

Abstract
Homozygous Mnx1 mutation causes permanent neonatal diabetes in humans, but via unknown mechanisms. Our systematic and longitudinal analysis of Mnx1 function during murine pancreas organogenesis and into the adult uncovered novel stage-specific roles for Mnx1 in endocrine lineage allocation and β-cell fate maintenance. Inactivation in the endocrine-progenitor stage shows that Mnx1 promotes β-cell while suppressing δ-cell differentiation programs, and is crucial for postnatal β-cell fate maintenance. Inactivating Mnx1 in embryonic β-cells (Mnx1Δbeta) caused β-to-δ-like cell transdifferentiation, which was delayed until postnatal stages. In the latter context, β-cells escaping Mnx1 inactivation unexpectedly upregulated Mnx1 expression and underwent an age-independent persistent proliferation. Escaper β-cells restored, but then eventually surpassed, the normal pancreatic β-cell mass, leading to islet hyperplasia in aged mice. In vitro analysis of islets isolated from Mnx1Δbeta mice showed higher insulin secretory activity and greater insulin mRNA content than in wild-type islets. Mnx1Δbeta mice also showed a much faster return to euglycemia after β-cell ablation, suggesting that the new β-cells derived from the escaper population are functional. Our findings identify Mnx1 as an important factor in β-cell differentiation and proliferation, with the potential for targeting to increase the number of endogenous β-cells for diabetes therapy.
KEY WORDS: β-cell versus δ-cell fate selection, Endocrine lineage diversification, β-cell fate maintenance, β-cell proliferation, Islet
Highlighted article: Mnx1 acts as a lineage specification factor in the mouse pancreas – promoting beta-cell fate and maintenance. It also has a role in compensatory beta-cell proliferation.
INTRODUCTION
Similar to most other organ systems, lineage determination and cell specification in the pancreas involve a highly ordered series of events, consisting of appropriately programmed extrinsic signaling pathways that regulate hierarchies of cell-intrinsic transcriptional networks. Various dynamically regulated genes encoding transcription factors have been linked to specific steps in endocrine cell subtype specification, differentiation and maturation (Pan and Wright, 2011). A comprehensive understanding of these processes could provide important advances in the current effort to develop β-cell replacement or regeneration therapies for patients with diabetes.
The homeodomain-protein-encoding gene motor neuron and pancreas homeobox 1 (Mnx1, formerly Hb9 or Hlxb9), encodes a transcription factor required for the development of multiple tissues, including pancreas. In the mouse, Mnx1 is expressed in the motor neurons, notochord, the entire dorsal endoderm and the ventral pre-pancreatic endoderm at the embryonic day 8 (E8) (Tanabe et al., 1998; Li et al., 1999). Endodermal Mnx1 expression is transient and forms a dorsal-ventral gradient at E9.5 (Sherwood et al., 2009), with expression persisting in both pancreatic buds at E10.5, and being downregulated between E10.5 (Li et al., 1999) and E12.5 (Harrison et al., 1999). In the adult mouse pancreas, Mnx1 is specifically expressed in mature β-cells (Harrison et al., 1999; Li et al., 1999).
Global inactivation of Mnx1 leads to dorsal pancreatic bud agenesis, while the ventral bud develops normally (Harrison et al., 1999; Li et al., 1999). By contrast, using Pdx1 cis-regulatory sequences to induce high-level Mnx1 mis-expression over the entire early pancreatic epithelium results in highly deficient pancreas organogenesis, and the pancreatic mesenchyme seems to adopt a stomach/intestinal mesenchymal state (Li and Edlund, 2001). Together, these studies emphasize that the early endodermal Mnx1 expression, in both timing and level, must be tightly controlled for proper dorsal pancreas specification.
In addition to its role in dorsal pancreas specification, global Mnx1 null mutants have a nearly threefold increase in δ-cells, and the remaining β-cells in the ventral pancreas are immature, with reduction or absence of β-cell maturation markers (Harrison et al., 1999; Li et al., 1999). Thus, these initial studies suggested that Mnx1 regulates β-cell differentiation and maturation. Furthermore, Mnx1 homozygous mutation was recently shown to cause permanent neonatal diabetes mellitus in humans (Bonnefond et al., 2013; Flanagan et al., 2014), suggesting a potentially conserved role of Mnx1 in β-cell function between mouse and human.
The limited number of studies on Mnx1 are mostly from over a decade ago, and, while indicating its essential nature in pancreas organogenesis, they did not focus on the endocrine progenitor or β-cell-specific requirements for this factor, or relate its activity to the more recent advances in our understanding of pancreatic endocrine-cell ontogeny and fate maintenance. Here, we report the inactivation of Mnx1 in distinct contexts using Cre driven from the endocrine-progenitor stage using transgenic gene-regulatory sequences from neurogenin-3 (Ngn3Cre), and in the β-cell itself using insulin (RIP2Cre). We performed an extensive, systematic and longitudinal analysis to characterize the resulting effects on endocrine cells from organogenesis on into adult life. We identify Mnx1 as an endocrine-precursor-stage instructor of β-cell lineage allocation, and it is crucial for maintaining the β-cell against conversion to a δ-like (somatostatin-producing) phenotype. The incomplete inactivation of Mnx1 within the insulin-producing cell pool led to the presence of escaper β-cells within islets populated with increased δ-like cell numbers. The escaper cells upregulated Mnx1 expression and displayed a large, persistent increase in proliferation lasting into aged mice. Our findings identify Mnx1 as another β-cell-programming factor that initiates and maintains β-cell-specific gene expression programs and represses alternative endocrine-lineage programs. These eminent functions in β-cell differentiation and proliferation render Mnx1 a potentially important therapeutic target, particularly in reprogramming other cell types into β-cells, or in stimulating β-cell proliferation.
RESULTS
Novel Mnx1 expression in Pax6+ endocrine precursors
Previous studies showed that early pancreatic Mnx1 expression is transient and temporally regulated (Harrison et al., 1999; Li et al., 1999), yet its expression pattern during organogenesis at that time was incompletely characterized, because it was not placed in reference to the many, more recently described, regulators of endocrine-lineage differentiation. We therefore re-examined Mnx1 expression, focusing on stages of early pancreas development between E10.5 and 14.5. Mnx1 protein was detected in essentially all cells of the dorsal and ventral pancreatic buds at E10.5, and excluded from the duodenum (red line, Fig. 1A). In E11.5 tissue, dorsal-bud Mnx1-positivity was notably heterogeneous compared with Pdx1, whereas ventral-bud expression was downregulated (Fig. 1B). In contrast to previous reports, Mnx1 was still detectable at E12.5, but was now restricted to tip domains of the pancreatic epithelium, as shown by co-labeling of Mnx1 against Ptf1a or Cpa1 (Fig. 1C). The numbers of Mnx1+Ptf1a+Cpa1+ cells, however, decreased over time to become relatively scattered among the tip epithelial domains. The distribution of these Mnx1+Ptf1a+Cpa1+ cells was similar to the distribution of tip multipotent progenitor cells (MPC) between E12.5 and E14.5, which are Ptf1a+Sox9+HNF1β+ (Pan et al., 2013). These data provide new insight by indicating that Mnx1 is not uniformly downregulated between E10.5 and 12.5, but continues to mark tip MPC upon tip-trunk compartmentalization, thus highlighting the dynamic expression of Mnx1 during early pancreas organogenesis.
Fig. 1.

Immunodetection of Mnx1 in developing and adult mouse and human pancreas. (A,B) Immunolabeling comparison of Pdx1 with Mnx1 in the embryonic mouse pancreas at E10.5 (A) and E11.5 (B). (C) Mnx1 detected in Ptf1a+ Cpa1+ tip MPC at E12.5. (D) Mnx1 is absent in Ngn3+ endocrine progenitors, but present in Pax6+ endocrine precursors (E). (F) Mnx1 can be found in insulin+ developing β-cells as early as E14.5. Arrows indicate Mnx1 expression in insulin-endocrine precursors. (G,H) Mnx1 protein was not detected in α-cells (G), δ-cells or in PP cells (H). (I) Mnx1 is restricted to β-cells in adult mouse pancreas. (J) In human fetal pancreas, Mnx1 is detected in the Sox9+ MPC population at gestational week 7 (G7w). (K,L) In G18w (K) and 5-year-old tissue (L), Mnx1 was immunodetected in β-cells and Sox9+ trunk/duct cells. Scale bars: 50 µm.
