasmic domain than for the full-length protein, however, consistent with the role of TM interactions in sAnk1-SERCA1 interactions noted in our experiments above.
We further confirmed interaction between the cytoplasmic domain of sAnk1 and SERCA1 using a bacterially expressed sAnk1(29–155) fused to MBP. We bound amylose resin to sAnk1(29–155)-MBP or MBP alone and incubated these samples with detergent extracts of COS7 cells transfected to express SERCA1. Pull-down of SERCA1 by the resins was assessed by immunoblot analysis. We found that the sAnk1(29–155)-MBP fusion protein pulled down ∼9-fold more SERCA1 than the MBP control (Fig. 7B, top).
Finally, we next performed blot overlay assays to determine whether SERCA1 interacts with the cytoplasmic domain of sAnk1 directly. SR vesicle proteins were separated by SDS-PAGE, transferred to PVDF membranes, and incubated with sAnk1(29–155)-MBP fusion protein or MBP protein alone. Probing the blots with anti-MBP revealed a band at ∼110 kDa only where sAnk1(29–155)-MBP was overlaid. Equivalent lanes were probed with anti-SERCA1 to show that this band corresponded with SERCA1 (Fig. 7C, left). Equal loading was demonstrated by staining with Ponceau Red, which also showed a distinct band for SERCA1 at ∼110 kDa. Taken together with our co-IP, Ca2+-ATPase assays, and AFRET data (Fig. 5), these results indicate that sAnk1 associates with SERCA1 directly and that both its TM and cytoplasmic domains mediate this association.
Discussion
Regulation of SERCA activity in both skeletal and cardiac muscle has been extensively investigated for over 20 years. The majority of these efforts have focused on the homologous proteins PLN and SLN. These small TM proteins of the SR are differentially expressed in different muscle fibers while exhibiting a similar, although distinct, ability to modulate SERCA activity, mediated in part by the binding of their TM domains to SERCA (14, 17, 20, 84, 85). Binding is thought to occur within a pocket made up of the SERCA TM helices M2, M4, M6, and M9 (26, 30) and leads to a reduction in the apparent Ca2+ affinity of SERCA (39). Our research has focused on sAnk1, a small protein encoded by the ANK1 gene that concentrates with SERCA in the nSR of skeletal muscle. Previous studies from our laboratory established that reduced expression of sAnk1 is accompanied by disruption of the nSR and the loss of SERCA protein. Here, we show that the TM domain of sAnk1 shares significant sequence similarity with SLN and mediates binding to SERCA and regulation of its enzymatic activity. We also show that, as in PLN-SERCA interactions (36, 84), the cytoplasmic domain of sAnk1 contributes to the binding of sAnk1 to SERCA and that this binding is direct. Our results indicate that sAnk1 is a novel regulator of SERCA1 activity.
sAnk1 was first identified as a small membrane-bound protein that co-purified with SERCA1 from enriched SR fractions of rabbit skeletal muscle (43). It localizes to the SR surrounding M-bands and Z-disks, where its C-terminal, cytoplasmic domain is able to interact with obscurin (present at M-bands and Z-disks (50, 51)) and titin (present at Z-disks (47)). Studies with mice lacking obscurin (55) or sAnk1 (56) as well as with mutant forms of sAnk1 expressed in skeletal myofibers, ongoing in our laboratory, are consistent with the idea that binding of sAnk1 to obscurin links the SR membrane to the underlying contractile apparatus. Perhaps more significantly, however, sAnk1 is crucial in maintaining integrity of the nSR. Using RNAi technology to reduce the expression of sAnk1 in flexor digitorum brevis myofibers, we found that sAnk1 knockdown disrupts the nSR and also reduces the amount of SERCA protein (49). These changes reduced SR Ca2+ load and the rate of Ca2+ clearance from the cytosol following a 4-CmC-induced Ca2+ transient. Furthermore, quantitative analysis of colocalization revealed that the remaining sAnk1 following knockdown was only ∼25% localized with SERCA1 compared with ∼50% in controls.
These observations raised the question of how sAnk1 influences the stability of the nSR and organization of proteins in the nSR membrane. Comparisons showed that the TM sequence of sAnk1 is very similar to the TM region of SLN. The total sequence similarity shared between both rodents and humans is >80%, with 29 and 24% sequence identity, respectively. Our analyses reveal that these similarities are highly significant. Because SLN interacts with SERCA via its TM domain (22), we predicted that sAnk1 would do so too. Co-immunoprecipitation experiments showed that sAnk1 and SERCA1 associated specifically in SR vesicles isolated from rabbit muscle and in membrane fractions from transfected COS7 cells. Similarly, AFRET experiments in COS7 cells co-expressing pairs of CFP and YFP fusion proteins showed energy transfer from SERCA1-CFP to sAnk1-YFP. This suggests that these proteins reside within 10 nm of one another in living cells (86) and are likely to interact directly. Furthermore, Ca2+-ATPase assays showed that co-expression of sAnk1-FLAG and SERCA1 significantly reduced the apparent Ca2+ affinity of SERCA1. Together, these data support our hypothesis that sAnk1 is able to interact with SERCA1 to regulate its activity similar to SLN. Moreover, our results showing that the cytoplasmic domain of sAnk1 binds to SERCA1 in blot overlay experiments indicate that the association of sAnk1 with SERCA1 is direct and not mediated by other proteins.
