

The title twisted thiosemicarbazone molecule has, respectively, anti- and syn-dispositions of the p-tolyl-N—H and imino-N—H groups with respect to the central thione-S atom allowing for the formation of an intramolecular p-tolyl-N—H⋯N(imino) hydrogen bond. The presence of N—H⋯S hydrogen bonds lead to layers in the bc plane which are connected by methyl-C—H⋯π interactions.
Keywords: crystal structure, hydrogen bonding, thiourea derivative, thiosemicarbazone, Hirshfeld surface analysis
Abstract
In the title thiosemicarbazone, C11H15N3S, the p-tolyl-N—H and imino-N—H groups are anti and syn, respectively, to the central thione-S atom. This allows for the formation of an intramolecular p-tolyl-N—H⋯N(imino) hydrogen bond. The molecule is twisted with the dihedral angle between the p-tolyl ring and the non-hydrogen atoms of the N=CMe2 residue being 29.27 (8)°. The crystal packing features supramolecular layers lying in the bc plane whereby centrosymmetric aggregates sustained by eight-membered thioamide {⋯HNCS}2 synthons are linked by further N—H⋯S hydrogen bonds. Layers are connected via methyl-C—H⋯π interactions. The supramolecular aggregation was further investigated by an analysis of the Hirshfeld surface and comparison made to related structures.
Chemical context
The reaction between an alcohol or amine (primary or secondary) with N-alkyl- or N-aryl-isothiocyanides usually results in the formation of thiocarbamides. For example, in the case of reactions involving a monofunctional alcohol, the reaction proceeds in the following manner: R—OH + R′N=C=S → ROC(=S)N(H)R′ (Ho et al., 2005 ▸). Often, reactions are facilitated by initially employing an alkali metal hydroxide as the base and later adding an acid, e.g. HCl (Ho et al., 2005 ▸). Such molecules are of interest as when deprotonated, they can function as effective thiolate ligands for phosphanegold(I) derivatives, which display biological activity. For example, Ph3PAu[SC(O–alkyl)=N(aryl)] compounds exhibit significant cytotoxicity against a variety of cancer cell lines and mechanistic studies show that they can kill cancer cells by initiating a variety of apoptotic pathways, both extrinsic and intrinsic (Yeo, Ooi et al., 2013 ▸; Ooi, Yeo et al., 2015 ▸). Related species, i.e. Ph3PAu[SC(O–alkyl)=N(p-tolyl)], exhibit potency against Gram-positive bacteria (Yeo, Sim et al., 2013 ▸). Over and above these considerations, systematic studies into the structural chemistry of these molecules, which have proven relatively easy to crystallize, have been of some interest in crystal engineering endeavours (Ho et al., 2006 ▸; Kuan et al., 2008 ▸). In the course of studies to increase the functionality of the thiocarbamide molecules, bipodal {1,4-[MeOC(=S)N(H)]2C6H4} was successfully synthesized along with binuclear phosphanegold(I) complexes (Yeo, Khoo et al., 2015 ▸). Recent attempts at expanding this chemistry by using thiourea as an amine donor have been undertaken. As reported very recently, 1:2 reactions between thiourea and R′N=C=S resulted in the isolation of salts containing 1,2,3-thiazole-based cations resulting from 1:1 cyclizations (Yeo, Tan et al., 2015 ▸). Herein, the product of an analogous reaction involving a bifunctional amine, i.e. H2NNH2 (hydrazine) with (p-tolyl)N=C=S, conducted in acetone solution, is described, namely the thiosemicarbazone, (I). Molecules related to (I) and especially their metal complexes continue to attract attention owing to potential biological activity (Dilworth & Hueting, 2012 ▸; Lukmantara et al., 2013 ▸; Su et al., 2013 ▸).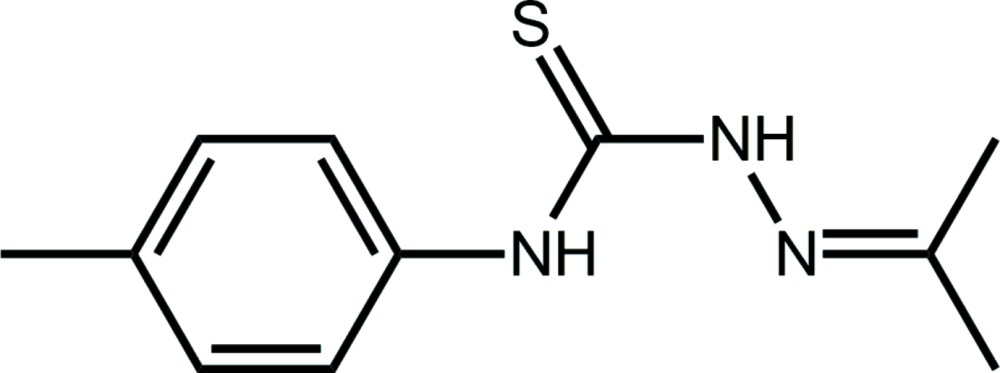
Structural commentary
The molecular structure of (I), Fig. 1 ▸, comprises three planar regions. The central NC(=S)N chromophore (the r.m.s. deviation of the fitted atoms is 0.0020 Å) has anti- and syn-dispositions of the N1- and N2-bound H atoms, respectively, with respect to the central thione-S1 atom. The N1-bound H atom is syn to the imino-N3 atom allowing for the formation of a five-membered loop via an N1—H⋯N3 hydrogen bond, Table 1 ▸. The central plane forms dihedral angles of 23.49 (4)° with the propan-2-ylideneamino residue (N=CMe2; r.m.s. deviation for the C3N atoms = 0.0002 Å) and 43.30 (5)° with the p-tolyl ring. Overall, the molecule is twisted as quantified by the dihedral angle between the outer planes of 29.27 (8)°.