At E14.5, Mnx1 was detected in cells that co-expressed the endocrine precursor marker Pax6 (Fig. 1E). This marking of the endocrine precursor population during the secondary transition was previously not described. In addition, Mnx1 was found in insulin+ β-cells as early as E14.5 (Fig. 1F; arrows indicate Mnx1+insulin− endocrine precursors). In no case did we detect Mnx1 expression in Neurog3+ cells (Fig. 1D), glucagon+ cells (Fig. 1G), somatostatin+ or pancreatic polypeptide+ (PP, also known as Ppy – Mouse Genome Informatics) cells (Fig. 1H), suggesting that Mnx1 is not expressed in the Neurog3+ endocrine progenitors, α-, δ- or PP-cells. In the adult pancreas, the vast majority of the β-cells was Mnx1-immunopositive. Only one or two insulin+ cells per islet were apparently Mnx1−, as is typical for many immunodetection analyses of transcription factors (TFs). If they are truly Mnx1−, the significance of this minute population is unknown.
We also studied human pancreatic tissue to gain insight into the level of functional conservation. In human pancreas, Mnx1 was detected in Sox9+ cells as early as gestational week 7 (G7w, equivalent to mouse E10.5, Fig. 1J; Pan and Wright, 2011), suggesting that Mnx1 also marks early MPC in human fetal pancreas. In later-stage tissue, Mnx1 was found in insulin+ β-cells and Sox9+ trunk epithelial cells at G18w (equivalent to mouse E16.5; Fig. 1K), and was maintained in β-cells and Sox9+ ducts in juvenile pancreas samples (5 years old; Fig. 1L). These are the first observations to provide insight into similarity and distinctions in Mnx1 expression patterns between human and mouse pancreas development.
Mnx1 suppresses δ-cell fate and promotes β-cell fate during endocrine differentiation
The multiphase and cell type-specific Mnx1 patterns described above suggested several possible functions during organogenesis. To begin to determine cell type-specific Mnx1 functions, we used a floxed allele (Mnx1fl) in which the homeodomain-encoding exon 3 was flanked by LoxP sites. We confirmed that Mnx1fl could produce functional nulls by crossing to the germ-line EIIACre deleter line to derive EIIACre;Mnx1fl/fl mice (Fig. S1A). These mice phenocopied the global null phenotypes (Harrison et al., 1999; Li et al., 1999), with dorsal pancreas agenesis and normal ventral pancreas formation (Fig. S1C) compared with Mnx1fl/fl (Fig. S1B).
Our new placement of Mnx1 in Pax6+ endocrine precursors led us to determine how Mnx1 integrates into the transcriptional hierarchies controlling lineage allocation during endocrine diversification. To determine Mnx1 function in the Pax6+ endocrine precursor state, we crossed Mnx1fl to a Ngn3-Cre BAC transgenic line to generate Ngn3-Cre;Mnx1fl/fl mice (hereafter denoted Mnx1Δendo; Fig. 2A). Because Neurog3 is expressed earlier than Pax6, this model inactivated Mnx1 function in all endocrine cells, including Pax6+ precursors, as early as E9.5 (Schonhoff et al., 2004). Pups and pancreata from Ngn3-Cre, Mnx1fl/+ and Mnx1fl/fl genotypes displayed similar blood-glucose levels, gross morphology and endocrine cell proportions; thus, in all data shown herein, Mnx1Δendo will be compared with their respective age-matched Mnx1fl/fl littermates. Mnx1Δendo mutant pups developed extreme hyperglycemia already at postnatal day 2 (P2) (Fig. 2B), which probably explains their inability to survive past P10, and suggests functional deficits in or decreased numbers of β cells.
Fig. 2.
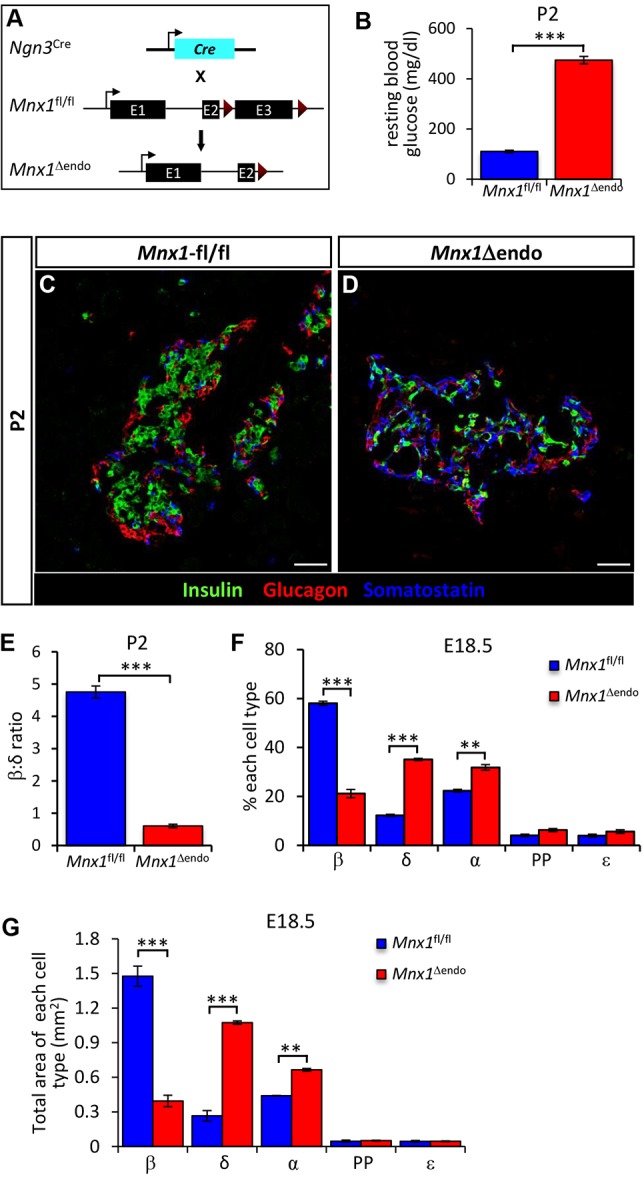
Dramatic increase in δ-cell numbers and significant decrease in β-cell numbers in Mnx1Δendo. (A) Schematic of conditional Mnx1 deletion in endocrine progenitors with Ngn3-Cre. (B) Random blood-glucose measurement showed that Mnx1Δendo mutant pups were hyperglycemic at P2 (n=10). (C,D) Immunolabeling with insulin, glucagon and somatostatin showed a dramatic increase in δ-cell number, concomitant with a decrease β-cell number at P2. (E) Significant decrease in β:δ cell ratio in Mnx1Δendo mutants at P2 (n=4). (F,G) Quantification of endocrine cell types fraction (F) and area (G) at E18.5, indicating significant increase in δ-cells at the expense of β-cells in Mnx1Δendo pancreas (n=4). Scale bars: 50 µm. Data shown as mean±s.e.m. *P<0.05, **P<0.005, ***P<0.001.
To uncover potential causes of the hyperglycemia in P2-P5 Mnx1Δendo mutants, we analyzed the islet hormone-expressing cell types using immunofluorescence. The Mnx1Δendo mutant pancreas at P2 displayed a significant decrease in insulin+ β-cell numbers, and a concomitant, striking increase in somatostatin+ cells compared with control Mnx1fl/fl (Fig. 2C,D; quantified in Fig. 2E). Analysis of endocrine-cell numbers at prenatal stages (E18.5) revealed a threefold decrease in the insulin+ β-cell population (∼58% in control, ∼21% in mutant), accompanied by a threefold increase in somatostatin+ cells (∼12% in control, ∼35% in mutant; Fig. 2F,G). These late-gestation tissues also showed a slight, but significant, increase in glucagon+ α-cells (∼22% in control, ∼31% in mutant), whereas PP and ε-cell numbers had not changed substantially (Fig. 2F).
