

Background: Protein trans-splicing as a molecular design tool has been demonstrated for soluble but not yet for membrane proteins.
Results: Two separate polypeptides have been spliced in vivo, yielding correctly folded and functional proteorhodopsin.
Conclusion: Trans-splicing of α-helical membrane proteins under native conditions is possible.
Significance: Our findings are important for the folding, assembly, and engineering of membrane proteins.
Keywords: seven-helix receptor, intein, membrane protein, photoreceptor, protein folding, protein splicing, proton pump, solid-state NMR
Abstract
Protein trans-splicing using split inteins is well established as a useful tool for protein engineering. Here we show, for the first time, that this method can be applied to a membrane protein under native conditions. We provide compelling evidence that the heptahelical proteorhodopsin can be assembled from two separate fragments consisting of helical bundles A and B and C, D, E, F, and G via a splicing site located in the BC loop. The procedure presented here is on the basis of dual expression and ligation in vivo. Global fold, stability, and photodynamics were analyzed in detergent by CD, stationary, as well as time-resolved optical spectroscopy. The fold within lipid bilayers has been probed by high field and dynamic nuclear polarization-enhanced solid-state NMR utilizing a 13C-labeled retinal cofactor and extensively 13C-15N-labeled protein. Our data show unambiguously that the ligation product is identical to its non-ligated counterpart. Furthermore, our data highlight the effects of BC loop modifications onto the photocycle kinetics of proteorhodopsin. Our data demonstrate that a correctly folded and functionally intact protein can be produced in this artificial way. Our findings are of high relevance for a general understanding of the assembly of membrane proteins for elucidating intramolecular interactions, and they offer the possibility of developing novel labeling schemes for spectroscopic applications.
Introduction
During the past years, protein trans-splicing (PTS)2 and related methods such as native chemical ligation or expressed chemical ligation have been explored as useful tools for protein engineering (1, 2). Ligating a protein from separate fragments offers the possibility of probing the structural and functional assembly of multidomain proteins. Furthermore, it allows the expression of proteins that would be toxic at full length (3), and it has been used for fluorescence (4, 5) or segmental isotope labeling (6, 7).
The most flexible approach to PTS is offered by split inteins, which are naturally occurring proteins undergoing an autocatalytic process during which a peptide bond between their flanking sequences (extein) is created, whereas the intein itself is excised (8). PTS can take place in vivo with both precursors encoded in two different vectors, which are induced separately, or in vitro after separate precursor expression and purification (6). The split intein DnaE from Nostoc punctiforme (Npu DnaE) PCC 73102 has been the most popular so far because of its robustness and high splicing rate compared with other inteins (9, 10).
So far, mainly soluble proteins have been targeted. Only one study recently demonstrated in vitro PTS of a small, insoluble protein fibril under denaturing conditions (11). In contrast, protein trans-splicing of membrane proteins has been rarely studied. The only example reported until now focused on in vitro splicing under denaturing conditions of the β barrel outer membrane protein from Escherichia coli (12). Of course, the possibility to splice a membrane protein from separate fragments is rather tempting, but its general applicability would require a first successful demonstration for an α-helical membrane protein under native conditions, i.e. without the need of additional unfolding/refolding steps. Such an endeavor, however, will lead directly to the fundamental question of whether a functional membrane protein can be assembled from separate fragments consisting of bundles of transmembrane α helices. A first hint that such an approach could be indeed possible is provided by the observation that proteolytically generated fragments of bacteriorhodopsin spontaneously cluster non-covalently into a retinal binding bundle in mixed micelles and lipid vesicles (13–15).
Here we demonstrate that the heptahelical membrane protein proteorhodopsin can be assembled from two separately expressed fragments using PTS on the basis of split inteins (Fig. 1A). The observation made for bacteriorhodopsin mentioned above led to our working hypothesis that proteorhodopsin (pR) fragments also associate with each other, which would support successful protein trans-splicing.
FIGURE 1.

A, in vivo protein trans-splicing of two α-helical fragments of pR on the basis of split inteins. B, design of the pR-intein segments. The N-terminal precursor contains helices A and B of pR, followed by DnaEN102 and a Strep tag. The C-terminal precursor consists of a Strep tag, an Smt3 solubility tag, NpuΔ36, and helices C, D, E, F, and G of pR, followed by an HSV-His6 tag. By protein trans-splicing, the solubility and both Strep tags are excised, resulting in full-length HSV-His6-tagged pR (pRLIG). C, topology plot of pRWT and pRLIG/pRSCFNG. To ensure an efficient splicing reaction, the BC loop amino acid sequence was modified as depicted. pSB, protonated Schiff base.
For a proof of concept, a retinal protein such as pR seems ideal because its functional integrity can be assessed by optical spectroscopy. Green-absorbing pR from Gamma-proteobacterium EBAC31A08 is a heptahelical membrane protein (27 kDa) functioning as light-driven proton pump (16). The pR protein family can be divided into green- and blue-absorbing subgroups (17). The backbone structure of green pR in detergent micelles is known from solution-state NMR (18), whereas the structure of blue pR was solved by x-ray crystallography (19). Furthermore, the functional mechanism of pR has been studied extensively by optical and infrared spectroscopy, electrophysiology, and solid-state NMR (20–26).
Here pR has been reconstructed by covalently linking two segments consisting of helices A and B and C, D, E, F, and G using protein trans-splicing in vivo (6) (Fig. 1A). We favored this approach over in vitro splicing to minimize the risk of potential stabilization problems by separate solubilization and purification steps of both segments. The ligation product (pRLIG) could be purified at quantitative amounts, and its structural and photophysical characteristics were analyzed in detergent micelles as well as in lipid bilayers using optical spectroscopy and high-field as well as DNP-enhanced solid-state NMR. For efficient protein trans-splicing, the splicing site in the BC loop was slightly modified. Therefore, pRLIG was compared with a pR mutant (pRSCFNG) with the same BC loop in addition to wild-type pR (pRWT). Our data show unambiguously that the ligation product, pRLIG, is identical to its non-ligated counterpart, pRSCFNG, in terms of photocycle characteristics, stability, fold, and structure. Our data prove unambiguously that a large α-helical membrane protein can be assembled correctly from individual segments using the split intein approach under native conditions. Furthermore, the role of the BC loop for the photodynamics of pR will be discussed on the basis of the observed differences between pRWT and pRSCFNG together with the relevance and significance of our findings in the context of membrane protein folding and possible applications.
Experimental Procedures
Vector Design and Protein Expression
The N segment containing the helices A and B of proteorhodopsin without its signal sequence (amino acids 1–20) and the DnaE NpuN102 (Addgene, pSABAD219) were cloned in a pRSF vector (Invitrogen). The C segment, constructed of an N-terminal Smt3 solubility tag (UniProt, Q12306), helices C, D, E, F, and G of pR, NpuΔ36, and a C-terminal HSV-His6 tag for purification, were cloned into a pBAD vector (Invitrogen). Both constructs have a C- or N-terminal tag, respectively, to separate unligated segments from the ligation product (Fig. 1B).
For expression, E. coli C43 (DE3) cells were cotransfected with both vectors. The cells were plated on agar plates containing 100 μg/ml ampicillin and 35 μg/ml kanamycin. Expression took place in 500-ml cultures in 2-liter flasks to provide enough oxygen for optimal cell growth. Luria broth (LB) medium containing the previously mentioned antibiotics was inoculated with 1% of an overnight LB preculture inoculated with a freshly transformed clone. Cells were grown at 37 °C and 220 rpm until an A600 of 0.7–0.8 was reached. Cells were spun down, washed with LB, and resuspended gently in 500 ml M9+ medium (6.78 g/liter K2HPO4, 3 g/liter KH2PO4, 1 g/liter NaCl, 0.25 g/liter adenine, 0.325 g/liter guanosin, 0.1 g/liter cytosine, 0.1 g/liter thymine, 0.25 g/liter uracil, 0.13 g/liter phenylalanine, 0.23 g/liter leucine, 0.17 g/liter tyrosine, 0.23 g/liter valine, and 0.05 g/liter tryptophan) containing 4 g/liter glucose, 1 g/liter NH4Cl (for labeled samples, 13C6-glucose and 15NH4Cl), 0.01 mm FeCl3, 2 mm MgSO4, 0.15 mg/ml vitamin mixture (Centrum), and antibiotics. The cells were adapted for 1 h at 30 °C and induced with 0.1% l-arabinose and 2.1 μm retinal. After 3 h, the temperature was reduced to 20 °C, and the second induction with 0.2 mm isopropyl 1-thio-β-d-galactopyranoside took place. Cells were grown for further 16–18 h.
