

Abstract
Historically, androgen-deprivation therapy has been the cornerstone for treatment of metastatic prostate cancer. Unfortunately, nearly majority patients with prostate cancer transition to the refractory state of castration-resistant prostate cancer (CRPC). Newer therapeutic agents are needed for treating these CRPC patients that are unresponsive to androgen deprivation and/or chemotherapy. The histone deacetylase (HDAC) family of enzymes limits the expression of genomic regions by improving binding between histones and the DNA backbone. Modulating the role of HDAC enzymes can alter the cell’s regulation of proto-oncogenes and tumor suppressor genes, thereby regulating potential neoplastic proliferation. As a result, histone deacetylase inhibitors (HDACi) are now being evaluated for CRPC or chemotherapy-resistant prostate cancer due to their effects on the expression of the androgen receptor gene. In this paper, we review the molecular mechanism and functional target molecules of different HDACi as applicable to CRPC as well as describe recent and current clinical trials involving HDACi in prostate cancer. To date, four HDAC classes comprising 18 isoenzymes have been identified. Recent clinical trials of vorinostat, romidepsin, and panobinostat have provided cautious optimism towards improved outcomes using these novel therapeutic agents for CPRC patients. Nevertheless, no phase III trial has been conducted to cement one of these drugs as an adjunct to androgen-deprivation therapy. Consequently, further investigation is necessary to delineate the benefits and drawbacks of these medications.
Keywords: castration-resistant prostate cancer, clinical trials, histone deacetylase inhibitors
Introduction
Prostate cancer is estimated to comprise 26% of newly diagnosed male cancers in the USA in 2015, making it the most commonly diagnosed cancer in men and the second leading cause of cancer-related death in men for this year [Siegel et al. 2015]. Androgen-deprivation therapy has been the mainstay treatment for advanced prostate cancer and induces remission in 80–90% of men with advanced disease, resulting in a median disease progression-free survival of 12–33 months. Unfortunately, in a majority of patients, neoplastic cells will subsequently continue to proliferate despite previous response to androgen deprivation. This progressive state is termed castration-resistant prostate cancer (CRPC), which carries a median overall survival of 23–37 months starting from the initial onset of androgen deprivation [Hellerstedt and Pienta, 2008]. Therefore, studies evaluating a newer generation of agents are necessary to prolong life expectancy and quality of life for patients suffering from CRPC.
The molecular mechanisms underlying the proliferation of prostate cancer cells under an androgen-deprivation environment are currently under investigation. One of these mechanisms is the covalent acetylation and deacetylation of histone proteins. These covalent modifications are important in regulating the transcription of proto-oncogenes and tumor suppressor genes. The binding and retraction of acetyl groups to histones are reversible and heritable from one generation to the next. These modifications are mediated by two sets of enzymes, histone deacetylase (HDAC) and histone acetyltransferase (HAT).
In particular, the HDAC family of enzymes is of current interest in urology because these proteins offer a novel therapeutic target to limit prostate cancer proliferation. HDAC regulates the expression of several functional genes, including the androgen receptor (AR) in prostate cells. Consequently, histone deacetylase inhibitors (HDACi) are now being evaluated for their effects on CRPC patients. The following is a discussion of the molecular mechanisms and functional target molecules of different HDACi as applicable to CRPC as well as a description of the current clinical trials involving HDACi in prostate cancer.
Epigenetics and prostate cancer
Epigenetics and the HDAC family of enzymes
Epigenetics is the study of heritable changes in gene expression that are not concomitantly accompanied by changes in DNA sequences. The key modifications of DNA involving epigenetics are the DNA methylation of CpG islands in the promoter region of genes and the covalent modifications involving the acetylation and deacetylation of histones [Bode and Dong, 2004]. Histones are proteins that form a scaffold allowing genomic DNA to wrap in a systematic fashion. The expression of genes in a particular genomic region is thereby regulated by its winding around histones. Modification of these histone proteins by acetylation and deacetylation controls the tightness of DNA winding around histones, and therefore, controls the expression of the genes at that histone’s location. HAT enzymes transfer acetyl moieties to lysines in the N-terminal histone tails through use of a cofactor, acetyl-coenzyme A. This results in the neutralization of the negative charge of the nitrogen in the ε-amino group of the lysine residue, which in turn, leads to a more open form of chromatin that is associated with activation of gene expression. Contrarily, the acetyl groups are in turn cleaved off by HDAC enzymes leading to a more condensed form of chromatin and gene silencing [Wagner et al. 2010]. In summation, HDAC represents a family of enzymes that cooperate with the HAT family of enzymes to modulate chromatin structure and transcriptional activity via changes in the acetylation status of nucleosomal histones.