Because previous studies suggested that the remaining β-cells in the ventral pancreas of Mnx1 global-null mice were immature – being Glut2− and Nkx6.1−, and Pdx1LO (Harrison et al., 1999; Li et al., 1999) – we further evaluated the remaining 20% Mnx1-deleted β-cells (Fig. S2H,I) in Mnx1Δendo tissue with respect to several informative β-cell-specific TFs. In contrast to previous findings in the global null, the nuclear Pdx1 and Nkx6.1 signal in Mnx1Δendo mutant β-cells at P2 was equivalent to control β-cells (Fig. S2A-D). Because MafA nuclear localization is usually well-established by P4, and is indicative of mature β-cell status (Guo et al., 2013), we tested for its presence and subcellular localization in mutant β-cells. MafA was cytoplasmic in the remaining insulin+somatostatin− cells, unlike the nuclear localization in control β-cells (Fig. S2E,F); thus, we concluded that the remaining insulin+ β-like cells in the Mnx1Δendo mutants were immature and probably poorly functional. The early-lethality phenotype precluded testing for the degree of maturity at a physiological level, as even normal β-cells are not fully mature at this age. Taken together, we conclude that Mnx1 promotes β-cell fate and suppresses δ-cell fate, with this allocation occurring in the Pax6+ endocrine-precursor stage.
Mnx1 loss in Pax6+ endocrine precursors leads to aberrant lineage allocation and postnatal β-to-δ cell transdifferentiation
The dramatic decrease in the β-to-δ cell proportion seen in Mnx1Δendo pancreatic tissue could result from either aberrant lineage allocation in the Pax6+ endocrine-precursor pool, or from β-to-δ cell transdifferentiation. To distinguish between these two possibilities, we examined β- and δ-cell numbers during relatively late stages of the secondary transition, when there is a large wave of β- and δ-cell commitment. We observed a dramatic decrease in β-cell number as early as E16.5 in Mnx1Δendo mutant pancreas (Fig. 3B). Conversely, at the same stage, both populations of HhexHIsomatostatin− cells (likely δ-cell precursors) and HhexHI somatostatin+ (δ- or δ-like) cells were markedly increased over control (Fig. 3A-D. Note: duct cells were HhexLO). We rarely found somatostatin+insulin+ double-positive cells at E16.5. We extended this analysis to P2 to determine if the HhexHIsomatostatin− δ-cell precursor number continued to increase over the perinatal period. Surprisingly, the number of HhexHIsomatostatin− δ-cell precursors was similar to the control at this stage, but the number of HhexHIsomatostatin+ δ cells was greatly increased, suggesting that a substantial proportion of the earlier HhexHIsomatostatin− δ-cell precursors (observed at E16.5) had differentiated into HhexHIsomatostatin+ cells (Fig. 3E,F). In addition, we also observed a population of cells that were insulin+Hhex+somatostatin+, suggestive of β-to-δ cell transdifferentiation. (Validating this potential transdifferentiation by lineage tracing would require dual-level lineage tracing, because endocrine cells would all be EYFP-labeled in the Mnx1Δendo;ROSA26REYFP pancreata.) These analyses suggest that the increased δ/δ-like cell number observed in Mnx1Δendo mutants occurred by both lineage reallocation from the Pax6+ endocrine precursors and by transdifferentiation of Mnx1− β-like cells into δ-like cells (Fig. 3G); these mechanisms are not mutually exclusive.
Fig. 3.

Embryonic endocrine lineage-allocation defect, followed by postnatal β-to-δ transdifferentiation as two major routes to increased δ-cell numbers. (A,B) Representative sections from E16.5 pancreata with immunodetection of Hhex, insulin and somatostatin illustrate the increase in Hhex+ somatostatin− δ-cell precursors in Mnx1Δendo mutants. (C,D) Quantitative analysis of E16.5 Mnx1fl/fl and Mnx1Δendo pancreata indicate a dramatic increase in total number Hhex+somatostatin− δ-cell precursors (C) and total Hhex+somatostatin+ δ-cell numbers (D) in Mnx1Δendo mutant. (E,F) Immunofluorescence analysis of Hhex, insulin and somatostatin on P2 pancreas demonstrates the presence of Hhex+insulin+somatostatin+ cells (white arrows) in Mnx1Δendo pancreas tissue, an indirect indicator of β-to-δ transdifferentiation. (G) Model showing early stage endocrine lineage-allocation defects and late-stage β-to-δ transdifferentiation when Mnx1 is inactivated in endocrine progenitors. (H-J) Quantification indicates that total endocrine area was reduced in Mnx1Δendo mutant (H), but total Pax6+ endocrine precursors were increased in Mnx1Δendo mutant at E18.5 (I), indicating failure of a subset of Pax6+ precursors to differentiate towards hormone-producing endocrine cells in the absence of Mnx1 (J). Scale bars: 50 µm. Data are shown as mean±s.e.m. **P<0.005, ***P<0.001.
We also noted an overall decrease in total hormone+ endocrine-cell area (Fig. 3H), although the total acinar area was not changed significantly in Mnx1Δendo mutants at E18.5 (Fig. S2G). Consistent with this observation, this stage also showed an overaccumulation of Pax6+ hormone-negative endocrine precursors (Fig. 3I). Because Ngn3+ cell numbers were unaffected (data not shown), this evidence suggests a significant block in the differentiation program of Mnx1-deficient endocrine-precursor cells (Fig. 3J).
Mnx1 is required for β-cell fate maintenance
The expression of Mnx1 in β-cells long after their specification suggests a continued important role in β-cell differentiation and function. To address this issue, we used RIP2-Cre transgenic mice to derive RIP2-Cre;Mnx1fl/fl mice (denoted as Mnx1Δbeta hereafter; Fig. 4A). The Mnx1Δbeta newborn pups had slightly, but noticeably, higher glycemia, persisting through 1 month of age, but returning to normal as mice aged (Fig. S3A-D). To explore the potential defects in β-cell differentiation or function in Mnx1Δbeta mutants, we tested for hormone-producing cell types in Mnx1Δbeta and control mice. Our analysis began with 4-month-old mice, in which the Mnx1Δbeta mutant islets had substantially increased δ-cell numbers (Fig. 4B-D). Glucagon+ α-cells were also slightly increased, but far less than the δ-cells. Although the proportion of β-cells was slightly reduced compared with Mnx1fl/fl control tissue, the number of β-cells quantified by β-cell area on extensive sectional analysis was similar to controls (Fig. 4B-D; Fig. S4D); the issue of β-cell repopulation from escaper β-cells is discussed below. PP and ε-cell numbers were also similar between mutant and controls (data not shown).
Fig. 4.

β-to-δ cell transdifferentiation in Mnx1Δbeta mutants. (A) Schematic showing β-cell-specific Mnx1 deletion using RIP2-Cre transgenics. (B,C) Immunolabeling with insulin, somatostatin and glucagon showed an increase in δ- and α-cells, as well as larger islet size in the Mnx1Δbeta islets. (D) Quantitative analysis of β-, δ- and α-cell fraction indicate an increased in δ- and α-cell populations concomitant with decreased in percentage of β-cells (n=3). (E,F) Lineage-tracing analysis using ROSA26REYFP reporter show co-expression of EYFP and somatostatin, indicating that a subset of δ-cells in the Mnx1Δbeta mutant are derived from β-cells. (G,H) β-to-δ transdifferentiation, as indicated by EYFP+somatostatin+insulin+ cells (arrows), can be detected as early as P5. (I,J) Measurement of islet somatostatin secretion (I) and total somatostatin content (J) in vitro indicate that mutant δ-cells hypersecrete somatostatin and actively synthesize more somatostatin compared with control islets (n=3). Somatostatin secretion was normalized to islet number. Scale bars: 50 µm. Data are shown as mean±s.e.m. **P<0.005, ***P<0.001, ****P<0.0001.
The gain in δ- and α-cell populations in Mnx1Δbeta mutants could have arisen by de novo neogenesis, or by transdifferentiation of Mnx1-inactivated β-cells. We generated Mnx1Δbeta;ROSA26REYFP mice in order to use lineage tracing to dissect the sequence of events leading up to the Mnx1Δbeta phenotype described above. At 2 months of age, EYFP labeling in RIP2-Cre;ROSA26REYFP control islets was restricted to β-cells, with none in somatostatin+ cells, whereas in Mnx1Δbeta islets, ∼56% of EYFP+ cells were somatostatin+ (δ-like) cells (Fig. 4E,F; Fig. S4E), thus derived from β-cells that lost Mnx1. A small proportion of glucagon+ α-cells were also EYFP+ (∼12% of total EYFP+ cells), indicative of their β-cell origin. We conclude that Mnx1 is required for β-cell fate maintenance, and that in its absence, β-cells lose their identity and transdifferentiate, predominantly into δ-cells and, to a lesser extent, into α-cells.
