

ABSTRACT
Vasculogenesis and angiogenesis are controlled by vascular endothelial growth factor A (VEGF-A). Dysregulation of these physiological processes contributes to the pathologies of heart disease, cancer and stroke. Rho GTPase proteins play an integral role in VEGF-mediated formation and maintenance of blood vessels. The regulatory functions of RhoA and RhoB in vasculogenesis and angiogenesis are well defined, whereas the purpose of RhoC remains poorly understood. Here, we describe how RhoC promotes vascular homeostasis by modulating endothelial cell migration, proliferation and permeability. RhoC stimulates proliferation of human umbilical vein endothelial cells (HUVECs) by stabilizing nuclear β-catenin, which promotes transcription of cyclin D1 and subsequently drives cell cycle progression. RhoC negatively regulates endothelial cell migration through MAPKs and downstream MLC2 signaling, and decreases vascular permeability through downregulation of the phospholipase Cγ (PLCγ)–Ca2+–eNOS cascade in HUVECs. Using a VEGF-inducible zebrafish (Danio rerio) model, we observed significantly less vascular permeability in RhoC morpholino (MO)-injected zebrafish than control MO-injected zebrafish. Taken together, our findings suggest that RhoC is a key regulator of vascular homeostasis in endothelial cells.
KEY WORDS: RhoC, VEGF, Endothelial cell, Migration, Proliferation, Permeability
Highlighted Article: RhoC maintains vascular homeostasis in endothelial cells yet is dispensable for vascular development. Inhibition of RhoC represents an attractive therapeutic approach to prevent cancer metastasis.
INTRODUCTION
The Rho family of small GTPases regulates diverse signaling effectors and cellular functions to control cellular adhesion, gene transcription and cell cycle progression by acting as a molecular switch that oscillates between an active GTP-bound state and an inactive GDP-bound form (Van Aelst and D'Souza-Schorey, 1997). For instance, RhoA activates at least 11 different effector molecules to modulate cell motility, cell morphology and cell–cell adhesion through control of actin cytoskeletal organization (Bishop and Hall, 2000; Paterson et al., 1990; Takaishi et al., 1994). Rho proteins also function in membrane ruffling, smooth muscle contraction, development of stress fibers and focal adhesions, neurite retraction in neuronal cells and cytokinesis (Hirata et al., 1992; Jalink et al., 1994; Nishiki et al., 1990; Nishiyama et al., 1994; Piekny et al., 2005; Ridley and Hall, 1992). Among Rho subfamily members (RhoA, RhoB, RhoC, RhoE and RhoG), the most well-studied proteins are RhoA, RhoB and RhoC. RhoC shares ∼93% amino acid identity with RhoA and 86% similarity with RhoB (Ridley, 1997; Wheeler and Ridley, 2004). Although these three Rho proteins are structurally similar, they are functionally distinct.
Rho proteins regulate nascent blood vessel formation, termed vasculogenesis, as well as the development of new capillaries from pre-existing ones, a process named angiogenesis (Carmeliet, 2000; Risau, 1997; Risau and Flamme, 1995). Vascular endothelial growth factor (VEGF) is a multifunctional cytokine, endothelial mitogen and permeability factor that plays a crucial role in both vasculogenesis and angiogenesis. The function of VEGF in endothelial cells is mediated primarily by two tyrosine kinase receptors, VEGFR-1 and VEGFR-2. Previously, we have shown that VEGFR-2 regulates endothelial cell migration through activation of RhoA (Zeng et al., 2002). Others have demonstrated that RhoA signaling is essential for various aspects of VEGF-induced vasculogenesis and angiogenesis, including endothelial cell motility, proliferation, survival and permeability (Bryan et al., 2010; van Nieuw Amerongen et al., 2003). Global knockout of RhoA in mice has not been reported, suggesting that it is embryonic lethal, whereas fibroblast-specific knockout of RhoA inhibits mitosis in mouse embryos (Melendez et al., 2011). RhoB- and RhoC-deficient mice are viable and have no significant developmental impairments (Hakem et al., 2005; Liu et al., 2001). Despite the structural similarity of the Rho isoforms, they likely function distinctly in endothelial cells due to varied tissue-specific and context-dependent expression, unique epigenetic and post-translational modifications, and differential molecular mechanisms of regulation. RhoB regulates stage-specific survival of endothelial cells during vascular development by controlling Akt trafficking, and RhoB-knockout mice exhibit retarded vascular development in the retina, which manifests as disrupted sprout morphology (Adini et al., 2003). Thus, RhoB regulates endothelial cell migration, vessel assembly and tube formation (Howe and Addison, 2012), cellular processes required for sprouting angiogenesis. One report has shown VE-cadherin signals through RhoC to regulate Rho kinase activity, myosin light chain 2 (MLC2) phosphorylation and actomyosin contractility during tube formation in endothelial cells co-cultured with human dermal fibroblasts (Abraham et al., 2009). However, little is known about the role of RhoC in VEGF-mediated signaling in endothelial cells and vascular development, as most studies of RhoB- and RhoC-deficient cells and mice have concentrated on their regulation of vesicular trafficking and cancer development.
We sought to determine how RhoC regulates the VEGF signaling pathway and to assess its role in vascular development as well as angiogenesis. Our studies suggest that RhoC is activated upon VEGF stimulation through VEGFR-2 to regulate endothelial cell proliferation, migration and permeability through modulation of diverse signaling cascades. We found that RhoC promotes human umbilical vein endothelial cell (HUVEC) proliferation by protecting β-catenin from proteosomal degradation and thereby stimulating cell cycle progression. By contrast, RhoC negatively regulates endothelial cell migration by decreasing downstream ERK1 and ERK2 (ERK1/2, also known as MAPK-42 and MAPK-44, and MAPK3 and MAPK1), p38 MAPK family and MLC2 phosphorylation events, and vascular permeability through downregulation of VEGF-mediated phospholipase C (PLC)-γ1 phosphorylation, intracellular Ca2+ mobilization and endothelial nitric oxide synthase (eNOS, also known as NOS3) activity. We corroborated the latter finding in vivo using a VEGF-inducible zebrafish model of vascular permeability and observed greater vascular permeability in RhoC morpholino (MO)-injected zebrafish than controls. Taken together, our data shows that RhoC represents an important molecular modulator of vascular homeostasis, which might have important clinical implications in the treatment of cancer and vascular diseases, including cardiac and cerebral infarctions.
RESULTS
VEGF stimulation activates RhoC
VEGF-A has been described to induce RhoA activity within 1 min post-stimulation in HUVECs (van Nieuw Amerongen et al., 2003; Zeng et al., 2002). VEGF-A induction results in increased expression but not activity of RhoB protein in HUVECs (Howe and Addison, 2012). Therefore, we sought to determine whether RhoC is activated upon VEGF stimulation. Serum-starved HUVECs were treated with VEGF-A for 1, 3 or 5 min and active GTP-bound RhoA and RhoC was immunoprecipitated from cell lysates. Like RhoA, RhoC also was activated within 1 min post-stimulation with VEGF-A (Fig. 1A).
Fig. 1.

RhoC promotes proliferation and negatively regulates migration through activation of VEGF. (A) Serum-starved HUVECs were stimulated with 10 ng/ml VEGF-A for 1, 3 and 5 min. Lysates were immunoprecipitated with the respective substrate GST-tagged beads, and GTP-bound RhoC and GTP-bound RhoA were detected by immunoblotting. (B) HUVECs were serum starved overnight and stimulated without (−V) or with VEGF-A for 2 or 5 min (+V2 and +V5, respectively). Lysates were immunoprecipitated with GST-tagged beads for the respective substrate, and GTP-bound RhoC and RhoA were detected by immunoblotting. A densitometry analysis of the depicted immunoblots was performed using ImageJ software and is shown in the graphs below the blots. (C) HUVECs were transfected with control or RhoC siRNA using Oligofectamine for 48 h. 4×104 cells were plated in a 24-well plate, serum starved (0.2%) overnight and treated with 10 ng/ml VEGF-A. Thymidine incorporation assays were performed. ***P≤0.0001, paired two-tailed Student's t-test (RhoC siRNA +VEGF treated group versus control siRNA +VEGF treated group); *P≤0.05 (RhoC −VEGF group versus control siRNA −VEGF group). (D) 5×104 serum-starved HUVECs treated with control or RhoC siRNA were seeded into collagen-coated Transwell chambers and inserted into 24-well plates containing low-serum EGM. 10 ng/ml VEGF-A was added in the lower chamber and a Transwell migration assay was performed for 4 h. ***P≤0.0001, paired two-tailed Student's t-test (RhoC siRNA +VEGF treated group versus control siRNA +VEGF treated group). Experiments were repeated at least three times, and graphs in C and D show the mean±s.d.
Relative expression of Rho family members and the effects of RhoC depletion
Given that Rho family members regulate endothelial cell function, we aimed to determine their relative expression in endothelial cells and the effect of RhoC depletion. As assessed by immunoblotting, RhoC knockdown by using a small interfering RNA (siRNA) did not change RhoB protein levels but increased RhoA protein expression (supplementary material Fig. S1A). Next, we examined RhoA activity in HUVECs by performing a pulldown assay after RhoC depletion. RhoC knockdown completely abrogated RhoC activity but had a minimal effect on RhoA activity (Fig. 1B). In summary, our results suggest that RhoC has no effect on RhoB protein expression, and although RhoC knockdown increases RhoA protein expression, it has little effect on RhoA activity.
RhoC promotes proliferation and negatively regulates migration in a VEGF-dependent manner
The role of RhoA in the regulation of VEGF-induced endothelial cell migration has been well documented, but RhoA has no effect on the proliferation of endothelial cells (Bryan et al., 2010; van Nieuw Amerongen et al., 2003; Zeng et al., 2002). Similarly, RhoB is required for endothelial cell migration but is dispensable for HUVEC viability (Howe and Addison, 2012). Based on these findings, we sought to determine whether RhoC regulates VEGF-stimulated proliferation and migration in endothelial cells.
