

Abstract
Argonaute 2 (Ago2) protein is a central effector of RNA interference (RNAi) pathways and regulates mammalian genes on a global level. The mechanisms of Ago2-mediated silencing are well understood, but less is known about its regulation. Recent reports indicate that phosphorylation significantly affects Ago2 activity. Here, we investigated the effect of mutating all known phospho-residues within Ago2 on its localization and activity. Ago2 associates with two different cytoplasmic RNA granules known as processing bodies (P-bodies) and stress granules, but the nature of this phenomenon is controversial. We report that replacing serine with a phospho-mimetic aspartic acid at position 798 completely abrogates association of Ago2 with P-bodies and stress granules. The effect of this mutation on its activity in gene silencing was modest, which was surprising because association of Ago2 with cytoplasmic RNA granules is thought to be a consequence of its role in RNAi. As such, our data indicate that targeting of Ago2 to P-bodies and stress granules is separable from its role in RNAi and likely requires dynamic phosphorylation of serine 798.
Keywords: Argonaute, P bodies, phosphorylation, RNAi, stress granules
INTRODUCTION
Argonaute proteins are critical for small RNA-dependent gene silencing in eukaryotes. Recent estimates suggest that in mammalian cells, Argonautes regulate expression of the majority of protein-encoding genes through post-transcriptional mechanisms (Friedman et al. 2009). As such, it is critical that these pathways are tightly controlled. Indeed, the number of identified trans-acting factors that directly or indirectly affect Argonaute protein activity is growing quickly. One of the first Argonaute modulators described is the molecular chaperone Hsp90 (Tahbaz et al. 2001; Pare et al. 2009). Subsequent studies revealed that Hsp90 activity is required for biogenesis of RNA-induced silencing complexes (RISCs), of which Argonautes are core components (Tahbaz et al. 2004; Iwasaki et al. 2010; Miyoshi et al. 2010; Iki et al. 2012; Izumi et al. 2013; Martinez et al. 2013; Pare et al. 2013). Moreover, recent evidence indicates that co-opting of Hsp90 as a RISC assembly factor occurred very early during evolution of the RNAi pathway (Wang et al. 2013).
Post-translational modification of Argonaute proteins can also modulate RISC formation and/or activity. For example, hydroxylation of specific proline residues is required for both Argonaute stability (Qi et al. 2008) and interaction with Hsp90 (Wu et al. 2011). Methylation of arginine residues in some Argonaute proteins, particularly those of the Piwi subfamily, have been shown to increase stability and consequently gene-silencing function (Kirino et al. 2009). Conversely, during virus infection or cellular stress, inhibition of small RNA-dependent gene silencing is correlated with an increase in poly(ADP)-ribosylation of Argonautes (Leung et al. 2011; Seo et al. 2013). Similarly, the expression profiles of miRNAs can be significantly affected by ubiquitylation of Argonaute proteins, which results in their destabilization (Rybak et al. 2009). Finally, similar to many other proteins in eukaryotic cells, Argonaute proteins are substrates for multiple protein kinases (Zeng et al. 2008; Rudel et al. 2011; Horman et al. 2013; Shen et al. 2013).
There are four members of the Argonaute subfamily in mammalian cells, but Ago2 is the only member that can cleave targeted mRNAs (Liu et al. 2004). Although Ago2 is the most widely expressed and predominant Argonaute isoform in mammalian somatic cells, the majority of endogenous miRNA-mediated gene silencing does not involve endonucleolytic cleavage of targeted mRNAs. Results from mass spectrometry analyses indicate the presence of at least seven phosphoamino acid residues in Ago2 (Rudel et al. 2011). To date, only three of these phosphoamino acid residues in Ago2 have been studied in a functional manner; however, it is evident that Argonaute phosphorylation affects small RNA-based gene silencing through multiple mechanisms. First, phosphorylation of tyrosine-393 negatively impacts the interaction between Ago2 and Dicer (Shen et al. 2013), the RNase responsible for processing duplex precursors into mature microRNAs (miRNAs). Disrupting this interaction affects miRNA maturation, particularly with respect to precursor miRNAs containing a long-loop structure. Once pre-miRNAs are processed into mature duplexes, the MID domains of Argonaute proteins anchor the 5′ phosphates of guide RNA strands in RISC. However, phosphorylation of tyrosine-529 in this domain of Ago2 prevents loading of small RNAs onto RISC (Rudel et al. 2011). As such, regulated phosphorylation of tyrosine-529 may be a critical step in RISC activation. Finally, the phosphorylation status of serine-387 has been reported to suppress the endonucleolytic cleavage activity of Ago2, while enhancing the silencing of the targeted mRNAs by translational repression (Horman et al. 2013). Phosphorylation of serine-387 by AKT reportedly stimulates Ago2 interaction with GW182, a P-body resident protein and critical component of the ribonucleoprotein complex that represses translation of miRNA-targeted mRNAs. Increased interaction between phosphorylated Ago2 and GW182 may underlie the change of silencing mechanism for this protein. Consistent with this scenario is the observation that activation of the PI3K-AKT-mTOR kinase pathway in hematopoietic cells increases both expression of GW182 and association of Argonaute proteins with mRNAs in high molecular weight complexes (La Rocca et al. 2015). The association of Argonautes with high molecular weight complexes correlated with improved efficiency of slicer-independent silencing by RISC loaded with miRNAs and provides strong evidence of an intricate kinase-mediated regulatory mechanism for RNAi involving both direct and indirect modification of Argonautes.