In earlier-stage analysis (E18.5), Mnx1 protein was diminished in all Cre-reporting β-cells of Mnx1Δbeta mice (EYFP+Mnx1− cells; Fig. S5C,D). Although the Cre activity from RIP2-Cre begins in β-cells formed as early as E13.5 (Gannon et al., 2000), we did not observe any β-to-δ or β-to-α transdifferentiation before birth (Fig. S5A,B). The earliest detection of such transdifferentiation was at P5, when a minor number of EYFP+somatostatin+ cells were detected (representative islet shown in Fig. 4H), as well as insulin+Hhex+ or insulin+Hhex+somatostatin+ transitional cell states (Fig. S5E,F). The proportional representation of insulin+ cells was similar to control at P5. Consistent with our previous findings in the context of forced TF expression within the embryonic endocrine lineages (Yang et al., 2011), our observations suggest that Mnx1-deficient β-cells receive some type of postnatal stimulus to allow their conversion toward other endocrine-hormone-expressing states.
The massively increased δ/δ-like cell number in Mnx1Δbeta mutants could result in overproduction of somatostatin, which is a well-known inhibitor of insulin secretion (Alberti et al., 1973). There are limited mature δ-cell markers, and the β-cell-derived δ-like cells all expressed Hhex (Zhang et al., 2014) and somatostatin (Fig. S5F). Quantitative analysis showed a 3.5-fold greater representation of δ-cells over controls, but in vitro assays on isolated islets showed that somatostatin content and secretion from mutant islets were >10-fold higher than controls, suggesting overproduction and hypersecretion of somatostatin (Fig. 4I,J). The somatostatin secretion in mutant islets was not regulated by glucose, unlike in controls, but was substantially potentiated by the cAMP analog 3-isobutyl-1-methylxanthine (IBMX) (Fig. 4I). Thus, the δ/δ-like cells in mutant islets are functionally distinct from wild-type δ-cells.
β-cells escaping Mnx1 inactivation in Mnx1Δbeta mutants repopulate the islet
We found evidence that incomplete Mnx1 inactivation across the β-cell population was a result of lack of Cre production in 15-20% of the β-cells in RIP2-Cre islets, which might be caused by the RIP2-Cre transgene being packaged in such a way that it cannot be activated, leading to the variegated expression (Fig. S6). This represents an important finding, because the lack of Cre production was not caused by an inability in some cells to express both insulin genes, as Ins1 and Ins2 mRNA were highly upregulated in these Cre− β-cells (Fig. 6C; Fig. S8C; see below). Such incomplete recombination was followed by subsequent islet repopulation by β-cells that did not undergo Mnx1 inactivation – hereafter ‘escaper β-cells’. At P15, insulin+somatostatin+ cells were much more pervasive, although the [insulin+somatostatin+ plus insulin+] pool was still approximately equivalent to the control insulin+ pool (data not shown). At 4 months of age, this double-positivity was vastly resolved to hormone monopositivity; that is, insulin+ or somatostatin+ cells (Fig. 4B,C and Fig. 5G-N). The number of insulin+ β-cells was very similar to that in controls in 4-month-old Mnx1Δbeta pancreas (Fig. S4D), and the vast majority were Cre− and EYFP−, suggesting derivation from escaper β-cells (Fig. 4E,F and Fig. 5A,B; Fig. S6A-C). RIP2-Cre functions in ∼85% of β-cells, as judged by our Cre immunolabeling (Fig. S6A) and by previously reported ROSA26-based β-galactosidase production after ROSA26R activation (Gannon et al., 2000), which agrees with our direct estimation of the efficiency of RIP2-Cre-inactivation of Mnx1 in Mnx1Δbeta mice, using Mnx1 immunodetection. At P5, around 85% of β-cells were Mnx1− and ∼15% were Mnx1+ (Fig. 5C,D; Fig. S5C,D); these data also indicate efficient deletion of Mnx1. At 2 and 4 months of age, almost all β-cells in Mnx1Δbeta mice were Cre− and Mnx1+ (Fig. 5E,F; Fig. S6C), and the β-cell proportional representation in islets was reproducibly similar to controls, suggesting that the initial 15% escaper β-cells had repopulated the islets (more directly addressed in the following section below). We propose that in the Mnx1Δbeta pancreas, β-cells gradually transdifferentiate into δ- (and α-) cells after birth, and escaper β-cells expand in number to repopulate the islets. The elevated ad-lib-feeding glycemia in mice between birth and 1 month of age occurs over the active β-to-δ transdifferentiation phase, wherein there are insufficient escaper β-cells to maintain euglycemia. As escaper β-cells repopulated the islets, ad-lib-feeding blood glucose improved with age (Fig. S3A-D).
Fig. 6.

Escaper β-cells secrete and produce more insulin, and continue to proliferate in Mnx1Δbeta aged mice via downregulation of menin. (A,B) Measurement of insulin secretion from isolated islets (A) and plasma insulin levels (B) indicate that Mnx1Δbeta β-cells secrete more insulin (n=3). Insulin secretion was normalized to islet number. (C,D) qRT-PCR analysis indicates significantly increased mRNA expression for Ins2 (C) and Mnx1 (D) in Mnx1Δbeta β-cells (n=4). (E-G) Mnx1Δbeta β-cells showed increased proliferation as determined by Ki67 at 1 month (n=4; E), 4 months (n=4; F) and 14 months (n=4; G). (H,I) qRT-PCR analysis show decreased expression of cell-cycle inhibitors Cdkn2a, Bmi1 and Cdkn1a (H), and increased expression of cell-cycle-positive regulators Cdk6 and Ccnd1 (I). (J) qRT-PCR analysis on 4-month-old islets RNA shows that menin mRNA expression was also downregulated in the Mnx1Δbeta islets. (K,L) Immunolabeling of menin showed that menin protein was significantly reduced in Mnx1Δbeta β-cells at 14 months. Menin protein level was similar in Mnx1fl/fl and RIP2-Cre β-cells, indicating no effect on Menin from the transgenic Cre driver. Scale bars: 50 µm. Data are shown as mean±s.e.m. *P<0.05, **P<0.005, ***P<0.001, ****P<0.0001.
Fig. 5.

β-cells escaping Mnx1 deletion in Mnx1Δbeta repopulate the islets. (A,B) Lineage-tracing analysis using the ROSA26REYFP reporter shows that the majority of Mnx1Δbeta β-cells at 2 months of age are escaper β-cells, as they are EYFP− compared with RIP2-Cre;ROSA26REYFP. (C,D) Immunolabeling with Mnx1, insulin and somatostatin show that the majority of the Mnx1Δbeta insulin+ β-cells are devoid of Mnx1 at P5. (E,F) Most of the Mnx1Δbeta insulin+ β-cells are Mnx1+. (G-N) The escaper β-cells in Mnx1Δbeta islets are Glut2+ (G,H), Pdx1+ (I,J), Nkx6.1+ (K,L) and MafA+ (M,N). (O,P) Intraperitoneal glucose tolerance tests indicate that Mnx1Δbeta mice are glucose intolerant at 4 months (O), but glucose clearance improved by 6 months of age (P). Scale bars: 50 µm. Data are shown as mean±s.e.m. *P<0.05, **P<0.005, ***P<0.001.
The putative escaper cells produced normal, mature β-cell-specific markers, such as Glut2 (Fig. 5G,H), Pdx1 (Fig. 5I,J), Nkx6.1 (Fig. 5K,L) and MafA (Fig. 5M,N) compared with 4-month-old controls. The 4-month-old Mnx1Δbeta mice did show abnormal glucose clearance by intraperitoneal glucose tolerance testing (IPGTT; Fig. 5O), but this improved by 6 months of age (Fig. 5P). The glucose intolerance was not a result of peripheral insulin resistance, because there was a normal insulin-tolerance response (Fig. S7A). We reasoned that the increase in δ-cell number and somatostatin secretion in Mnx1Δbeta islets caused impaired insulin secretion and the glucose-intolerance phenotype. We tested this idea by insulin secretion assays on in vitro-cultured islets from 6-month-old mice. We chose this stage to assure the completion of all β-to-δ transdifferentiation, and to allow sufficient time for the functional maturation of the repopulating escaper β-cells. Our data demonstrated that even in the presence of high somatostatin levels (Fig. 4I), insulin secretion was not blocked. Instead, more insulin was being produced in the mutant islets, and then secreted in response to glucose or glucose + IBMX, as shown by insulin secretion assay (Fig. 6A) and total plasma insulin levels in vivo (Fig. 6B). Consistent with these results, Ins1 and Ins2 mRNAs were highly upregulated in mutant islets (Fig. 6C; Fig. S8C), possibly as a result of increased Mnx1 mRNA and protein levels (Fig. 6D; Fig. S8A,B), because Mnx1 is a potent regulator of insulin transcription and secretion (Shi et al., 2013). Altogether, escaper β-cells in the Mnx1Δbeta islets were mature, they produced and secreted more insulin, and became insensitive to the inhibitory effect of somatostatin on insulin secretion.