To date, four HDAC classes comprising 18 isoenzymes have been identified (Table 1). Class I HDAC is primarily localized in the nucleus and ubiquitously expressed in all tissues. Class I consists of HDACs 1, 2, 3, and 8. Class I HDACs have the deacetylase domain located at their N-terminal and carry a variable Carbon-terminal (C-terminal) depending on the specific HDAC of the class. Class II HDACs are localized both in the nucleus as well as the cytoplasm. Class II consists of HDACs 4, 5, 6, 7, 9, and 10. Class II HDACs have the deacetylase domain at the C-terminal with the exception of HDAC 6, which contains two acetylase domains at both the N- and C-terminals. Class III HDACs are homologues of yeast silent information regulator 2 proteins and consist of sirtuins 1–7. Class III HDACs are distinct from Class I and II HDACs due to their enzymatic dependence on coenzyme nicotinamide adenine dinucleotide for deacetylase activity. Contrarily, Class I and II HDACs have a zinc coordinated active site. Class IV HDAC has the property of both class I and class II, and consists of HDAC 11 [Perry et al. 2010; Shankar and Srivastava, 2008].
Table 1.
Current histone deacetylase classification.
| HDAC class | Members | Localization | Activity domain |
|---|---|---|---|
| Class I | HDAC 1 | N | Zinc-coordinated active site, N-terminal HDAC activity |
| HDAC 2 | N | ||
| HDAC 3 | N | ||
| HDAC 8 | N/C | ||
| Class II | HDAC 4 | N/C | Zinc-coordinated active site, C-terminal HDAC activity |
| HDAC 5 | N/C | ||
| HDAC 6 | C | ||
| HDAC 7 | N/C | ||
| HDAC 9 | N/C | ||
| HDAC 10 | C | ||
| Class III | SIRT 1 | N | Coenzyme nicotinamide adenine dinucleotide-dependent active site |
| SIRT 2 | C | ||
| SIRT 3 | N/C | ||
| SIRT 4 | C | ||
| SIRT 5 | C | ||
| SIRT 6 | N | ||
| SIRT 7 | N | ||
| Class IV | HDAC 11 | N/C | N- and C-terminal HDAC activity |
C, cytoplasmic; HDAC, histone deacetylase; N, nuclear; SIRT, sirtuin.
In cancer, uncontrolled HDAC expression results in the deacetylation of histone proteins. Deacet-ylation causes DNA to be wrapped tightly by histones, thereby inhibiting gene expression. If tightly wound DNA incorporates tumor suppressor genes, a neoplastic proliferation of cells may indefinitely result [Abbas and Gupta, 2008].
HDAC in prostate cancer
Recently, it has been shown that a majority of recurrent prostate cancers are neither hormone refractory nor androgen independent, but are rather dependent on the AR signaling axis [Montgomery et al. 2008]. The AR is a cytoplasmic protein that binds to testosterone or dihydrotestosterone before entering the nucleus leading to the alteration of gene transcription. Several mechanisms exist for the increased signaling of the AR. These include the amplification and overexpression of the AR, enhanced AR signal transduction through alterations in coactivators/corepressors, and activation of the AR or downstream regulatory molecules by cross-talk with other signaling pathways [Feldman and Feldman, 2001].