Given the large number of genes targeted by small RNA-based gene silencing, these pathways must be regulated at multiple levels. Evidence to date indicates that phosphorylation of serine-387, tyrosine-393, and tyrosine-529 regulate Argonaute activity by three different mechanisms. However, the role of phosphorylation at other sites in Ago2 has not been reported. Here, we used site-directed mutagenesis to change every known phosphoamino acid residue in Ago2 to residues that either prevent or mimic constitutive phosphorylation. The subcellular localizations as well as endonuclease-dependent and -independent gene-silencing activities of these mutants were then analyzed. Whereas most Ago2 mutants did not exhibit significant changes in localization or RNAi activity, substitution of aspartic acid for serine at position 798 elicited modest but significant changes in silencing activity. Intriguingly, association of this Ago2 mutant with P-bodies and stress granules was completely abolished. Whereas other studies have shown that microscopic P-bodies are not required for RNAi (Behm-Ansmant et al. 2006; Chu and Rana 2006; Eulalio et al. 2007b), this is the first report indicating that the association of Ago2 with cytoplasmic RNA granules can be uncoupled from its role in gene silencing.
RESULTS AND DISCUSSION
Previous studies revealed that mammalian Ago2 is phosphorylated on at least seven amino acid residues (Fig. 1A), but the functional consequences of only three of these events have been studied in any significant detail. To further explore the role of phosphorylation at each of these sites, we used site-directed mutagenesis to create a panel of 14 mutants in which each phosphoamino acid residue was converted to either a nonphosphorylatable amino acid residue or a mimic of a constitutively phosphorylated amino acid residue. Serines and threonines were substituted with alanines (nonphosphorylatable) or aspartic acid (phospho-mimetic), whereas tyrosine residues were changed to either phenylalanine (nonphosphorylatable) or glutamic acid (phospho-mimetic).
FIGURE 1.

Localization of Ago2 phosphorylation mutants. (A) Schematic representation of Ago2 domains and known phosphoamino acid residues. (B) HeLa cells transiently transfected with plasmids expressing wild-type or mutant Ago2 were processed for indirect immunofluorescence (IIF) microscopy. Dcp1a-positive P-bodies were analyzed for Ago2 signal using quantitative laser scanning cytometry. The colocalization between Ago2 and Dcp1a for all mutants is relative to wild type, which was set to 1.0. (C) HeLa cells were transiently cotransfected with plasmids expressing wild-type or mutant Ago2. Before processing for IIF microscopy, cells were treated with 500 µM sodium arsenite for 45 min. Stress granules positive for endogenous TIA-1 were analyzed for Ago2 signal using quantitative laser scanning cytometry. The colocalization between Ago2 and TIA-1 for all mutants is relative to wild type, which was set to 1.0. Paired Student's two-tailed t-test were used to compare relative colocalization of mutant Ago2 and RNA granule to that of wild-type Ago2. Error bars indicate SE. (D) HeLa cells transiently transfected with plasmids expressing myc-tagged Ago2 (wild type, PAZ9, or S798D) were processed for IIF microscopy. Exogenously expressed Ago2 was detected using an anti-myc antibody. Dcp1a-positive P-bodies are marked by arrows. (E) HeLa cells were transiently cotransfected with plasmids expressing myc-tagged Ago2 (wild type, PAZ9, or S798D) and DsRed-tagged TIA-1. Following treatment with 500 µM sodium arsenite for 1 h, cells were processed for IIF microscopy. Exogenously expressed Ago2 was detected using an anti-myc antibody. TIA-1-positive stress granules are marked by arrowheads. Outlined regions are expanded in inset. Scale bars, 10 µm. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001; (****) P < 0.0001.
Mutation of specific phospho-residues of Ago2 alters its subcellular localization
Earlier reports have drawn a correlation between Ago2 activity and its interaction with cytoplasmic ribonucleoprotein granules such as P-bodies and stress granules (Chu and Rana 2006; Eulalio et al. 2007a,b; Franks and Lykke-Andersen 2008). The relationship between RNAi-mediated silencing activity and localization to P-bodies is not fully understood, but targeting of Ago2 to these granules is dependent on small RNA binding (Pare et al. 2011). This observation was consistent with the idea that Ago2 localizes to RNA granules as a consequence of its mRNA silencing activity, as opposed to a previous theory that espoused that RNAi activity was confined to these structures. To determine whether any of the 14 phospho-mutants had altered subcellular localizations, we performed a high-content, unbiased, automated image analysis using laser scanning cytometry. HeLa cells were transfected with plasmids encoding myc-tagged versions of each of the 14 phospho-mutants, wild type, as well as Ago2 PAZ9 and Ago2 H634P, which served as controls. The Ago2 PAZ9 mutant has been previously characterized (Liu et al. 2005) and, because of multiple site-directed mutations the PAZ domain, cannot bind to small RNAs, and it is therefore inactive in both slicer-mediated and slicer-independent silencing. Conversely, the Ago2 H634P mutant retains its small RNA-binding and slicer-independent silencing activities, but it is catalytically inactivated by a mutation in the endonuclease domain active site (Liu et al. 2004). As such, it is unable to cleave targeted mRNAs. At 24 h post-transfection, cells were processed for indirect immunofluorescence microscopy using antibodies to myc and Dcp1a, a well-characterized P-body component (van Dijk et al. 2002). At least 500 cells were analyzed for each sample (n = 3), after which the mean colocalizations between Ago2 mutants and Dcp1a were plotted relative to that observed for wild type (myc-Ago2 WT; Fig. 1B). As expected, Ago2 PAZ9 and Ago2 Y529E, two mutants that cannot bind miRNAs, did not colocalize with P-bodies. Decreased colocalization was also observed for Ago2 Y529F, an observation that is consistent with a previous report from this laboratory (Pare et al. 2011). Interestingly, very little of the Ago2 S798D mutant was found to associate with P-bodies, whereas the Ago2 T303D mutant showed a small, yet significant, increase in colocalization with P-bodies compared with wild-type Ago2 (Fig. 1B). In contrast to a previous report (Zeng et al. 2008), we found that changing serine 387 to alanine (Ago2 S387A) had no effect on localization of the Ago2 to P-bodies. This may be due to the fact that different methodologies were used to determine the extent of colocalization to P-bodies. Zeng et al. (2008) used the H1299 cell line and manually counted “Flag-Ago2-stained” P-bodies in 40 cells. In contrast, we used HeLa cells, and relied on automated analysis software to count the number of myc-Ago2-positive P-bodies (as indicated by colocalization with costained Dcp1a) in a minimum of 500 cells.