Continued proliferation of escaper β-cells and islet hyperplasia in aged Mnx1Δbeta mice
During the course of analysis, we realized that average islet size seemed slightly larger in Mnx1Δbeta mutants at 4 months (Fig. 5H,J,L,N) and 6 months of age (data not shown), suggesting possible increased β-cell replication, transdifferentiation of β-cells from other endocrine lineages, or β-cell neogenesis. The latter two possibilities would need to be determined by a currently unavailable two-tier lineage tracing strategy, presumably including a non-Cre method. Nevertheless, our detection of ∼twofold increase in β-cell proliferation at 1 and 4 months of age (Fig. 6E,F) was consistent with the time when active β-to-δ transdifferentiation occurred, and the Cre− escaper β-cells had started to repopulate the islet (Fig. S6). Interestingly, the absence of Cre (the RIP2-Cre remained inactive) and the increased proliferation of escaper β-cells in Mnx1Δbeta mutants were persistent, with ∼5-fold increase in Ki67+ β-cells in 14-month-old mutant islets (Fig. 6G; Fig. S6D-F). These data were supported by qRT-PCR analysis showing decreased expression of several cell-cycle inhibitors (Cdkn2a, Bmi1, Cdkn1a), whereas positive cell-cycle regulators (Cdk6 and Ccnd1) were markedly increased (Fig. 6H,I). In addition, menin, a β-cell proliferation inhibitor and tumor suppressor, was downregulated at both mRNA (Fig. 6J) and protein levels (Fig. 6K,L) in escaper β-cells in Mnx1Δbeta mice as early as 4 months of age, and its decreased expression was maintained in 14-month-old mice. This finding is consistent with a recent report that menin interacts with and inhibits Mnx1 in regulating MIN6 cell line proliferation (Shi et al., 2013). The increased β-cell proliferation in Mnx1Δbeta mice ultimately led to islet hyperplasia (Fig. 7E,F), and doubling of endocrine and β-cell numbers at 14 months (Fig. 7A,B). The observed differential in proliferation index is sufficient to cover the increased β-cell number occurring over the prolonged period from 4 to 14 months in Mnx1Δbeta islets. The differential in β-cell proliferation could have been even greater at the untested intermediate time points. Both δ- and α-cell numbers were also increased because of the β-cell transdifferentiation (Fig. 7C,D). Considering that only ∼15% of β-cells retained Mnx1-positivity at P5 in Mnx1Δbeta mutants, there was a remarkable ∼13-fold increase in β-cell number (Mnx1+insulin+) between P5 and 14 months of age compared with controls. Even with the doubled β-cell number, the fraction of β-cells within islets remained similar to control because of the increase in other cell types (Fig. S8C). Thus, persistent replication of escaper β-cells, likely involving downregulation of menin and bypassing the progressive inhibition of β-cell proliferation in normally aging mice, eventually led to islet hyperplasia (Fig. 7H).
Fig. 7.

Sustained proliferation of escaper β-cells leads to islet hyperplasia in Mnx1Δbeta aged mice, and alleviates hyperglycemia following STZ treatment. (A-D) Quantification of total area of hormone+ (A), insulin+ (B), somatostatin+ (C) and glucagon+ (D) area demonstrate a twofold increase in total endocrine, insulin+ and glucagon+ area, in addition to the approximately fourfold increase in somatostatin+ area. (E,F) Immunolabeling with insulin shows the presence of hyperplastic islet in Mnx1Δbeta pancreata. (G) Measurement of resting blood glucose over 6-month period upon streptozotocin (STZ) treatment showed the return of blood glucose to normal levels 2 months post-STZ treatment compared with Mnx1fl/fl mice. (H) Summary of processes occurring in Mnx1Δbeta mutant. (I) Model showing novel Mnx1 function in promoting β-cell fate and suppressing δ-cell fate in the Pax6+ endocrine precursors. Mnx1 is continuously required to maintain β-cell fate in the developing β-cells. When Mnx1− β-cells transdifferentiate into δ-cells, a subset of Mnx1+ β-cells start to repopulate islets by proliferation, leading to islet hyperplasia in aged mice. Scale bars: 100 µm. Data are shown as mean±s.e.m. *P<0.05, **P<0.005.
Escaper β-cells alleviate hyperglycemia upon STZ treatment at 6 months of age
We reasoned that, if the cell progeny from escaper β-cells were functionally mature and maintained such proliferative capability in older mice, they might also be capable of repopulating the islet and restoring euglycemia in the experimental setting of ablating most pre-existing escaper β-cells in aged mice. We killed β-cells with two high doses of streptozotocin (STZ) in 6-month-old control and mutant mice and monitored blood glucose over six months. As in our previous study (Pan et al., 2013), mice with 70-80% STZ-mediated β-cell ablation often exhibited hyperglycemia (of >350 mg/dl) within one week post-injection. STZ-injected control mice remained hyperglycemic for up to 24 weeks post-STZ treatment. By contrast, Mnx1Δbeta mice returned to euglycemia within 8 weeks post-STZ (Fig. 7G). These data indicate that functional escaper β-cells in aged mice were more capable than controls in repopulating the islets and alleviating hyperglycemia.
DISCUSSION
Here, we present evidence on the differential function of Mnx1 across several development stages in different pancreatic islet cell types. Apart from its initial function in dorsal pancreas specification, Mnx1 appears to be a lineage-allocation factor, regulating cell-fate choice in Pax6+ endocrine precursors to become β-cells instead of δ-cells. As development progresses, Mnx1 has an essential role in β-cell fate maintenance. We also discovered a previously unanticipated, non-autonomous role of Mnx1 in β-cell proliferation. These unexpected results not only allow us to begin to elucidate the molecular mechanisms regulating β-cell proliferation via the Mnx1-menin route, but also highlight the potential implication of δ-cells and intra-islet communication in β-cell proliferation.
Mnx1 as a β-cell lineage-allocation factor
Loss of Mnx1 function in endocrine progenitors resulted in mutant pups with hyperglycemia at early postnatal stages. This phenotype is similar to the recently reported human homozygous Mnx1 mutation that resulted in permanent neonatal diabetes, suggesting a highly conserved Mnx1 function between mouse and human. The presence of normal exocrine mass in this human with Mnx1 mutation strongly suggested that the dorsal pancreas still formed, and that the Mnx1 requirement was at either the endocrine-precursor stage or in the Sox9+ bipotent progenitor pool, showing an equivalent role in β-cell specification in human and mouse (Bonnefond et al., 2013). It is noteworthy that the persistent Mnx1 expression we found in human fetal and adult duct cells implies a possible function in maintaining the ductal lineage. This hypothesis awaits future testing, probably in vitro.
Our data demonstrated a marked increase in δ-cell numbers concomitant with reduced β-cell numbers in Mnx1Δendo mutants that was caused by defective early-lineage allocation in the endocrine precursors and postnatal β-to-δ cell transdifferentiation. These data extend the findings of Li et al. (1999) in the global Mnx1 null mutants, and show that Mnx1 is required in the endocrine precursor, but not MPC population for β-cell lineage allocation and differentiation. Our results are in contrast, however, to findings in zebrafish, in which Mnx1 is required to suppress α-cell fate while promoting β-cell fate, with the most important difference being that δ-cell numbers were distinctly unaffected in morphants (Dalgin et al., 2011). This discrepancy is probably due to a slight difference in gene regulatory modules controlling endocrine-cell differentiation between mouse and zebrafish. Nevertheless, in both species, Mnx1 is a novel lineage-allocation factor for β-cells.