To this end, we knocked down RhoC in endothelial cells using two unique RhoC siRNAs and confirmed effective RhoC protein knockdown by immunoblotting (Fig. 1B; supplementary material Fig. S1B). To assess proliferation, HUVECs were transfected with control or RhoC siRNA, stimulated with VEGF-A and subjected to thymidine incorporation assays. RhoC knockdown significantly inhibited VEGF-induced proliferation of HUVECs (P=0.00018; Fig. 1C). Basal proliferation of HUVECs was also decreased upon RhoC knockdown (P=0.032; Fig. 1C), albeit to a lesser extent than in the presence of VEGF. We performed Boyden chamber migration assays and demonstrated that VEGF-dependent HUVEC migration was significantly increased upon RhoC knockdown (Fig. 1D). RhoC knockdown had no effect on apoptosis (data not shown).
We next sought to determine whether RhoC promotes proliferation and decreases migration in other endothelial cell types or if these effects of RhoC are specific to HUVECs. Thus, we knocked down RhoC in human lymphatic microvascular endothelial cells (LyECs) and human brain microvascular endothelial cells (HBMVECs) using siRNA (Fig. 2A). Similar to our results in HUVECs, we observed increased migration upon RhoC knockdown in HBMVECs (Fig. 2B) and decreased proliferation in LyECs treated with RhoC siRNA (Fig. 2C). Our results suggest that RhoC promotes proliferation and negatively regulates migration in distinct populations of human endothelial cells; however, RhoC knockdown had no effect on HBMVEC proliferation (Fig. 2D) or LyEC migration (data not shown).
Fig. 2.

RhoC knockdown decreases LyEC proliferation and cyclin D expression in LyECs and HBMVECs, as well as increasing HBMVEC migration. (A) HBMVECs were transfected with control or RhoC siRNA (si) for 48 h, serum-starved overnight, and treated with VEGF-A (+V) for 16 h. Cyclin D1, RhoC and β-actin (loading control) were detected by western blotting of cell lysates. (B) 5×104 serum-starved HUVECs treated with control or RhoC siRNA were seeded into collagen-coated Transwell chambers overnight and inserted into 24-well plates containing low-serum EGM. 10 ng/ml VEGF-A was added in the lower chamber and a Transwell migration assay was performed for 4 h. ***P<0.0001, paired two-tailed Student's t-test. (C,D) LyECs (C) or HBMVECs (D) were transfected with control or RhoC siRNA using Oligofectamine for 48 h. 4×104 cells were plated in a 24-well plate, serum-starved (0.2%) overnight and treated with 10 ng/ml VEGF. Thymidine incorporation assays were performed. **P<0.05, paired two-tailed Student's t-test (RhoC siRNA −VEGF treated group versus control siRNA −VEGF treated group); *P<0.10 (RhoC +VEGF group versus control siRNA +VEGF group); NS, not significant. Results in B (n≥7) and C,D (n=2) are mean±s.d.
Interestingly, inhibition of RhoC by siRNA in MDA-MB-231 breast cancer cells led to decreased proliferation and less invasion (supplementary material Fig. S2A–C). Taken together, our findings suggest that RhoC promotes endothelial cell proliferation and negatively regulates endothelial cell migration, whereas RhoC promotes both proliferation and migration in breast tumor cells.
Involvement of RhoC in VEGF-mediated downstream signaling cascades
We sought to determine the signaling pathways through which RhoC negatively regulates VEGF-mediated endothelial cell migration. The Ras–Raf–MEK–ERK1/2 pathway has a well-documented role in endothelial cell function. ERK1/2 regulates endothelial cell migration as well as proliferation in vivo and in vitro (Srinivasan et al., 2009). Serum-starved HUVECs treated with either control or RhoC siRNA were administered 10 ng/ml VEGF-A for 5 or 10 min and immunoblotted for phosphorylated ERK1/2 (pERK1/2). Upon RhoC knockdown, pERK1/2 was detected after 5 min of VEGF stimulation compared to 10 min in the control siRNA-treated HUVECs (Fig. 3A; supplementary material Fig. S3A). RhoC depletion also led to increased VEGF-induced phosphorylation of stress-induced protein kinases like the p38 MAPK family (Fig. 3A; supplementary material Fig. S3B) and JNK (also known as SAPK) family (Fig. 3A; supplementary material Fig. S3D). We observed little to no change in phosphorylation of the pro-survival molecule Akt (isoforms 1, 2 and 3) at serine 473 (Fig. 3A; supplementary material Fig. S3C). Phosphorylation of Src has been shown to regulate migration of endothelial cells in response to VEGF through binding with T-cell-specific adapter (TSAd, also known as SH2D2A) (Matsumoto et al., 2005). However, we did not observe any change in Src phosphorylation upon RhoC knockdown in HUVECs (supplementary material Fig. S2D).
Fig. 3.

RhoC regulates migration through ERK1/2. HUVECs were transfected with control or RhoC siRNA for 48 h, serum-starved overnight, and treated with VEGF-A for 5, 10, 15 or 20 min (+V5, +V10, +V5 and +V20, respectively). (A) Cell lysates were collected and immunoblotted (IB) with antibodies against phosphorylated ERK1/2 (pERK1/2), total ERK1/2, phosphorylated p38 MAPKs (pP38MAPK), phosphorylated Akt1, Akt and Akt3 (pAkt1/2/3), total Akt1, Akt and Akt3 (Akt1/2/3), phosphorylated JNK family proteins (pSAPK/JNK) and α-tubulin (loading control). (B) After serum starvation, cells were treated with 10 or 20 µM of MEK1 inhibitor for 1 h and 5×104 cells were seeded into collagen-coated Transwell chambers and were then inserted into 24-well plates containing low-serum EGM. VEGF-A (10 ng/ml) was added in the lower chamber and a Transwell migration assay was performed for 4 h. Results are mean±s.d. (experiments were repeated at least three times in triplicates). *P≤0.05 and **P≤0.001 (paired two-tailed Student's t-test). (C) Cell lysates were collected and western blotted with antibodies against phosphorylated LIMK1/2 (pLIMK), total LIMK1, total LIMK2, phosphorylated MLC2 (pMLC-2), RhoC and β-actin (loading control). Vertical lines indicate where lanes were removed and composite images were generated from the same immunoblot. Please see supplementary material Fig. S3 for densitometry plots of the blots shown in A and C.
RhoC regulates migration through ERK1/2
MEK1 (also known as MAP2K1) is upstream of ERK1/2 in the Ras–Raf–MEK–ERK1/2 signaling pathway. To confirm the role of ERK1/2 in the RhoC-mediated negative regulation of endothelial cell migration, we repeated the migration assay with control or RhoC-depleted HUVECs that were pre-treated with inhibitor against MEK1 for 1 h, seeded into collagen-coated Transwell chambers and incubated for another 4 h in the presence or absence of VEGF-A. We observed a significant increase in VEGF-induced cell migration after RhoC siRNA treatment compared to controls (Fig. 3B). As expected, this RhoC-knockdown-mediated increase in endothelial cell migration was blocked in presence of 10 and 20 µM MEK1 inhibitor (Fig. 3B), whereas MEK1 inhibitor had no effect on the migration of control siRNA-treated HUVECs (supplementary material Fig. S1D). This result suggests that RhoC signals through MEK1 and downstream of ERK1/2 in a VEGF-dependent manner to negatively regulate HUVEC migration. Rho family members activate Rho kinases like ROCK1 and ROCK2 (Riento and Ridley, 2003), which phosphorylate downstream LIM kinases and regulatory myosin light chains (MLCs) (Leung et al., 1996; Uehata et al., 1997). Moreover, MAPKs themselves influence cell motility by regulating MLC kinase activity and finally phosphorylation of MLCs (Klemke et al., 1997). Therefore, we examined the phosphorylation status of LIMK1 and LIMK2 (LIMK1/2), and MLC2 upon VEGF-A stimulation and siRNA-mediated RhoC knockdown in HUVECs. RhoC depletion did not affect LIMK1/2 phosphorylation (Fig. 3C; supplementary material Fig. S3E,F) but increased MLC2 phosphorylation (Fig. 3C; supplementary material Fig. S3G). Although focal adhesion kinase (FAK) activity has also been shown to play a role in VEGF-induced endothelial cell migration (Rousseau et al., 2000), RhoC knockdown did not affect VEGF-stimulated FAK phosphorylation (supplementary material Fig. S2E). Taken together, our findings suggest RhoC negatively regulates endothelial cell migration through a cascade that includes VEGF-mediated MAPK phosphorylation and downstream MLC2 phosphorylation, but excludes signaling through LIMK1/2 and FAK.
RhoC promotes proliferation through cell cycle progression
RhoC knockdown decreases VEGF-induced endothelial cell proliferation yet increases the phosphorylation of pro-proliferative molecules, such as ERK1/2. Given these seemingly disparate results, we hypothesized that RhoC promotes proliferation through regulation of cell cycle progression. Indeed, we observed a significantly greater percentage of cells in the G1 phase and a concomitant lower percentage in S and G2 phases following RhoC knockdown and VEGF stimulation, thus suggesting that RhoC controls VEGF-induced proliferation through cell cycle regulatory mechanisms (Fig. 4A). RhoC knockdown using two distinct siRNAs in HUVECs inhibited the expression of cyclin D1 (Fig. 4B; supplementary material Fig. S1B). RhoC knockdown in LyECs and HBMVECs also led to decreased VEGF-dependent and -independent expression of cyclin D1 (Fig. 2A). Cyclin D1 is known to promote cell cycle passage through the G0 to S phase; therefore, inhibition of cyclin D through RhoC knockdown likely blocks cell cycle progression through G0 to S and causes accumulation of cells in G1 phase as we observed. Correspondingly, upon RhoC knockdown and VEGF-A induction in HUVECs, we saw no upregulation of cyclin A and B (Fig. 4B), which control mitosis. Finally, we assessed expression of the Cip and Kip family members p21Cip1 (p21, also known as CDKN1C) and p27Kip1 (p27, also known as CDKN1B), which regulate cell cycle progression by binding to a variety of cyclin–CDK complexes and inhibiting their kinase activity (Johnson and Walker, 1999). Depletion of RhoC in HUVECs increased both VEGF-dependent and -independent expression of the cell cycle inhibitor p27 but had no effect on p21 (Fig. 4B). Our results suggest that RhoC promotes endothelial cell proliferation by stimulating cycle cell progression through upregulation of cyclin D1 and negative regulation of the cell cycle inhibitor p27.