We used a combination of co-IP, AFRET, and ATPase assays to elucidate the role of the sAnk1 TM domain in its ability to bind to SERCA1 and inhibit its activity. We found that the TM domain of sAnk1 is important in mediating its interaction with SERCA1. sAnk1 (all-L) and SERCA1 were found to co-immunoprecipitate together only ∼50% as efficiently as WT sAnk1 and SERCA1. Similarly, AFRET analysis revealed a decrease in the Δrmean between SERCA1-CFP and sAnk1 (all-L)-YFP compared with WT. The reduced interaction between SERCA1 and sAnk1 (all-L) suggests that sAnk1 may dock to SERCA1 in the same pocket, composed of several of the TM helices of SERCA1, used by SLN and PLN. We speculate that the binding that persists between SERCA1 and sAnk1 (all-L) is due to the sAnk1 cytoplasmic domain. The most interesting effect of mutating the sAnk1 TM domain was the inability of the mutant to shift the apparent Ca2+ affinity of SERCA. This observation suggests that specific amino acids within the TM region of WT sAnk1 are important for its inhibitory function.
Recently, structural modeling was used to analyze the similarities between the α-helical TM region of SLN and MLN, another small protein of the SR membrane that can inhibit SERCA (41). We used the same software to compare the TM regions of sAnk1 and SLN and found that several of the conserved TM residues of sAnk1 shared similar spatial orientations with several identical and conserved residues of SLN (Fig. 8A). In agreement with our hypothesis and our experimental observations, automated docking simulations predicted that the TM region of sAnk1 binds the same region of SERCA as SLN (Fig. 8B). Closer examination of the predicted docking site of sAnk1 shows amino acid side chains protruding into the binding pocket, where SLN binds SERCA1 (22). When compared with a published crystal structure of the SERCA1-SLN complex, it is clear that several of these residues are shared between sAnk1 and SLN (Fig. 8C). These models are consistent with sAnk1 binding to SERCA1 in the same pocket as SLN, PLN, and MLN to modulate SERCA activity.
FIGURE 8.
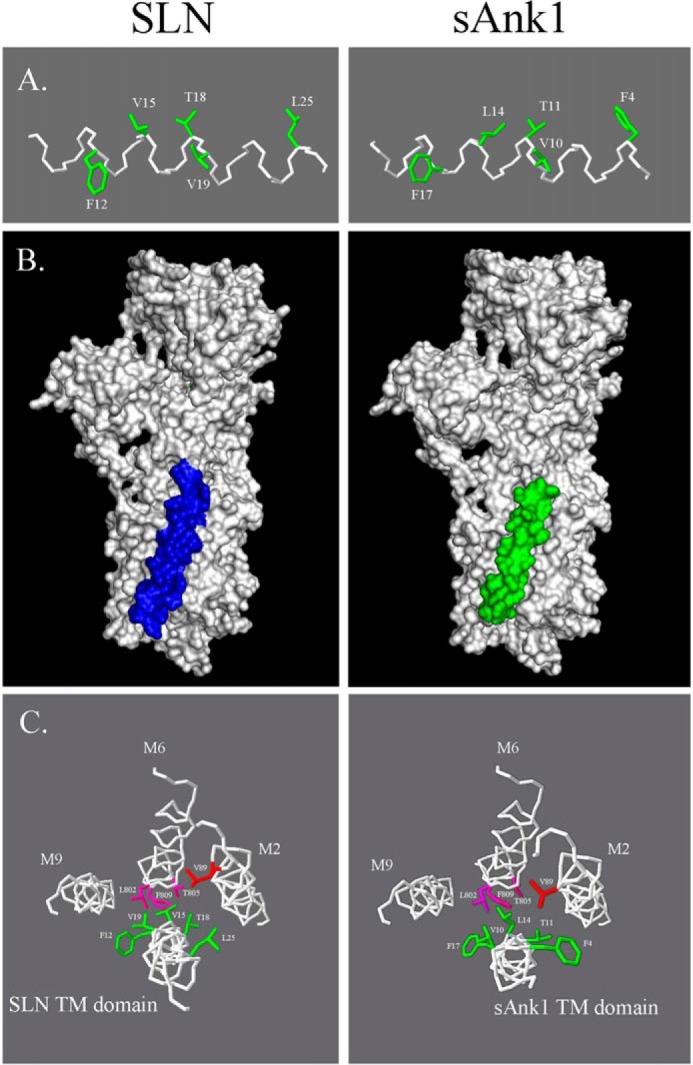
Modeling the sAnk1 TM domain. A, the TM domains of SLN (top) and sAnk1 (bottom) were modeled with I-TASSER software. The results show that five side chains in the TM α-helix of sAnk1 occupy very similar positions along the helix as their counterparts in the TM domain of SLN (highlighted in green). B, ClusPro version 2.0 software was used to determine the predicted docking site of sAnk1 to SERCA1. Comparison of this model with the published crystal structure of rabbit SERCA1a docked to SLN was performed with PyMOL version 1.3. The results show that sAnk1 is predicted to dock to SERCA1 in a similar, but not identical, position to SLN. C, backbone helices of SERCA and sAnk1 are shown in white. SERCA residues highlighted in magenta and red are reported to be important for SLN-mediated SERCA inhibition. Similar residues shared between sAnk1 and SLN are highlighted in green. The docking model shows that the sAnk1 residues that extend into the TM binding pocket of SERCA1 are positioned similarly to those of SLN. Orientation is from inside the SR lumen, looking down into the cytoplasm.