For the most part, expression of each of the 14 phospho-mutants was comparable to that of exogenously expressed wild-type myc-Ago2 and, in all cases, higher than endogenous Ago2 (Supplemental Fig. 1). As such, the observed decreased P-body localization of Ago2 S798D was not due to lower expression. Moreover, expression levels of two other myc-tagged mutants (Ago2 S798A and Ago2 Y393E) were routinely lower than Ago2 S798D; however, no appreciable decrease in granule localization was observed in these cases.
Similar to P-bodies, localization of Ago2 to stress granules requires small RNA binding (Pare et al. 2011). It is therefore thought that targeting of Ago2 to these structures is also a consequence of its role in RNAi and is mediated by its interaction with targeted mRNAs. We next sought to determine whether recruitment of Ago2 to stress granules was affected by mutation of phosphoamino acid residues. Transfected HeLa cells expressing myc-tagged Ago2 mutants were used in a similar approach as above except that 45 min before fixation cells were treated with sodium arsenite (500 µM), an oxidative stressor, to induce the formation of stress granules. Cells were then processed for indirect fluorescence with antibodies to myc and TIA-1, a stress granule resident protein (Kedersha et al. 1999). Computational analysis of the data showed that, as with targeting to P-bodies, recruitment of Ago2 PAZ9, Ago2 Y529E, Ago2 Y529F, and the Ago2 S798D mutants to stress granules was significantly impaired (Fig. 1C). Similar to what was observed for Ago2 T303D and P-bodies, targeting of Ago2 S387D to TIA-1-positive stress granules was slightly, but significantly, increased compared with wild-type Ago2. We were able to rule out the trivial possibility that decreased localization of Ago2 mutants to RNA granules resulted from loss of these structures because the total number of granules was unchanged in cells overexpressing wild-type or mutant Ago2 (Supplemental Fig. 2).
The aberrant localization of Ago2 S798D (Fig. 1B,C) was confirmed using confocal microscopy. HeLa cells were transfected with plasmids expressing myc-Ago2 WT, 3xmyc-Ago2 PAZ9, or myc-Ago2 S798D. A plasmid expressing DsRed-tagged TIA-1 was cotransfected into cells treated with arsenite for 45 min before fixation, while endogenous Dcp1a was used as a marker for P-bodies. Ago2 PAZ9 and Ago2 S798D were distributed diffusely throughout the cytoplasm and did not overlap with Dcp1a-positive P-bodies or TIA-1-positive stress granules (Fig. 1D,E). We also validated the localization data of the remaining mutants by transfecting plasmids expressing all 14 myc-tagged Ago2 mutants into HeLa cells that were subsequently treated with or without arsenite before fixation. Endogenous Dcp1a was used as a marker for P-bodies, whereas endogenous TIA-1 was used to detect stress granules. Together, results from these experiments confirmed that Y529F, Y529E, and S798D are the only phospho-mutants of Ago2 with marked localization defects (Supplemental Fig. 3).
Phospho-mimetic substitution at position 798 does not affect interaction of Ago2 with mature miRNAs or RISC-loading complex components Dicer and Hsp90
In some cases, point mutations can have profound effects on the folding of proteins, resulting in altered three-dimensional structures. To address the possibility that loss of RNA granule targeting was due to Ago2 S798D being misfolded, we assessed its ability to interact with well-characterized protein (Hsp90 and Dicer) and nucleic acid (miRNAs) binding partners. HeLa cells were transfected with plasmids encoding myc-tagged Ago2 (wild type, PAZ9, S798A, or S798D) and then processed for coimmunoprecipitation and immunoblotting. Data in Figure 2A show that both Hsp90 and Dicer coimmunoprecipitated with the Ago2 S798D mutant. Levels of coimmunoprecipitated Hsp90 and Dicer were normalized to the amount of Ago2 bound by the anti-myc beads. The relative amounts of Hsp90 and Dicer detected in the coimmunoprecipitates are shown in Figure 2B. Similar to Ago2 PAZ9, significantly more Hsp90 and Dicer were bound to Ago2 S798D than to wild-type Ago2. Moreover, we demonstrated that wild type Ago2, Ago2 S798A, and Ago2 S798D all formed stable complexes with Dcp1a. (Supplemental Fig. 4). However, the significance of this interaction remains unclear as Ago2 PAZ9, which does not associate with P-bodies, also binds Dcp1a (Supplemental Fig. 4; Liu et al. 2005). Furthermore, it has been shown that Ago2 Y529E, despite being functionally inactive and not localized to granules, interacts with GW182, a P-body resident protein and critical component to the RNAi-mediated silencing pathway (Rudel et al. 2011). One interpretation of the results in Figure 2, A and B, is that the transfer of the miRNA from Dicer to Argonaute triggers the dissolution of the RISC-loading complex; therefore, Ago2 mutants that cannot be efficiently loaded with miRNA, dwell in complex with Dicer and the loading machinery. In contrast to Ago2 S798D, the amount of Dicer and Hsp90 associated with Ago2 S798A was not significantly different from wild-type Ago2. Given that Ago2 S798D interacts with two critical proteins (Dicer and Hsp90) involved in RISC-loading, as well as Dcp1a, we conclude that this mutant is not misfolded.