Figure 1.

The molecular structure of (I) showing displacement ellipsoids at the 70% probability level.
Table 1. Hydrogen-bond geometry (, ).
Cg1 is the centroid of the C2C7 ring.
| DHA | DH | HA | D A | DHA |
|---|---|---|---|---|
| N1H1NN3 | 0.88(1) | 2.09(2) | 2.572(2) | 114(1) |
| N1H1NS1i | 0.88(1) | 2.87(2) | 3.5618(17) | 137(2) |
| N2H2NS1ii | 0.88(2) | 2.57(2) | 3.4373(16) | 169(2) |
| C8H8C Cg1iii | 0.98 | 2.81 | 3.716(2) | 154 |
Symmetry codes: (i)  ; (ii)
; (ii)  ; (iii)
; (iii)  .
.
Two P21/c polymorphs have been reported for the parent compound, i.e. having a phenyl rather than a p-tolyl substituent (Jian et al., 2005 ▸; Venkatraman et al., 2005 ▸); the structure of (I) also crystallizes in the P21/c space group. As revealed from the data collated in Table 2 ▸, there is a high degree of concordance in the key bond lengths and angles for the three molecules, as might be expected. However, there are some notable differences in the torsion-angle data as well as in the dihedral angles between the three least-squares planes discussed above, Table 2 ▸. From these and the overlay diagram shown in Fig. 2 ▸, it is apparent that the molecular structure of (I) more closely matches that observed in the polymorph reported by Venkatraman et al. (2005 ▸) rather than that described by Jian et al. (2005 ▸). This conclusion is also vindicated in the unit cell data, i.e. a = 12.225 (3), b = 7.618 (2), c = 11.639 (3) Å, β = 102.660 (4)° reported for the former (Venkatraman et al., 2005 ▸).
Table 2. Geometric data (, ) for (I) and two polymorphs of (II).
| Parameter | (I) | Form a of (II)a | Form b of (II)b |
|---|---|---|---|
| N2N3 | 1.397(2) | 1.386(2) | 1.392(2) |
| C1S1 | 1.6873(18) | 1.6816(17) | 1.6826(17) |
| C1N1 | 1.350(2) | 1.337(2) | 1.343(2) |
| C1N2 | 1.350(2) | 1.359(2) | 1.348(2) |
| C2N1 | 1.422(2) | 1.420(2) | 1.425(2) |
| C9N3 | 1.280(2) | 1.279(2) | 1.276(2) |
| C1N1C2 | 127.79(16) | 129.98(14) | 127.97(14) |
| C1N2N3 | 117.72(15) | 118.17(14) | 118.33(14) |
| N2N3C9 | 117.91(15) | 118.85(15) | 117.73(14) |
| S1C1N1 | 125.45(14) | 126.00(13) | 125.75(13) |
| S1C1N2 | 120.00(14) | 119.37(13) | 119.50(12) |
| N1C1N2 | 114.54(16) | 114.63(15) | 114.74(15) |
| S1C1N2N3 | 169.57(12) | 177.46(12) | 170.56(12) |
| C1N1C2C3 | 132.2(2) | 153.87(18) | 131.10(19) |
| C1N2N3C9 | 165.78(16) | 168.43(16) | 165.85(15) |
| CN2S / C3N | 23.49(4) | 13.19(8) | 22.42(9) |
| CN2S / aryl | 43.30(5) | 39.26(6) | 43.90(6) |
| C3N / aryl | 29.27(8) | 40.15(8) | 30.18(8) |
Figure 2.

Overlay diagram of the molecules in (I), red image, and (II), forms a (green) and b (blue). The molecules have been overlapped so that the central NC(=S)N chromophores are coincident.