The changes in binding and the ability of sAnk1 to inhibit SERCA1 activity when the TM domain is mutated to all leucines are particularly interesting, considering the similarity between sAnk1 WT and sAnk1 (all-L). The TM domain of WT sAnk1 contains 6 leucine residues, and 8 of the 11 remaining TM residues are hydrophobic. Therefore, only 3 of the TM amino acid residues between positions 4 and 20 represent non-conservative mutations. The 3 hydrophilic residues within the TM domain are Thr-6, Glu-7, and Thr-11. We speculate that these residues may contribute to the ability of sAnk1 to inhibit SERCA1 activity, but we recognize that several specific hydrophobic residues may also play a role in modulating SERCA activity, as with SLN and PLN (20, 24, 60, 87, 88), including Val-10 and Leu-8 residues, which are conserved between sAnk1 and SLN. Future studies to determine the specific TM residues necessary for sAnk1 to exhibit its inhibitory effect will therefore be important.
In addition to TM contacts, cytoplasmic and lumenal interactions are significant for PLN and SLN to regulate SERCA, respectively (36). It was shown recently that mutation or deletion of the C-terminal residues of PLN led to improper localization and impaired regulatory activity (83). Similarly, the highly conserved C terminus of SLN is important for SLN function. Gorski et al. (37) showed that the lumenal tail of SLN (RSYQY) was required for SLN to inhibit SERCA1 activity maximally and that a truncated SLN mutant lacking the 4 C-terminal amino acid residues inhibited SERCA1 activity less effectively than full-length SLN (37, 38). These data are in agreement with earlier mutagenic studies that identified Tyr-29, Gln-30, and Tyr-31 as residues important for the inhibitory effect of SLN (20). The significance of the SLN lumenal tail was further indicated with a chimeric PLN mutant which had these residues from SLN added to its C terminus. This chimeric protein was superinhibitory and may help to explain the differences observed in the relative abilities of sAnk1 and SLN to inhibit SERCA1 activity (37). Future studies using chimeric variants of sAnk1 can address this possibility.
Because a significant portion of the sAnk1 polypeptide extends into the cytosol (45), we sought to determine whether, like PLN and SLN, sAnk1 could interact with SERCA1 independently of the TM domain. This was addressed using the cytoplasmic domain of sAnk1 (residues 29–155) in co-IP, pull-down, and blot overlay assays. The results show that the cytoplasmic domain of sAnk1 is also able to interact with SERCA1, although less efficiently than full-length sAnk1. This would be consistent with a model in which sAnk1-SERCA1 interactions are mediated by a combination of interactions occurring in the TM and cytoplasmic domains of both proteins. More extensive structural analyses and mutagenesis studies are under way to test this idea.
Although sAnk1 interacts directly with SERCA1, it is not clear how, or even if, this interaction helps to stabilize or regulate the function of the nSR in muscle. Short term incubation of myofibers with siRNA to reduce sAnk1 expression and elimination of sAnk1 completely by homologous recombination both lead to loss of the nSR and associated reductions in the rate of Ca2+ clearance from the myoplasm (49, 56). Decreased levels of sAnk1 should increase SERCA1 activity, leading to increased rates of Ca2+ clearance, but the effects of any such increase may be overridden by the loss of the nSR itself. They may also be altered by changes in other proteins, such as SLN, that regulate SERCA activity but that may also interact with sAnk1 and the sAnk1-SERCA1 complex. These interactions may influence the stability of the nSR membrane either directly, through altered membrane curvature or changes in protein-protein or protein-lipid binding, or indirectly, through changes in the handling of Ca2+. Alternatively, the ability of sAnk1 to bind to cytoskeletal proteins, including obscurin (44, 48, 50) (see also Ref. 52), may influence the stability of the nSR. It will be of considerable interest to learn whether or not the sAnk1 cytoplasmic domain can bind obscurin and SERCA1 simultaneously. Alterations in expression and activity of SERCA in skeletal and cardiac muscle are linked to several forms of muscular dystrophy and cardiomyopathies, including heart failure (5, 7–10). In addition, age-related alterations in SERCA levels have been observed in both animal models and senescent human myocardium, suggesting that it may be relevant to the aging process (6). SERCA activity has also been shown to play a critical role in the pathogenesis of Alzheimer disease, exemplifying the broad implications of understanding SERCA regulation (89–91) and the significance of discovering potential targets for manipulating SERCA activity.
Here we have demonstrated that sAnk1 is able to interact with SERCA1 in skeletal muscle in a way much like SLN. Furthermore, this interaction results in SERCA1 inhibition, as measured by a reduction in the apparent Ca2+ affinity of SERCA1. The possibility that the expression of sAnk1 is not limited to striated muscle suggests that it may play a more universal role as a regulator of SERCA activity in other tissues. Co-expression of sAnk1 with either SLN, PLN, or MLN in various tissues also suggests a more intricate level of co-regulation by multiple micropeptides and small proteins. Future studies will aim to determine whether sAnk1 can interact with SLN and superinhibit SERCA activity.
Author Contributions
P. F. D. and R. J. B. designed the study and wrote the paper. P. F. D. performed and analyzed experiments shown in Figs. 2, 3, 4, 6, 7, and the computer simulations shown in Fig. 8. J. M. designed and prepared fluorescent constructs used for AFRET studies in COS7 cells. M. A. R. contributed expertise in AFRET to aid in the design and execution of AFRET experiments. AFRET data were collected by M. L. M. and analyzed by P. F. D.
Acknowledgment
SERCA1 and FLAG-SLN cDNA constructs were generous gifts from Dr. David MacLennan (University of Toronto).