Intracrine androgen production may play a critical role in maintaining tumoral androgen levels and development of CRPC [Stanbrough et al. 2006]. We have previously shown that prostate-derived factor promotes AR-positive prostate-tumor progression through autocrine and paracrine stimulation by upregulating cell proliferation via the extracellular signal-regulated kinase 1/2 signal pathway [Chen et al. 2007]. Dillard and colleagues have shown that advanced prostate cancer cells are able to synthesize their own testosterone from cholesterol indicating supporting intracrine function seen in advanced CRPC. One of the key players for this autonomous testosterone synthesis is the AR, which may undergo changes leading to its subsequent deregulation [Dillard et al. 2008]. A portion of this deregulation of the AR is often governed at the epigenetic level. Epigenetic changes are potentially reversible, or in other words, they may be amenable to pharmacologic interventions. Therefore, a lot of work in recent years has focused on the development of inhibitors of histone-modifying enzymes.
Several lines of evidence have shown that HDACs are abundantly expressed and upregulated in prostate cancer [Waltregny et al. 2004; Weichert et al. 2008]. Weichert and colleagues studied the expression patterns of HDACs 1, 2, and 3 in prostate cancer using a patient cohort of 192 patients who underwent radical prostatectomy. Their data showed that HDACs 1 and 2 correlated positively with Gleason scores, with high-grade tumors expressing both isoforms at higher rates. In addition, expression of HDACs 1, 2, and 3 correlated significantly with the Ki-67-positive proliferative fraction of prostate cancer cells, indicating increased cellular proliferation. They also demonstrated a significantly lower probability of disease-free survival based on the Kattan nomogram for patients with high-HDAC 2 tumors compared with patients with low-HDAC 2 tumors [Weichert et al. 2008].
HDAC plays a critical role in the regulation of the AR, therefore, HDACi are an important therapeutic consideration in CRPC. In the following sections, we will discuss some important molecular pathways where HDACi impact cellular signaling and cancer cell growth. The effects of HDACi on various enzymes are depicted in Figure 1.
Figure 1.
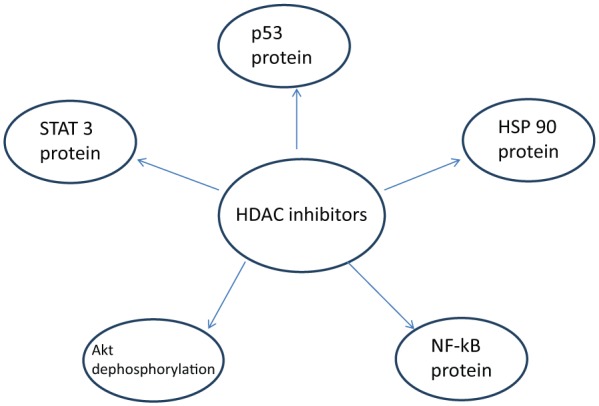
The unique proteins modifying cell survival and proliferation that HDAC inhibitors have been to shown to affect: Akt, protein kinase B; Hsp90, heat shock protein-90; NF-kB, nuclear factor-kappa B; STAT 3, signal transducers and activators of transcription.
Molecular mechanism of action of HDACi
Effect of HDACi on the AR and heat shock protein-90
The AR is bound to the heat shock protein-90 (Hsp90) molecular chaperone complex, which is essential for AR stability, activation, and maturation [Fang et al. 1996; Neckers and Ivy, 2003].
Hsp90 clients include several proteins of potential importance in mediating prostate cancer progression, including the wild-type and mutated AR, human epidermal growth factor receptor 2, and protein kinase B (Akt). Therefore, the inhibition of Hsp90 function causes the proteasomal degradation of proteins that require this chaperone for maturation and stability. Hence, Hsp90 has been targeted in prostate-cancer therapy [Solit et al. 2003].
Modification of Hsp90 by acetylation has been reported by Yu and colleagues [Yu et al. 2002]. It was observed that a depsipeptide HDAC inhibitor, romidepsin (FK228), could deplete Hsp90 client proteins Raf-1, ErbB1, and ErbB2. They further showed that romidepsin induced Hsp90 acetylation in nonsmall-cell lung cancer cell lines, which correlated with the loss of its binding activities to adenosine triphosphate (ATP) and client proteins.