FIGURE 2.
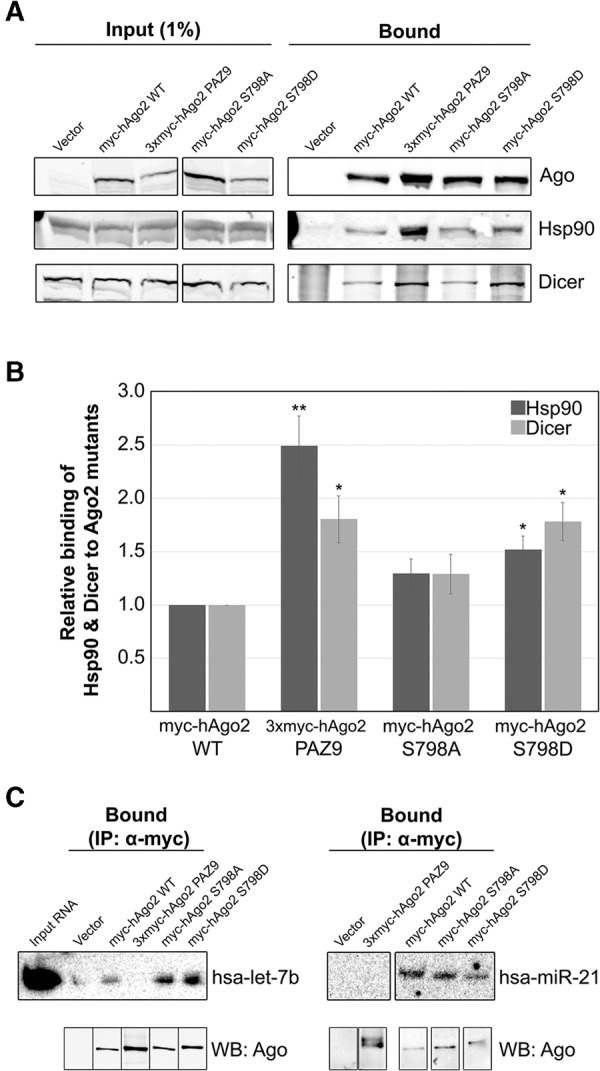
Interaction of myc-hAgo2 S798D with components of the RISC-loading complex and small RNAs. (A) HeLa cells transiently transfected with plasmids encoding wild-type or mutant Ago2 were lysed 16 h post-transfection and subjected to immunoprecipitation using sepharose beads coated with anti-myc antibodies. Total cell lysate (Input) and bound fractions were subjected to SDS-PAGE and immunoblotted for Ago2, Hsp90, and Dicer as indicated. (B) Signal intensities for Hsp90 and Dicer were quantitated and normalized to level of immunoprecipitated Ago2. The bar graph indicates the fold change in the amount of each coimmunoprecipitated Ago2-binding partner relative to the amount associated with wild-type Ago2. Paired Student's two-tailed t-test was used to compare amounts of coimmunoprecipitated Hsp90 and Dicer to amounts bound to wild-type Ago2, (*) P < 0.05; (**) P < 0.01. Error bars indicate SE. (C) HeLa cells transiently transfected with plasmids expressing wild-type or mutant Ago2 were lysed 16 h post-transfection and subjected to immunoprecipitation using sepharose beads coated with monoclonal anti-myc antibodies. RNA was extracted from the bound fractions (IP α-myc) and subjected to UREA-PAGE. Cells transfected with empty vector were used as the negative control for immunoprecipitation (labeled “Vector”). Loading of RNA was normalized to level of bound Ago2 (immunoblot of 3% of immunoprecipitated protein shown below). MiRNAs detected by Northern blotting using 32P-labeled oligonucleotide probes. Total RNA isolated from cell lysate included as a control (Input RNA).
The localization defect associated with Ago2 S798D (Fig. 1) closely resembles that of two other Ago2 mutants (Ago2 PAZ9 and Ago2 Y529E), which are unable to bind to small RNAs. Moreover, both S798D and PAZ9 Ago2 mutants bind significantly more Hsp90 and Dicer than wild-type Ago2. Analysis of the crystal structure of Ago2 in complex with a miRNA suggests that serine-798 interacts with the phosphate backbone at position 5 of the miRNA (Schirle and MacRae 2012). According to this structure, serine-798 is predicted to have limited surface availability, which would restrict access by protein kinases. However, tyrosine-529 has similar predicted surface availability and increasing evidence indicates that it is phosphorylated in vivo (Rudel et al. 2011; Mazumder et al. 2013). This may indicate that Ago2 adopts a conformation where one or both of these amino acid residues are accessible to kinases. Based on these data, we predicted that Ago2 S798D would not bind small RNAs because of electrostatic repulsion. To test this, the relative levels of two endogenous miRNAs (hsa-let-7b and hsa-miR-21) bound to myc-tagged wild-type and mutant Ago2 proteins were determined by coimmunoprecipitation and Northern blotting. Data in Figure 2C revealed, unexpectedly, that Ago2 S798D behaves similarly to wild-type Ago2 in binding endogenous mature miRNAs. These observations appear to rule out the possibility that at position 798 (unlike at position 529) the presence of a large, negative charge serves to prevent binding of the small RNA. Although it seems unlikely, we cannot rule out the possibility that the substituted aspartic acid at position 798 is differentially aligned within the structure of Ago2 than a phosphoserine at this position, thereby allowing for the interaction with the negatively charged phosphate backbone of the small RNA, whereas phosphorylation of the native serine residue may have more significant effects on small RNA binding. Irrespective of the mechanism, to our knowledge, this is the first mutation of Ago2 that uncouples miRNA binding from targeting to cytoplasmic RNA granules.