Fig. 4.

RhoC promotes proliferation through cell cycle progression. HUVECs were transfected with control or RhoC siRNA for 48 h, serum-starved overnight, and treated with VEGF-A for 16 h (A,B; +V16h) or 6 or 12 h (C, +V6h and +V12h, respectively). Experiments were repeated at least three times. (A) The cells were fixed, stained with propidium iodide, and analyzed by FACS. The mean±s.d. percentage of cells with DNA content in each of the three phases of the cell cycle is shown over three independent determinations. *P≤0.05, and **P≤0.001 (paired two-tailed Student's t-test). (B,D) Cyclin D1, cyclin A, cyclin B1, p27, p21, RhoC and β-actin (loading control) were detected by immunoblotting (IB) of cell lysates (B). Vertical lines indicate where lanes were removed and composite images were generated from the same immunoblot. Densitometry of the indicated immunoblots was performed using ImageJ software (D). (C) Nuclear fractions were collected and subjected to western blotting using an anti-β-catenin antibody.
RhoC upregulates β-catenin
We next sought to further investigate the molecular mechanism through which RhoC regulates cell cycle progression. Because active nuclear β-catenin directly interacts with Lef1 and Tcf transcription factors to promote cyclin D1 expression, we hypothesized that RhoC might upregulate cyclin D1 and stimulate cell cycle progression through β-catenin. Indeed, RhoC knockdown decreased nuclear β-catenin protein expression in HUVECs in the presence and absence of VEGF-A stimulation (Fig. 4C). To confirm these findings in vivo, we knocked down the zebrafish RhoC homologues, Rhoad and Rhoae (supplementary material Fig. S4A,B), by injecting MOs into transgenic fli1:EGFP embyros in which EGFP is expressed in the vasculature. Notably, we did not observe any defects or delays in zebrafish vascular development upon MO-mediated knockdown of Rhoad and Rhoae (data not shown). At 3 days post-fertilization, we sectioned the zebrafish embryos and performed immunofluorescence staining for β-catenin. As expected, we observed less β-catenin in the vessels of zebrafish injected with Rhoad and Rhoae MO compared to siblings administered control MO (supplementary material Fig. S4C). Our data implies a scenario in which RhoC promotes proliferation by upregulating β-catenin, which in turn, promotes cyclin D1 expression to subsequently drive cell cycle progression.
VEGF activates RhoC through VEGFR-2
We have shown that RhoC regulates endothelial cell migration through the MAPK pathway and regulates proliferation through control of the cell cycle; therefore, we sought to investigate the upstream signaling events that initiate VEGF-dependent RhoC signaling in endothelium. VEGFR-1 and VEGFR-2 are expressed on the cell surface of most vascular endothelial cells, and VEGF-A binds to both VEGFR-1 and VEGFR-2 to mediate downstream signaling. We aimed to determine which receptor is involved in VEGF-induced RhoC activation. HUVECs were treated with control, VEGFR-1, or VEGFR-2 siRNA and, after 48 h, cell lysates were collected and an activated RhoC pulldown assay was performed. RhoC activation was completely blocked upon VEGFR-2 knockdown (Fig. 5A) but not following VEGFR-1 knockdown (supplementary material Fig. S1E), which indicates that VEGF-A activates RhoC through VEGFR-2 signaling.
Fig. 5.
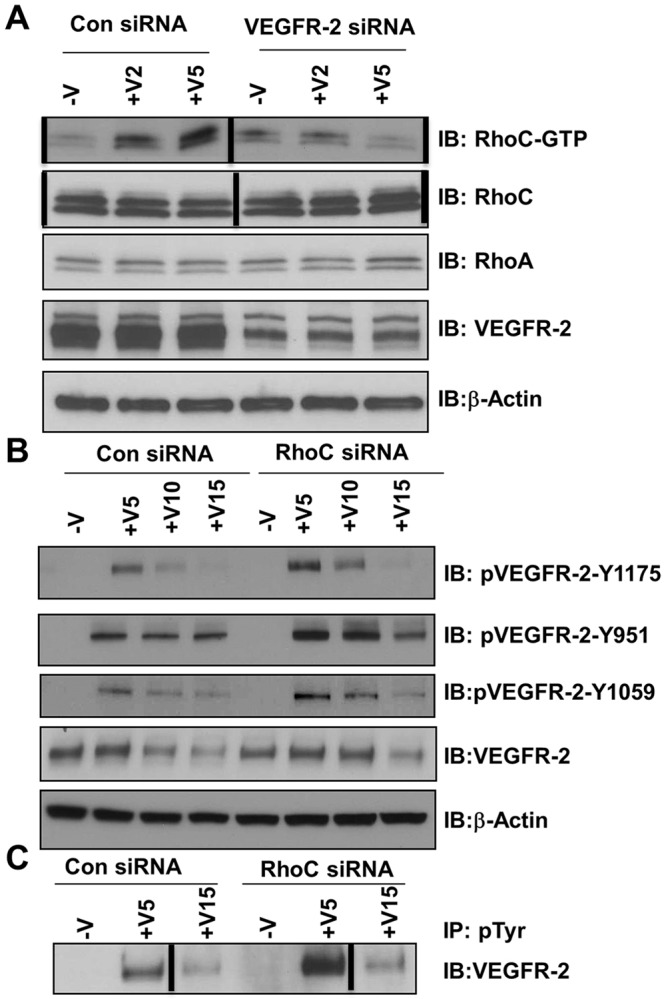
VEGFR-2 is required for RhoC signaling, but VEGFR-2 phosphorylation is negatively regulated by RhoC. HUVECs were transfected with control or VEGFR-2 siRNA, serum-starved overnight, and treated with 10 ng/ml VEGF-A for 2, 5, 10 or 15 min (+V2, +V5, +V10 and +V15, respectively). (A) Lysates were immunoprecipitated with GST beads and GTP-bound RhoC was detected by immunoblotting (IB). Levels of total RhoC, RhoA, VEGFR-2 and β-actin (loading control) are shown. (B) Lysates were immunoblotted for phosphorylated VEGFR-2 (pVEGR-2) Y1175, pVEGFR-2 Y951, pVEGFR-2 Y1059, total VEGFR-2 and β-actin (loading control). (C) Lysates were immunoprecipitated (IP) with anti-phosphorylated-tyrosine (pTyr) antibody and immunoblotted with antibody against VEGFR-2. Vertical lines indicate where lanes were removed and composite images were generated from the same immunoblot. Experiments were repeated at least three times.
RhoC depletion affects VEGF-mediated VEGFR-2 phosphorylation
We next examined the phosphorylation of VEGFR-2 at different tyrosine residues. RhoC depletion led to a significant increase in VEGF-induced VEGFR-2 phosphorylation at tyrosine residues 951, 1059 and 1175 (Fig. 5B), as well as total tyrosine phosphorylation (Fig. 5C). Upon binding VEGF, VEGFR-2 undergoes endocytosis through a classical clathrin-mediated pathway and either is targeted for recycling back to the plasma membrane or for sequential proteasome and lysosomal degradation (Eichmann and Simons, 2012). We also evaluated total VEGFR-2 levels in RhoC-depleted cells at 5, 10 and 15 min post-stimulation with VEGF. Interestingly, RhoC knockdown delayed the degradation of VEGFR-2 compared to control siRNA-treated cells (Fig. 5B), thus implicating RhoC in VEGFR-2 trafficking.
RhoC controls phosphorylation of PLC-γ1 and eNOS and induces intracellular Ca+2 release
VEGF was originally described as vascular permeability factor based on its property of increasing vessel wall permeability (Senger et al., 1983). VEGF regulates vascular permeability by binding to VEGFR-2 and stimulating PLCγ-dependent inositol (1,4,5)-trisphosphate (IP3) production, increasing the cytosolic Ca2+ concentration and leading to greater eNOS production (Brock et al., 1991; Jho et al., 2005; Wu et al., 1999). In light of our results suggesting that VEGF activates RhoC through VEGFR-2, we sought to determine whether RhoC regulates vascular permeability through the VEGFR-2–PLCγ–Ca2+–eNOS cascade. VEGF stimulation is known to activate PLC-γ1 through phosphorylation of Y783 (Tahir et al., 2009). Correspondingly, RhoC knockdown in VEGF-stimulated HUVECs significantly increased PLC-γ1 phosphorylation at Y783 (Fig. 6A). RhoC siRNA treatment increased basal and VEGF-stimulated eNOS phosphorylation at serine 1177 (Fig. 6B), the most thoroughly studied activation site of eNOS (Granger et al., 1994). We performed an intracellular Ca2+ release assay and demonstrated that endothelial cells expressing RhoC siRNA exhibited increased Ca2+ flux compared with controls (Fig. 6C), indicating that RhoC does indeed negatively regulate Ca2+ flux. Taken together, our findings suggest that RhoC negatively regulates VEGF-induced vascular permeability.
Fig. 6.