This work was supported in part by National Institutes of Health Grants RO1 DK077140 (to M. A. R.) and RO1 AR056330 (to R. J. B.).
- SR
- sarcoplasmic reticulum
- nSR
- network compartment of the SR
- SERCA
- sarco(endo)plasmic reticulum Ca2+-ATPase
- TM
- transmembrane
- PLN
- phospholamban
- SLN
- sarcolipin
- MLN
- myoregulin
- sAnk1
- small ankyrin 1
- RyR1
- ryanodine receptor-1
- JPH1
- junctophilin-1
- pAb
- polyclonal antibody
- MBP
- maltose-binding protein
- CFP
- cyan fluorescent protein
- AFRET
- anisotropy-based fluorescence resonance energy transfer
- IP
- immunoprecipitation
- ER
- endoplasmic reticulum.
References
- 1.Ziman A. P., Ward C. W., Rodney G. G., Lederer W. J., and Bloch R. J. (2010) Quantitative measurement of Ca2+ in the sarcoplasmic reticulum lumen of mammalian skeletal muscle. Biophys. J. 99, 2705–2714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Launikonis B. S., Zhou J., Royer L., Shannon T. R., Brum G., and Ríos E. (2005) Confocal imaging of [Ca2+] in cellular organelles by SEER, shifted excitation and emission ratioing of fluorescence. J. Physiol. 567, 523–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kabbara A. A., and Allen D. G. (1999) Measurement of sarcoplasmic reticulum Ca2+ content in intact amphibian skeletal muscle fibres with 4-chloro-m-cresol. Cell Calcium 25, 227–235 [DOI] [PubMed] [Google Scholar]
- 4.Somlyo A. V., Gonzalez-Serratos H. G., Shuman H., McClellan G., and Somlyo A. P. (1981) Calcium release and ionic changes in the sarcoplasmic reticulum of tetanized muscle: an electron-probe study. J. Cell Biol. 90, 577–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brini M., and Carafoli E. (2009) Calcium pumps in health and disease. Physiol. Rev. 89, 1341–1378 [DOI] [PubMed] [Google Scholar]
- 6.Periasamy M., and Kalyanasundaram A. (2007) SERCA pump isoforms: their role in calcium transport and disease. Muscle Nerve 35, 430–442 [DOI] [PubMed] [Google Scholar]
- 7.Adachi T. (2010) Modulation of vascular sarco/endoplasmic reticulum calcium ATPase in cardiovascular pathophysiology. Adv. Pharmacol. 59, 165–195 [DOI] [PubMed] [Google Scholar]
- 8.Díaz M. E., Graham H. K., O'neill S. C., Trafford A. W., and Eisner D. A. (2005) The control of sarcoplasmic reticulum Ca content in cardiac muscle. Cell Calcium 38, 391–396 [DOI] [PubMed] [Google Scholar]
- 9.Schmidt A. G., Zhai J., Carr A. N., Gerst M. J., Lorenz J. N., Pollesello P., Annila A., Hoit B. D., and Kranias E. G. (2002) Structural and functional implications of the phospholamban hinge domain: impaired SR Ca2+ uptake as a primary cause of heart failure. Cardiovasc. Res. 56, 248–259 [DOI] [PubMed] [Google Scholar]
- 10.Vangheluwe P., Tjwa M., Van Den Bergh A., Louch W. E., Beullens M., Dode L., Carmeliet P., Kranias E., Herijgers P., Sipido K. R., Raeymaekers L., and Wuytack F. (2006) A SERCA2 pump with an increased Ca2+ affinity can lead to severe cardiac hypertrophy, stress intolerance and reduced life span. J. Mol. Cell. Cardiol. 41, 308–317 [DOI] [PubMed] [Google Scholar]
- 11.Koss K. L., Ponniah S., Jones W. K., Grupp I. L., and Kranias E. G. (1995) Differential phospholamban gene expression in murine cardiac compartments. Molecular and physiological analyses. Circ. Res. 77, 342–353 [DOI] [PubMed] [Google Scholar]
- 12.Tada M., and Toyofuku T. (1998) Molecular regulation of phospholamban function and expression. Trends Cardiovasc. Med. 8, 330–340 [DOI] [PubMed] [Google Scholar]
- 13.Briggs F. N., Lee K. F., Wechsler A. W., and Jones L. R. (1992) Phospholamban expressed in slow-twitch and chronically stimulated fast-twitch muscles minimally affects calcium affinity of sarcoplasmic reticulum Ca2+-ATPase. J. Biol. Chem. 267, 26056–26061 [PubMed] [Google Scholar]
- 14.Odermatt A., Taschner P. E., Scherer S. W., Beatty B., Khanna V. K., Cornblath D. R., Chaudhry V., Yee W. C., Schrank B., Karpati G., Breuning M. H., Knoers N., and MacLennan D. H. (1997) Characterization of the gene encoding human sarcolipin (SLN), a proteolipid associated with SERCA1: absence of structural mutations in five patients with Brody disease. Genomics 45, 541–553 [DOI] [PubMed] [Google Scholar]
- 15.Vangheluwe P., Schuermans M., Zádor E., Waelkens E., Raeymaekers L., and Wuytack F. (2005) Sarcolipin and phospholamban mRNA and protein expression in cardiac and skeletal muscle of different species. Biochem. J. 389, 151–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Babu G. J., Zheng Z., Natarajan P., Wheeler D., Janssen P. M., and Periasamy M. (2005) Overexpression of sarcolipin decreases myocyte contractility and calcium transient. Cardiovasc. Res. 65, 177–186 [DOI] [PubMed] [Google Scholar]