Chen and colleagues described the HDAC inhibitor LAQ824 and its role [Chen et al. 2005]. LAQ824 affects all Hsp90 client proteins, including the depletion of AR in LNCaP cells by inducing Hsp90 acetylation. This results in the inhibition of its ATP-binding activity and induces the dissociation of the Hsp90–AR complex leading to AR degradation.
Effect of HDACi on p53
The p53 tumor suppressor exerts antiproliferative effects, including growth arrest, apoptosis, and cell senescence, in response to various types of stress. It is well known that phosphorylation and acetylation are involved in the posttranslational modifications of p53 [Bode and Dong, 2004; Appella and Anderson, 2001; Roy and Tenniswood, 2007].
It has been shown that the HAT p300/CREB binding protein (CBP) strongly potentiates p53-dependent transcriptional activation, and that acetylation of p53 by p300 markedly stimulates its sequence-specific DNA-binding activity and transcriptional activity [Gu and Roeder 1997; Gu et al. 1997]. Therefore, HDACi may be used to potentiate the biological effects of p53 on growth suppression.
Effect of HDACi on the signal transducers and activators of transcription family of proteins
The signal transducers and activators of transcription (STAT) family of proteins are self-signaling transcription factors in cytoplasm. They are utilized by most cytokine receptors to rapidly turn on genetic expression in nuclei. Function-related posttranslational modifications of STAT proteins in response to treatment with cytokine or growth factor include phosphorylation of a single tyrosine residue. Yuan and colleagues reported that STAT proteins undergo acetylation of a single amino-acid residue, lysine 685, in response to cytokine stimulation. They concluded that this modification is essential for STAT proteins to form stable dimers and to activate transcription [Yuan et al. 2005].
Effect of HDACi on Akt dephosphorylation
Akt is a serine/threonine kinase that functions as a regulator of cell survival and metabolism through effects on several downstream proteins [Favard et al. 2010]. Chen and colleagues demonstrated that HDACi facilitate dephosphorylation of Akt by altering the dynamics of HDAC–protein phosphatase 1 (PP1) complexes. The group used U87MG human glioblastoma cells and PC3 prostate cancer cell lines to reveal the novel histone acetylation-independent mechanism by which HDACi mediate the dephosphorylation of Akt through the disruption of HDAC–PP1 complexes. HDACi selectively target HDACs 1 and 6 to disrupt the respective HDAC–PP1complexes resulting in an increased association of PP1 with Akt [Chen et al. 2005].
Effect of HDACi on nuclear factor-kappa B
Nuclear factor-kappa B (NF-kB) corresponds to an inducible transcription factor complex that plays an important role in anti-apoptotic responses in mammals. It has a subunit named ReIA and phosphorylation of an N-terminal site on this subunit by protein kinase A facilitates NF-kB assembly with CBP/p300. Both CBP and p300 are coactivators and contain HAT activity that has been implicated in the regulation of gene expression. This subunit can be deacetylated through interaction with HDAC 3 [Chen et al. 2001].
Recent clinical trials of HDACi in CRPC
Vorinostat
Vorinostat is a small molecular inhibitor of class I and II HDACs. It has been shown to inhibit the growth of PC-3, DU-145, LNCaP human prostate cancer cell lines, and suppression of PC-3 xenograft tumors. It mediates Akt dephosphorylation through the disruption of the complexes of HDAC bound to PP1, leading to more PP1–Akt association complexes, resulting in increased PP1–Akt association [Kulp et al. 2006]. The drug recently underwent a phase II clinical trial [ClinicalTrials.gov identifier: NCT00330161] as described in Table 2 [Bradley et al. 2009]. Patients received open-label oral vorinostat 400 mg daily continuously. The primary endpoint was the proportion of patients who did not demonstrate disease progression at 6 months. A total of 27 patients were enrolled, and all of them had received previous chemotherapy. A total of 11 patients (41%) discontinued therapy because of toxicity and 13 patients (48%) were removed because of disease progression. The best objective response obtained was stable disease in two patients (7%). No declines in prostate-specific antigen (PSA) of more than 50% were observed [Bradley et al. 2009].