RhoC controls phosphorylation of PLC-γ1 and eNOS and induces intracellular Ca+2 release. HUVECs were transfected with control or RhoC siRNA, serum starved overnight, and treated with 10 ng/ml VEGF for 5, 10 or 15 min (+V5, +V10 and +V15, respectively). (A) Cell lysates were immunoblotted with antibodies against phosphorylated PLC-γ1 (pPLC-γ1) (Y783) and total PLC-γ-1. The corresponding densitometry graph is shown to the right. (B) Cell lysates were immunoblotted for phosphorylated eNOS (peNOS) (S1177), total eNOS and β-actin (loading control). The corresponding densitometry graph is shown to the right. (C) HUVECs transfected with control or RhoC siRNA were serum-starved overnight, loaded with Fura-2 AM and then stimulated with VEGF-A (10 ng/ml) at 50 s. All experiments were repeated at least three times.
Given our in vitro observations, we sought to determine whether RhoC controls permeability in vivo. We have previously described a heat-inducible VEGF zebrafish model used in conjunction with MO-mediated protein knockdown that can assess genetic regulation of vascular permeability in real time (Hoeppner et al., 2012). In Danio rerio, five Rho gene isoforms exist, Rhoaa, Rhoab, Rhoac, Rhoad and Rhoae, and the protein similarity of all human and zebrafish Rho subfamily members is 79.1% with 97.4% overall similarity (Salas-Vidal et al., 2005). Phylogenetic studies have suggested that human RhoC is most similar to zebrafish Rhoad and Rhoae (Salas-Vidal et al., 2005). First, we performed in situ hybridization to demonstrate that RhoC is expressed within the vasculature of zebrafish (Fig. 7A,B). We then designed Rhoad- and Rhoae-specific MOs and achieved knockdown of both genes after co-microinjection of the MOs in one- to two-cell stage zebrafish embryos (supplementary material Fig. S4A,B). At 3 days post-fertilization, we performed microangiography in the MO-injected embryos with fluorophore-conjugated dextrans, induced VEGF through heat induction and immediately performed live imaging of extravasated red tracer as a measure of vascular permeability. In the presence of VEGF-induction, we observed significantly greater vascular permeability in zebrafish injected with Rhoad and Rhoae MO than control MO-injected zebrafish (Fig. 7C,D). Interestingly, RhoC knockdown also increased the vascular permeability in the absence of VEGF induction; however, physiological levels of VEGF are likely to be similar to those observed experimentally (Fig. 7C,D). Taken together with the finding that RhoC knockdown promotes PLCγ–Ca2+–eNOS signaling, these in vivo data suggest that RhoC negatively regulates VEGF-induced vascular permeability.
Fig. 7.

RhoC negatively regulates VEGF-induced vascular permeability in zebrafish. (A,B) Using zebrafish cDNA, a probe to zebrafish RhoC (Rhoad) was created and in situ hybridization was performed on 24 hpf zebrafish embryos. Multiple images were captured using a Zeiss Axioplan 2 microscope and overlayed in Photoshop, such that areas of focus were unmasked, to generate a composite image. Lateral (A) and superior (B) views are shown. DA, dorsal aorta; PCV, posterior cardinal vein; ISVs, anterior intersomitic vessels; NT, neural tubes. (C) Microangiography was performed on anesthetized 3 dpf zebrafish embryos by injecting FITC–dextran (2000 kDa) and Texas-Red–dextran (70 kDa), VEGF was induced through heat exposure (when applicable), and extravasation of red tracer as a measure of zebrafish vascular permeability was live imaged using a ZEISS LSM 780 confocal microscope. Control, no MO injection and no VEGF induction; Cont MO, control MO injection; RhoC MO, Rhoad and Rhoae MO injection; Unind, no VEGF induction; VEGF, heat induction of VEGF transgene. (D) Quantification of extravasated red tracer. **P<0.05 (RhoC MO, VEGF Induced versus Control MO, VEGF Induced); *P<0.05 (RhoC MO, Uninduced versus Control; Control MO, VEGF Induced versus Control) (paired two-tailed Student's t-test). Results are mean±s.d. (n=??).
DISCUSSION
During the initial phases of vascular development, mesodermal precursors differentiate into endothelial cells to form nascent blood vessels, a process termed vasculogenesis (Risau and Flamme, 1995). Subsequently, blood vessels become capable of additional expansion through two forms of angiogenesis: enlargement of pre-existing vessels or formation of new vessels from pre-existing ones. Both processes require endothelial cell proliferation, whereas the latter expansion, known as sprouting angiogenesis, is also dependent upon endothelial cell migration, vessel assembly and tube formation (Risau, 1997).
RhoA, RhoB and RhoC are members of the Rho family of small GTPases. Rho proteins act as molecular switches, alternating between an inactive GDP-bound and an active GTP-bound state to relay signals from cell surface receptors associated with growth factors, adhesion molecules, cytokines and G-protein-coupled receptors (Van Aelst and D'Souza-Schorey, 1997). RhoA regulates endothelial cell proliferation, migration, vessel assembly and tube formation, and thus plays an indispensable role in vasculogenesis and angiogenesis (Bryan et al., 2010; van Nieuw Amerongen et al., 2003). RhoB deficiency has been shown to result in the apoptosis of primary endothelial cells during sprouting angiogenesis in vivo and tube formation in vitro through a mechanism in which RhoB regulates the nuclear trafficking of AKT to control endothelial cell survival (Adini et al., 2003). Correspondingly, the finding that RhoB-knockout mice are smaller than wild-type mice might reflect an angiogenesis defect (Liu et al., 2001), as delays in retinal vascular development have been observed (Adini et al., 2003). The function of RhoC in vasculogenesis and angiogenesis is less defined.
Here, we demonstrate that VEGF signals through VEGFR-2 to activate RhoC (Fig. 8). We show that RhoC negatively regulates global VEGFR-2 tyrosine phosphorylation and, more precisely phosphorylation of VEGFR-2 tyrosine residues Y951, Y1059 and Y1175. Negative feedback inhibition of VEGFR-2 by RhoC is a novel finding that warrants further future investigation. VEGFR-2 has been shown to undergo endocytosis and subsequent recycling or degradation following binding of VEGF ligand (Eichmann and Simons, 2012). VEGF-induced vascular permeability is stimulated in endothelial cells through a signaling cascade involving TSAd and Src tyrosine kinase, which is activated by tyrosine phosphorylation of VEGFR-2 at Y951 (Sun et al., 2012). Phosphorylation at Y1175 of VEGFR-2 also promotes vascular permeability by enabling VEGF binding, phosphorylation and activation of PLCγ1 (Takahashi et al., 2001), which ultimately leads to Ca2+ influx. Correspondingly, our results indicate that knockdown of RhoC causes phosphorylation of PLCγ1 and increased Ca2+ flux in endothelial cells as well as increased VEGF-induced vascular permeability in zebrafish. We have previously demonstrated that tyrosine residues Y951 and Y1059 are required for VEGF-induced endothelial cell migration and proliferation, respectively (Zeng et al., 2001). Mutational analysis has revealed that MAPK activation requires phosphorylation of Y1059, but not Y951 (Zeng et al., 2001). Here, we observed increased endothelial cell migration and greater phosphorylation of VEGFR-2 Y951 and Y1059 upon RhoC knockdown, which suggests that RhoC negatively regulates migration through decreased activation of Y951 and inhibition of MAPK signaling mediated by reduced Y1059 phosphorylation. Given that VEGFR-2 Y1059 activation promotes proliferation and that MAPK activation also has a pro-proliferative effect, we expected RhoC knockdown to stimulate endothelial cell proliferation. However, RhoC knockdown decreased proliferation despite increased Y1059 phosphorylation and MAPK activity. Given these seemingly disparate results, we sought an alternative mechanism through which RhoC could promote proliferation and found that RhoC stabilized nuclear β-catenin, which stimulated transcription of cyclin D1 and subsequently drove cell cycle progression and proliferation. Taken together, our results suggest that the anti-proliferative effects mediated by decreased MAPK activity and VEGFR-2 Y1059 phosphorylation are overridden by the ability of active nuclear β-catenin to stimulate cyclin D1, cell cycle progression and proliferation in endothelial cells.
Fig. 8.
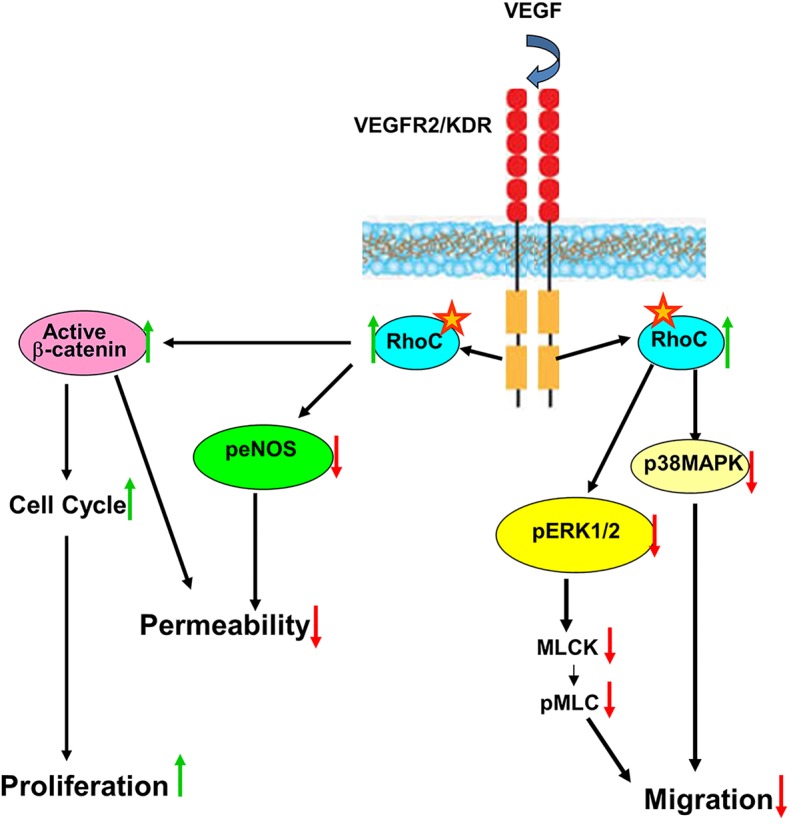
RhoC maintains vascular homeostasis through regulation of endothelial VEGF signaling. VEGF signals through VEGFR-2 to activate the GTP-bound form of RhoC. Active RhoC promotes nuclear stabilization of β-catenin, which acts as a transcription factor to increase the expression of cyclin D1, drive cell cycle progression, and stimulate endothelial cell proliferation. RhoC negatively regulates VEGF-induced vascular permeability by decreasing phosphorylation of PLC-γ1 and phosphorylation of eNOS (peNOS) to reduce intracellular cellular Ca2+ release. Stress-induced protein kinase p38 MAPKs are also downregulated by RhoC. Endothelial cell migration is negatively controlled by RhoC through a decreased activation of the Ras–Raf–MEK–MAPK signaling pathway. Specifically, RhoC reduces phosphorylation of p38 MAPKs and ERK1/2 along with that of downstream MLC2 to negatively modulate endothelial cell proliferation. Our data suggests RhoC regulates the phosphorylation of VEGFR-2.