The dramatic increase in δ-cell numbers in the Mnx1Δendo mutants suggests that Mnx1 is required to suppress the δ-cell program in endocrine precursors (Fig. 7I). Recent studies from Zhang et al. (2014) demonstrated that loss of Hhex in endocrine progenitors and adult δ-cells led to a complete loss of δ-cells, indicating that Hhex is a principal determinant of δ-cell fate during endocrine lineage allocation and postnatal δ-cell fate maintenance. Prior studies in Drosophila and mouse reported that Mnx1, via its tinman repressor domain, acts predominantly as a repressor in neuron subtype determination (Broihier and Skeath, 2002; William et al., 2003; Lacin et al., 2014). These studies, together with our observation of a substantial increase in Hhex+ δ-cell precursor numbers in the Mnx1Δendo mutants, lead us to hypothesize that Mnx1 might also act as a repressor in the pancreas, suppressing δ-cell programming through direct or indirect inhibition of Hhex expression in endocrine precursors and β-cells. In addition, the presence of potential Hhex binding sites in Mnx1 regulatory regions (Klaus Kaestner, personal communication) further suggests that cross-repressive interactions establish and maintain their mutually exclusive expression in the β- versus δ-cell. A similar mutual inhibition of TFs in endocrine cell fate determination was previously reported for Pax4 and Arx in β- versus α-cell differentiation (Collombat et al., 2003).
A remarkable increase in δ-cell numbers at the expense of β-cells, which occurs in the Mnx1Δendo mutant, was also observed in Pax4−/−Arx−/− and Arx−/−Nkx2.2−/− double-null mutants (Collombat et al., 2005; Kordowich et al., 2011; Mastracci et al., 2011), suggesting possible genetic interaction between Mnx1, Pax4, Arx and Nkx2.2. In the Pax4−/− pancreas, Mnx1 expression is undetectable in β-cells (Wang et al., 2004), indicating that Mnx1 might act downstream of Pax4 in promoting β-cell fate. Furthermore, we observed a slight increase in α-cell numbers in the Mnx1Δendo mutants, suggesting that Mnx1 also functions to suppress Arx and prevent α-cell program activation in endocrine progenitors or precursors (Fig. 7I). Future valuable insight could be gleaned from elucidating the epistatic relationship of Mnx1 with these TF genes, and how it fits into the transcriptional hierarchy controlling endocrine lineage diversification.
Mnx1 in β-cell fate maintenance
Inactivation of Mnx1 in the developing β-cells leads to postnatal β-to-δ-cell, and, to a lesser extent, β-to-α-cell, transdifferentiation, indicating that Mnx1 is required continuously to maintain β-cell fate after specification. The activation of Hhex and Arx (not shown) in β-cells upon Mnx1 deletion suggests that Mnx1 uses a repressive mechanism similar to those described above in endocrine precursors to suppress δ- and α-cell programs in β-cells. Future identification of the types of co-factors that form the Mnx1 repressor complex will provide insights on the epigenetic regulation and molecular mechanisms of how Mnx1 maintains β-cell fate. Likewise, inactivating Mnx1 in mature β-cells with inducible systems will help addressing Mnx1 function in this population in the future.
We noticed that the Mnx1Δbeta mice still had impaired IPGTT at 4 months old, although, by this age, β-cell mass was already restored to normal. In addition, the defect in glucose clearance in this mutant was not a result of peripheral insulin resistance (Fig. S7A). Thus, the newly formed escaper β-cells might not be fully functional at this stage, a suggestion consistent with the finding of lower MafA levels in an appreciable number of these escaper β-cells. An alternative explanation is that the newly formed escaper β-cells within the islet (at 4 months) are relatively sensitive to the somatostatin-based inhibition of insulin secretion but, with aging, develop increased resistance to the inhibitory effect after long-term adaptation to high somatostatin levels. Nonetheless, these escaper β-cells apparently became fully functional by 6 months of age, as indicated by in vitro insulin secretory assay, and based on their ability to restore euglycemia after STZ-induced β-cell ablation in aged mice.
Unexpected β-cell compensatory growth in Mnx1Δbeta: β-cell proliferation-promoting signals
One surprising finding was the compensatory islet repopulation and eventual β-cell hyperplasia, which we propose arose from the Cre− β-cells escaping Mnx1 inactivation. We do not think that these large numbers of insulin-producing cells arose by transdifferentiation from an alternate minor endocrine lineage, namely the δ-cell, such as reported by Chera et al. (2014). There are differences between our study and theirs. Postnatal δ-to-β transdifferentiation observed by Chera et al. occurred under extreme (95-95%) β-cell diphtheria toxin-based ablation. The presence of substantial numbers of functional β-cells in the Mnx1Δbeta mice (15% escaper β-cells plus 85% of still-transdifferentiating insulin+somatostatin+ β-cells) should be insufficient to generate the signals that stimulate δ-to-β transdifferentiation. All other β-cell-directed Cre tools (Pdx1-CreER, MIP-CreER, RIP-CreER) have at best 80-90% recombination efficiency and therefore also create a situation in which the β-cell pool could include cells derived from, for example, wild-type δ-cells. Detecting such a potential contribution would require a currently unavailable second level of non-Cre-based δ-cell-specific lineage tracing. Conversely, strong evidence in favor of escaper β-cells being the primary repopulating pool is the doubling of β-cell proliferation observed when β-to-δ cell transdifferentiation was occurring at 1 month, and at 4 months, when the process was slowing down. This β-cell proliferation was even more pronounced (four- to fivefold) at 14 months in association with the β-cell hyperplasia in aged Mnx1Δbeta mice.
Similar β-cell compensatory growth occurs in mice double-heterozygous for insulin receptor and insulin receptor substrate 1 (IR+/−IRS1+/−), and in liver-specific insulin-receptor knockout (LIRKO) mice (Michael et al., 2000; Kido et al., 2000). Whereas IR+/−IRS1+/− and LIRKO mice develop hyperglycemia and peripheral insulin resistance at 6 months, the Mnx1Δbeta mutants remained euglycemic and only developed mild insulin resistance and glucose intolerance at 20 months of age (Fig. S7B,C). Thus, Mnx1Δbeta mutants serve as a new model to study the mechanisms of compensatory and persistent β-cell proliferation without hyperglycemia and insulin resistance.
It will take much more work to identify the exact signal(s) that stimulated β-cell expansion in the Mnx1Δbeta mutants, which comprises compensatory growth to restore the β-cell mass towards normal over the first 16 weeks of life, and later-stage islet hyperplasia associated with persistent β-cell proliferation (Fig. 7H). It is possible that the large numbers of δ-cells derived from β-cell transdifferentiation, and which seem substantially different from normal δ-cells, based on their hypersecretory state, produce novel signals to stimulate β-cell proliferation by acting locally (intra-islet) or systemically. Future co-transplantation of mutant δ-cells together with wild-type islets under the kidney capsule could be one way of addressing this possibility.
Previous studies also suggest several other possible influences. The hyperglycemic state in P5- to 1-month-old Mnx1Δbeta mutant mice might stimulate an initial compensatory phase of β-cell proliferation, because elevated glucose levels trigger modest β-cell replication in young mice (Salpeter et al., 2010, 2011). Nevertheless, as glycemia in Mnx1Δbeta mice had reproducibly returned to normal by 2 months of age, we presume that the longer-term sustenance of escaper β-cell proliferation arises from different stimuli.
The lower level of menin (an inhibitor of β-cell proliferation; Karnik et al., 2007) could directly or indirectly contribute to the persistent β-cell proliferation of escaper β-cells in older Mnx1Δbeta mice. Aged Men1+/− mice have hyperplastic islets and develop insulinomas (Shi et al., 2013; Desai et al., 2014). Menin interacts physically with Mnx1 to inhibit β-cell proliferation, and menin downregulation in MIN6 or human insulinoma cells causes Mnx1 accumulation, with the stabilized phospho-Mnx1 having anti-apoptotic characteristics and positively regulating genes that modulate insulin levels (Shi et al., 2013; Desai et al., 2014). Thus, menin downregulation probably increases Mnx1 mRNA and protein levels, and the increased Mnx1 then feeds forward to increase insulin expression, as well as inducing cell-cycle positive-acting factors and reducing cell-cycle-inhibitor expression. It will be valuable in the future to elucidate better which signal(s) downregulate(s) menin expression and linkages with how Mnx1 regulates β-cell proliferation. Prior findings showed that insulin signaling via the Foxo1/Pdx1/insulin pathway is a predominant influence in compensatory β-cell growth in LIRKO mice (Okada et al., 2007). Therefore, the elevated insulin production and secretion in Mnx1Δbeta mutant escaper β-cells could act in an autocrine or paracrine manner to increase β-cell proliferation. Together, the combined menin downregulation, elevated Mnx1 and increased insulin production might all contribute to causing the persistent proliferation of escaper β-cells. Understanding how these factors interact to drive the cell-cycle forward and bypass the age-inhibitory effect on β-cell proliferation could help to discover ways to restore endogenous β-cells or β-cell function in diabetic patients.