NMR invesitgations
The conformation of (I) was also investigated in CDCl3 solution by NMR methods. Assignments were made with the aid of the interpretative program, Chemdraw Ultra (CambridgeSoft Corporation, 2002 ▸). On the basis of multiple 1H and 13C{1H} resonances for the methyl groups of the propan-2-ylideneamino residue, it appears that the (propan-2-ylideneamino)thiourea residue has a locked configuration, consistent with the persistence of the intramolecular N1—H⋯N3 hydrogen bond in CDCl3 solution.
Supramolecular features
In the crystal, N—H⋯S and C—H⋯π interactions provide identifiable points of contact between molecules; these interactions are quantified in Table 1 ▸. Centrosymmetrically related molecules are connected by pairs of amide-N2—H⋯S1 hydrogen bonds, forming eight-membered thioamide {⋯HNCS}2 synthons. These are connected into supramolecular layers in the bc plane by amide-N1—H⋯S1 hydrogen bonds so that the S1 atom accepts two hydrogen bonds, Fig. 3 ▸. The p-tolyl groups protrude to either side of each layer and inter-digitate along the a axis with adjacent layers allowing for the formation of methyl-C8—H⋯π(C2–C7) interactions, thereby consolidating the three-dimensional architecture, Fig. 4 ▸.
Figure 3.

Supramolecular layer in the bc plane in the crystal packing of (I). Centrosymmetric aggregates mediated by eight-membered thioamide {⋯HNCS}2 synthons (shown as orange dashed lines) are linked by additional amide-N—H⋯S hydrogen bonds, shown as blue dashed lines.
Figure 4.

A view of the unit cell contents of (I) shown in projection down the b axis. Supramolecular layers, illustrated in Fig. 3 ▸, are linked via C—H⋯π interactions, shown as purple dashed lines, leading to a three-dimensional architecture.
Analysis of the Hirshfeld surfaces
The crystal packing was further investigated by an analysis of the Hirshfeld surface (Spackman & Jayatilaka, 2009 ▸) employing CrystalExplorer (Wolff et al., 2012 ▸). Fingerprint plots (Rohl et al., 2008 ▸) were calculated, as were the electrostatic potentials using TONTO (Spackman et al., 2008 ▸; Jayatilaka et al., 2005 ▸) integrated into CrystalExplorer; the electrostatic potentials were mapped on the Hirshfeld surfaces using the STO–3G basis set at the level of Hartree–Fock theory over a range of ±0.075 au.
Two views of the Hirshfeld surface mapped over d norm are shown in Fig. 5 ▸ a and b. The deep-red depressions at the S1 and N2 atoms (Fig. 5 ▸ a) confirm their role as an acceptor and donor in the hydrogen-bonding scheme, respectively. This is also evident from the dark-red and blue regions, respectively, on the Hirshfeld surface mapped over the calculated electrostatic potential (Fig. 5 ▸ c). The diminutive red spots near S1 and N1 (Fig. 5 ▸ b) indicate their involvement in the intermolecular N—H⋯S hydrogen bond.
Figure 5.

Views of the Hirshfeld surface for (I): (a) and (b) mapped over d norm, and (c) mapped over the calculated electrostatic potential.
The overall two-dimensional fingerprint plot (Fig. 6 ▸ a) and those delineated into H⋯H, S⋯H/H⋯S, N⋯H/H⋯N and C⋯H/H⋯C H⋯H (Fig. 6 ▸ b–d, respectively) interactions operating in the crystal structure of (I) are illustrated in Fig. 6 ▸; the relative contributions are summarized in Table 3 ▸. The prominent pair of sharp spikes of equal length (d e + d i = 2.45 Å; Fig. 6 ▸ b) with a 15.2% contribution due to S⋯H/H⋯S contacts to the Hirshfeld surfaces also suggest the presence of these interactions in the crystal packing. The light-red region near N3 (Fig. 5 ▸ a) and diminutive red spot near N1—H (Fig. 5 ▸ b) are consistent with relatively smaller contributions from N⋯H/H⋯N contacts, i.e. 2.5 and 3.0%, respectively, and are indicative of the weak intramolecular hydrogen bond. The strength of such an interaction can be visualized from the 2D fingerprint plot corresponding to N⋯H/ H⋯N contacts (Fig. 6 ▸ c). The bright-orange spot in the surface mapped with d e (within a red circle in Fig. 7 ▸) about the aryl ring and a light-blue region around the tolyl-hydrogen atom, H8C (Fig. 7 ▸), suggest a contribution from the C—H⋯π interaction (Table 1 ▸). This is also evident through distinct pair of ‘wings’ in the fingerprint plot corresponding to C⋯H/H⋯C contacts (Fig. 6 ▸ d). The wing at the top, left belongs to C—H donors, while that at the bottom, right corresponds to the surface around π-acceptors with 11.3 and 7.8% contribution from C⋯H and H⋯C contacts, respectively. The H⋯H contacts reflected in the middle of scattered points in Fig. 6 ▸ e provide the most significant contribution, i.e. 57.0%, to the Hirshfeld surface arising from a side-ways approach. The small, flat segments delineated by the blue outline in the surface mapped with curvedness (Fig. 8 ▸) and the small (i.e. 0.7%) contribution from C⋯C contacts to the surface indicates the absence of π–π stacking interactions in the structure.