RhoC clearly regulates processes involved in angiogenesis. However, we did not observe any defects or delays in zebrafish vascular development upon MO-mediated knockdown of Rhoad and Rhoae. Previous studies have demonstrated that RhoC-null mice are viable and do not exhibit post-natal developmental defects (Hakem et al., 2005), which corresponds with our finding that RhoC knockdown does not adversely affect zebrafish blood vessel formation. Although RhoC is dispensable for vascular development, it likely acts a molecular switch to modulate angiogenesis in processes such as wound healing, tumor growth, diabetic retinopathy and macular degeneration by controlling endothelial cell proliferation, migration and permeability. Correspondingly, our studies suggest that RhoC helps maintain vascular homeostasis – the delicate balance between vascular injury and repair. Vascular injury is usually trigged by cytokines (Nakagawa et al., 2004), hypoxia (Voelkel and Tuder, 2000), shear stress (Dimmeler et al., 1999; Malek and Izumo, 1994) and/or oxidative stress (Treins et al., 2001). The ability of RhoC to sustain homeostasis in pathological settings is exemplified by our observation that RhoC prevents acute endothelial hyperpermeability in zebrafish. VEGF-dependent RhoC signaling promotes endothelial cell proliferation and negatively regulates migration and permeability to repair and compensate for endothelial cell loss from the vascular wall, an effect of vascular injury. Various vascular beds require unique VEGF signaling modulation for survival and normal turnover of blood vessels (Lazarus and Keshet, 2011). As a small GTPase, the ability of RhoC to act as a molecular switch enables it to play a pivotal role in the maintenance of vascular homeostasis in normal adult physiology as well as pathologic conditions. The precise vascular regulation exerted by RhoC is exemplified by its distinct tissue-specific control. For example, Wang and colleagues have observed that RhoC knockdown decreases migration and invasion of human mammary endothelial cells (Wang et al., 2008), whereas our results suggest that RhoC knockdown increases HUVEC migration. Previous HUVEC studies have demonstrated that RhoC deletion promotes vascular sprouting twofold (Del Galdo et al., 2013), and that RhoC knockdown results in a loss-of-directionality migration phenotype and inefficient migration from the point of origin in single-cell tracking experiments (Mitin et al., 2013). The discrepancy in HUVEC migration might stem from the different assays utilized and inherent differences in the migratory properties of a single cell versus numerous cells. To validate our HUVEC findings and further investigate endothelial cells with various tissue origins, we evaluated the effect of RhoC knockdown in LyECs and HBMVECs. Similar to our results in HUVECs, we observed increased migration upon RhoC knockdown in HBMVECs and decreased proliferation in RhoC-siRNA-treated LyECs. Furthermore, we also saw decreased expression of cyclin D1 in RhoC-knockdown HUVECs, HBMVECs and LyECs, suggesting that RhoC promotes cyclin D1 expression to drive proliferation in these endothelial cells.
Although the studies presented here focus on the function of RhoC in endothelial cells, we speculate that RhoC has a distinct role in cancer cells. We show that RhoC promotes proliferation and invasion of MDA-MB-231 breast cancer cells. Similarly, a recent report has demonstrated that MDA-MB-231 cells with reduced levels of RhoC exhibit decreased migration (Willmer et al., 2013). Overexpression of RhoC has been shown to increase angiogenic factors in breast epithelial cells (Kawata et al., 2014). The creation of RhoC-knockout mice confirms these in vitro findings, as loss of RhoC decreases tumor cell motility and metastatic cell survival leading to inhibition of metastasis (Hakem et al., 2005). Furthermore, another in vivo study has determined that RhoC promotes the ability of melanoma cells to extravasate from blood vessels and invade the lungs (van Golen et al., 2000). Hence, numerous studies support the notion that RhoC stimulates tumor cell migration and invasion. Conversely, in endothelial cells RhoC negatively regulates migration. These opposite functions of RhoC likely reflect differences in endothelial cells versus cancer cells of epithelial origin. RhoC has been shown to promote epithelial–mesenchymal transition (EMT) in breast, colon, prostate and ovarian cancer cells (Bellovin et al., 2006; Gou et al., 2014; Kawata et al., 2014; Sequeira et al., 2008). EMT is a process through which epithelial cells transform from cells with tight cell–cell junctions, definitive basal and apical polarity and a sheet-like growth phenotype to spindle-like and motile cells, which have been linked to chemotherapeutic resistance, cancer progression, formation of subpopulations of cancer stem-like cells and cell invasion (Savagner, 2010). Given the finding that RhoC is required for tumor metastasis, inhibition of RhoC represents an attractive therapeutic approach to prevent cancer metastasis. The finding that RhoC is not required for embryonic or postnatal development in mice (Hakem et al., 2005) or zebrafish, as shown here, bodes well for therapeutically targeting RhoC in cancer cells; however, our studies suggest that RhoC inhibition increases endothelial cell migration and permeability while decreasing proliferation, and the clinical significance of these effects on the endothelium should be considered upon therapeutic targeting of RhoC.
MATERIALS AND METHODS
Reagents
VEGF-A protein was obtained from R&D Systems (Minneapolis, MN). Antibodies against VEGFR-2, phosphorylated VEGFR-2 (951), phosphorylated Akt 1, 2 and 3, total Akt 1, 2 and 3, PLC-γ1, cyclin B1, cyclin D1, p21, p27, total LIMK2, β-catenin and Src were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies against phosphorylated tyrosine (clone 4G10) and phosphorylated VEGFR-2 (1059) were purchased from Upstate (Lake Placid, NY, USA). Antibodies against phosphorylated VEGFR-2 (1175), pERK1/2, total ERK1/2, RhoA, RhoB, RhoC, the p38 MAPK family, the phosphorylated p38 MAPK family, the phosphorylated JNK family, phosphorylated FAK (Tyr397), total FAK, phosphorylated LIMK1/2, total LIMK1, phosphorylated MLC2, phosphorylated PLC-γ1 (Tyr783) and cyclin A were obtained from Cell Signaling (Danvers, MA). The antibody against phosphorylated Src (Tyr418) antibody was from BioSource International (Camarillo, CA). Antibodies against phosphorylated eNOS, total eNOS and β-actin were from BD Biosciences (San Jose, CA) and the anti-α-tubulin antibody was from Abcam (Cambridge, UK). siRNAs against VEGFR-1 and VEGFR-2 were from Santa Cruz Biotechnology. RhoC siRNA #1 was from Sigma-Aldrich (St Louis, MO) and RhoC siRNA #2 was purchased from Qiagen (Venlo, Limburg, The Netherlands). Control siRNA was purchased from Dharmacon. The Rho activation kit was purchased from Millipore (Lake Placid, NY). E64d was from Sigma-Aldrich and MG132 was from Boston Biochem (Cambridge, MA). MEK1 inhibitor was purchased from Cell Signaling.
Cell culture
HUVECs were purchased from Lonza Group (Basel, Switzerland) and passaged in EGM growth medium (Lonza). Primary human brain microvascular endothelial cells (HBMVECs) were purchased from Cell Systems (Kirkland, WA) and cultured in CSC serum-containing medium (Cell Systems). Human lymphatic microvascular endothelial cells (LyECs) were purchased from Lonza and cultured in EBM-2 medium with EGM-2 MV Bullet Kit. MDA-MB-231 (human breast adenocarcinoma) cells were purchased from ATCC (Manassas, VA) and were cultured in RPMI 1640 medium from Life Technologies (Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS) and penicillin-streptomycin.
siRNA transfection
105 HUVECs were seeded in 60-mm plates and cultured for 24 h in EGM. Cells were washed with OPTI-MEM reduced serum medium and transfected with 100 nM RhoC, VEGFR-1 or VEGFR2 siRNA or control siRNA using Oligofectamine (Life Technologies). After 4 h, antibiotic-free EGM was added and cell lysates were prepared. MDA-MB-231 cells were transfected with 100 nM control or RhoC siRNA using DharmaFECT 4 purchased from GE Dharmacon (Lafayette, CO).