Cells were harvested, resuspended in 300 mm NaCl, 50 mm HEPES (pH 7.0) (containing MgCl2, protease inhibitor mixture from Roche (1 tablet for each 50 ml), and DNase) and disrupted using a Constant System cell disrupter at 1.85 kbar. The membrane was pelleted by ultracentrifugation at 55,000 rpm for 1 h. The membrane was homogenized in 300 mm NaCl, 50 mm MES, and 5 mm imidazole (pH 6) and solubilized overnight in 1.5% dodecyl-β-d-maltoside (DDM) at 4 °C. The next day, unsolubilized material was spun down in an ultracentrifuge for 1 h. The supernatant was incubated with 1.5 ml nickel-nitrilotriacetic acid per liter of culture in the presence of 20 mm imidazole for several hours at 4 °C. The resin was washed with 10 column values of washing buffers with increasing imidazole concentrations (300 mm NaCl, 50 mm MES (pH 6), and 0.15% DDM) containing 20, 50, mm and 80 mm imidazole, respectively. Protein was eluted with 500 mm imidazole and 0.05% DDM (pH 7). Next the protein was concentrated in a Centricon (50-kDa cutoff), and the pH value was adjusted to 8. The protein was incubated overnight at 4 °C with ∼0.5 ml Strep resin per liter of culture. The flow through of the Strep purification contains the purified ligation product. About 2–3 mg per liter of ligated pR were obtained.
Expression, purification, and reconstitution into liposomes of pRWT were done as described previously (22). In addition, the following mutations were introduced into the pRWT template by site-directed mutagenesis using primers from Eurofins MWG Operon: the single mutants pRD88S, pRS89C, and pRP90F; the insertion mutant pRInsNG, in which residues Asn and Gly were inserted following Pro90; and pRSCFNG, with residues SCFNG replacing Asp88-Ser89-Pro90. Mutated sequences were verified by Eurofins MWG Operon. All mutants were expressed, purified, and reconstituted as pRWT. Reconstitution was carried out into 1,2-dimyristoyl-sn-glycero-3-phosphocholine/1,2-dimyristoyl-sn-glycero-3-phosphate (9:1 mol/mol) liposomes at a lipid-to-protein ratio of 1:2 (w/w). Optical spectroscopy was applied to samples solubilized in 0.05% DDM. All NMR experiments were carried out on proteoliposomes.
SDS-PAGE and Western Blot Analysis
At several time points during expression, samples of the culture were taken, and the reaction was stopped by adding SDS buffer. Samples were diluted to an optical density of ∼1 and boiled for 10 min at 95 °C before loading on precast gels from Expedeon. The gels ran for about 35 min at 180 V, followed by staining with Coomassie Brilliant Blue. For immunodetection, proteins were blotted on PVDF membranes using a wet blotting system. Membranes were blocked with 3% BSA and incubated with anti-Strep or anti-His antibody in TBS or TBS-TT (Tris-buffered saline, pH 7.5, 0.2% Tween 20, 0.05% Triton X-100), respectively, containing 0.3% BSA. Signals were detected by chemiluminescence.
Preparation of 14,15-13C All-trans-retinal Proteorhodopsin Samples for DNP-enhanced NMR
All-trans-retinal 13C-labeled at positions 14 and 15 was synthesized as described previously and was provided by Prof. Richard Brown (University of Southampton) (22). Retinal was added directly to the E. coli cultures for incorporation into proteo-opsin. For DNP enhancement, the reconstituted samples were incubated with 10% H2O, 30% d8-glycerol, 60% D2O, and 20 mm TOTAPOL (27) overnight at 4 °C as described before (22). The solution was removed completely before the sample was packed into a 3.2-mm ZrO2 rotor.
CD Spectroscopy
Circular dichroism was measured with a J-815 spectrometer (Jasco) using a protein concentration of 7–15 μm in a buffer containing 2 mm NaCl, 2 mm HEPES, and 0.05% DDM (pH 7). CD spectra were measured from 190–250 nm. Melting curves were recorded at 222 nm from 20–110 °C.
pH Titration of λmax
The protein was titrated from pH 4 to 11 using 50 mm sodium acetate, MES, HEPES, Tris, or K2HPO4 as buffer. For each pH value, an absorption spectrum from 250–600 nm was recorded. The wavelength of the bound retinal was plotted against the pH values, and the resulting curves were fit using a Boltzmann function.
Time-resolved Optical Spectroscopy
The flash photolysis experiments were performed with an Nd:YAG laser (Spitlight 600, Innolas Laser GmbH) as the pump source. We used a Xenon flash lamp generating microsecond white light pulses of low intensity for probing the absorbance of the sample. For detection, an intensified charge-coupled device camera (PI-MAX 3, Princeton Instruments) was deployed.
DNP-enhanced MAS NMR
DNP-enhanced 13C spectra were recorded on a Bruker 400 DNP system consisting of a 400-MHz wide bore Avance II NMR spectrometer, a 263-GHz Gyrotron as the microwave source, and a 3.2-mm HCN cryo-MAS probe. All experiments were conducted with a sample rotation rate of 8 kHz and microwave power at the probe of 10.5 W. During DNP experiments, the temperature was kept at around 110 K. For all experiments, 100-kHz decoupling using SPINAL-64 (28) was applied during acquisition. 13C magnetization was created by a ramped cross-polarization step with 0.8-ms contact time. 13C-double quantum filter experiments were obtained using the POST-C7 (29) sequence for double quantum excitation and reconversion.
High-field MAS NMR
High-field spectra were recorded using a Bruker Avance III 850 MHz spectrometer equipped with a triple-resonance DVT 3.2-mm HCN probe. All spectra were recorded with a MAS rate of 14 kHz at 270 K. Chemical shift referencing was carried out with respect to 4,4-dimethyl-4-silapentane-1-sulfonic acid through alanine (179.85 ppm). For all experiments, 83.3-kHz decoupling using SPINAL-64 (28) was applied during evolution and acquisition. 1H and 13C 90° pulses were set to 3 and 4 μs, respectively. 15N CP MAS NMR spectra were recorded using a contact time of 1 ms and an acquisition time of 15 ms. The initial 13C CP step for the proton-driven spin diffusion experiment was achieved using a contact time of 1.5 ms. A mixing time of 20 ms was used, and a recycle delay of 3 ms was applied. In the indirect dimension, an acquisition time of 6 ms, 600 increments, and 128 scans/increment were used. In case of the NCA experiments, a CP contact time of 1.25 ms for the first H-N CP transfer step and 3.5 ms for the second N-C CP transfer were applied. In the indirect dimension, an acquisition time of 10 ms, 80 increments, and 128 scans/increment were used. Spectra were recorded at pH 9 with ∼6 mg of pRLIG and 10 mg of pRWT and pRSCFNG.
Results
In Vivo Protein Trans-splicing
Proteorhodopsin was divided into an N-terminal segment consisting of helices A and B and a C-terminal segment containing helices C, D, E, F and G. The first 102 amino acids of the Npu DnaE intein were fused to the C terminus of the N segment. This construct is hereafter referred to as the N precursor. Consequently, the C precursor consists of the last 36 C-terminal intein residues followed by the C segment of pR (Fig. 1B). Ideally, the location of the splicing site should have only minimal effects on protein structure or function. We therefore selected the BC loop because no distinct functional role was known so far, and only a small β turn in contrast to the full β sheet, as in bacteriorhodopsin, has been observed (18, 19, 30, 31). Other regions, such as the EF loop, are known to affect color and protein stability (22, 32), none of which has been reported for the BC loop. For an effective splicing reaction, the amino acid sequence of the protein segments flanking the inteins has been modified according to the native extein sequence, as described previously (9). Therefore, residues Asp88-Ser89-Pro90 in the BC loop of pRWT are replaced by SCFNG in the ligated protein pRLIG (Fig. 1C). For comparison, a mutant pRSCFNG with the same BC loop was prepared.