Figure 6.

2D Fingerprint plots for (I): (a) full, (b) delineated to show S⋯H/H⋯S, (c) N⋯H/H⋯N, (d) C⋯H/H⋯C, and (e) H⋯H interactions.
Table 3. Relative contribution (%) to intermolecular interactions calculated from the Hirshfeld surface.
| Contact | Contribution |
|---|---|
| HH | 57.0 |
| SH/HS | 15.2 |
| NH/HN | 5.5 |
| CH/HC | 19.1 |
| CC | 0.7 |
| NN | 1.4 |
| CN | 0.8 |
| CS | 0.2 |
| others | 0.1 |
Figure 7.

View of the Hirshfeld surface for (I) mapped over d e.
Figure 8.

Hirshfeld surfaces for (I) mapped over curvedness.
Database survey
According to a search of the Cambridge Structural Database (Groom & Allen, 2014 ▸), there are no direct analogues of (I), either in the coordinated or uncoordinated form. As mentioned in the Structural commentary, the parent compound has been characterized in two polymorphic forms (Jian et al., 2005 ▸; Venkatraman et al., 2005 ▸). The parent compound, LH, has also been observed to coordinate metal centres. Thus, monodentate coordination via the thione-S atom was observed in a neutral complex [ZnCl2(LH)2] (Bel’skii et al., 1987 ▸). By contrast, a chelating mode via thione-S and imino-N atoms was observed in each of the charged complexes [CoBr(LH)2]Br (Dessy et al., 1978 ▸) and [(η6-p-cymene)RuCl(LH)]Cl (Su et al., 2013 ▸). The most closely related structure having the p-tolyl substituent but variations at the imino-N atom is one where one methyl group has been substituted by a phenyl (Zhang et al., 2011 ▸). This is also a twisted molecule with the dihedral angle between the p-tolyl and NC3 residues being 65.44 (7)°.
Synthesis and crystallization
To p-tolyl isothiocyanate (Sigma–Aldrich; 10 mmol, 1.49 g) in acetone (20 ml) was added hydrazine monohydrate (Sigma–Aldrich; 10 mmol, 0.76 ml). The resulting mixture was stirred for 4 h at room temperature. Both chloroform (20 ml) and acetonitrile (20 ml) were then added, and the resulting mixture left for slow evaporation. Light-brown crystals were obtained after 2 weeks. Yield: 2.012 g (91%). M.p. 412–413 K. 1H NMR (400 MHz, CDCl3, 298 K): 9.19 (s, br, 1H, NH—N), 8.56 (s, br, 1H, NH), 7.49 (d, 2H, m-aryl, J = 8.28 Hz), 7.17 (d, 2H, o-aryl, J = 8.24 Hz), 2.34 (s, 3H, aryl-CH3), 2.05 (s, 3H, CH3), 1.94 (s, 3H, CH3). 13C NMR (400 MHz, CDCl3, 298 K): 176.4 [CS], 149.6 [C(CH3)2], 135.8 [Cipso], 135.4 [Cpara], 129.3 [Cmeta], 124.5 [Cortho], 25.3 [CH3], 21.0 [aryl-CH3], 16.9 [CH3, syn to N—H]. IR (cm−1): ν(N—H) 3240, 3168 (m), ν(C=N) 1514 (vs), ν(C—N) 1267 (s), ν(C=S) 1188 (vs).
Refinement
Crystal data, data collection and structure refinement details are summarized in Table 4 ▸. Carbon-bound H-atoms were placed in calculated positions (C—H = 0.95–0.98 Å) and were included in the refinement in the riding model approximation, with U iso(H) set to 1.2–1.5U eq(C). The N-bound H atoms were located in a difference Fourier map but were refined with a distance restraint of N—H = 0.88±0.01 Å, and with U iso(H) set to 1.2U eq(N).