- 17.Gayan-Ramirez G., Vanzeir L., Wuytack F., and Decramer M. (2000) Corticosteroids decrease mRNA levels of SERCA pumps, whereas they increase sarcolipin mRNA in the rat diaphragm. J. Physiol. 524, 387–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stammers A. N., Susser S. E., Hamm N. C., Hlynsky M. W., Kimber D. E., Kehler D. S., and Duhamel T. A. (2015) The regulation of sarco(endo)plasmic reticulum calcium-ATPases (SERCA). Can. J. Physiol. Pharmacol. 93, 843–854 [DOI] [PubMed] [Google Scholar]
- 19.Hellstern S., Pegoraro S., Karim C. B., Lustig A., Thomas D. D., Moroder L., and Engel J. (2001) Sarcolipin, the shorter homologue of phospholamban, forms oligomeric structures in detergent micelles and in liposomes. J. Biol. Chem. 276, 30845–30852 [DOI] [PubMed] [Google Scholar]
- 20.Odermatt A., Becker S., Khanna V. K., Kurzydlowski K., Leisner E., Pette D., and MacLennan D. H. (1998) Sarcolipin regulates the activity of SERCA1, the fast-twitch skeletal muscle sarcoplasmic reticulum Ca2+-ATPase. J. Biol. Chem. 273, 12360–12369 [DOI] [PubMed] [Google Scholar]
- 21.Afara M. R., Trieber C. A., Glaves J. P., and Young H. S. (2006) Rational design of peptide inhibitors of the sarcoplasmic reticulum calcium pump. Biochemistry 45, 8617–8627 [DOI] [PubMed] [Google Scholar]
- 22.Asahi M., Sugita Y., Kurzydlowski K., De Leon S., Tada M., Toyoshima C., and MacLennan D. H. (2003) Sarcolipin regulates sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA) by binding to transmembrane helices alone or in association with phospholamban. Proc. Natl. Acad. Sci. U.S.A. 100, 5040–5045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hutter M. C., Krebs J., Meiler J., Griesinger C., Carafoli E., and Helms V. (2002) A structural model of the complex formed by phospholamban and the calcium pump of sarcoplasmic reticulum obtained by molecular mechanics. Chembiochem 3, 1200–1208 [DOI] [PubMed] [Google Scholar]
- 24.Morita T., Hussain D., Asahi M., Tsuda T., Kurzydlowski K., Toyoshima C., and Maclennan D. H. (2008) Interaction sites among phospholamban, sarcolipin, and the sarco(endo)plasmic reticulum Ca2+-ATPase. Biochem. Biophys. Res. Commun. 369, 188–194 [DOI] [PubMed] [Google Scholar]
- 25.Seidel K., Andronesi O. C., Krebs J., Griesinger C., Young H. S., Becker S., and Baldus M. (2008) Structural characterization of Ca2+-ATPase-bound phospholamban in lipid bilayers by solid-state nuclear magnetic resonance (NMR) spectroscopy. Biochemistry 47, 4369–4376 [DOI] [PubMed] [Google Scholar]
- 26.Toyoshima C., Asahi M., Sugita Y., Khanna R., Tsuda T., and MacLennan D. H. (2003) Modeling of the inhibitory interaction of phospholamban with the Ca2+ ATPase. Proc. Natl. Acad. Sci. U.S.A. 100, 467–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Traaseth N. J., Ha K. N., Verardi R., Shi L., Buffy J. J., Masterson L. R., and Veglia G. (2008) Structural and dynamic basis of phospholamban and sarcolipin inhibition of Ca2+-ATPase. Biochemistry 47, 3–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Asahi M., Kurzydlowski K., Tada M., and MacLennan D. H. (2002) Sarcolipin inhibits polymerization of phospholamban to induce superinhibition of sarco(endo)plasmic reticulum Ca2+-ATPases (SERCAs). J. Biol. Chem. 277, 26725–26728 [DOI] [PubMed] [Google Scholar]
- 29.Autry J. M., Rubin J. E., Pietrini S. D., Winters D. L., Robia S. L., and Thomas D. D. (2011) Oligomeric interactions of sarcolipin and the Ca-ATPase. J. Biol. Chem. 286, 31697–31706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Buffy J. J., Buck-Koehntop B. A., Porcelli F., Traaseth N. J., Thomas D. D., and Veglia G. (2006) Defining the intramembrane binding mechanism of sarcolipin to calcium ATPase using solution NMR spectroscopy. J. Mol. Biol. 358, 420–429 [DOI] [PubMed] [Google Scholar]
- 31.Harrer J. M., and Kranias E. G. (1994) Characterization of the molecular form of cardiac phospholamban. Mol. Cell. Biochem. 140, 185–193 [DOI] [PubMed] [Google Scholar]
- 32.Li M., Reddy L. G., Bennett R., Silva N. D. Jr, Jones L. R., and Thomas D. D. (1999) A fluorescence energy transfer method for analyzing protein oligomeric structure: application to phospholamban. Biophys. J. 76, 2587–2599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Simmerman H. K., Kobayashi Y. M., Autry J. M., and Jones L. R. (1996) A leucine zipper stabilizes the pentameric membrane domain of phospholamban and forms a coiled-coil pore structure. J. Biol. Chem. 271, 5941–5946 [DOI] [PubMed] [Google Scholar]
- 34.Traaseth N. J., Verardi R., Torgersen K. D., Karim C. B., Thomas D. D., and Veglia G. (2007) Spectroscopic validation of the pentameric structure of phospholamban. Proc. Natl. Acad. Sci. U.S.A. 104, 14676–14681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Watanabe Y., Kijima Y., Kadoma M., Tada M., and Takagi T. (1991) Molecular weight determination of phospholamban oligomer in the presence of sodium dodecyl sulfate: application of low-angle laser light scattering photometry. J. Biochem. 110, 40–45 [DOI] [PubMed] [Google Scholar]