MATERIALS AND METHODS
Mice
The Mnx1 floxed allele was described in Harrison et al. (1999). Ngn3-Cre (Schonhoff et al., 2004), RIP2-Cre (Gannon et al., 2000), EIIaCre (Lakso et al., 1996) and ROSA26REYFP (Srinivas et al., 2001) mice were described previously. Animals and embryos were PCR-genotyped. All experiments were under protocols approved by Vanderbilt University IACUC.
Tissue preparation and immunostaining
Embryonic or adult mouse pancreas and human fetal pancreas were fixed (2-4 h, 4% paraformaldehyde, 4°C), and, for cryosections, were washed twice in cold PBS, sucrose-equilibrated (30%, 4°C, overnight) and OCT-embedded (Tissue-Tek, Sakura). Immunofluorescence staining was performed on 10-µm cryosections (Kawaguchi et al., 2002). Antibodies used are shown in Table S1.
Data collection, morphometric and statistical analysis
Images from Zeiss confocal (LSM 510 META upright) or Apotome were analyzed with LSM Image Browser and Zeiss Axiovision 4.8 software, respectively. As there was no difference in cell size between endocrine cell types in control and mutant, measurements of endocrine cell area were performed based on whole-section imaging with scanning (ScanScope FL; Aperio Technologies) followed by systematic Quantification analysis performed by the NIH ImageJ software. For endocrine cell number and area at E16.5 and E18.5, every sixth section (at least 60 μm separated; ∼20 sections per tissue) was counted. For adult pancreas, every twentieth section (at least 200 μm apart; ∼15-20 sections per tissue) was counted. β-cell proliferation was assessed by either Ki67 labeling (6000-8000 β-cells/tissue analyzed). For all quantitative analyses, n≥3 mice were used per group. Comparisons between two groups were carried out by Student's t-test. Data are mean±s.e.m. Statistical significance was assigned when P<0.05.
RNA extraction and qRT-PCR
RNA isolation (Trizol, Invitrogen), DNase treatment (Ambion), cDNA synthesis and qPCR (SYBR Green, Bio-Rad) were performed using GAPDH as housekeeping gene and the primer-probe sets shown in Table S2. Three to four samples per genotype per stage were collected, and qPCR was performed at least twice on each sample to determine ΔCT. Results were subjected to Student's t-test to determine significance (P<0.05).
Glucose tolerance testing, islet insulin and somatostatin secretion
Intraperitoneal glucose-tolerance testing (2.0 g glucose injected per kg body weight) was performed after a 14- to 16-h fast as described (Fujitani et al., 2006; Brissova et al., 2014). Islets were isolated by collagenase P digestion of the pancreas and handpicked under microscopic guidance to nearly 100% purity. Analysis of islet function using static incubation system was performed by the Vanderbilt Islet Procurement Core (P30 DK020593) as described previously (Dai et al., 2012). Insulin and somatostatin concentration in the culture medium, and insulin and somatostatin content in islet extracts, were determined by radioimmunoassay (insulin, RI-13K, Millipore; somatostatin, RK-060-14, Phoenix Pharmaceuticals).
Acknowledgements
We are grateful to Anastasia Coldren and Jeff Duryea for technical assistance. We thank Roland Stein, Anna Means, Guoqiang Gu and Wright Lab members for discussions.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
F.C.P. developed the concept, designed and performed experiments and data analysis, and wrote the manuscript. M.B. performed experiments and data analysis and edited the manuscript. A.C.P. performed manuscript editing. S.P. provided the published Mnx1 floxed allele. C.V.E.W. developed the concept, performed manuscript writing and editing.
Funding
We acknowledge the Vanderbilt Center for Imaging Shared Resource, supported in part through Vanderbilt University Medical Center's Digestive Disease Research Center, Diabetes Research Training Center, and Vanderbilt Ingram Cancer Center, supported by National Institutes of Health (NIH) grants [CA68485, DK20593, DK58404 and DK59637]. This work was also supported by grants from the Department of Veterans Affairs, the NIH [DK72473, DK89572, DK104211]; the Juvenile Diabetes Research Foundation (JDRF) and the Vanderbilt Diabetes Research and Training Center [DK20593]. Further support was from the NIH [U19 DK 042502 and U01 DK 089570 to F.C.P. and C.V.E.W.]. Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.126011/-/DC1
References
- Alberti K. G. M. M., Christensen N. J., Christensen S. E., Hansen A. A. P., Iversen J., Lundbaek K., Seyer-Hansen K. and Orskov H. (1973). Inhibition of insulin secretion by somatostatin. Lancet 302, 1299-1301. 10.1016/S0140-6736(73)92873-0 [DOI] [PubMed] [Google Scholar]
- Bonnefond A., Vaillant E., Philippe J., Skrobek B., Lobbens S., Yengo L., Huyvaert M., Cavé H., Busiah K., Scharfmann R. et al. (2013). Transcription factor gene MNX1 is a novel cause of permanent neonatal diabetes in a consanguineous family. Diabetes Metab. 39, 276-280. 10.1016/j.diabet.2013.02.007 [DOI] [PubMed] [Google Scholar]
- Brissova M., Aamodt K., Brahmachary P., Prasad N., Hong J.-Y., Dai C., Mellati M., Shostak A., Poffenberger G., Aramandla R. et al. (2014). Islet microenvironment, modulated by vascular endothelial growth factor-A signaling, promotes β cell regeneration. Cell Metab. 19, 498-511. 10.1016/j.cmet.2014.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broihier H. T. and Skeath J. B. (2002). Drosophila homeodomain protein dHb9 directs neuronal fate via crossrepressive and cell-nonautonomous mechanisms. Neuron 35, 39-50. 10.1016/S0896-6273(02)00743-2 [DOI] [PubMed] [Google Scholar]
- Chera S., Baronnier D., Ghila L., Cigliola V., Jensen J. N., Gu G., Furuyama K., Thorel F., Gribble F. M., Reimann F. and Herrera P. L. (2014). Diabetes recovery by age-dependent conversion of pancreatic δ-cells into insulin producers. Nature 514, 503-507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collombat P., Mansouri A., Hecksher-Sorensen J., Serup P., Krull J., Gradwohl G. and Gruss P. (2003). Opposing actions of Arx and Pax4 in endocrine pancreas development. Genes Dev. 17, 2591-2603. 10.1101/gad.269003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collombat P., Hecksher-Sorensen J., Broccoli V., Krull J., Ponte I., Mundiger T., Smith J., Gruss P., Serup P. and Mansouri A. (2005). The simultaneous loss of Arx and Pax4 genes promotes a somatostatin-producing cell fate specification at the expense of the alpha- and beta-cell lineages in the mouse endocrine pancreas. Development 132, 2969-2980. 10.1242/dev.01870 [DOI] [PubMed] [Google Scholar]
- Dai C., Brissova M., Hang Y., Thompson C., Poffenberger G., Shostak A., Chen Z., Stein R. and Powers A. C. (2012). Islet-enriched gene expression and glucose-induced insulin secretion in human and mouse islets. Diabetologia 55, 707-718. 10.1007/s00125-011-2369-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalgin G., Ward A. B., Hao L. T., Beattie C. E., Nechiporuk A. and Prince V. E. (2011). Zebrafish mnx1 controls cell fate choice in the developing endocrine pancreas. Development 138, 4597-4608. 10.1242/dev.067736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai S. S., Modali S. D., Parekh V. I., Kebebew E. and Agarwal S. K. (2014). GSK-3β protein phosphorylates and stabilizes HLXB9 protein in insulinoma cells to form a targetable mechanism of controlling insulinoma cell proliferation. J. Biol. Chem. 289, 5386-5398. 10.1074/jbc.M113.533612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanagan S. E., De Franco E., Allen H. L., Zerah M., Abdul-Rasoul M. M., Edge J. A., Stewart H., Alamiri E., Hussain K., Wallis S. et al. (2014). Analysis of transcription factors key for mouse pancreatic development establishes NKX2-2 and MNX1 mutations as causes of neonatal diabetes in man. Cell Metab. 19, 146-154. 10.1016/j.cmet.2013.11.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujitani Y., Fujitani S., Boyer D. F., Gannon M., Kawaguchi Y., Ray M., Shiota M., Stein R. W., Magnuson M. A. and Wright C. V. E. (2006). Targeted deletion of a cis-regulatory region reveals differential gene dosage requirements for Pdx1 in foregut organ differentiation and pancreas formation. Genes Dev. 20, 253-266. 10.1101/gad.1360106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gannon M., Shiota C., Postic C., Wright C. V. E. and Magnuson M. (2000). Analysis of the Cre-mediated recombination driven by rat insulin promoter in embryonic and adult mouse pancreas. Genesis 26, 139-142. [DOI] [PubMed] [Google Scholar]