Table 4. Experimental details.
| Crystal data | |
| Chemical formula | C11H15N3S |
| M r | 221.32 |
| Crystal system, space group | Monoclinic, P21/c |
| Temperature (K) | 100 |
| a, b, c () | 13.7289(13), 7.4341(7), 11.5757(11) |
| () | 102.690(1) |
| V (3) | 1152.58(19) |
| Z | 4 |
| Radiation type | Mo K |
| (mm1) | 0.25 |
| Crystal size (mm) | 0.12 0.05 0.03 |
| Data collection | |
| Diffractometer | Bruker SMART APEX CCD |
| Absorption correction | Multi-scan (SADABS; Sheldrick, 1996 ▸) |
| T min, T max | 0.970, 0.993 |
| No. of measured, independent and observed [I > 2(I)] reflections | 10739, 2646, 2052 |
| R int | 0.050 |
| (sin /)max (1) | 0.650 |
| Refinement | |
| R[F 2 > 2(F 2)], wR(F 2), S | 0.041, 0.098, 1.02 |
| No. of reflections | 2646 |
| No. of parameters | 147 |
| No. of restraints | 2 |
| H-atom treatment | H atoms treated by a mixture of independent and constrained refinement |
| max, min (e 3) | 0.27, 0.27 |
Supplementary Material
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S2056989015017624/hb7507sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989015017624/hb7507Isup2.hkl
Supporting information file. DOI: 10.1107/S2056989015017624/hb7507Isup3.cml
CCDC reference: 1425975
Additional supporting information: crystallographic information; 3D view; checkCIF report
Acknowledgments
This research was supported by the Trans-disciplinary Research Grant Scheme (TR002-2014A) provided by the Ministry of Education, Malaysia. The intensity data set was provided by the University of Malaya Crystallographic Laboratory.
supplementary crystallographic information
Crystal data
| C11H15N3S | F(000) = 472 |
| Mr = 221.32 | Dx = 1.275 Mg m−3 |
| Monoclinic, P21/c | Mo Kα radiation, λ = 0.71073 Å |
| a = 13.7289 (13) Å | Cell parameters from 1590 reflections |
| b = 7.4341 (7) Å | θ = 3.0–25.5° |
| c = 11.5757 (11) Å | µ = 0.25 mm−1 |
| β = 102.690 (1)° | T = 100 K |
| V = 1152.58 (19) Å3 | Prism, light-brown |
| Z = 4 | 0.12 × 0.05 × 0.03 mm |
Data collection
| Bruker SMART APEX CCD diffractometer | 2646 independent reflections |
| Radiation source: fine-focus sealed tube | 2052 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.050 |
| φ and ω scans | θmax = 27.5°, θmin = 3.0° |
| Absorption correction: multi-scan (SADABS; Sheldrick, 1996) | h = −17→17 |
| Tmin = 0.970, Tmax = 0.993 | k = −9→9 |
| 10739 measured reflections | l = −13→15 |
Refinement
| Refinement on F2 | 2 restraints |
| Least-squares matrix: full | Hydrogen site location: mixed |
| R[F2 > 2σ(F2)] = 0.041 | H atoms treated by a mixture of independent and constrained refinement |
| wR(F2) = 0.098 | w = 1/[σ2(Fo2) + (0.0332P)2 + 0.7356P] where P = (Fo2 + 2Fc2)/3 |
| S = 1.02 | (Δ/σ)max < 0.001 |
| 2646 reflections | Δρmax = 0.27 e Å−3 |
| 147 parameters | Δρmin = −0.27 e Å−3 |
Special details
| Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| S1 | 0.14991 (3) | 0.10760 (6) | 0.53754 (4) | 0.01865 (13) | |
| N1 | 0.17657 (11) | 0.1754 (2) | 0.31707 (14) | 0.0179 (3) | |
| H1N | 0.1468 (14) | 0.175 (3) | 0.2419 (9) | 0.023 (6)* | |