Table 2.
Current and recent trials of histone deacetylase inhibitors in prostate cancer.
| Histone deacetylase inhibitor | Trial phase | Study design/results | ClinicalTrials.gov identifier |
|---|---|---|---|
| Vorinostat | Phase II | Trial of 400 mg daily PO vorinostat. Disease progression measured at 6 months. A total of 27 patients: 41% removed due to toxicity; 48% had disease progression; 7% did not progress. | NCT00330161 |
| Vorinostat | Phase I | Trial of PO vorinostat and IV temsirolimus. Currently ongoing. | NCT01174199 |
| Vorinostat | Phase II | Trial of PO vorinostat and PO bicalutamide with either IM leuprolide or SC goserelin acetate followed by radical prostatectomy. Currently ongoing. | NCT00589472 |
| Romidepsin | Phase II | Trial of IV romidepsin. Disease progression measured at 6 months. A total of 35 patients: 31% removed due to toxicity; 6% filled RECIST criteria; 6% had more than 50% PSA decline. | NCT00106418 |
| Pracinostat SB939 | Phase II | Trial of PO pracinostat SB939. A total of 32 patients: 25% with PSA response; 6.3% with more than 50%; 22% with objectively stable disease at 1.6–8 months. | NCT01075308 |
| Panobinostat | Phase I | Trial of IV panobinostat versus IV panobinostat, docetaxel, and prednisone. Disease progression measured at 6 months. A total of 16 patients, 8 in each arm. No apparent synergistic effect of combination. | NCT00663832 |
| Panobinostat | Phase II | Trial of IV panobinostat. Disease progression measured at 24 weeks. A total of 35 patients: 14% had PSA decrease (none more than 50%). | NCT00667862 |
| Panobinostat | Phase I/II | Trial of PO panobinostat with bicalutimide measured at 9 months. A total of 9 patients: 22% with more than 50% PSA reduction; 33% with stable PSA. Limited toxicity. | NCT00878436 |
IM, intramuscular; IV, intravenous; PO, by mouth; PSA, prostate-specific antigen; RECIST, response evaluation criteria in solid tumors; SC, subcutaneous.
Two other clinical trials are concurrently evaluating oral vorinostat. One is a phase I trial [ClinicalTrials.gov identifier: NCT01174199] with the Roswell Park Cancer Institute evaluating vorinostat with intravenous temsirolimus for patients with metastatic disease. In addition to evaluating clinical outcomes and PSA changes, the study will also interestingly use positron emission tomography/computed tomography to monitor decrease in interval tumor size. The other current ongoing trial is a National Cancer Institute phase II study [ClinicalTrials.gov identifier: NCT00589472] known as the ‘Total Androgen-Receptor Gene Expression Targeted Therapy (TARGET) trial’, which evaluates neoadjuvant vorinostat with oral bicalutamide and intramuscular leuprolide acetate or subcutaneous goserelin acetate 4–8 weeks prior to radical prostatectomy. This is an exciting phase II study combining androgen-deprivation therapy with an HDAC inhibitor to weigh outcomes in the neoadjuvant setting. Such a trial may start to shift our view of the clinical roles for vorinostat and other HDACi.
Romidepsin
Romidepsin (depsipeptide, FK228, FR901228, NSC630176) is a member of the cyclic depsipeptide class of HDACi. Romidepsin can induce the hyperacetylation of Hsp90, disrupting the complex between Hsp90 and its client proteins, inhibiting their synthesis and function [Yu et al. 2002].
A phase II study [ClinicalTrials.gov identifier: NCT00106418] of 35 patients with metastatic CRPC revealed minimal clinical activity of romidepsin is also described in Table 2. Two patients had a partial response that lasted longer than 6 months with a PSA decline of over 50%, but 11 patients (31%) had to discontinue the medication due to toxicity [Molife et al. 2010]. In total, two patients fulfilled response evaluation criteria in solid tumors. We could not identify any other ongoing clinical trials that are studying romidepsin.