Proliferation assay
Control or RhoC siRNA-treated HUVECs (4×104/ml) were seeded in 24-well plates, cultured for 24 h in EGM, and subsequently serum starved (0.2%) in the presence of VEGF (10 ng/ml). The following day, 1 µCi (0.000037 GBq) of [3H]thymidine was added to each well; 4 h later, cells were washed with chilled PBS, fixed with 100% cold methanol and trichloroacetic-acid-precipitable radioactivity was measured. For the thymidine incorporation assay using cancer cells, 6×104 MDA-MB-231 cells/ml were used. Proliferation assays using HBMVECs and LyECs were performed as described for HUVECs except HBMVECs and LyECs were cultured in their appropriate medium.
Migration assay
5×104 control- or RhoC-siRNA-treated serum-starved HUVECs were seeded into collagen-coated Transwell chambers with a diameter of 6.5 mm and a pore size of 8 μm (Corning CoStar Corporation, Cambridge, MA) and inserted into 24-well plates containing serum-starved EGM. After incubation at 37°C for 1 h, 10 ng/ml VEGF-A was added to the lower chamber. To determine the effect of ERK1/2 on VEGF-induced cell migration, siRNA-treated HUVECs were pre-treated with 10 or 20 μM of MEK1 inhibitor for 1 h and then cells were seeded into collagen coated Transwell chambers. Following incubation for 4 h with or without VEGF-A at 37°C, cells that remained in the upper chamber were gently removed with a cotton swab. Cells that had invaded through the filter were fixed in 4% paraformaldehyde (PFA) and then stained with 0.2% Crystal Violet dissolved in 2% ethanol. Migration was quantified by counting the number of cells on the filter using bright-field optics with a Nikon Diaphot microscope equipped with a 16-square reticule (1 mm2). Four separate fields were counted for each filter. Three separate experiments were analyzed and the mean is reported. Migration assays using HBMVECs and LyECs were performed as described for HUVECs except HBMVECs and LyECs were cultured in their appropriate medium and cultured in Transwell chambers 12 h prior to initial serum starvation.
Cell cycle analysis
DNA content was measured after staining cells with propidium iodide. HUVECs transfected with control or RhoC siRNA were serum starved (0.2%) for 18 h. Following starvation, the cells were treated or not with VEGF-A (10 ng/ml) and collected at 16 h after treatment. The cells were trypsinized, washed in PBS and fixed in 95% ethanol for 1 h. Cells were rehydrated, washed in PBS and treated with RNaseA (1 mg/ml) followed by staining with propidium iodide (100 mg/ml). Flow cytometric quantification of DNA was performed with a FACScan (Becton Dickinson, San Jose, CA) and data analysis was performed using the Modfit software (Verity Software House Inc., Topsham, ME). Experiments were repeated at least three times.
Immunoprecipitation and western blot analysis
Control or RhoC siRNA-treated, serum-starved (0.2% serum for 24 h) HUVECs were pre-treated with VEGF (10 ng/ml) for indicated times. Whole-cell lysates from HUVECs were prepared in RIPA buffer supplemented with protease inhibitor and phosphatase inhibitor. Following centrifugation at 20,000 g for 10 min at 4°C, 250 µg of protein lysate was incubated with 2 µg respective antibody for 1 h and 50 µl of protein-A/G-conjugated agarose beads overnight at 4°C. Beads were washed with RIPA buffer three times, immunoprecipitates were resuspended in SDS sample buffer, electrophoresis was performed, and proteins were transferred to polyvinyl difluoride membranes and immunoblotted. Antibody-reactive bands were detected by enzyme-linked chemiluminescence (Amersham, Piscataway, NJ). These experiments were repeated at least three times.
Intracellular Ca2+ release
HUVECs transfected with control or RhoC siRNA, were serum starved (0.2% serum) overnight, loaded with Fura-2 AM and then stimulated with VEGF-A (10 ng/ml). Intracellular Ca2+ concentrations were measured with the DeltaScan illumination system using Felix software (Photon Technology International, Edison, NJ).
Whole-mount in situ hybridization
536 bases of the open reading frame of zebrafish RhoC (Rhoad) were amplified and cloned into a vector derived from pCR-BluntIITOPO (Life Technologies, Inc.) using zebrafish cDNA. Rhoad primers used were 5′-GGTGATTGTGGGAGATGGAG-3′ and 5′-TCTTCTTGCGCTTACGGACT-3′. To generate digoxigenin (DIG)-labeled antisense probes, the DNA plasmid was linearized with Not1 (New England BioLabs, Ipswich, MA), followed by transcription using SP6 polymerase and the 10 DIG RNA labeling mix from Roche (Indianapolis, IN). In situ hybridization was then performed according to a published procedure (Thisse and Thisse, 2008).
Microinjections and assessment of vascular permeability in VEGF-inducible zebrafish
One-cell stage transgenic VEGF-inducible zebrafish (Hoeppner et al., 2012) embryos were arrayed in an agarose microinjection template and 1.5 nl of Cre mRNA (12.5 ng/μl) was microinjected into the cell of the embryo. Rhoad (5′-TCCACCTGCAGATCATAATTGGGTA-3′) and Rhoae (5′-TCCACCTGCAGATCATAATTGGGTA-3′) MOs were designed, purchased from Gene Tools, LLC (Philomath, OR), and microinjected into embryos at the one- or two-cell stage. 9.25 ng Rhoad and Rhoae (2.2 nl of 500 µM) or 9.25 ng (2.2 nl of 500 µM) of nonspecific control MO were microinjected. Zebrafish expressing the VEGF-inducible transgene were selected by monitoring expression of eGFP in their eyes. Microangiography was performed on anesthetized embryos, which had been placed in an agarose microinjection template, at 3 days post fertilization by inserting a glass microneedle through the pericardium directly into the ventricle. FITC–dextran with a molecular mass of 2000 kDa and Texas-Red–dextran with a molecular mass of 70 kDa were used (Life Technologies, Inc.). The dextran was solubilized in embryo medium at 2 mg/ml concentration. Heat-shock induction of VEGF was performed by transferring the zebrafish from 28.5°C to 37°C embryo water for 5 min immediately prior to imaging. The visualization and real-time imaging was performed using the previously described SCORE methodology (Petzold et al., 2010) on a ZEISS LSM 780 confocal microscope using standard FITC and dsRed filter sets. Zebrafish were used and maintained according to Institutional Animal Care and Use Committee guidelines at Mayo Clinic.
Small GTPase pulldown assay
RhoA, RhoB and RhoC activation assay kits from Millipore were used to perform these assays. Magnesium lysis buffer (MLB) was made by diluting 5× MLB purchased from EMD Millipore (Billerica, MA) by adding sterile distilled water containing 10% glycerol. To the 1× MLB diluted buffer, protease inhibitor cocktail and phosphatase inhibitor were added. The cells were rinsed twice with ice-cold PBS and an appropriate amount of ice-cold 1× MLB was added. The lysates were transferred to microfuge tubes and the manufacturer's protocol was followed. Rho immunoblotting was performed with anti-RhoA, anti-RhoB and anti-RhoC antibodies.
Invasion assay
100 µl of 3 mg/ml Matrigel solution (BD Biosciences) was overlaid on the upper surface of Transwell chambers with a diameter of 6.5 mm and a pore size of 8 µm (Corning CoStar Corporation). The Matrigel was allowed to solidify by incubating the plates for ∼1 h at 37°C. Medium (0.6 ml) containing 10% FBS was then added to the bottom chamber of the Transwells. MDA-MB-231 cells that had been treated with siRNA were trypsinized and resuspended in corresponding serum-free medium. Subsequently, 2×105 cells/ml in a volume of 200 µl of medium were added to the upper chamber of each well. Cells were incubated for 8 h at 37°C. Cells that remained in the upper chamber were removed by gently scraping with a cotton swab. Cells that had invaded through the filter were fixed in 100% methanol and stained with 0.2% Crystal Violet dissolved in 2% ethanol. Invasion was quantified by counting the number of cells on the filter using bright-field optics with a Nikon Diaphot microscope equipped with a 16-square reticule (1 mm2). Four separate fields were counted for each filter. The average of three separate experiments has been documented.
In vitro apoptosis assay
105 HUVECs were seeded in 60-mm plates and cultured for 24 h in EGM. The next day, cells were washed with OPTI-MEM reduced serum medium and transfected with 100 nM control or RhoC siRNA. After 48 h, cells were serum starved (0.2%) and the treated with VEGF (10 ng/ml) for 16 h. Annexin-V–FITC (Biovision, Mountain View, CA) was used to assess apoptosis through flow cytometry. Additional exposure to propidium iodide made it possible to differentiate early apoptotic cells (Annexin-positive and propidium-iodide-negative) from late apoptotic cells (Annexin- and propidium-iodide-positive). The average of three separate experiments has been documented.
Nuclear extract preparation
Control- or RhoC-siRNA-treated, serum-starved (0.2% serum, for 24 h) HUVEC suspensions were incubated in a hypotonic buffer [10 mM HEPES pH 7.8, 10 mM KCl, 2 mM MgCl2, 0.1 mM EDTA, 10 μg/ml aprotinin, 3 mM dithiothreitol (DTT), and 0.2 mM phenylmethylsulfonyl fluoride (PMSF)] for 15 min on ice. Nonionic detergent IGE-PAL (Sigma Aldrich) (10%) was then added to the cell suspension and mixed vigorously. Thereafter, the whole mixture was centrifuged at 14,000 rpm for 5 min. The pellets were again suspended in a hypertonic buffer solution (50 mM HEPES pH 7.8, 50 mM KCl, 300 mM NaCl, 0.1 mM EDTA, 10 μg/ml aprotinin, 3 mM DTT, and 0.2 mmol/l PMSF) and mixed on a rotating rack for 25 min at 4°C. Finally, the sample was centrifuged at 20,000 g for 10 min, and the supernatant was collected as nuclear extract.
Statistical analysis
The independent-sample Student's t-test was used to test the probability of significant differences between two groups. Statistical significance was defined as *P≤0.05 and **P≤0.001, and ***P≤0.0001. Error bars show calculated s.d. values.