- 36.Toyofuku T., Kurzydlowski K., Tada M., and MacLennan D. H. (1994) Amino acids Glu2 to Ile18 in the cytoplasmic domain of phospholamban are essential for functional association with the Ca2+-ATPase of sarcoplasmic reticulum. J. Biol. Chem. 269, 3088–3094 [PubMed] [Google Scholar]
- 37.Gorski P. A., Glaves J. P., Vangheluwe P., and Young H. S. (2013) Sarco(endo)plasmic reticulum calcium ATPase (SERCA) inhibition by sarcolipin is encoded in its luminal tail. J. Biol. Chem. 288, 8456–8467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gramolini A. O., Kislinger T., Asahi M., Li W., Emili A., and MacLennan D. H. (2004) Sarcolipin retention in the endoplasmic reticulum depends on its C-terminal RSYQY sequence and its interaction with sarco(endo)plasmic Ca2+-ATPases. Proc. Natl. Acad. Sci. U.S.A. 101, 16807–16812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.MacLennan D. H., Asahi M., and Tupling A. R. (2003) The regulation of SERCA-type pumps by phospholamban and sarcolipin. Ann. N.Y. Acad. Sci. 986, 472–480 [DOI] [PubMed] [Google Scholar]
- 40.Fajardo V. A., Bombardier E., Vigna C., Devji T., Bloemberg D., Gamu D., Gramolini A. O., Quadrilatero J., and Tupling A. R. (2013) Co-expression of SERCA isoforms, phospholamban and sarcolipin in human skeletal muscle fibers. PLoS ONE 8, e84304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Anderson D. M., Anderson K. M., Chang C. L., Makarewich C. A., Nelson B. R., McAnally J. R., Kasaragod P., Shelton J. M., Liou J., Bassel-Duby R., and Olson E. N. (2015) A micropeptide encoded by a putative long noncoding RNA regulates muscle performance. Cell 160, 595–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Birkenmeier C. S., White R. A., Peters L. L., Hall E. J., Lux S. E., and Barker J. E. (1993) Complex patterns of sequence variation and multiple 5′ and 3′ ends are found among transcripts of the erythroid ankyrin gene. J. Biol. Chem. 268, 9533–9540 [PubMed] [Google Scholar]
- 43.Zhou D., Birkenmeier C. S., Williams M. W., Sharp J. J., Barker J. E., and Bloch R. J. (1997) Small, membrane-bound, alternatively spliced forms of ankyrin 1 associated with the sarcoplasmic reticulum of mammalian skeletal muscle. J. Cell Biol. 136, 621–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Armani A., Galli S., Giacomello E., Bagnato P., Barone V., Rossi D., and Sorrentino V. (2006) Molecular interactions with obscurin are involved in the localization of muscle-specific small ankyrin1 isoforms to subcompartments of the sarcoplasmic reticulum. Exp. Cell Res. 312, 3546–3558 [DOI] [PubMed] [Google Scholar]
- 45.Porter N. C., Resneck W. G., O'Neill A., Van Rossum D. B., Stone M. R., and Bloch R. J. (2005) Association of small ankyrin 1 with the sarcoplasmic reticulum. Mol. Membr. Biol. 22, 421–432 [DOI] [PubMed] [Google Scholar]
- 46.Hopitzan A. A., Baines A. J., and Kordeli E. (2006) Molecular evolution of ankyrin: gain of function in vertebrates by acquisition of an obscurin/titin-binding-related domain. Mol. Biol. Evol. 23, 46–55 [DOI] [PubMed] [Google Scholar]
- 47.Bagnato P., Barone V., Giacomello E., Rossi D., and Sorrentino V. (2003) Binding of an ankyrin-1 isoform to obscurin suggests a molecular link between the sarcoplasmic reticulum and myofibrils in striated muscles. J. Cell Biol. 160, 245–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Borzok M. A., Catino D. H., Nicholson J. D., Kontrogianni-Konstantopoulos A., and Bloch R. J. (2007) Mapping the binding site on small ankyrin 1 for obscurin. J. Biol. Chem. 282, 32384–32396 [DOI] [PubMed] [Google Scholar]
- 49.Ackermann M. A., Ziman A. P., Strong J., Zhang Y., Hartford A. K., Ward C. W., Randall W. R., Kontrogianni-Konstantopoulos A., and Bloch R. J. (2011) Integrity of the network sarcoplasmic reticulum in skeletal muscle requires small ankyrin 1. J. Cell Sci. 124, 3619–3630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kontrogianni-Konstantopoulos A., Jones E. M., Van Rossum D. B., and Bloch R. J. (2003) Obscurin is a ligand for small ankyrin 1 in skeletal muscle. Mol. Biol. Cell 14, 1138–1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kontrogianni-Konstantopoulos A., and Bloch R. J. (2003) The hydrophilic domain of small ankyrin-1 interacts with the two N-terminal immunoglobulin domains of titin. J. Biol. Chem. 278, 3985–3991 [DOI] [PubMed] [Google Scholar]