For dual expression of both segments, E. coli C43 cells were cotransfected with one vector coding for the N and a second for the C precursor, controlled by a T7 and an araBAD promoter, respectively. The design of our constructs had to be optimized to achieve efficient in vivo trans-splicing. First, expression at high concentration is necessary because the splicing efficiency depends on a sufficient concentration of both precursors. Second, the precursor expressed first has to be stable enough to last in the cytoplasm until the second precursor is available for splicing. Third, we assumed that the splicing reaction might be more efficient in the cytosol because both intein segments might have a higher degree of freedom, which would be beneficial to achieve the best orientation with respect to each other. We therefore tried to reduce insertion of the precursors into the membrane directly after translation. After trans-splicing, the ligated full-length proteorhodopsin, pRLIG, was obtained (Fig. 1B).
To optimize expression, precursors were tested independently and screened for best expression temperature and inductor concentration. The C precursor showed the highest expression rates and was therefore cloned in the pBAD vector to be induced first. To further enhance expression and stability, a Smt3 solubility tag was added (33). To inhibit fast membrane insertion of the N precursor, the signal peptide of pRWT was omitted. Nevertheless, both precursors insert into the membrane, as judged by Western blot analysis of the membrane fraction (data not shown). For purification of the ligated protein, an HSV-His6 tag, as for pRWT, was added at the C terminus. To separate the unligated precursors from the ligated pR after the splicing reaction, a second Strep tag at both precursors was introduced (Fig. 1B).
To follow the ongoing splicing reaction producing pRLIG, expression levels were analyzed by SDS-PAGE and Western blotting at certain time points after the first induction with l-arabinose (Fig. 2). After 4 h, the C precursor (37.3 kDa) was detected by anti-Strep as well as anti-His Western blotting (Figs. 2, A and B). Induction with isopropyl 1-thio-β-d-galactopyranoside took place after 3 h, resulting in detection of the N precursor (20.8 kDa) 1 h later by the anti-Strep antibody (Fig. 2A). The amount of both precursors increased with time, and pRLIG (30.5 kDa) started to appear after 4 h (Fig. 2B). After 20 h, two further bands were observed in the anti-His blot at ∼20 kDa and in the anti-Strep blot at ∼10 kDa. The first band is most likely the result of a C-terminal cleavage product in which the Smt3 tag is also degraded (anti-His blot). The second band could be an excised intein without the Smt3 tag (anti-Strep blot). After 24 h, precursors and ligated protein were also detectable in SDS-PAGE (Fig. 2C).
FIGURE 2.

A–C, Western blots (A and B) and SDS-PAGE (C) of the protein trans-splicing reaction. 4 h after the first and 1 h after the second induction, the presence of N and C precursors as well as ligated protein, pRLIG, was detected in the anti-Strep (A) and anti-His6 blot (B). After 20 h, a C-terminal cleavage product (C-precursorΔSmt3) and the excised intein (inteinΔSmt3) became visible. After 24 h, precursors and pRLIG were also detectable in SDS-PAGE (C).
The ligation product pRLIG was purified by two affinity chromatography steps. pRLIG was first separated from other proteins cosolubilized from the E. coli membrane via its His6 tag, and non-spliced precursors were removed via their Strep tag. With the protocol introduced here, up to 3 mg/liter of ligated protein could be produced, which amounts to almost 20% of conventional pR expression (15–20 mg/liter), enabling a comprehensive biophysical analysis as described below. All three proteins were studied solubilized in 0.05% DDM and l-α-dimyristoylphosphatidylcholine/1,2-dimyristoyl-sn-glycero-3-phosphate proteoliposomes with a lipid-to-protein ratio of 1:2 (w/w).
Stationary Optical and CD Spectroscopy of Ligated Proteorhodopsin
The optical absorption spectrum of pR is a sensitive indicator for its correct fold because of the characteristic opsin shift of the retinal cofactor. The protein pRLIG created here by protein trans-splicing shows the expected absorption maxima at 280 and 522 nm at pH 7 (Fig. 3A). The ratio of A280/A520 of 1.9 is close to the value expected from the molar extinction coefficients (ϵ280 = 75,860 m−1cm−1, ϵ520 = 44,500 m−1cm−1), indicating high sample purity. The retinal absorption, λmax, at pH 7 is almost identical for pRLIG (522 nm) and pRSCFNG (523 nm), but both are slightly blue-shifted with respect to pRWT (529 nm). Comparing the pH dependence of λmax further corroborates the similarity between pRLIG and pRSCFNG (Fig. 3B). Both curves are blue-shifted but less sigmoidal with respect to pRWT and show a smaller total change in λmax. The pKa of the primary proton acceptor Asp97, as derived from the inflection point of these curves, is similar in all three cases and agrees with earlier reports (22).
FIGURE 3.
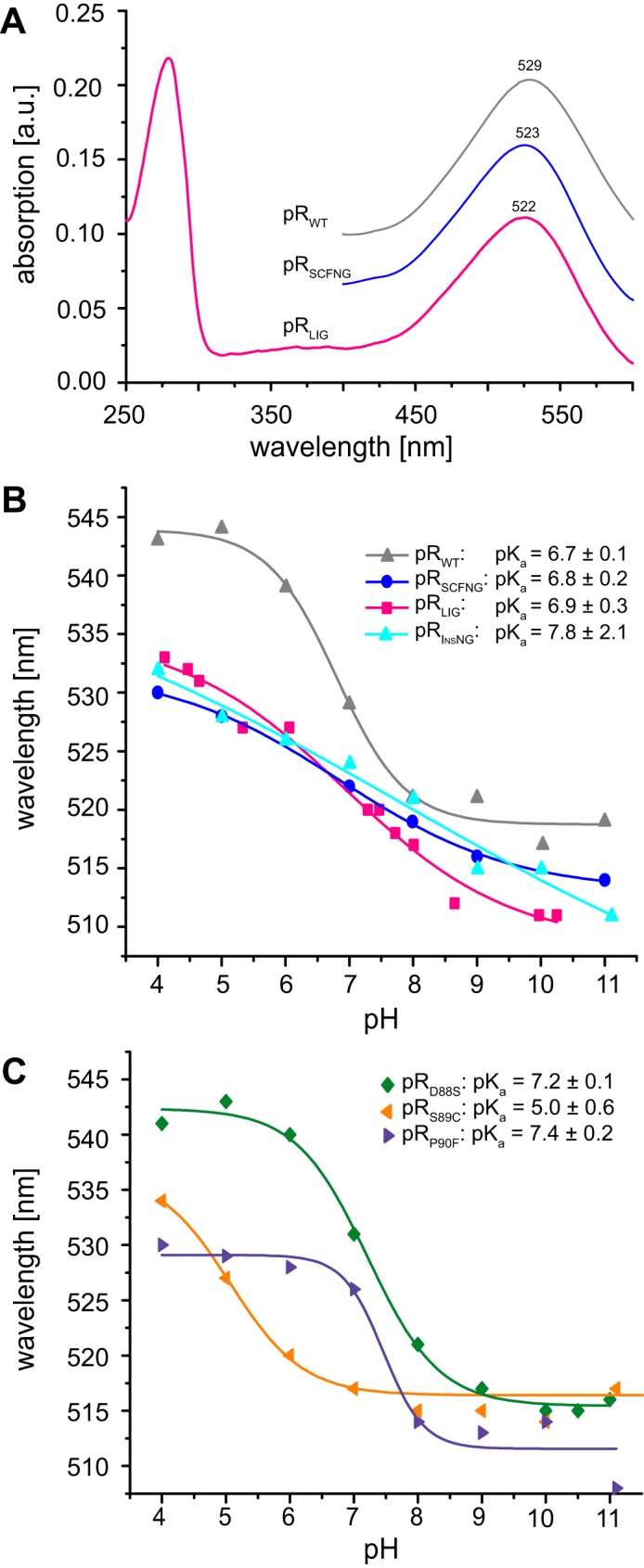
A, stationary light absorption spectrum of pRLIG at pH 7. The ratio of the absorption at 280 nm was twice as strong compared with 520 nm, demonstrating high sample purity. The values of λmax of pRSCFNG and pRLIG were nearly identical, but they were blue-shifted by 5 nm with respect to pRWT. a.u., arbitrary units. B, pH titration of max for pRLIG, pRSCFNG, pRWT, and pRInsNG in the range of pH 4–11. The pKa values of the primary proton acceptor Asp97 for each sample were determined from the inflections points of the curve. C, pH titration of λmax for pRD88S, pRS89C, and pRP90F. The pKa of Asp97 increases upon introduction of D88S and P90F but is reduced with the S89C mutation. The observation that the pKa is not altered in pRLIG and pRSCFNG might be explained by compensating effects when replacing the residue triplet Asp88-Ser89-Pro90 with Ser88-Cys89-Phe90.