Silencing activity of Ago2 is mildly affected by phospho-mimetic mutation of Serine-798
Given the evidence linking Ago2 localization to cytoplasmic RNA granules and gene-silencing function, we next explored the consequence of mutating phosphoamino acid residues on the activity of Ago2. To measure endonuclease-dependent silencing activity, we constructed a reporter (dsGFP-miR21) encoding destabilized GFP (dsGFP) and a single perfectly complementary binding site to mouse miR-21 (Fig. 3A), a miRNA that is highly expressed in MEFs. A plasmid encoding dsGFP and an untargeted 3′ UTR site (dsGFP-nsc), was used as a negative control. MEF cells lacking the gene encoding Ago2 (Liu et al. 2004) were electroporated with plasmids encoding either wild-type Ago2 or the phospho-mutants; DsRed-C1-Monomer; and dsGFP-miR21 reporter (or dsGFP-nsc control). The molar ratios of plasmids in the electroporation ensured that all DsRed-expressing cells were cotransfected with Ago2 and the dsGFP-based reporters (data not shown). Expression of dsGFP and DsRed was determined by flow cytometry 24 h after electroporation, followed by normalization of dsGFP against DsRed. GFP expression is represented as a percent of the total expression of the dsGFP-nsc control. As expected, cotransfection of the plasmid expressing wild-type Ago2 caused a 91% reduction in the fluorescence of cells expressing dsGFP-miR21 (Fig. 3B). Levels of dsGFP expression in cells transfected with plasmids encoding the slicer-dead (Ago2 H634P) or small RNA-binding mutants (Ago2 PAZ9 and Ago2 Y529E) of Ago2 were not significantly different from those expressing vector alone. Paired sample t-tests were performed, confirming that the difference in GFP expression, relative to wild-type Ago2, was significant for Ago2 H634P, Ago2 PAZ9, Ago2 Y529E, and Ago2 S798D. However, although the decrease in slicer-dependent silencing efficiency of Ago2 S798D was significantly different from wild-type Ago2, it was not as impaired as the Ago2 H634P, Ago2 PAZ9, or Ago2 Y529E mutants. That is, silencing activity of Ago2 S798D was reduced less than twofold compared with approximately sixfold for the silencing null mutants, Ago2 PAZ9, and Ago2 H634P (Fig. 3B). The differences in silencing activity between wild-type Ago2 and Ago2 S798D proteins were not specific to myc-tagged variants, as we observed similar differences with DsRed-tagged variants (data not shown).
FIGURE 3.

Efficiency of myc-Ago2 phospho-mutants in slicer-dependent and slicer-independent silencing assays. (A) Schematic representation of dsGFP-miR21 reporter used to assay slicer-dependent silencing activity. (B) Ago2−/− mouse embryonic fibroblast cells were transiently electroporated with plasmids expressing wild-type or mutant Ago2 alongside dsGFP-miR21 (or dsGFP-nsc as a control), and pDsRed-C1-monomer. GFP expression was determined by flow cytometry and normalized against DsRed expression. The bar graph represents the expression of dsGFP-miR21 relative to dsGFP-nsc for each Ago2 construct. (C) Schematic representation of dsGFP-Nonslicer reporter used to assay slicer-independent silencing activity. (D) Ago2−/− mouse embryonic fibroblast cells were transiently electroporated with plasmids expressing wild-type or mutant Ago2 alongside dsGFP-NonSlicer (or dsGFP-nsc as a control), and pDsRed-C1-monomer. Cells were then transfected with Dicer-substrate siRNA. GFP expression was determined by flow cytometry and normalized against DsRed expression. The bar graph represents the expression of dsGFP-NonSlicer relative to dsGFP-nsc for each Ago2 construct. Paired Student's two-tailed t-test was used to compare amounts of dsGFP expression in cells cotransfected with mutant Ago2 constructs to those cotransfected with wild-type Ago2, (*) P < 0.05; (**) P < 0.01. Error bars indicate SE.
We also constructed a reporter plasmid containing a synthetic, 107-bp 3′ UTR (dsGFP-Nonslicer; Fig. 3C) containing three binding sites for a Dicer-substrate siRNA engineered not to target any mammalian (including murine) genomic sequences. To prevent Ago2-mediated cleavage of the mRNA target, the binding sites within the synthetic 3′ UTR were mismatched at positions 9 and 10 (Bartel 2004). Finally, the binding sites within the 3′ UTR were spaced in a manner to bias nonslicer silencing by Ago2 (Broderick et al. 2011).
Using the same experimental conditions described above, followed by transfection of the Dicer-substrate siRNA, we assessed the ability of the Ago2 phospho-mutants to repress translation without cleaving the targeted mRNA. Following normalization to DsRed expression, expression of dsGFP from the dsGFP-Nonslicer was determined relative to expression from the nonsilenced control plasmid (Fig. 3D). Consistent with our expectations, coexpression of Ago2 PAZ9 or Ago2 Y529E did not cause an appreciable decrease in fluorescence compared with vector alone. That is, Ago2 PAZ9 and Ago2 Y529E were relatively ineffective in suppressing dsGFP-Nonslicer translation, in contrast to exogenously expressed wild-type Ago2 (Fig. 3D). The mean GFP fluorescence in cells expressing the Ago2 phospho-mutants was somewhat variable, likely because of silencing functions of the three remaining endogenous Ago proteins 1, 3, and 4. Despite this variability, Ago2 S798D was the only other mutant whose silencing activity was statistically different from wild-type Ago2.