- Guo S., Dai C., Guo M., Taylor B., Harmon J. S., Sander M., Robertson R. P., Powers A. C. and Stein R. (2013). Inactivation of specific β cell transcription factors in type 2 diabetes. J. Clin. Invest. 123, 3305-3316. 10.1172/JCI65390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison K. A., Thaler J., Pfaff S. L., Gu H. and Kehrl J. H. (1999). Pancreas dorsal lobe agenesis and abnormal islets of Langerhans in Hlxb9-deficient mice. Nat. Genet. 23, 71-75. 10.1038/12674 [DOI] [PubMed] [Google Scholar]
- Karnik S. K., Chen H., McLean G. W., Heit J. J., Gu X., Zhang A. Y., Fontaine M., Yen M. H. and Kim S. K. (2007). Menin controls growth of pancreatic beta-cells in pregnant mice and promotes gestational diabetes mellitus. Science 318, 806-809. 10.1126/science.1146812 [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y., Cooper B., Gannon M., Ray M., MacDonald R. J. and Wright C. V. E. (2002). The role of the transcriptional regulator Ptf1a in converting intestinal to pancreatic progenitors. Nat. Genet. 32, 128-134. 10.1038/ng959 [DOI] [PubMed] [Google Scholar]
- Kido Y., Burks D. J., Withers D., Bruning J. C., Kahn C. R., White M. F. and Accili D. (2000). Tissue-specific insulin resistance in mice with mutations in the insulin receptor, IRS-1, and IRS-2. J. Clin. Invest. 105, 199-205. 10.1172/JCI7917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kordowich S., Collombat P., Mansouri A. and Serup P. (2011). Arx and Nkx2.2 compound deficiency redirects pancreatic alpha- and beta-cell differentiation to a somatostatin/ghrelin co-expressing cell lineage. BMC Dev. Biol. 11, 52 10.1186/1471-213X-11-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacin H., Rusch J., Yeh R. T., Fujioka M., Wilson B. A., Zhu Y., Robie A. A., Mistry H., Wang T., Jaynes J. B. et al. (2014). Genome-wide identification of Drosophila Hb9 targets reveals a pivotal role in directing the transcriptome within eight neuronal lineages, including activation of nitric oxide synthase and Fd59a/Fox-D. Dev. Biol. 388, 117-133. 10.1016/j.ydbio.2014.01.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakso M., Pichel J. G., Gorman J. R., Sauer B., Okamoto Y., Lee E., Alt F. W. and Westphal H. (1996). Efficient in vivo manipulation of mouse genomic sequences at the zygote stage. Proc. Natl. Acad. Sci. USA 93, 5860-5865. 10.1073/pnas.93.12.5860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. and Edlund H. (2001). Persistent expression of Hlxb9 in the pancreatic epithelium impairs pancreatic development. Dev. Biol. 240, 247-253. 10.1006/dbio.2001.0440 [DOI] [PubMed] [Google Scholar]
- Li H., Arber S., Jessell T. M. and Edlund H. (1999). Selective agenesis of the dorsal pancreas in mice lacking homeobox gene Hlxb9. Nat. Genet. 23, 67-70. 10.1038/126694 [DOI] [PubMed] [Google Scholar]
- Mastracci T. L., Wilcox C. L., Arnes L., Panea C., Golden J. A., May C. L. and Sussel L. (2011). Nkx2.2 and Arx genetically interact to regulate pancreatic endocrine cell development and endocrine hormone expression. Dev. Biol. 359, 1-11. 10.1016/j.ydbio.2011.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael M. D., Kulkarni R. N., Postic C., Previs S. F., Shulman G. I., Magnuson M. A. and Kahn C. R. (2000). Loss of insulin signaling in hepatocytes leads to severe insulin resistance and progressive hepatic dysfunction. Mol. Cell 6, 87-97. 10.1016/S1097-2765(05)00015-8 [DOI] [PubMed] [Google Scholar]
- Okada T., Liew C. W., Hu J., Hinault C., Michael M. D., Krtzfeldt J., Yin C., Holzenberger M., Stoffel M. and Kulkarni R. N. (2007). Insulin receptors in beta-cells are critical for islet compensatory growth response to insulin resistance. Proc. Natl. Acad. Sci. USA 104, 8977-8982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan F. C. and Wright C. V. (2011). Pancreas organogenesis: from bud to plexus to gland. Dev. Dyn. 240, 530-565. 10.1002/dvdy.22584 [DOI] [PubMed] [Google Scholar]
- Pan F. C., Bankaitis E. D., Boyer D., Xu X., Van de Casteele M., Magnuson M. A., Heimberg H. and Wright C. V. E. (2013). Spatiotemporal patterns of multipotentiality in Ptf1a-expressing cells during pancreas organogenesis and injury-induced facultative restoration. Development 140, 751-764. 10.1242/dev.090159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salpeter S. J., Klein A. M., Huangfu D., Grimsby J. and Dor Y. (2010). Glucose and aging control the quiescence period that follows pancreatic beta cell replication. Development 137, 3205-3213. 10.1242/dev.054304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salpeter S. J., Klochendler A., Weinberg-Corem N., Porat S., Granot Z., Shapiro A. M. J., Magnuson M. A., Eden A., Grimsby J., Glaser B. et al. (2011). Glucose regulates cyclin D2 expression in quiescent and replicating pancreatic β-cells through glycolysis and calcium channels. Endocrinology 152, 2589-2598. 10.1210/en.2010-1372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonhoff S. E., Giel-Moloney M. and Leiter A. B. (2004). Neurogenin 3-expressing progenitor cells in the gastrointestinal tract differentiate into both endocrine and non-endocrine cell types. Dev. Biol. 270, 443-454. 10.1016/j.ydbio.2004.03.013 [DOI] [PubMed] [Google Scholar]
- Sherwood R. I., Chen T.-Y. A. and Melton D. A. (2009). Transcriptional dynamics of endodermal organ formation. Dev. Dyn. 238, 29-42. 10.1002/dvdy.21810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi K., Parekh V. I., Roy S., Desai S. S. and Agarwal S. K. (2013). The embryonic transcription factor Hlxb9 is a menin interacting partner that controls pancreatic β-cell proliferation and the expression of insulin regulators. Endocr. Relat. Cancer 20, 111-122. 10.1530/ERC-12-0077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivas S., Watanabe T., Lin C.-S., William C. M., Tanabe Y., Jessell T. M. and Costantini F. (2001). Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev. Biol. 1, 4 10.1186/1471-213X-1-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe Y., William C. and Jessell T. M. (1998). Specification of motor neuron identity by the MNR2 homeodomain protein. Cell 95, 67-80. 10.1016/S0092-8674(00)81783-3 [DOI] [PubMed] [Google Scholar]
- Wang J., Elghazi L., Parker S. E., Kizilocak H., Asano M., Sussel L. and Sosa-Pineda B. (2004). The concerted activities of Pax4 and Nkx2.2 are essential to initiate pancreatic beta-cell differentiation. Dev. Biol. 266, 178-189. 10.1016/j.ydbio.2003.10.018 [DOI] [PubMed] [Google Scholar]
- William C. M., Tanabe Y. and Jessell T. M. (2003). Regulation of motor neuron subtype identity by repressor activity of Mnx class homeodomain proteins. Development 130, 1523-1536. 10.1242/dev.00358 [DOI] [PubMed] [Google Scholar]
- Yang Y.-P., Thorel F., Boyer D. F., Herrera P. L. and Wright C. V. E. (2011). Context-specific α- to-β-cell reprogramming by forced Pdx1 expression. Genes Dev. 25, 1680-1685. 10.1101/gad.16875711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., McKenna L. B., Bogue C. W. and Kaestner K. H. (2014). The diabetes gene Hhex maintains δ-cell differentiation and islet function. Genes Dev. 28, 829-834. 10.1101/gad.235499.113 [DOI] [PMC free article] [PubMed] [Google Scholar]