Pracinostat
Pracinostat (SB939) is an orally active hydroxamic acid-based HDAC inhibitor. The pharmacokinetic profile has been excellent in both mouse models and calculated extrapolation for humans [Javaraman et al. 2011; Novotny-Diermayr et al. 2010]. In a recent phase II trial [ClinicalTrials.gov identifier: NCT01075308] of 32 patients, two patients achieved greater than 50% PSA reduction, and seven patients had objectively stable disease at 1.6–8 months. Five patients suffered from fatigue and four patients had neutropenia as the most common side effect of treatment [Eigl et al. 2012]. No current trials of pracinostat are ongoing.
Panobinostat
Similar to pracinostat, panobinostat belongs to the structurally novel cinnamic hydroxamic acid class of compounds. Treatment of LNCaP, an AR-positive prostate cancer cell line, with less than 100 nM panobinostat resulted in significant degradation of the AR as well as inhibition of cell growth, through acetylation and subsequent inhibition of the Hsp90 chaperone function [Chen et al. 2005].
In a combination study [ClinicalTrials.gov identifier: NCT00663832] of oral panobinostat versus oral panobinostat plus docetaxel in patients with advanced prostate cancer, all patients in the panobinostat monotherapy arm progressed under therapy, despite evidence of detectable levels of histone hyperacetylation in peripheral blood mononuclear cells as described in Table 2. Contrarily, 63% (5/8) of patients in the combination arm showed a PSA decline of greater than 50% [Rathkopf et al. 2008]. Furthermore, intravenous panobinostat was studied alone in a phase II trial [ClinicalTrials.gov identifier: NCT00667862] involving 35 patients. In this trial 14% of patients had a PSA decrease, but none of these decreases were greater than 50%. Fatigue and thrombocytopenia were the major adverse effects [Rathkopf et al. 2013]. Lastly, a phase I/II trial of oral panobinostat of nine patients was conducted with bicalutamide with outcomes measured at 9 months. Three patients had stable PSA and two patients had a greater than 50% reduction in PSA. The most concerning side effect was only thrombocytopenia without bleeding noted in three patients [Ferrari et al. 2011].
Conclusions and prospective
Together, these findings indicate the complexity in the mechanism of HDACi that underlies their high potency in suppressing tumor growth in vitro and in vivo. These agents offer a possible means towards treating patients suffering from CRPC. Further phase II trials are needed to identify individual CRPC subsets that may particularly benefit from these medications based on either epidemiologic or genetic criteria. Such investigations will offer insight towards not only the benefits and adverse effects of HDACi but also our overall understanding of these novel therapeutic agents.
Footnotes
Funding: This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement: The authors declare that there is no conflict of interest to disclose.
Contributor Information
Dharam Kaushik, Department of Urology, University of Texas Health Science Center and Cancer Therapy and Research Center, 7703 Floyd Curl Drive, San Antonio, TX 78229-3900, USA.
Vishal Vashistha, Department of Internal Medicine, Cleveland Clinic Foundation, Cleveland, OH, USA.
Sudhir Isharwal, Section of Urology, University of Nebraska Medical Center, Omaha, NE, USA.
Soud A. Sediqe, Department of Internal Medicine, MetroHealth Medical Center, Cleveland, OH, USA
Ming-Fong Lin, Section of Urology, and Department of Biochemistry, University of Nebraska Medical Center, Omaha, NE, USA.