| N2 | 0.01790 (11) | 0.1178 (2) | 0.33437 (14) | 0.0174 (3) | |
| H2N | −0.0236 (13) | 0.073 (3) | 0.3744 (17) | 0.032 (6)* | |
| N3 | −0.00516 (11) | 0.1118 (2) | 0.21071 (13) | 0.0177 (3) | |
| C1 | 0.11491 (13) | 0.1348 (2) | 0.38949 (16) | 0.0158 (4) | |
| C2 | 0.28274 (13) | 0.1793 (2) | 0.34532 (16) | 0.0174 (4) | |
| C3 | 0.33249 (14) | 0.0928 (3) | 0.26883 (17) | 0.0218 (4) | |
| H3 | 0.2955 | 0.0313 | 0.2013 | 0.026* | |
| C4 | 0.43587 (14) | 0.0956 (3) | 0.29054 (17) | 0.0240 (4) | |
| H4 | 0.4688 | 0.0357 | 0.2374 | 0.029* | |
| C5 | 0.49234 (14) | 0.1841 (3) | 0.38822 (17) | 0.0218 (4) | |
| C6 | 0.44118 (15) | 0.2713 (3) | 0.46362 (18) | 0.0240 (4) | |
| H6 | 0.4781 | 0.3329 | 0.5311 | 0.029* | |
| C7 | 0.33772 (14) | 0.2703 (3) | 0.44266 (17) | 0.0221 (4) | |
| H7 | 0.3046 | 0.3319 | 0.4949 | 0.027* | |
| C8 | 0.60488 (15) | 0.1844 (3) | 0.4134 (2) | 0.0316 (5) | |
| H8A | 0.6299 | 0.3006 | 0.4475 | 0.047* | |
| H8B | 0.6271 | 0.1650 | 0.3395 | 0.047* | |
| H8C | 0.6307 | 0.0880 | 0.4695 | 0.047* | |
| C9 | −0.09645 (13) | 0.1333 (2) | 0.15732 (16) | 0.0163 (4) | |
| C10 | −0.18214 (14) | 0.1717 (3) | 0.21420 (17) | 0.0220 (4) | |
| H10A | −0.1571 | 0.2260 | 0.2922 | 0.033* | |
| H10B | −0.2169 | 0.0592 | 0.2233 | 0.033* | |
| H10C | −0.2285 | 0.2549 | 0.1643 | 0.033* | |
| C11 | −0.11855 (14) | 0.1185 (3) | 0.02523 (16) | 0.0201 (4) | |
| H11A | −0.0561 | 0.1262 | −0.0024 | 0.030* | |
| H11B | −0.1631 | 0.2166 | −0.0096 | 0.030* | |
| H11C | −0.1509 | 0.0028 | 0.0011 | 0.030* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| S1 | 0.0168 (2) | 0.0218 (2) | 0.0167 (2) | −0.00133 (19) | 0.00219 (18) | 0.00207 (18) |
| N1 | 0.0133 (8) | 0.0252 (8) | 0.0149 (8) | −0.0017 (6) | 0.0020 (6) | 0.0013 (7) |
| N2 | 0.0148 (8) | 0.0226 (8) | 0.0149 (8) | −0.0018 (6) | 0.0033 (6) | 0.0014 (6) |
| N3 | 0.0182 (8) | 0.0196 (8) | 0.0152 (8) | −0.0022 (6) | 0.0037 (6) | 0.0000 (6) |
| C1 | 0.0149 (9) | 0.0129 (8) | 0.0196 (9) | 0.0003 (7) | 0.0041 (7) | 0.0007 (7) |
| C2 | 0.0151 (9) | 0.0177 (8) | 0.0194 (9) | −0.0003 (7) | 0.0040 (7) | 0.0046 (7) |
| C3 | 0.0200 (10) | 0.0272 (10) | 0.0181 (10) | −0.0009 (8) | 0.0043 (8) | 0.0008 (8) |
| C4 | 0.0194 (10) | 0.0308 (11) | 0.0239 (11) | 0.0016 (8) | 0.0092 (8) | 0.0014 (8) |
| C5 | 0.0171 (10) | 0.0233 (9) | 0.0247 (10) | −0.0006 (8) | 0.0038 (8) | 0.0080 (8) |
| C6 | 0.0203 (10) | 0.0258 (10) | 0.0245 (11) | −0.0051 (8) | 0.0018 (8) | −0.0010 (8) |
| C7 | 0.0206 (10) | 0.0224 (9) | 0.0251 (11) | −0.0026 (8) | 0.0087 (8) | −0.0040 (8) |
| C8 | 0.0172 (10) | 0.0364 (12) | 0.0408 (13) | −0.0008 (9) | 0.0053 (9) | 0.0085 (10) |
| C9 | 0.0182 (9) | 0.0121 (8) | 0.0186 (9) | −0.0004 (7) | 0.0041 (7) | 0.0009 (7) |
| C10 | 0.0178 (10) | 0.0288 (10) | 0.0179 (10) | 0.0056 (8) | 0.0009 (8) | 0.0009 (8) |
| C11 | 0.0199 (10) | 0.0228 (9) | 0.0167 (9) | −0.0008 (8) | 0.0020 (8) | 0.0004 (7) |
Geometric parameters (Å, º)
| S1—C1 | 1.6873 (18) | C5—C8 | 1.508 (3) |
| N1—C1 | 1.350 (2) | C6—C7 | 1.387 (3) |
| N1—C2 | 1.422 (2) | C6—H6 | 0.9500 |
| N1—H1N | 0.876 (9) | C7—H7 | 0.9500 |