In contrast to what we observed in the slicer-dependent silencing assay, the activity of Ago2 S798D in the slicer-independent silencing assay was slightly increased relative to wild-type Ago2 (Fig. 3D). A recent study showed that phosphorylation at tyrosine-393 impairs maturation of long-loop miRNAs (Shen et al. 2013). However, because neither miR-21 nor the Dicer-substrate siRNA are of this type, it was not unexpected that Ago2 Y393E exhibited normal silencing activity in our assays. Despite the low electroporation efficiency and the presence of Agos 1, 2, and 3 in MEFs, we were able to detect the gene-silencing activity of ectopically expressed Ago2 in our assays (Supplemental Fig. 5).
The opposing effect of substituting an aspartic acid for serine-798 on slicer-dependent versus slicer-independent silencing somewhat mirrors, albeit with lessened effect, the observations of Zeng et al. (2008) for serine-387 mutations. Phosphorylation of serine-798 could be a mechanism by which the silencing preference of Ago2 is shifted toward translational repression rather than slicer activity. However, the effect of the S798D substitution on the silencing activity was relatively small. Whether this is due to its altered subcellular localization and/or subtle changes in small RNA-binding activity is not known at this point. It is possible that the high concentration of the small RNAs used in the reporter assays (i.e., miR-21 and the synthetic Dicer-substrate siRNA) partially compensated for subtle RNA binding, or slicer-mediated silencing, defects of Ago2 S798D. Nevertheless, the striking difference in localization of Ago2 S798D compared with wild-type Ago2 makes it tempting to speculate that dynamic phosphorylation of serine-798 is a mechanism for regulating the association of Ago2 with cytoplasmic RNA granules, either directly or by promoting interaction with binding partners that prevent its association with those structures. To this end, future work in identifying the kinase(s) responsible for the phosphorylation of serine-798 will undoubtedly improve our understanding of this means of regulating Argonaute activity. Analysis of the amino acid sequences surrounding serine-798, which is highly conserved among vertebrates, suggests that this amino acid residue is a substrate for AKT and/or protein kinase A (http://scansite.mit.edu/). The observation that Ago2 S798D binds miRNAs normally and maintains translational repression activity suggests that neither miRNA-binding nor translational suppression activity is sufficient for targeting Ago2 to cytoplasmic RNA granules. Indeed, association with these granules may reflect a mechanism that negatively regulates the translational repression activity of Ago2, and that phosphorylation of serine-798 prevents this inhibition, allowing the protein to remain active in the cytoplasm.
MATERIALS AND METHODS
Antibodies
Mouse monoclonal anti-myc (9E10) was prepared from a hybridoma cell line obtained from the American Type Culture Collection (ATCC). The rabbit polyclonal antibody (2D4) was generated against the PAZ domain of Ago2 in this laboratory (Tahbaz et al. 2004). Mouse monoclonal anti-Dicer (ab14601) and rabbit monoclonal anti-Dcp1-a (ab47811) were from Abcam. Goat polyclonal anti-HSP90 (sc-1055) and goat anti-T-cell-restricted intracellular antigen (TIA-1) (sc-1751) were from Santa Cruz Biotechnology. Donkey anti-mouse conjugated to Alexa 680 (A10038), donkey anti-goat conjugated to Alexa 680 (A21084), donkey anti-mouse conjugated to Alexa 488 (A21202), donkey anti-rabbit conjugated to Alexa 647 (A31573), and chicken anti-goat conjugated to Alexa 647 (A21469) were purchased from Life Technologies. Donkey anti-rabbit conjugated to IR-Dye 800 was purchased from Li-Cor Biosciences.
Plasmid construction
Myc-Ago2 WT construction was described previously in Pare et al. (2011). The Ago2 mutants H634P and PAZ9 were obtained from Dr. G.J. Hannon (Cold Spring Harbor Laboratory). All 14 Ago2 phospho-mutants were created from a myc-tagged Ago2 wild-type construct using the QuickChange site-directed mutagenesis kit from Agilent technologies using primers described in Supplemental Table 1.
The plasmids pcDNA-5/TO and pDsRed-Monomer-C1 were purchased from Life Technologies and Clontech Laboratories, respectively. Construction of pDsRed-TIA-1 was described previously (Pare et al. 2009).
Construction of the plasmid encoding destabilized GFP (dsGFP), a gift from Dr. C.S. Sullivan, was previously described (Sullivan and Ganem 2005). dsGFP-mir21 was constructed by ligating the duplex (5′-AATTCTCAACATCAGTCTGATAAGCTAGA; 5′-AATTCTAGCTTATCAGACTGATGTTGAG) into dsGFP digested with EcoR1. dsGFP-nsc was constructed by ligating the duplex (5′-AATTCGTCGTTAATCGCGTATAATACAGATCTG; 5′-AATTCAGATCTGTATTATACGCGATTAACGACG) into dsGFP digested with EcoR1. dsGFP-Nonslicer was constructed by ligating the duplex (5′-AATTCCGTATTATATTGATTAACGAAACGGGACGGCGCACGCGCGTATTATATTGATTAACGAAACGGGACGGCGCACGCGCGTATT-ATATTGATTAACGGGGCCAGATCTG; 5′-AATTCAGATCTGGCCCCGTTAATCAATATAATACGCGCGTGCGCCGTCCCGTTT-CGTTAATCAATATAATACGCGCGTGCGCCGTCCCGTTTCGTTAATCAATATAATACGG) into dsGFP digested with EcoR1.