References
- Abbas A., Gupta S. (2008) The role of histone deacetylases in prostate cancer. Epigenetics 3: 300–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appella E., Anderson C. (2001) Post-translational modifications and activation of p53 by genotoxic stresses. Eur J Biochem 268: 2764–2772. [DOI] [PubMed] [Google Scholar]
- Bode A., Dong Z. (2004) Post-translational modification of p53 in tumorigenesis. Nat Rev Cancer 4: 793–805. [DOI] [PubMed] [Google Scholar]
- Bradley D., Rathkopf D., Dunn R., Stadler W., Liu G., Smith D., et al. (2009) Vorinostat in advanced prostate cancer patients progressing on prior chemotherapy (NCI Trial # 6862): trial results and IL-6 analysis. A study by the DOD Prostate Cancer Clinical Trial Consortium and University of Chicago Phase II Consortium. Cancer 115: 5541–5549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C., Wang Y., Yang H., Huang P., Kulp S., Yang C., et al. (2007) Histone deacetylase inhibitors sensitize prostate cancer cells to agents that produce DNA double-strand breaks by targeting Ku70 acetylation. Cancer Res 67: 5318–5327. [DOI] [PubMed] [Google Scholar]
- Chen L., Fischle W., Verdin E., Greene W. (2001) Duration of nuclear NF-κB action regulated by reversible acetylation. Science 293: 1653–1657. [DOI] [PubMed] [Google Scholar]
- Chen L., Meng S., Wang H., Bali P., Bai W., Li B., et al. (2005) Chemical ablation of androgen receptor in prostate cancer cells by the histone deacetylase inhibitor LAQ824. Mol Cancer Ther 4: 1311–1319. [DOI] [PubMed] [Google Scholar]
- Dillard P., Ming-Fong L., Khan S. (2008) Androgen-independent prostate cancer cells acquire the complete steroidogenic potential of synthesizing testosterone from cholesterol. Mol Cell Endocrinol 295: 115–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eigl B., North S., Winquist E., Powers J., Good J., Sharma M., et al. (2012) A phase II study of SB939 in patients with castration resistant prostate cancer (CRPC). AACR-NCI-EORTC International Conference. Abstract A221. [Google Scholar]
- Fang Y., Fliss A., Robins D., Caplan A. (1996) Hsp90 regulates androgen receptor hormone binding affinity in vivo. J Biol Chem 271: 28697–28702. [DOI] [PubMed] [Google Scholar]
- Favard E., Xue G., Parcellier A., Bozulic L., Hemmings B. (2010) Protein kinase B (PKB/Akt), a key mediator of the P13K signaling pathway. Curr Top Microbiol Immunol 346: 31–56. [DOI] [PubMed] [Google Scholar]
- Feldman B., Feldman D. (2001) The development of androgen-independent prostate cancer. Nat Rev Cancer 1: 34–45. [DOI] [PubMed] [Google Scholar]
- Ferrari A., Stein M., Alumkal J., Gomez-Pinillos A., Catamero D., Mayer T., et al. (2011) A phase I/II randomized study of panobinostat and bicalutamide in castration-resistant prostate cancer (CRPC) patients progressing on second-line hormone therapy. J Clin Oncol 29: 156. [Google Scholar]
- Gu W., Roeder R. (1997) Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 90: 595–606. [DOI] [PubMed] [Google Scholar]
- Gu W., Xi X., Roeder R. (1997) Synergistic activation of transcription by CBP and p53. Nature 387: 819–823. [DOI] [PubMed] [Google Scholar]
- Hellerstedt B., Pienta K. (2008) The current state of hormonal therapy for prostate cancer. CA Cancer J Clin 52: 154–179. [DOI] [PubMed] [Google Scholar]
- Javaraman R., Pilla Reddy V., Pasha M., Wang H., Sangthongpitag K., Yeo P., et al. (2011) Preclinical metabolism and disposition of SB939 (Pracinostat), an orally active histone deacetylase inhibitor, and prediction of human pharmacokinetics. Drug Metabo Dispos 39: 2219–2232. [DOI] [PubMed] [Google Scholar]
- Kulp S., Chen C., Wang D., Chen C., Chen C. (2006) Antitumor effects of a novel phenylbutyrate-based histone deacetylase inhibitor, (S)-HDAC-42, in prostate cancer. Clin Cancer Res 12: 5199–5206. [DOI] [PubMed] [Google Scholar]