- 52.Gokhin D. S., and Fowler V. M. (2011) Cytoplasmic γ-actin and tropomodulin isoforms link to the sarcoplasmic reticulum in skeletal muscle fibers. J. Cell Biol. 194, 105–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kontrogianni-Konstantopoulos A., Ackermann M. A., Bowman A. L., Yap S. V., and Bloch R. J. (2009) Muscle giants: molecular scaffolds in sarcomerogenesis. Physiol. Rev. 89, 1217–1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kontrogianni-Konstantopoulos A., Catino D. H., Strong J. C., Randall W. R., and Bloch R. J. (2004) Obscurin regulates the organization of myosin into A bands. Am. J. Physiol. Cell Physiol. 287, C209–C217 [DOI] [PubMed] [Google Scholar]
- 55.Lange S., Ouyang K., Meyer G., Cui L., Cheng H., Lieber R. L., and Chen J. (2009) Obscurin determines the architecture of the longitudinal sarcoplasmic reticulum. J. Cell Sci. 122, 2640–2650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Giacomello E., Quarta M., Paolini C., Squecco R., Fusco P., Toniolo L., Blaauw B., Formoso L., Rossi D., Birkenmeier C., Peters L. L., Francini F., Protasi F., Reggiani C., and Sorrentino V. (2015) Deletion of small ankyrin 1 (sAnk1) isoforms results in structural and functional alterations in aging skeletal muscle fibers. Am. J. Physiol. Cell Physiol. 308, C123–C138 [DOI] [PubMed] [Google Scholar]
- 57.Rizzo M. A., Springer G., Segawa K., Zipfel W. R., and Piston D. W. (2006) Optimization of pairings and detection conditions for measurement of FRET between cyan and yellow fluorescent proteins. Microsc. Microanal. 12, 238–254 [DOI] [PubMed] [Google Scholar]
- 58.Markwardt M. L., Kremers G. J., Kraft C. A., Ray K., Cranfill P. J., Wilson K. A., Day R. N., Wachter R. M., Davidson M. W., and Rizzo M. A. (2011) An improved cerulean fluorescent protein with enhanced brightness and reduced reversible photoswitching. PLoS One 6, e17896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fu M. H., and Tupling A. R. (2009) Protective effects of Hsp70 on the structure and function of SERCA2a expressed in HEK-293 cells during heat stress. Am. J. Physiol. Heart Circ. Physiol. 296, H1175–H1183 [DOI] [PubMed] [Google Scholar]
- 60.Asahi M., Kimura Y., Kurzydlowski K., Tada M., and MacLennan D. H. (1999) Transmembrane helix M6 in sarco(endo)plasmic reticulum Ca2+-ATPase forms a functional interaction site with phospholamban. Evidence for physical interactions at other sites. J. Biol. Chem. 274, 32855–32862 [DOI] [PubMed] [Google Scholar]
- 61.Eletr S., and Inesi G. (1972) Phase changes in the lipid moieties of sarcoplasmic reticulum membranes induced by temperature and protein conformational changes. Biochim. Biophys. Acta 290, 178–185 [DOI] [PubMed] [Google Scholar]
- 62.Kosk-Kosicka D. (2013) Measurement of Ca2+-ATPase activity (in PMCA and SERCA1). Methods Mol. Biol. 937, 343–356 [DOI] [PubMed] [Google Scholar]
- 63.Maruyama K., and MacLennan D. H. (1988) Mutation of aspartic acid-351, lysine-352, and lysine-515 alters the Ca2+ transport activity of the Ca2+-ATPase expressed in COS-1 cells. Proc. Natl. Acad. Sci. U.S.A. 85, 3314–3318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lencesova L., O'Neill A., Resneck W. G., Bloch R. J., and Blaustein M. P. (2004) Plasma membrane-cytoskeleton-endoplasmic reticulum complexes in neurons and astrocytes. J. Biol. Chem. 279, 2885–2893 [DOI] [PubMed] [Google Scholar]
- 65.Bolte S., and Cordelières F. P. (2006) A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 224, 213–232 [DOI] [PubMed] [Google Scholar]
- 66.Piston D. W., and Rizzo M. A. (2008) FRET by fluorescence polarization microscopy. Methods Cell Biol. 85, 415–430 [DOI] [PubMed] [Google Scholar]
- 67.Gade P., Ramachandran G., Maachani U. B., Rizzo M. A., Okada T., Prywes R., Cross A. S., Mori K., and Kalvakolanu D. V. (2012) An IFN-γ-stimulated ATF6-C/EBP-β-signaling pathway critical for the expression of death associated protein kinase 1 and induction of autophagy. Proc. Natl. Acad. Sci. U.S.A. 109, 10316–10321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rizzo M. A., and Piston D. W. (2005) High-contrast imaging of fluorescent protein FRET by fluorescence polarization microscopy. Biophys. J. 88, L14–L16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kontrogianni-Konstantopoulos A., Huang S. C., and Benz E. J. Jr (2000) A nonerythroid isoform of protein 4.1R interacts with components of the contractile apparatus in skeletal myofibers. Mol. Biol. Cell 11, 3805–3817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang Y. (2008) I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Comeau S. R., Gatchell D. W., Vajda S., and Camacho C. J. (2004) ClusPro: a fully automated algorithm for protein-protein docking. Nucleic Acids Res. 32, W96–W99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Comeau S. R., Gatchell D. W., Vajda S., and Camacho C. J. (2004) ClusPro: an automated docking and discrimination method for the prediction of protein complexes. Bioinformatics 20, 45–50 [DOI] [PubMed] [Google Scholar]