| N2—C1 | 1.350 (2) | C8—H8A | 0.9800 |
| N2—N3 | 1.397 (2) | C8—H8B | 0.9800 |
| N2—H2N | 0.875 (9) | C8—H8C | 0.9800 |
| N3—C9 | 1.280 (2) | C9—C11 | 1.496 (2) |
| C2—C7 | 1.387 (3) | C9—C10 | 1.496 (3) |
| C2—C3 | 1.389 (3) | C10—H10A | 0.9800 |
| C3—C4 | 1.386 (3) | C10—H10B | 0.9800 |
| C3—H3 | 0.9500 | C10—H10C | 0.9800 |
| C4—C5 | 1.388 (3) | C11—H11A | 0.9800 |
| C4—H4 | 0.9500 | C11—H11B | 0.9800 |
| C5—C6 | 1.395 (3) | C11—H11C | 0.9800 |
| C1—N1—C2 | 127.79 (16) | C2—C7—C6 | 119.87 (18) |
| C1—N1—H1N | 113.3 (14) | C2—C7—H7 | 120.1 |
| C2—N1—H1N | 117.3 (14) | C6—C7—H7 | 120.1 |
| C1—N2—N3 | 117.72 (15) | C5—C8—H8A | 109.5 |
| C1—N2—H2N | 118.4 (15) | C5—C8—H8B | 109.5 |
| N3—N2—H2N | 120.2 (15) | H8A—C8—H8B | 109.5 |
| C9—N3—N2 | 117.91 (15) | C5—C8—H8C | 109.5 |
| N1—C1—N2 | 114.54 (16) | H8A—C8—H8C | 109.5 |
| N1—C1—S1 | 125.45 (14) | H8B—C8—H8C | 109.5 |
| N2—C1—S1 | 120.00 (14) | N3—C9—C11 | 116.21 (16) |
| C7—C2—C3 | 119.20 (17) | N3—C9—C10 | 126.34 (17) |
| C7—C2—N1 | 122.98 (17) | C11—C9—C10 | 117.46 (16) |
| C3—C2—N1 | 117.79 (17) | C9—C10—H10A | 109.5 |
| C4—C3—C2 | 120.30 (18) | C9—C10—H10B | 109.5 |
| C4—C3—H3 | 119.9 | H10A—C10—H10B | 109.5 |
| C2—C3—H3 | 119.9 | C9—C10—H10C | 109.5 |
| C3—C4—C5 | 121.42 (18) | H10A—C10—H10C | 109.5 |
| C3—C4—H4 | 119.3 | H10B—C10—H10C | 109.5 |
| C5—C4—H4 | 119.3 | C9—C11—H11A | 109.5 |
| C4—C5—C6 | 117.52 (17) | C9—C11—H11B | 109.5 |
| C4—C5—C8 | 121.55 (18) | H11A—C11—H11B | 109.5 |
| C6—C5—C8 | 120.92 (18) | C9—C11—H11C | 109.5 |
| C7—C6—C5 | 121.69 (18) | H11A—C11—H11C | 109.5 |
| C7—C6—H6 | 119.2 | H11B—C11—H11C | 109.5 |
| C5—C6—H6 | 119.2 | ||
| C1—N2—N3—C9 | −165.78 (16) | C3—C4—C5—C6 | −0.4 (3) |
| C2—N1—C1—N2 | −171.29 (17) | C3—C4—C5—C8 | 178.82 (19) |
| C2—N1—C1—S1 | 9.3 (3) | C4—C5—C6—C7 | 0.0 (3) |
| N3—N2—C1—N1 | 11.0 (2) | C8—C5—C6—C7 | −179.16 (18) |
| N3—N2—C1—S1 | −169.57 (12) | C3—C2—C7—C6 | −1.1 (3) |
| C1—N1—C2—C7 | −50.1 (3) | N1—C2—C7—C6 | −178.81 (17) |
| C1—N1—C2—C3 | 132.2 (2) | C5—C6—C7—C2 | 0.7 (3) |
| C7—C2—C3—C4 | 0.8 (3) | N2—N3—C9—C11 | −177.62 (15) |
| N1—C2—C3—C4 | 178.60 (17) | N2—N3—C9—C10 | 2.4 (3) |
| C2—C3—C4—C5 | 0.0 (3) |
Hydrogen-bond geometry (Å, º)
Cg1 is the centroid of the C2–C7 ring.
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1N···N3 | 0.88 (1) | 2.09 (2) | 2.572 (2) | 114 (1) |
| N1—H1N···S1i | 0.88 (1) | 2.87 (2) | 3.5618 (17) | 137 (2) |
| N2—H2N···S1ii | 0.88 (2) | 2.57 (2) | 3.4373 (16) | 169 (2) |
| C8—H8C···Cg1iii | 0.98 | 2.81 | 3.716 (2) | 154 |
Symmetry codes: (i) x, −y+1/2, z−1/2; (ii) −x, −y, −z+1; (iii) −x+1, −y, −z+1.
References
- Bel’skii, V. K., Prisyazhnyuk, A. I., Kolchinskii, E. V. & Koksharova, T. V. (1987). J. Struct. Chem. 27, 808–811.
- Brandenburg, K. (2006). DIAMOND. Crystal Impact GbR, Bonn, Germany.
- Bruker (2008). APEX2 and SAINT. Bruker AXS Inc., Madison, Wisconsin, USA.
- CambridgeSoft Corporation (2002). CHEMDRAW Ultra. Cambridge, USA.
- Dessy, G., Fares, V., Scaramuzza, L., Tomlinson, A. A. B. & De Munno, G. (1978). J. Chem. Soc. Dalton Trans. pp. 1549–1554.