The similar pH dependence of λmax of pRLIG and pRSCFNG and their difference to pRWT indicates that not the protein trans-splicing process but the modified BC loop affects the photophysical properties of pR. To rationalize this assumption, we created an insertion mutant, pRInsNG, containing residues Asn and Gly after position 90 as well as the single mutants pRD88S, pRS89C, and pRP90F. The pH dependence of λmax of pRInsNG (Fig. 3B) shows similar characteristics as pRLIG and pRSCFNG, demonstrating that extending the BC loop by both residues, asparagine and glycine, is sufficient to cause the observed effect. The curve is even less sigmoidal and shows an almost linear slope. Interestingly, each of the single point mutations, D88S, S89C, and P90F, of the three preceding residues shifts the pKa of Asp97 in different directions (Fig. 3C). We therefore conclude that the opposite effects of these mutations cancel each other when all of them are present simultaneously as in pRLIG and pRSCFNG.
The stability and fold of pRLIG was analyzed by CD spectroscopy. Spectra were recorded from 190–240 nm and compared with the data of pRSCFNG and pRWT. All three CD spectra showed the characteristics of a predominately α-helical fold with minima at 208 and 222 nm for all samples (34) (Fig. 4A). The temperature dependence of the α-helical bands was followed at 222 nm. The melting curves of both PRLIG and PRSCFNG showed a rather similar behavior, with transition temperatures of ∼80 °C and 88 °C, respectively. A slightly more sigmoidal behavior was observed for pRWT, with a transition temperature of 100 °C (Fig. 4B).
FIGURE 4.
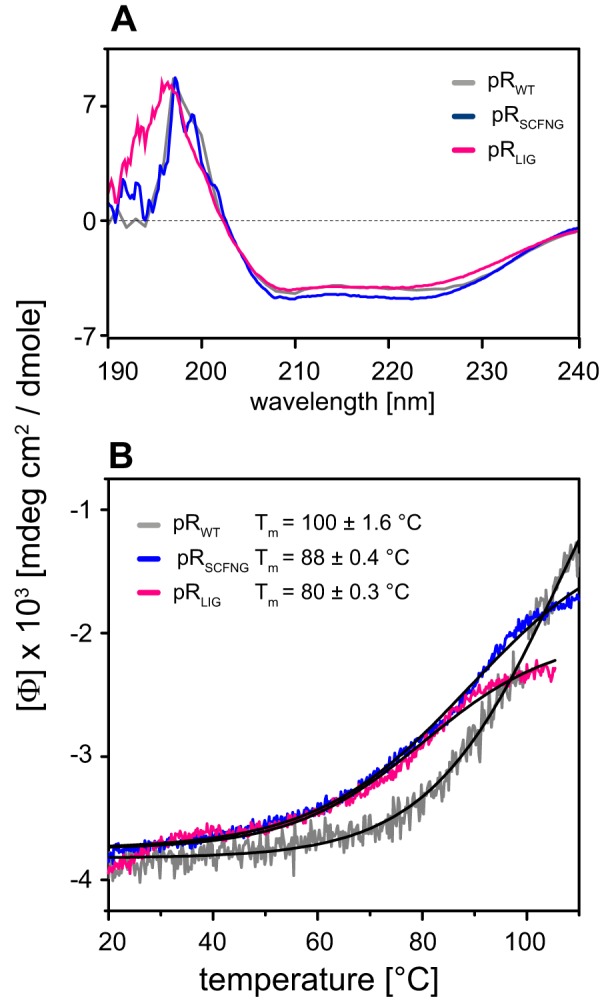
A, CD spectra of pRWT, pRSCFNG, and pRLIG showing similarly shaped spectra, indicating a primarily α-helical fold. B, the temperature dependence of the α-helical band was followed at 222 nm. A melting temperature of 100 °C was determined for pRWT, which is slightly lower for pRSCFNG (88 °C) and pRLIG (80 °C).
Photodynamics Determined by Time-resolved Optical Spectroscopy
The photoactivity of the ligated protein was characterized by optical flash photolysis experiments in the microsecond to second time range. Transient absorption changes recorded at three characteristic wavelengths were compared for pRLIG, pRWT, and pRSCFNG (Fig. 5). The transient absorption at 500 nm describes the photodynamics of the ground-state bleach and the decay of the K intermediate. The accumulation of the late intermediates was detected at 590 nm, and the formation and decay of the M state were monitored at 400 nm. In all three cases, a full photocycle was observed. The transients for pRLIG and pRSCFNG were almost indistinguishable from each other, illustrating that both proteins are functionally identical. In comparison to pRWT, the kinetics of the K and M intermediates are slightly slower, and the accumulation of M and the late N/O intermediates is decreased significantly and they return faster to the ground state. These observations lead to the conclusion that protein trans-splicing does not alter the activity of pR but also indicate that the BC loop affects the photodynamics.
FIGURE 5.
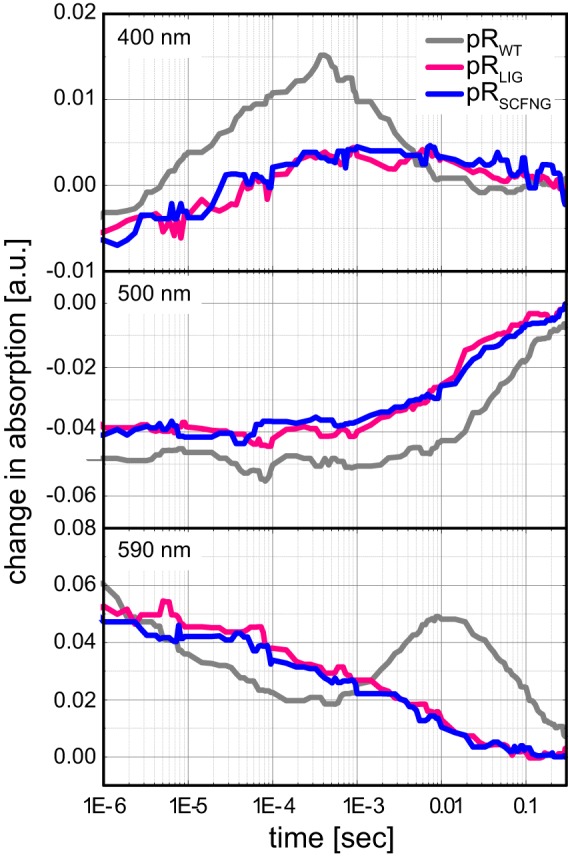
Flash photolysis experiments on pRLIG, pRSCFNG and pRWT. The formation and decay of the M state were monitored at 400 nm, the ground-state bleach at 500 nm, and the decay of the K state and buildup of late intermediates were observed at 590 nm. pRLIG and pRSCFNG are indistinguishable from each other but show an accelerated photocycle compared with pRWT. a.u., arbitrary units.