Acknowledgements
The authors thank Dr Noriko Umemoto for experimental validation of zebrafish in situ hybridization studies and the Mayo Clinic zebrafish core facility for their assistance.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
S.S., Y.W., R.B. and S.D. performed the in vitro HUVECs experiments. L.H.H. developed the zebrafish permeability model and performed zebrafish experiments and imaging. Y.W. sectioned zebrafish embryos. V.M.B. performed the zebrafish in situ hybridization. S.S. and C.z.C. validated MO-mediated RhoC knockdown in zebrafish. L.H.H. and S.S. prepared the manuscript. R.R., S.C.E. and D.M. supervised the project. D.M. developed the original hypothesis and directed the entire project.
Funding
This work was supported by National Institutes of Health [grant numbers CA78383, CA150190, HL70567 to D.M. and CA187035 to L.H.H.]; and the American Heart Association [grant numbers 13POST14510025 to L.H.H. and 14POST20390029 to Y.W.]. X.G. is a fellow of National Cancer Institute [grant number NCI-T32 CA148073]. Deposited in PMC for release after 12 months.
Supplementary material
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.167601/-/DC1
References
- Abraham S., Yeo M., Montero-Balaguer M., Paterson H., Dejana E., Marshall C. J. and Mavria G. (2009). VE-Cadherin-mediated cell-cell interaction suppresses sprouting via signaling to MLC2 phosphorylation. Curr. Biol. 19, 668-674. 10.1016/j.cub.2009.02.057 [DOI] [PubMed] [Google Scholar]
- Adini I., Rabinovitz I., Sun J. F., Prendergast G. C. and Benjamin L. E. (2003). RhoB controls Akt trafficking and stage-specific survival of endothelial cells during vascular development. Genes Dev. 17, 2721-2732. 10.1101/gad.1134603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellovin D. I., Simpson K. J., Danilov T., Maynard E., Rimm D. L., Oettgen P. and Mercurio A. M. (2006). Reciprocal regulation of RhoA and RhoC characterizes the EMT and identifies RhoC as a prognostic marker of colon carcinoma. Oncogene 25, 6959-6967. 10.1038/sj.onc.1209682 [DOI] [PubMed] [Google Scholar]
- Bishop A. L. and Hall A. (2000). Rho GTPases and their effector proteins. Biochem. J. 348, 241-255. 10.1042/bj3480241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brock T. A., Dvorak H. F. and Senger D. R. (1991). Tumor-secreted vascular permeability factor increases cytosolic Ca2+ and von Willebrand factor release in human endothelial cells. Am. J. Pathol. 138, 213-221. [PMC free article] [PubMed] [Google Scholar]
- Bryan B. A., Dennstedt E., Mitchell D. C., Walshe T. E., Noma K., Loureiro R., Saint-Geniez M., Campaigniac J.-P., Liao J. K. and D'Amore P. A. (2010). RhoA/ROCK signaling is essential for multiple aspects of VEGF-mediated angiogenesis. FASEB J. 24, 3186-3195. 10.1096/fj.09-145102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P. (2000). Mechanisms of angiogenesis and arteriogenesis. Nat. Med. 6, 389-395. 10.1038/74651 [DOI] [PubMed] [Google Scholar]
- Del Galdo S., Vettel C., Heringdorf D. M. and Wieland T. (2013). The activation of RhoC in vascular endothelial cells is required for the S1P receptor type 2-induced inhibition of angiogenesis. Cell. Signal. 25, 2478-2484. 10.1016/j.cellsig.2013.08.017 [DOI] [PubMed] [Google Scholar]
- Dimmeler S., Fleming I., Fisslthaler B., Hermann C., Busse R. and Zeiher A. M. (1999). Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature 399, 601-605. 10.1038/21224 [DOI] [PubMed] [Google Scholar]
- Eichmann A. and Simons M. (2012). VEGF signaling inside vascular endothelial cells and beyond. Curr. Opin. Cell Biol. 24, 188-193. 10.1016/j.ceb.2012.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gou W.-F., Zhao Y., Lu H., Yang X.-F., Xiu Y.-L., Zhao S., Liu J.-M., Zhu Z.-T., Sun H.-Z., Liu Y.-P. et al. (2014). The role of RhoC in epithelial-to-mesenchymal transition of ovarian carcinoma cells. BMC Cancer 14, 477 10.1186/1471-2407-14-477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granger H. J., Ziche M., Hawker J. R. Jr, Meininger C. J., Czisny L. E. and Zawieja D. C. (1994). Molecular and cellular basis of myocardial angiogenesis. Cell. Mol. Biol. Res. 40, 81-85. [PubMed] [Google Scholar]
- Hakem A., Sanchez-Sweatman O., You-Ten A., Duncan G., Wakeham A., Khokha R. and Mak T. W. (2005). RhoC is dispensable for embryogenesis and tumor initiation but essential for metastasis. Genes Dev. 19, 1974-1979. 10.1101/gad.1310805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata K., Kikuchi A., Sasaki T., Kuroda S., Kaibuchi K., Matsuura Y., Seki H., Saida K. and Takai Y. (1992). Involvement of rho p21 in the GTP-enhanced calcium ion sensitivity of smooth muscle contraction. J. Biol. Chem. 267, 8719-8722. [PubMed] [Google Scholar]
- Hoeppner L. H., Phoenix K. N., Clark K. J., Bhattacharya R., Gong X., Sciuto T. E., Vohra P., Suresh S., Bhattacharya S., Dvorak A. M. et al. (2012). Revealing the role of phospholipase Cbeta3 in the regulation of VEGF-induced vascular permeability. Blood 120, 2167-2173. 10.1182/blood-2012-03-417824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe G. A. and Addison C. L. (2012). RhoB controls endothelial cell morphogenesis in part via negative regulation of RhoA. Vasc. Cell 4, 1 10.1186/2045-824X-4-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalink K., van Corven E. J., Hengeveld T., Morii N., Narumiya S. and Moolenaar W. H. (1994). Inhibition of lysophosphatidate- and thrombin-induced neurite retraction and neuronal cell rounding by ADP ribosylation of the small GTP-binding protein Rho. J. Cell Biol. 126, 801-810. 10.1083/jcb.126.3.801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jho D., Mehta D., Ahmmed G., Gao X.-P., Tiruppathi C., Broman M. and Malik A. B. (2005). Angiopoietin-1 opposes VEGF-induced increase in endothelial permeability by inhibiting TRPC1-dependent Ca2 influx. Circ. Res. 96, 1282-1290. 10.1161/01.RES.0000171894.03801.03 [DOI] [PubMed] [Google Scholar]
- Johnson D. G. and Walker C. L. (1999). Cyclins and cell cycle checkpoints. Annu. Rev. Pharmacol. Toxicol. 39, 295-312. 10.1146/annurev.pharmtox.39.1.295 [DOI] [PubMed] [Google Scholar]
- Kawata H., Kamiakito T., Omoto Y., Miyazaki C., Hozumi Y. and Tanaka A. (2014). RhoC upregulation is correlated with reduced E-cadherin in human breast cancer specimens after chemotherapy and in human breast cancer MCF-7 cells. Hormones Cancer 5, 414-423. 10.1007/s12672-014-0199-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemke R. L., Cai S., Giannini A. L., Gallagher P. J., de Lanerolle P. and Cheresh D. A. (1997). Regulation of cell motility by mitogen-activated protein kinase. J. Cell Biol. 137, 481-492. 10.1083/jcb.137.2.481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarus A. and Keshet E. (2011). Vascular endothelial growth factor and vascular homeostasis. Proc. Am. Thorac. Soc. 8, 508-511. 10.1513/pats.201102-021MW [DOI] [PubMed] [Google Scholar]
- Leung T., Chen X. Q., Manser E. and Lim L. (1996). The p160 RhoA-binding kinase ROK alpha is a member of a kinase family and is involved in the reorganization of the cytoskeleton. Mol. Cell. Biol. 16, 5313-5327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu A.-X., Rane N., Liu J.-P. and Prendergast G. C. (2001). RhoB is dispensable for mouse development, but it modifies susceptibility to tumor formation as well as cell adhesion and growth factor signaling in transformed cells. Mol. Cell. Biol. 21, 6906-6912. 10.1128/MCB.21.20.6906-6912.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malek A. M. and Izumo S. (1994). Molecular aspects of signal transduction of shear stress in the endothelial cell. J. Hypertens. 12, 989-999. 10.1097/00004872-199409000-00001 [DOI] [PubMed] [Google Scholar]
- Matsumoto T., Bohman S., Dixelius J., Berge T., Dimberg A., Magnusson P., Wang L., Wikner C., Qi J. H., Wernstedt C. et al. (2005). VEGF receptor-2 Y951 signaling and a role for the adapter molecule TSAd in tumor angiogenesis. EMBO J. 24, 2342-2353. 10.1038/sj.emboj.7600709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melendez J., Stengel K., Zhou X., Chauhan B. K., Debidda M., Andreassen P., Lang R. A. and Zheng Y. (2011). RhoA GTPase is dispensable for actomyosin regulation but is essential for mitosis in primary mouse embryonic fibroblasts. J. Biol. Chem. 286, 15132-15137. 10.1074/jbc.C111.229336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitin N., Rossman K. L., Currin R., Anne S., Marshall T. W., Bear J. E., Bautch V. L. and Der C. J. (2013). The RhoGEF TEM4 regulates endothelial cell migration by suppressing actomyosin contractility. PLoS ONE 8, e66260 10.1371/journal.pone.0066260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T., Li J. H., Garcia G., Mu W., Piek E., Böttinger E. P., Chen Y., Zhu H. J., Kang D.-H., Schreiner G. F. et al. (2004). TGF-beta induces proangiogenic and antiangiogenic factors via parallel but distinct Smad pathways. Kidney Int. 66, 605-613. 10.1111/j.1523-1755.2004.00780.x [DOI] [PubMed] [Google Scholar]