- Dilworth, J. R. & Hueting, R. (2012). Inorg. Chim. Acta, 389, 3–15.
- Farrugia, L. J. (2012). J. Appl. Cryst. 45, 849–854.
- Gans, J. & Shalloway, D. (2001). J. Mol. Graphics Modell. 19, 557–559. [DOI] [PubMed]
- Groom, C. R. & Allen, F. H. (2014). Angew. Chem. Int. Ed. 53, 662–671. [DOI] [PubMed]
- Ho, S. Y., Bettens, R. P. A., Dakternieks, D., Duthie, A. & Tiekink, E. R. T. (2005). CrystEngComm, 7, 682–689.
- Ho, S. Y., Cheng, E. C.-C., Tiekink, E. R. T. & Yam, V. W.-W. (2006). Inorg. Chem. 45, 8165–8174. [DOI] [PubMed]
- Jayatilaka, D., Grimwood, D. J., Lee, A., Lemay, A., Russel, A. J., Taylo, C., Wolff, S. K., Chenai, C. & Whitton, A. (2005). TONTO – A System for Computational Chemistry. Available at: http://hirshfeldsurface.net/
- Jian, F.-F., Bai, Z.-S., Xiao, H.-L. & Li, K. (2005). Acta Cryst. E61, o653–o654.
- Kuan, F. S., Yei Ho, S., Tadbuppa, P. P. & Tiekink, E. R. T. (2008). CrystEngComm, 10, 548–564.
- Lukmantara, A. Y., Kalinowski, D. S., Kumar, N. & Richardson, D. R. (2013). Org. Biomol. Chem. 11, 6414–6425. [DOI] [PubMed]
- Ooi, K. K., Yeo, C. I., Ang, K.-P., Akim, A. Md., Cheah, Y.-K., Halim, S. N. A., Seng, H.-L. & Tiekink, E. R. T. (2015). J. Biol. Inorg. Chem. 20, 855–873. [DOI] [PubMed]
- Rohl, A. L., Moret, M., Kaminsky, W., Claborn, K., McKinnon, J. J. & Kahr, B. (2008). Cryst. Growth Des. 8, 4517–4525.
- Sheldrick, G. M. (1996). SADABS. University of Göttingen, Germany.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Sheldrick, G. M. (2015). Acta Cryst. C71, 3–8.
- Spackman, M. A. & Jayatilaka, D. (2009). CrystEngComm, 11, 19–32.
- Spackman, M. A., McKinnon, J. J. & Jayatilaka, D. (2008). CrystEngComm, 10, 377–388.
- Su, W., Qian, Q., Li, P., Lei, X., Xiao, Q., Huang, S., Huang, C. & Cui, J. (2013). Inorg. Chem. 52, 12440–12449. [DOI] [PubMed]
- Venkatraman, R., Swesi, A. T. & Yamin, B. M. (2005). Acta Cryst. E61, o3914–o3916.
- Westrip, S. P. (2010). J. Appl. Cryst. 43, 920–925.
- Wolff, S. K., Grimwood, D. J., McKinnon, J. J., Turner, M. J., Jayatilaka, D. & Spackman, M. A. (2012). Crystal Explorer. The University of Western Australia.
- Yeo, C. I., Khoo, C.-H., Chu, W.-C., Chen, B.-J., Chu, P.-L., Sim, J.-H., Cheah, Y.-K., Ahmad, J., Halim, S. N. A., Seng, H.-L., Ng, S., Otero-de-la-Roza, A. & Tiekink, E. R. T. (2015). RSC Adv 5, 41401–41411.
- Yeo, C. I., Ooi, K. K., Akim, A. Md., Ang, K. P., Fairuz, Z. A., Halim, S. N. B. A., Ng, S. W., Seng, H.-L. & Tiekink, E. R. T. (2013). J. Inorg. Biochem. 127, 24–38. [DOI] [PubMed]
- Yeo, C. I., Sim, J.-H., Khoo, C.-H., Goh, Z.-J., Ang, K.-P., Cheah, Y.-K., Fairuz, Z. A., Halim, S. N. B. A., Ng, S. W., Seng, H.-L. & Tiekink, E. R. T. (2013). Gold Bull. 46, 145–152.
- Yeo, C. I., Tan, Y. S. & Tiekink, E. R. T. (2015). Acta Cryst E71, 1159–1164. [DOI] [PMC free article] [PubMed]
- Zhang, Y.-L., Wu, C.-Z. & Zhang, F.-J. (2011). Acta Cryst. E67, o1547. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S2056989015017624/hb7507sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989015017624/hb7507Isup2.hkl
Supporting information file. DOI: 10.1107/S2056989015017624/hb7507Isup3.cml
CCDC reference: 1425975
Additional supporting information: crystallographic information; 3D view; checkCIF report