Solid-state NMR on pRLIG Reconstituted in Lipid Bilayers
Optical spectroscopy, as described above for all three proteins solubilized in detergent micelles, has shown a high functional similarity between pRLIG and its non-ligated counterpart, pRSCFNG. For a deeper structural analysis, solid-state NMR has been applied, which enabled us to probe key features of pR directly within the lipid bilayer.
First, the retinal itself was investigated to verify whether it assumes the same configuration in pRLIG as in pRWT. Therefore, samples containing 14,15-13C-all-trans retinal were prepared (Fig. 6A). Using double quantum filter experiments, both 13C resonances could be detected at 120.2 and 161.9 ppm for C14 and C15 in pRLIG, which is almost identical to the chemical shifts of 120.2 and 161.1 ppm observed for pRWT (Fig. 6B). Because no significant difference between pRLIG and pRWT was detected, additional experiments on pRSCFNG were omitted. These data show that the chromophore remains in a 100% all-trans configuration and is not affected by the splicing procedure. Because of the small number of spins and the small amount of labeled retinal available, signal enhancement on the basis of DNP was applied. Using TOTAPOL (27) as a polarizing agent, a 20-fold signal enhancement was achieved, as reported previously (21, 22).
FIGURE 6.

A, isotope labeling of the retinal-Schiff base complex. B, DNP-enhanced 13C MAS NMR spectrum of 14,15-13C-all-trans-retinal bound to pRWT and pRLIG. The natural abundance of 13C signals was suppressed by a double quantum filter so that only signals C14 and C15 of the retinal cofactor remained. The chemical shift of resonances in pRLIG was identical to pRWT. C, 15N MAS NMR spectra of [U-15N]pRWT, pRSCFNG, and pRLIG as well as of [15N-Lys]pRSCFNG and of the mutants [U-15N]pRS89C, pRP90F, and pRInsNG. The signal of His75 was shifted from 162 ppm in pRWT to 172 ppm in pRLIG and pRSCFNG. The signal of the protonated Schiff base (pSB) was broadened but not shifted in comparison with pRWT. The spectrum of [15N-Lys]pRSCFNG confirmed the assignment of both Schiff base and His75 resonances. The spectrum of the pRS89C mutant was identical to that of pRWT. In contrast, the P90F mutant also showed a broad peak at 182 ppm and two broad peaks at 172 and 162 ppm. The spectrum of pRInsNG was very similar to the ligation product. It also showed two broad peaks at 182 and 172 ppm. The spectra in A were recorded at 400 MHz/263 GHz at 115 K. The spectra in B were acquired at 850 MHz at 275 K. The proteins were reconstituted into l-α-dimyristoylphosphatidylcholine/1,2-dimyristoyl-sn-glycero-3-phosphate lipid bilayers. The pH was adjusted to 9.
Next, the 15N resonances of the protonated Schiff base and of residue His75 were compared. The Schiff base chemical shift is a sensitive indicator for altered Schiff base-counterion interactions or alterations in the H-bonding network within the chromophore binding pocket. Residue His75, located in helix B, has been shown to interact with Asp97 in helix C and should therefore be sensitive to the interaction between both helices (23, 35). The 15N spectrum of [U-15N]pRWT showed both resonances at 182 ppm (protonated Schiff base) and 162 ppm (Nϵ2, His75), as reported previously (23, 36) (Fig. 6C). The spectra of pRSCFNG and pRLIG were almost identical with respect to each other, but their line shapes were different in comparison with pRWT. The Schiff base resonance was broadened but remained at 182 ppm, which was in line with the almost unaltered retinal chemical shifts, but the His75 resonance appeared to be shifted to 172 ppm. Their assignment was confirmed by a selectively 15Nϵ Lys-labeled sample of pRSCFNG. To identify which of the introduced mutations are responsible for these effects, spectra of U-15N labeled samples of the single mutants pRS89C and pRP90F as well as for pRInsNG were recorded (Fig. 6C). Because the D88S mutation had been shown to affect the absorption characteristics of pR only very little (Fig. 3C), this mutant was not investigated further. We found that both the P90F mutation and the insertion of two additional amino acids in the BC loop cause the broadening of the Schiff base signal and the shift of the His75 signal observed in the spectra of pRLIG and pRSCFNG. In contrast, the S89C mutation had no effect.
To assess the fold of ligated pR within the lipid bilayer in comparison with a non-spliced sample, NCA and CC correlation spectra, which represent a structural protein fingerprint, were recorded for pRWT, pRSCFNG, and pRLIG (Fig. 7). Resolution and line width were comparable between all three samples, and the majority of cross peaks overlapped. This demonstrates that producing pR from two separately expressed fragments via protein trans-splicing results in a membrane protein of correct fold and structure. Upon closer inspection, using the known resonance assignment (31), a high degree of overlap between pRWT, pRLIG, and pRSCFNG could be confirmed as highlighted for some exemplarily residues in Fig. 7, but also some differences could be identified with respect to pRWT. As expected, cross-peaks of residues Asp88, Ser89, and Pro90 were missing in pRLIG and pRSCFNG because of the required alteration of the BC loop. The absence or shifting of the signals of Thr86, Gly87, and Thr91 is therefore not surprising because they are located directly before or behind the mutation site. Further effects have been observed for residues Thr29, Asp97, Glu142, Lys172, Asn224, and Asp227, which are further away from the BC loop.
FIGURE 7.
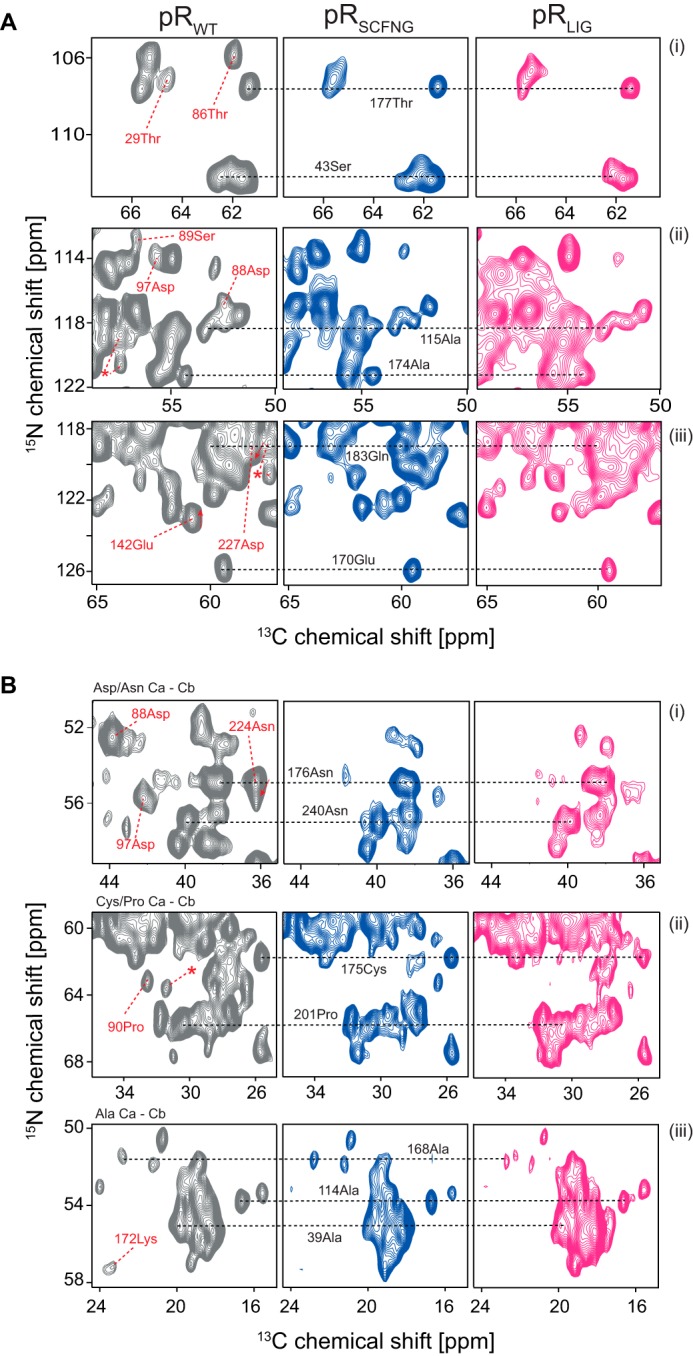
A and B, comparison of (A) 15N-13C (NCA) and (B) 13C-13C (proton-driven spin diffusion) correlation spectra of pRWT, pRSCFNG, and pRLIG. Resolution and line width were comparable between all three samples. Most peak positions were conserved, as shown for some selected resonances, illustrating that assembling a membrane protein via protein trans-splicing does not alter its structure. Because of the alteration in the BC loop, some resonances disappeared in pRLIG and pRSCFNG compared with pRWT (Asp88 in Aii/Bi, Ser89 in Aii, and Pro90 in Bii). A few other peaks in pRWT were shifted or broadened in pRSCFNG and pRLIG (Thr29, Thr86, Asp97, Glu142, Lys172, Asn224, and an unassigned cross peak (asterisk)). The signal-to-noise ratio for the pRLIG spectra was slightly reduced because of the lower amount of available protein. As for Fig. 6C, spectra were acquired at 850 MHz at 270 K. The proteins were reconstituted into l-α-dimyristoylphosphatidylcholine/1,2-dimyristoyl-sn-glycero-3-phosphate lipid bilayers. The pH was adjusted to 9.