- Nishiki T., Narumiya S., Morii N., Yamamoto M., Fujiwara M., Kamata Y., Sakaguchi G. and Kozaki S. (1990). ADP-ribosylation of the rho/rac proteins induces growth inhibition, neurite outgrowth and acetylcholine esterase in cultured PC-12 cells. Biochem. Biophys. Res. Commun. 167, 265-272. 10.1016/0006-291X(90)91760-P [DOI] [PubMed] [Google Scholar]
- Nishiyama T., Sasaki T., Takaishi K., Kato M., Yaku H., Araki K., Matsuura Y. and Takai Y. (1994). rac p21 is involved in insulin-induced membrane ruffling and rho p21 is involved in hepatocyte growth factor- and 12-O-tetradecanoylphorbol-13-acetate (TPA)-induced membrane ruffling in KB cells. Mol. Cell. Biol. 14, 2447-2456. 10.1128/MCB.14.4.2447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson H. F., Self A. J., Garrett M. D., Just I., Aktories K. and Hall A. (1990). Microinjection of recombinant p21rho induces rapid changes in cell morphology. J. Cell Biol. 111, 1001-1007. 10.1083/jcb.111.3.1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petzold A. M., Bedell V. M., Boczek N. J., Essner J. J., Balciunas D., Clark K. J. and Ekker S. C. (2010). SCORE imaging: specimen in a corrected optical rotational enclosure. Zebrafish 7, 149-154. 10.1089/zeb.2010.0660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piekny A., Werner M. and Glotzer M. (2005). Cytokinesis: welcome to the Rho zone. Trends Cell Biol. 15, 651-658. 10.1016/j.tcb.2005.10.006 [DOI] [PubMed] [Google Scholar]
- Ridley A. J. (1997). The GTP-binding protein Rho. Int. J. Biochem. Cell Biol. 29, 1225-1229. 10.1016/S1357-2725(97)00052-6 [DOI] [PubMed] [Google Scholar]
- Ridley A. J. and Hall A. (1992). The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell 70, 389-399. 10.1016/0092-8674(92)90163-7 [DOI] [PubMed] [Google Scholar]
- Riento K. and Ridley A. J. (2003). Rocks: multifunctional kinases in cell behaviour. Nat. Rev. Mol. Cell Biol. 4, 446-456. 10.1038/nrm1128 [DOI] [PubMed] [Google Scholar]
- Risau W. (1997). Mechanisms of angiogenesis. Nature 386, 671-674. 10.1038/386671a0 [DOI] [PubMed] [Google Scholar]
- Risau W. and Flamme I. (1995). Vasculogenesis. Annu. Rev. Cell Dev. Biol. 11, 73-91. 10.1146/annurev.cb.11.110195.000445 [DOI] [PubMed] [Google Scholar]
- Rousseau S., Houle F., Kotanides H., Witte L., Waltenberger J., Landry J. and Huot J. (2000). Vascular endothelial growth factor (VEGF)-driven actin-based motility is mediated by VEGFR2 and requires concerted activation of stress-activated protein kinase 2 (SAPK2/p38) and geldanamycin-sensitive phosphorylation of focal adhesion kinase. J. Biol. Chem. 275, 10661-10672. 10.1074/jbc.275.14.10661 [DOI] [PubMed] [Google Scholar]
- Salas-Vidal E., Meijer A. H., Cheng X. and Spaink H. P. (2005). Genomic annotation and expression analysis of the zebrafish Rho small GTPase family during development and bacterial infection. Genomics 86, 25-37. 10.1016/j.ygeno.2005.03.010 [DOI] [PubMed] [Google Scholar]
- Savagner P. (2010). The epithelial-mesenchymal transition (EMT) phenomenon. Ann. Oncol. 21 Suppl. 7, vii89-vii92. 10.1093/annonc/mdq292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senger D. R., Galli S. J., Dvorak A. M., Perruzzi C. A., Harvey V. S. and Dvorak H. F. (1983). Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science 219, 983-985. 10.1126/science.6823562 [DOI] [PubMed] [Google Scholar]
- Sequeira L., Dubyk C. W., Riesenberger T. A., Cooper C. R. and van Golen K. L. (2008). Rho GTPases in PC-3 prostate cancer cell morphology, invasion and tumor cell diapedesis. Clin. Exp. Metastasis 25, 569-579. 10.1007/s10585-008-9173-3 [DOI] [PubMed] [Google Scholar]
- Srinivasan R., Zabuawala T., Huang H., Zhang J., Gulati P., Fernandez S., Karlo J. C., Landreth G. E., Leone G. and Ostrowski M. C. (2009). Erk1 and Erk2 regulate endothelial cell proliferation and migration during mouse embryonic angiogenesis. PLoS ONE 4, e8283 10.1371/journal.pone.0008283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z., Li X., Massena S., Kutschera S., Padhan N., Gualandi L., Sundvold-Gjerstad V., Gustafsson K., Choy W. W., Zang G. et al. (2012). VEGFR2 induces c-Src signaling and vascular permeability in vivo via the adaptor protein TSAd. J. Exp. Med. 209, 1363-1377. 10.1084/jem.20111343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahir S. A., Park S. and Thompson T. C. (2009). Caveolin-1 regulates VEGF-stimulated angiogenic activities in prostate cancer and endothelial cells. Cancer Biol. Ther. 8, 2284-2294. 10.4161/cbt.8.23.10138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T., Yamaguchi S., Chida K. and Shibuya M. (2001). A single autophosphorylation site on KDR/Flk-1 is essential for VEGF-A-dependent activation of PLC-gamma and DNA synthesis in vascular endothelial cells. EMBO J. 20, 2768-2778. 10.1093/emboj/20.11.2768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaishi K., Sasaki T., Kato M., Yamochi W., Kuroda S., Nakamura T., Takeichi M. and Takai Y. (1994). Involvement of Rho p21 small GTP-binding protein and its regulator in the HGF-induced cell motility. Oncogene 9, 273-279. [PubMed] [Google Scholar]
- Thisse C. and Thisse B. (2008). High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat. Protoc. 3, 59-69. 10.1038/nprot.2007.514 [DOI] [PubMed] [Google Scholar]
- Treins C., Giorgetti-Peraldi S., Murdaca J. and Van Obberghen E. (2001). Regulation of vascular endothelial growth factor expression by advanced glycation end products. J. Biol. Chem. 276, 43836-43841. 10.1074/jbc.M106534200 [DOI] [PubMed] [Google Scholar]
- Uehata M., Ishizaki T., Satoh H., Ono T., Kawahara T., Morishita T., Tamakawa H., Yamagami K., Inui J., Maekawa M. et al. (1997). Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature 389, 990-994. 10.1038/40187 [DOI] [PubMed] [Google Scholar]
- Van Aelst L. and D'Souza-Schorey C. (1997). Rho GTPases and signaling networks. Genes Dev. 11, 2295-2322. 10.1101/gad.11.18.2295 [DOI] [PubMed] [Google Scholar]
- van Golen K. L., Wu Z.-F., Qiao X. T., Bao L. and Merajver S. D. (2000). RhoC GTPase overexpression modulates induction of angiogenic factors in breast cells. Neoplasia 2, 418-425. 10.1038/sj.neo.7900115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Nieuw Amerongen G. P., Koolwijk P., Versteilen A. and van Hinsbergh V. W. M. (2003). Involvement of RhoA/Rho kinase signaling in VEGF-induced endothelial cell migration and angiogenesis in vitro. Arterioscler. Thromb. Vasc. Biol. 23, 211-217. 10.1161/01.ATV.0000054198.68894.88 [DOI] [PubMed] [Google Scholar]
- Voelkel N. F. and Tuder R. M. (2000). Hypoxia-induced pulmonary vascular remodeling: a model for what human disease? J. Clin. Invest. 106, 733-738. 10.1172/JCI11144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W., Wu F., Fang F., Tao Y. and Yang L. (2008). RhoC is essential for angiogenesis induced by hepatocellular carcinoma cells via regulation of endothelial cell organization. Cancer Sci. 99, 2012-2018. 10.1111/j.1349-7006.2008.00902.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler A. P. and Ridley A. J. (2004). Why three Rho proteins? RhoA, RhoB, RhoC, and cell motility. Exp. Cell Res. 301, 43-49. 10.1016/j.yexcr.2004.08.012 [DOI] [PubMed] [Google Scholar]
- Willmer T., Contu L., Blatch G. L. and Edkins A. L. (2013). Knockdown of Hop downregulates RhoC expression, and decreases pseudopodia formation and migration in cancer cell lines. Cancer Lett. 328, 252-260. 10.1016/j.canlet.2012.09.021 [DOI] [PubMed] [Google Scholar]
- Wu H. M., Yuan Y., Zawieja D. C., Tinsley J. and Granger H. J. (1999). Role of phospholipase C, protein kinase C, and calcium in VEGF-induced venular hyperpermeability. Am. J. Physiol. 276, H535-H542. [DOI] [PubMed] [Google Scholar]
- Zeng H., Sanyal S. and Mukhopadhyay D. (2001). Tyrosine residues 951 and 1059 of vascular endothelial growth factor receptor-2 (KDR) are essential for vascular permeability factor/vascular endothelial growth factor-induced endothelium migration and proliferation, respectively. J. Biol. Chem. 276, 32714-32719. 10.1074/jbc.M103130200 [DOI] [PubMed] [Google Scholar]
- Zeng H., Zhao D. and Mukhopadhyay D. (2002). KDR stimulates endothelial cell migration through heterotrimeric G protein Gq/11-mediated activation of a small GTPase RhoA. J. Biol. Chem. 277, 46791-46798. 10.1074/jbc.M206133200 [DOI] [PubMed] [Google Scholar]