Cell culture and transfections
HeLa cells (ATCC) and mouse embryonic fibroblasts (MEFs) containing a homozygous deletion for Ago2 (a gift from Greg Hannon, Cold Spring Harbor) were cultured in Dulbecco's Modified Eagle Medium containing 10% heat-inactivated fetal bovine serum, 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), and 2.5 µg/mL Plasmocin (InvivoGen) at 37°C in a 5% CO2 atmosphere.
Electroporation was used to introduce plasmid DNA into Ago2−/− MEF cells. Briefly, 1 × 106 cells were resuspended in 25 mM HEPES (pH 7.6), 10 mM KH2PO4, 120 mM KCl, 0.15 mM CaCl2, 2 mM ethylene glycol tetraacetic acid (EGTA), 5 mM MgCl2, 5 mM adenosine triphosphate (ATP), and 2 mM l-glutamine and electroporated using 2 mm cuvettes (Bio-Rad) at 260 V, 500 μF. DsiRNA was transfected using RNAiMax (Life Technologies) following manufacturer's instructions.
HeLa cells were transfected with plasmid DNA using Lipofectamine 2000 (Life Technologies) following manufacturer's protocol in OptiMEM media (Life Technologies).
Immunofluorescence
Transfected cells were grown on coverslips in six-well plates or clear-bottom 96 µClear Greiner plates (Sigma-Aldrich), and then washed twice with 2 mL or 200 µL of phosphate-buffered saline (PBS), respectively. Cells were fixed using 2% paraformaldehyde for 10 min, and then permeabilized with 0.2% Triton X-100 in PBS for 4 min. Samples were blocked for 30 min using 5% skim milk in PBS before incubation with primary antibodies overnight at 4°C. Following two 10-min washes with PBS, slides were incubated with secondary antibodies for 1 h. 4′,6-diamidino-2-phenylindole (DAPI) (Life Technologies) was then added at 1 μg/mL in PBS for 10 min followed by mounting with ProLong Antifade Kit (Life Technologies). Samples were imaged on an Olympus IX-81 motorized microscope (Olympus) equipped with a spinning disk confocal unit (CSU X1, Yokogawa Electric Corporation) Images were acquired with a Hamamatsu EMCCD (C9100-13) (Hamamatsu Photonics) digital camera and analyzed using Volocity software (PerkinElmer Life and Analytical Sciences).
Laser scanning cytometry and image analysis
High-content quantitative imaging of Ago2 mutants in transfected HeLa cells was performed using a laser scanning cytometer system equipped with 405-, 488-, 561-, and 640-nm lasers, iBrowser analysis software (Thorlabs Inc.), and a 40× objective (0.45 NA) at a step size of 0.25 µm and a composite image composed of either 81 (9 × 9) or 100 (10 × 10) fields. Voltages were set allowing for a background pixel intensity of between 200 and 500 to limit saturation of the images. The cells were identified using the nuclear stain DAPI and the localization of myc-tagged Ago2 relative to P-bodies and stress granules was determined using object-based segmentation using iCys software (Thorlabs Inc.). Data were represented by plotting the integrated signals of myc-Ago2 integral versus that of P-bodies and stress granules. Background fluorescence levels were set by measuring the signals from cells transfected with the empty vector pcDNA-5/TO. At least 500 transfected cells were analyzed for each mutant or control. Paired sample t-test was used to determine the statistical significance of differences.
Flow cytometry
Ago2−/− MEFs were electroporated with DsRed-Monomer, myc-Ago2, and dsGFP reporter plasmids using a molar ratio of 1:4:16. Twenty-four hours post-electroporation, cells were harvested from plates by trypsinization followed by centrifugation at 1000g after which the cells were resuspended in PBS with 10% FBS and 5 mM EDTA. Samples were analyzed using a BD LSRFortessa cell analyzer and FACSDiva Software (BD Biosciences). After gating a minimum of 1000 DsRed-monomer-positive cells, the mean GFP fluorescence of this population was recorded. Background fluorescence was set using cells electroporated with the empty vector (pcDNA-5/TO). Cell doublets were excluded from the analyses by using a forward-scatter-width-discrimination.
Immunoprecipitation and immunoblotting
Protein G-Sepharose 4 fast flow beads (GE Healthcare) were washed twice with PBS, and then incubated at room temperature for 1 h with anti-myc (9E10) antibody followed by washing once with 10 volumes of 100 mM sodium borate. The cross-linking reagent dimethylpimelimidate (20 mM) was added to the beads for 30 min at room temperature, followed by two washes with 10 volumes of 200 mM ethanolamine. Beads were then incubated at room temperature with 200 mM ethanolamine for 2 h before washing three times with 10 volumes of PBS. Finally, beads were stored as a 50% slurry in PBS at 4°C until use in immunoprecipitation experiments.
Transfected cells were lysed for 10 min at 4°C with rotation in IP lysis buffer: 1% Triton X-100 buffer containing 1 mM MgCl2, 20 mM NaCl, and 50 mM Tris–HCl (pH 7.2) supplemented with 1× Complete protease inhibitor (Roche). Following centrifugation at 15,000g, anti-myc beads were added to the supernatants and rotated at 4°C for 1 h. Beads were then washed once with lysis buffer and Ago2-complexes were eluted by boiling in 2× Laemmli sample buffer (4% SDS, 20% glycerol, and 120 mM TRIS–HCl [pH 6.8]). Samples were resolved on 8% acrylamide gels by SDS-PAGE and transferred to nitrocellulose membranes (Bio-Rad). Membranes were blocked in 5% skim milk in PBS and then incubated with primary antibodies diluted in 1% bovine serum albumin in PBS containing 0.1% Tween-20 (PBS-T) overnight at 4°C. After several washes with PBS-T, blots were incubated with fluorophore-conjugated secondary antibodies for 1 h and washed in PBS-T. Membranes were scanned using an Odyssey 9120 infrared imaging system (Li-Cor Biosciences).