Discussion
Successful in Vivo Protein Trans-splicing of a Membrane Protein under Native Conditions
We demonstrated that an α-helical membrane protein can be covalently assembled from two separate segments via protein trans-splicing using split inteins.
One might expect that the application of protein trans-splicing to membrane proteins leads to misfolded or partially folded products, which is not the case, as demonstrated here. It is, however, important to disentangle whether potential modifications in structure and function arise from the protein trans-splicing procedure itself, i.e. by altered helix-helix interactions within the protein or from mutations, which are introduced to achieve a high trans-splicing efficiency. We therefore compared pRLIG with both pRWT and pRSCFNG. CD spectroscopy confirmed a global α-helical fold of pRLIG (Fig. 4A). Melting curves further demonstrate the stability of the protein (Fig. 4B). Correct folding and integrity of the ligated protein was further characterized by CC and NCA correlation spectra, which overlap with those of pRSCFNG. Some differences are observed compared with pRWT that are, however, directly related to the mutations in the BC loop and are not caused by the trans-splicing reaction.
The absorption color of retinal proteins is very tightly regulated by the conformation and orientation of the chromophore and residues in the retinal-binding pocket (37, 38), making the absorption characteristics a sensitive indicator for a correct functional fold. Here the absorption of pRLIG over a wide pH range was found to be identical to PRSCFNG, whereas a small blue shift compared with pRWT was observed (Fig. 3B), which, again, underlines that the observed differences are caused by the BC loop mutations alone. Most importantly, the pKa of the primary proton acceptor Asp97 is almost unaffected. Previous studies have shown that the protonation of Asp97 in helix C is stabilized by an interaction with His75 in helix B (23, 35). In our case, helices B and C belong to two separately expressed fragments that are connected through the trans-splicing reaction. The identical pKa value of Asp97 indicates that both helices are oriented correctly with respect to each other within the ligation product. Further evidence for a correctly folded retinal binding pocket comes from the retinal and Schiff base chemical shifts, which are identical for pRLIG, pRWT, and pRSCFNG (Fig. 5). The increased line width and altered 15N chemical shift of His75 is directly connected to the BC loop mutation, as all other differences observed here. Our flash photolysis data show that pRLIG undergoes a complete photocycle identical to pRSCFNG, but mutation-induced differences are observed compared with pRWT, which are discussed separately below.
Although its yield is lower compared with wild-type proteorhodopsin, the possibility to record multidimensional MAS NMR spectra of a membrane protein prepared by trans-splicing demonstrates that quantitative protein amounts can be prepared. The actual yield is affected by different factors, such as the trans-splicing efficiency of duration of expression. Although it takes no longer than for the wild-type, three steps instead of two are needed (expression of fragment 1, expression of fragment 2, and ligation). In addition, two inductions instead of one cause addition stress to the cells, which also decreases the protein yield.
The trans-splicing efficiency is determined to a great extent by the probability of both segments coming into close spatial proximity at the right orientation with respect to each other. Whether the reaction takes place at or within the membrane or within the cytosol cannot be directly concluded from our data. On one hand, both precursors insert into the membrane. On the other hand, increasing the solubility of the C precursor by omitting the signal peptide and by adding a solubility tag increases ligation efficiency, which indicates that this reaction takes place, at least partially, in the cytosol. As shown in Fig. 2, sufficient amounts of each precursor are produced, but only a part of these seems to assemble into the ligated protein. This could be caused by a generally low efficiency of the intein trans-splicing or by an unfavorable spatial separation of both fragments because the splicing reaction itself is known to be very fast and efficient (9, 10). Therefore, it seems more likely that only a fraction of the fragments are close enough to each other to ligate.
In summary, functional and structural data clearly prove that a correctly folded and active α-helical membrane protein, pRLIG, can be assembled from separate fragments via protein trans-splicing and that the resulting ligation product is indistinguishable from its non-ligated counterpart, pRSCFNG. Our data show unambiguously that all differences to pRWT are related to the mutations in the BC loop alone.
Data in the Context of Folding and Assembly of α-helical Membrane Proteins
The observation that pRLIG is assembled correctly via a ligation site in the BC loop indicates important helix-helix interactions between both fragments. In fact, helix-helix interactions within α-helical membrane proteins are essential for folding, stability, and function (39, 40). Evidence for their importance along other folding constraints imposed by loop-helix and helix-lipid interactions comes from a number of studies in which α-helical fragments of integral membrane proteins have been observed to cluster spontaneously within the membrane to form complexes functionally related to the original protein. This has been shown first for proteolytically generated fragments of bacteriorhodopsin (13–15) as well as in a number of other cases (41, 42–44). In the absence of structural data, these studies concluded a dominant role of helix-helix interactions within the membrane on the basis of the observation of partially recovered ligand binding upon fragment clustering. Here we go two steps further and demonstrate full recovery of functional activity as well as the correct structure of proteorhodopsin upon assembly.
The Role of the BC Loop
The BC loop was initially selected as a ligation site because no effects on structure and function were known. However, our data show that replacing Asp88-Ser89-Pro90 in the BC loop with SCFNG causes a slight blue shift, renders the pH dependence of λmax less sigmoidal, modifies the kinetics of the photocycle, and causes some subtle differences in the NMR spectra. To identify the individual mutations that are responsible for the observed effects, spectral data for pRD88S, pRS89C, pRP90F, and pRInsNG were determined (Figs. 3 and 6C). Interestingly, the insertion of two additional residues in the BC loop causes the same pH dependence of λmax as observed for pRLIG and pRSCFNG, whereas all other mutations show different pKa values but sigmoidal curves (Fig. 3C). The elongation of the BC loop seems to result in a smoother, less cooperative transition from the protonated to the unprotonated state of the primary proton acceptor and causes less well defined photocycle intermediates (Fig. 5). The most likely explanation is an altered interplay between the primary proton acceptor in helix C and His75 in helix B. It has been shown that a pH-dependent hydrogen bond between both residues stabilizes the high pKa of the primary proton acceptor (23). A weakening of this interaction might explain the less pronounced pH sensitivity of pRLIG and pRSCFNG. This conclusion is also supported by the altered 15N chemical shift of His75 upon modification of the BC loop, which indicates protonated His-Nϵ2 (23) and break of the H-bond (Fig. 6C).