- Molife L., Attard G., Fong P., Karavasilis V., Reid A., Patterson S., et al. (2010) Phase II, two-stage, single-arm trial of the histone deacetylase inhibitor (HDACi) romidepsin in metastatic castration-resistant prostate cancer (CRPC). Ann Oncol 21: 109–113. [DOI] [PubMed] [Google Scholar]
- Montgomery R., Mostaghel E., Vessella R., Hess D., Kalhorn T., Higano C., et al. (2008) Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res 68: 4447–4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neckers L., Ivy S. (2003) Heat shock protein 90. Curr Opin Oncol 15: 419–424. [DOI] [PubMed] [Google Scholar]
- Novotny-Diermayr V., Sangthongpitag K., Hu C., Wu X., Sausgruber N., Yeo P., et al. (2010) SB939, a novel potent and orally active histone deacetylase inhibitor with high tumor exposure and efficacy in mouse models of colorectal cancer. Mol Cancer Ther 9: 642–652. [DOI] [PubMed] [Google Scholar]
- Perry A., William R., Watson G., Lawler M., Hollywood D. (2010) The epigenome as a therapeutic target in prostate cancer. Nat Rev Urol 7: 668–680. [DOI] [PubMed] [Google Scholar]
- Rathkopf D., Picus J., Hussain A., Ellard S., Chi K., Nydam T., et al. (2013) A phase 2 study of intravenous panobinostat in patients with castration-resistant prostate cancer. Cancer Chemother Phamacol 72: 537–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathkopf D., Wong B., Ross R., George D., Picus J., Tanaka E., et al. (2008) A phase I study of oral panobinostat (LBH589) alone and in combination with docetaxel (Doc) and prednisone in castration-resistant prostate cancer (CRPC). J Clin Oncol 26: 5152. [Google Scholar]
- Roy S., Tenniswood M. (2007) Site-specific acetylation of p53 directs selective transcription complex assembly. J Biol Chem 282: 4765–4771. [DOI] [PubMed] [Google Scholar]
- Shankar S., Srivastava R. (2008) Histone deacetylase inhibitors: mechanisms and clinical significance in cancer: HDAC inhibitor-induced apoptosis. Adv Exp Med Biol 615: 261–298. [DOI] [PubMed] [Google Scholar]
- Siegel R., Miller K., Jemal A. (2015) Cancer statistics, 2015. CA Cancer J Clin 65: 5–29. [DOI] [PubMed] [Google Scholar]
- Solit D., Scher H., Rosen N. (2003) Hsp90 as a therapeutic target in prostate cancer. Semin Oncol 30: 709–716. [DOI] [PubMed] [Google Scholar]
- Stanbrough M., Bubley G., Ross K., Golub T., Rubin M., Penning T., et al. (2006) Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res 66: 2815–2825. [DOI] [PubMed] [Google Scholar]
- Wagner J., Hackanson B., Lübbert M., Jung M. (2010) Histone deacetylase (HDAC) inhibitors in recent clinical trials for cancer therapy. Clin Epigenetics 1: 117–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waltregny D., North B., Van Mellaert F., de Leval J., Verdin E., Castronovo V. (2004) Screening of histone deacetylases (HDAC) expression in human prostate cancer reveals distinct class I HDAC profiles between epithelial and stromal cells. Eur J Histochem 48: 273–290. [PubMed] [Google Scholar]
- Weichert W., Röske A., Gekeler V., Beckers T., Stephan C., Jung K., et al. (2008) Histone deacetylases 1, 2 and 3 are highly expressed in prostate cancer and HDAC2 expression is associated with shorter PSA relapse time after radical prostatectomy. Br J Cancer 98: 604–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X., Guo Z., Marcu M., Neckers L., Nguyen D., Chen G., et al. (2002) Modulation of p53, ErbB1, ErbB2, and Raf-1 expression in lung cancer cells by depsipeptide FR901228. J Natl Cancer Inst 94: 504–513. [DOI] [PubMed] [Google Scholar]
- Yuan Z., Guan Y., Chatterjee D., Chin Y. (2005) Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science 307: 269–273. [DOI] [PubMed] [Google Scholar]