- 73.Kozakov D., Brenke R., Comeau S. R., and Vajda S. (2006) PIPER: an FFT-based protein docking program with pairwise potentials. Proteins 65, 392–406 [DOI] [PubMed] [Google Scholar]
- 74.Kozakov D., Beglov D., Bohnuud T., Mottarella S. E., Xia B., Hall D. R., and Vajda S. (2013) How good is automated protein docking? Proteins 81, 2159–2166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Guex N., and Peitsch M. C. (1997) SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18, 2714–2723 [DOI] [PubMed] [Google Scholar]
- 76.Bombardier E., Smith I. C., Vigna C., Fajardo V. A., and Tupling A. R. (2013) Ablation of sarcolipin decreases the energy requirements for Ca2+ transport by sarco(endo)plasmic reticulum Ca2+-ATPases in resting skeletal muscle. FEBS Lett. 587, 1687–1692 [DOI] [PubMed] [Google Scholar]
- 77.Sahoo S. K., Shaikh S. A., Sopariwala D. H., Bal N. C., Bruhn D. S., Kopec W., Khandelia H., and Periasamy M. (2015) The N terminus of sarcolipin plays an important role in uncoupling sarco-endoplasmic reticulum Ca2+ ATPase (SERCA) ATP hydrolysis from Ca2+ transport. J. Biol. Chem. 290, 14057–14067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Senes A., Gerstein M., and Engelman D. M. (2000) Statistical analysis of amino acid patterns in transmembrane helices: the GxxxG motif occurs frequently and in association with beta-branched residues at neighboring positions. J. Mol. Biol. 296, 921–936 [DOI] [PubMed] [Google Scholar]
- 79.Altschul S. F., Gish W., Miller W., Myers E. W., and Lipman D. J. (1990) Basic local alignment search tool. J. Mol. Biol. 215, 403–410 [DOI] [PubMed] [Google Scholar]
- 80.Gyorke I., Hester N., Jones L. R., and Györke S. (2004) The role of calsequestrin, triadin, and junctin in conferring cardiac ryanodine receptor responsiveness to luminal calcium. Biophys. J. 86, 2121–2128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Shen X., Franzini-Armstrong C., Lopez J. R., Jones L. R., Kobayashi Y. M., Wang Y., Kerrick W. G., Caswell A. H., Potter J. D., Miller T., Allen P. D., and Perez C. F. (2007) Triadins modulate intracellular Ca2+ homeostasis but are not essential for excitation-contraction coupling in skeletal muscle. J. Biol. Chem. 282, 37864–37874 [DOI] [PubMed] [Google Scholar]
- 82.Kerr J. P., Ward C. W., and Bloch R. J. (2014) Dysferlin at transverse tubules regulates Ca2+ homeostasis in skeletal muscle. Front. Physiol. 5, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Abrol N., Smolin N., Armanious G., Ceholski D. K., Trieber C. A., Young H. S., and Robia S. L. (2014) Phospholamban C-terminal residues are critical determinants of the structure and function of the calcium ATPase regulatory complex. J. Biol. Chem. 289, 25855–25866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Toyofuku T., Kurzydlowski K., Tada M., and MacLennan D. H. (1994) Amino acids Lys-Asp-Asp-Lys-Pro-Val402 in the Ca2+-ATPase of cardiac sarcoplasmic reticulum are critical for functional association with phospholamban. J. Biol. Chem. 269, 22929–22932 [PubMed] [Google Scholar]
- 85.Toyofuku T., Kurzydlowski K., Tada M., and MacLennan D. H. (1993) Identification of regions in the Ca2+-ATPase of sarcoplasmic reticulum that affect functional association with phospholamban. J. Biol. Chem. 268, 2809–2815 [PubMed] [Google Scholar]
- 86.Förster T. (1948) Zwischenmolekulare Energiewanderung und Fluoreszenz. Ann. Physik. 437, 55–75 [Google Scholar]
- 87.Kimura Y., Kurzydlowski K., Tada M., and MacLennan D. H. (1997) Phospholamban inhibitory function is activated by depolymerization. J. Biol. Chem. 272, 15061–15064 [DOI] [PubMed] [Google Scholar]
- 88.Kimura Y., Asahi M., Kurzydlowski K., Tada M., and MacLennan D. H. (1998) Phospholamban domain Ib mutations influence functional interactions with the Ca2+-ATPase isoform of cardiac sarcoplasmic reticulum. J. Biol. Chem. 273, 14238–14241 [DOI] [PubMed] [Google Scholar]
- 89.Green K. N., Demuro A., Akbari Y., Hitt B. D., Smith I. F., Parker I., and LaFerla F. M. (2008) SERCA pump activity is physiologically regulated by presenilin and regulates amyloid β production. J. Cell Biol. 181, 1107–1116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Honarnejad K., and Herms J. (2012) Presenilins: role in calcium homeostasis. Int. J. Biochem. Cell Biol. 44, 1983–1986 [DOI] [PubMed] [Google Scholar]
- 91.Yu J. T., Chang R. C., and Tan L. (2009) Calcium dysregulation in Alzheimer's disease: from mechanisms to therapeutic opportunities. Prog. Neurobiol. 89, 240–255 [DOI] [PubMed] [Google Scholar]