Structural data of pR (18, 19, 31) indicate a β turn motif in the BC loop. Although different studies suggest contradictory data for the exact position of this β turn (Trp83-Gly87 (19) and Gly87-Pro90 (18)), the introduced mutations are in or next to the turn, respectively. We hypothesize that mutating the amino acids forming this secondary structure perturbs or even disrupts the β turn. Together with the elongations of the loop, this might lead to increased flexibility, which affects the His-Asp cluster formed between helices B and C. The observed blue shift of λmax could be caused by alteration of the β-ionine ring orientation induced by neighboring helices B or C.
Conclusion and Outlook
In this study, we show unambiguously that protein trans-splicing with split inteins in vivo can be applied to a membrane protein without losing secondary structure, stability, or function. The ligated protein behaves exactly the same as its non-ligated counterpart (pRSCFNG). All major differences observed in comparison with the wild type are due to the mutations introduced in the BC loop. NMR spectra are fully comparable in terms of dispersion, line width, and peak positions. Our data also reveal an unexpected effect of extending the BC loop by just two residues onto the photophysical properties of pR, which has not been reported before. Our observations support the previously discussed importance of interactions between transmembrane helices within membrane proteins and will trigger further developments toward segmental labeling for spectroscopic applications. It is worth mentioning that a high splicing efficiency does not exclusively depend on the native extein sequence used here, which might eventually enable protein trans-splicing with significantly less modified splicing sites (45). Proteorhodopsin is a typical α-helical membrane protein in terms of fold and size. Therefore, optimism seems justified that PTS can also be applied to many other membrane proteins.
Author Contributions
C. G. conceived and coordinated the study. M. M. and C. G. designed the study and wrote the paper. M. M. carried out and analyzed all experiments, except for those shown in Figs. 3C and 5. J. M. and M. M. carried out the experiments shown in Fig. 3C. C. E. E. and J. W. designed, performed and analyzed the experiments shown in Fig. 5. V. D. and A. B. provided knowledge on the use of split-inteins. A. B. and J. K. designed and constructed vectors for expression. J. B. B. provided assistance with the NMR experiments and NMR data analysis. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Prof. Hideo Iwai (University of Helsinki) for the plasmids for the split inteins used here and Prof. Richard C. D. Brown (University of Southampton) for 13C-labeled retinal. We also thank Dr. Jörn Plackmeier (University of Frankfurt) for TOTAPOL.
This work was supported by Deutsche Forschungsgemeinschaft (DFG)/Sonderforschungsbereich Grant 807 “Transport and Communication across Membranes.” The DNP experiments were supported by an equipment grant provided by DFG Grant GL 307/4-1 and Cluster of Excellence Macromolecular Complexes Frankfurt. The Centre for Biomolecular Magnetic Resonance is funded through the State of Hesse. The authors declare that they have no conflicts of interest with the contents of this article.
- PTS
- protein trans-splicing
- pR
- proteorhodopsin
- DDM
- dodecyl-β-d-maltoside
- MAS
- magic angle spinning
- DNP
- dynamic nuclear polarization
- CP
- cross-polarization.
References
- 1.Volkmann G., and Iwaï H. (2010) Protein trans-splicing and its use in structural biology: opportunities and limitations. Mol. Biosyst. 6, 2110–2121 [DOI] [PubMed] [Google Scholar]
- 2.Muir T. W., Sondhi D., and Cole P. A. (1998) Expressed protein ligation: a general method for protein engineering. Proc. Natl. Acad. Sci. U. S. A. 95, 6705–6710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Al-Ali H., Ragan T. J., Gao X., and Harris T. K. (2007) Reconstitution of modular PDK1 functions on trans-splicing of the regulatory PH and catalytic kinase domains. Bioconjugate Chem. 18, 1294–1302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang J.-Y., and Yang W. Y. (2009) Site-Specific Two-Color Protein Labeling for FRET Studies Using Split Inteins. J. Am. Chem. Soc. 131, 11644–11645 [DOI] [PubMed] [Google Scholar]
- 5.Ozawa T., and Umezawa Y. (2001) Detection of protein-protein interactions in vivo based on protein splicing. Curr. Opin. Chem. Biol. 5, 578–583 [DOI] [PubMed] [Google Scholar]
- 6.Züger S., and Iwai H. (2005) Intein-based biosynthetic incorporation of unlabeled protein tags into isotopically labeled proteins for NMR studies. Nat. Biotechnol. 23, 736–740 [DOI] [PubMed] [Google Scholar]
- 7.Busche A. E., Aranko A. S., Talebzadeh-Farooji M., Bernhard F., Dötsch V., and Iwaï H. (2009) Segmental isotopic labeling of a central domain in a multidomain protein by protein trans-splicing using only one robust DnaE intein. Angew. Chem. Int. Ed. Engl. 48, 6128–6131 [DOI] [PubMed] [Google Scholar]
- 8.Perler F. B., Davis E. O., Dean G. E., Gimble F. S., Jack W. E., Neff N., Noren C. J., Thorner J., and Belfort M. (1994) Protein splicing elements: inteins and exteins: a definition of terms and recommended nomenclature. Nucleic Acids Res. 22, 1125–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iwai H., Züger S., Jin J., and Tam P.-H. (2006) Highly efficient protein trans-splicing by a naturally split DnaE intein from Nostoc punctiforme. FEBS Lett. 580, 1853–1858 [DOI] [PubMed] [Google Scholar]
- 10.Zettler J., Schütz V., and Mootz H. D. (2009) The naturally split Npu DnaE intein exhibits an extraordinarily high rate in the protein trans-splicing reaction. FEBS Lett. 583, 909–914 [DOI] [PubMed] [Google Scholar]
- 11.Schubeis T., Lührs T., and Ritter C. (2015) Unambiguous assignment of short- and long-range structural restraints by solid-state NMR spectroscopy with segmental isotope labeling. Chem. Bio. Chem. 16, 51–54 [DOI] [PubMed] [Google Scholar]
- 12.Brenzel S., Cebi M., Reiss P., Koert U., and Mootz H. D. (2009) Expanding the scope of protein trans-splicing to fragment ligation of an integral membrane protein: towards modulation of porin-based ion channels by chemical modification. Chembiochem 10, 983–986 [DOI] [PubMed] [Google Scholar]
- 13.Popot J.-L., Gerchman S.-E., and Engelman D. M. (1987) Refolding of bacteriorhodopsin in lipid bilayers: a thermodynamically controlled two-stage process. J. Mol. Biol. 198, 655–676 [DOI] [PubMed] [Google Scholar]
- 14.Huang K. S., Bayley H., Liao M. J., London E., and Khorana H. G. (1981) Refolding of an integral membrane-protein: denaturation, renaturation, and reconstitution of intact bacteriorhodopsin and two proteolytic fragments. J. Biol. Chem. 256, 3802–3809 [PubMed] [Google Scholar]
- 15.Marti T. (1998) Refolding of bacteriorhodopsin from expressed polypeptide fragments. J. Biol. Chem. 273, 9312–9322 [DOI] [PubMed] [Google Scholar]
- 16.Béjà O., Aravind L., Koonin E. V., Suzuki M. T., Hadd A., Nguyen L. P., Jovanovich S. B., Gates C. M., Feldman R. A., Spudich J. L., Spudich E. N., and DeLong E. F. (2000) Bacterial Rhodopsin: evidence for a new type of phototrophy in the sea. Science 289, 1902–1906 [DOI] [PubMed] [Google Scholar]
- 17.Sabehi G., Massana R., Bielawski J. P., Rosenberg M., Delong E. F., and Béjà O. (2003) Novel Proteorhodopsin variants from the Mediterranean and Red Seas. Environ. Microbiol. 5, 842–849 [DOI] [PubMed] [Google Scholar]
- 18.Reckel S., Gottstein D., Stehle J., Löhr F., Verhoefen M. K., Takeda M., Silvers R., Kainosho M., Glaubitz C., Wachtveitl J., Bernhard F., Schwalbe H., Güntert P., and Dötsch V. (2011) Solution NMR structure of proteorhodopsin. Angew. Chem. Int. Ed. Engl. 50, 11942–11946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ran T., Ozorowski G., Gao Y., Sineshchekov O. A., Wang W., Spudich J. L., and Luecke H. (2013) Cross-protomer interaction with the photoactive site in oligomeric proteorhodopsin complexes. Acta Crystallogr. D. 69, 1965–1980 [DOI] [PubMed] [Google Scholar]
- 20.Friedrich T., Geibel S., Kalmbach R., Chizhov I., Ataka K., Heberle J., Engelhard M., and Bamberg E. (2002) Proteorhodopsin is a light-driven proton pump with variable vectoriality. J. Mol. Biol. 321, 821–838 [DOI] [PubMed] [Google Scholar]
- 21.Mao J., Do N.-N., Scholz F., Reggie L., Mehler M., Lakatos A., Ong Y.-S., Ullrich S. J., Brown L. J., Brown R. C., Becker-Baldus J., Wachtveitl J., and Glaubitz C. (2014) Structural basis of the green-blue color switching in proteorhodopsin as determined by NMR spectroscopy. J. Am. Chem. Soc. 136, 17578–17590 [DOI] [PubMed] [Google Scholar]
- 22.Mehler M., Scholz F., Ullrich S. J., Mao J., Braun M., Brown L. J., Brown R. C., Fiedler S. A., Becker-Baldus J., Wachtveitl J., and Glaubitz C. (2013) The EF loop in green proteorhodopsin affects conformation and photocycle dynamics. Biophys. J. 105, 385–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hempelmann F., Hölper S., Verhoefen M.-K., Woerner A. C., Köhler T., Fiedler S.-A., Pfleger N., Wachtveitl J., and Glaubitz C. (2011) His75-Asp97 cluster in green proteorhodopsin. J. Am. Chem. Soc. 133, 4645–4654 [DOI] [PubMed] [Google Scholar]
- 24.Verhoefen M.-K., Schäfer G., Shastri S., Weber I., Glaubitz C., Mäntele W., and Wachtveitl J. (2011) Low temperature FTIR spectroscopy provides new insights in the pH-dependent proton pathway of proteorhodopsin. Biochim. Biophys. Acta 1807, 1583–1590 [DOI] [PubMed] [Google Scholar]
- 25.Dioumaev A. K., Brown L. S., Shih J., Spudich E. N., Spudich J. L., and Lanyi J. K. (2002) Proton Transfers in the photochemical reaction cycle of proteorhodopsin. Biochemistry 41, 5348–5358 [DOI] [PubMed] [Google Scholar]
- 26.Váró G., Brown L. S., Lakatos M., and Lanyi J. K. (2003) Characterization of the photochemical reaction cycle of proteorhodopsin. Biophys. J. 84, 1202–1207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Song C., Hu K. N., Joo C. G., Swager T. M., and Griffin R. G. (2006) TOTAPOL: a biradical polarizing agent for dynamic nuclear polarization experiments in aqueous media. J. Am. Chem. Soc. 128, 11385–11390 [DOI] [PubMed] [Google Scholar]
- 28.Fung B. M., Khitrin A. K., and Ermolaev K. (2000) An improved broadband decoupling sequence for liquid crystals and solids. J. Magn. Reson. 142, 97–101 [DOI] [PubMed] [Google Scholar]
- 29.Hohwy M., Jakobsen H. J., Eden M., Levitt M. H., and Nielsen N. C. (1998) Broadband dipolar recoupling in the nuclear magnetic resonance of rotating solids: a compensated C7 pulse sequence. J. Chem. Phys. 108, 2686–2694 [Google Scholar]
- 30.Luecke H., Schobert B., and Richter H.-T. (1999) Structure of bacteriorhodopsin at 1.55 A resolution. J. Mol. Biol. 291, 899–911 [DOI] [PubMed] [Google Scholar]
- 31.Shi L., Ahmed M. A., Zhang W., Whited G., Brown L. S., and Ladizhansky V. (2009) Three-dimensional solid-state NMR study of a seven-helical integral membrane proton pump: structural insights. J. Mol. Biol. 386, 1078–1093 [DOI] [PubMed] [Google Scholar]
- 32.Yoshitsugu M., Shibata M., Ikeda D., Furutani Y., and Kandori H. (2008) Color change of proteorhodopsin by a single amino acid replacement at a distant cytoplasmic loop. Angew. Chem. Int. Ed. Engl. 47, 3923–3926 [DOI] [PubMed] [Google Scholar]
- 33.Malakhov M. P., Mattern M. R., Malakhova O. A., Drinker M., Weeks S. D., and Butt T. R. (2004) SUMO fusions and SUMO-specific protease for efficient expression and purification of proteins. J. Struct. Funct. Genomics 5, 75–86 [DOI] [PubMed] [Google Scholar]
- 34.Hennessey J. P. Jr., and Johnson W. C. Jr. (1981) Information-content in the circular-dichroism of proteins. Biochemistry 20, 1085–1094 [DOI] [PubMed] [Google Scholar]
- 35.Bergo V. B., Sineshchekov O. A., Kralj J. M., Partha R., Spudich E. N., Rothschild K. J., and Spudich J. L. (2009) His-75 in proteorhodopsin, a novel component in light-driven proton translocation by primary pumps. J. Biol. Chem. 284, 2836–2843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pfleger N., Lorch M., Woerner A. C., Shastri S., and Glaubitz C. (2008) Characterisation of Schiff base and chromophore in green proteorhodopsin by solid-state NMR. J. Biomol. NMR 40, 15–21 [DOI] [PubMed] [Google Scholar]
- 37.Hoffmann M., Wanko M., Strodel P., König P. H., Frauenheim T., Schulten K., Thiel W., Tajkhorshid E., and Elstner M. (2006) Color tuning in rhodopsins: the mechanism for the spectral shift between bacteriorhodopsin and sensory rhodopsin II. J. Am. Chem. Soc. 128, 10808–10818 [DOI] [PubMed] [Google Scholar]
- 38.Lin S. W., Kochendoerfer G. G., Carroll K. S., Wang D., Mathies R. A., and Sakmar T. P. (1998) Mechanisms of spectral tuning in blue cone visual pigments: visible and raman spectroscopy of blue-shifted rhodopsin mutants. J. Biol. Chem. 273, 24583–24591 [DOI] [PubMed] [Google Scholar]
- 39.Popot J. L., and Engelman D. M. (2000) Helical membrane protein folding, stability, and evolution. Annu. Rev. Biochem. 69, 881–922 [DOI] [PubMed] [Google Scholar]
- 40.Neumann J., Klein N., Otzen D. E., and Schneider D. (2014) Folding energetics and oligomerization of polytopic α-helical transmembrane proteins. Arch. Biochem. Biophys. 564, 281–296 [DOI] [PubMed] [Google Scholar]
- 41.Lee W. K., Han J. J., Jin B. S., Boo D. W., and Yu Y. G. (2009) Functional reconstitution of the human serotonin receptor 5-HT6 using synthetic transmembrane peptides. Biochem. Biophys. Res. Commun. 390, 815–820 [DOI] [PubMed] [Google Scholar]
- 42.Wrubel W., Stochaj U., and Ehring R. (1994) Construction and in vivo analysis of new split lactose permeases. FEBS Lett. 349, 433–438 [DOI] [PubMed] [Google Scholar]
- 43.Ridge K. D., Lee S. S., and Yao L. L. (1995) In vivo assembly of rhodopsin from expressed polypeptide fragments. Proc. Natl. Acad. Sci. U. S. A. 92, 3204–3208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nielsen S. M., Elling C. E., and Schwartz T. W. (1998) Split-receptors in the tachykinin neurokinin-1 system. Eur. J. Biochem. 251, 217–226 [DOI] [PubMed] [Google Scholar]
- 45.Cheriyan M., Pedamallu C. S., Tori K., and Perler F. (2013) Faster protein splicing with the Nostoc punctiforme DnaE intein using non-native extein residues. J. Biol. Chem. 288, 6202–6211 [DOI] [PMC free article] [PubMed] [Google Scholar]