Northern blots
Total RNA was resolved by electrophoresis using 15% acrylamide gels (19:1 acrylamide/bisacrylamide) containing 8 M urea before transfer to GeneScreen Plus membrane (PerkinElmer). The membranes were probed in UltraHyb Oligo buffer (Life Technologies) at 42°C with 5′-[32P]-end-labeled DNA oligonucleotides with exact complementarity to the mature miRNA sequences. U6 was probed as a loading control. The expression of miRNAs was quantified using ImageQuant software (GE Health Sciences).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
Technical support was provided by Eileen Reklow and Valeria Mancinelli. This work was funded by grants from the Canadian Breast Cancer Foundation (Prairies/NWT Region) and the Alberta Cancer Foundation awarded to T.C.H. T.C.H. is a Canada Research Chair. J.L.-O. was supported by a graduate studentship award from Alberta Innovates-Health Solutions.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.053207.115.
REFERENCES
- Bartel DP. 2004. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116: 281–297. [DOI] [PubMed] [Google Scholar]
- Behm-Ansmant I, Rehwinkel J, Doerks T, Stark A, Bork P, Izaurralde E. 2006. mRNA degradation by miRNAs and GW182 requires both CCR4:NOT deadenylase and DCP1:DCP2 decapping complexes. Genes Dev 20: 1885–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broderick JA, Salomon WE, Ryder SP, Aronin N, Zamore PD. 2011. Argonaute protein identity and pairing geometry determine cooperativity in mammalian RNA silencing. RNA 17: 1858–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu CY, Rana TM. 2006. Translation repression in human cells by microRNA-induced gene silencing requires RCK/p54. PLoS Biol 4: e210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eulalio A, Behm-Ansmant I, Izaurralde E. 2007a. P bodies: at the crossroads of post-transcriptional pathways. Nat Rev Mol Cell Biol 8: 9–22. [DOI] [PubMed] [Google Scholar]
- Eulalio A, Behm-Ansmant I, Schweizer D, Izaurralde E. 2007b. P-body formation is a consequence, not the cause, of RNA-mediated gene silencing. Mol Cell Biol 27: 3970–3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franks TM, Lykke-Andersen J. 2008. The control of mRNA decapping and P-body formation. Mol Cell 32: 605–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman RC, Farh KK, Burge CB, Bartel DP. 2009. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 19: 92–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horman SR, Janas MM, Litterst C, Wang B, MacRae IJ, Sever MJ, Morrissey DV, Graves P, Luo B, Umesalma S, et al. 2013. Akt-mediated phosphorylation of argonaute 2 downregulates cleavage and upregulates translational repression of MicroRNA targets. Mol Cell 50: 356–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iki T, Yoshikawa M, Meshi T, Ishikawa M. 2012. Cyclophilin 40 facilitates HSP90-mediated RISC assembly in plants. EMBO J 31: 267–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki S, Kobayashi M, Yoda M, Sakaguchi Y, Katsuma S, Suzuki T, Tomari Y. 2010. Hsc70/Hsp90 chaperone machinery mediates ATP-dependent RISC loading of small RNA duplexes. Mol Cell 39: 292–299. [DOI] [PubMed] [Google Scholar]
- Izumi N, Kawaoka S, Yasuhara S, Suzuki Y, Sugano S, Katsuma S, Tomari Y. 2013. Hsp90 facilitates accurate loading of precursor piRNAs into PIWI proteins. RNA 19: 896–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedersha NL, Gupta M, Li W, Miller I, Anderson P. 1999. RNA-binding proteins TIA-1 and TIAR link the phosphorylation of eIF-2 α to the assembly of mammalian stress granules. J Cell Biol 147: 1431–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirino Y, Kim N, de Planell-Saguer M, Khandros E, Chiorean S, Klein PS, Rigoutsos I, Jongens TA, Mourelatos Z. 2009. Arginine methylation of Piwi proteins catalysed by dPRMT5 is required for Ago3 and Aub stability. Nat Cell Biol 11: 652–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Rocca G, Olejniczak SH, Gonzalez AJ, Briskin D, Vidigal JA, Spraggon L, DeMatteo RG, Radler MR, Lindsten T, Ventura A, et al. 2015. In vivo, Argonaute-bound microRNAs exist predominantly in a reservoir of low molecular weight complexes not associated with mRNA. Proc Natl Acad Sci 112: 767–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung AK, Vyas S, Rood JE, Bhutkar A, Sharp PA, Chang P. 2011. Poly(ADP-ribose) regulates stress responses and microRNA activity in the cytoplasm. Mol Cell 42: 489–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Carmell MA, Rivas FV, Marsden CG, Thomson JM, Song JJ, Hammond SM, Joshua-Tor L, Hannon GJ. 2004. Argonaute2 is the catalytic engine of mammalian RNAi. Science 305: 1437–1441. [DOI] [PubMed] [Google Scholar]
- Liu J, Valencia-Sanchez MA, Hannon GJ, Parker R. 2005. MicroRNA-dependent localization of targeted mRNAs to mammalian P-bodies. Nat Cell Biol 7: 719–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez NJ, Chang HM, Borrajo Jde R, Gregory RI. 2013. The co-chaperones Fkbp4/5 control Argonaute2 expression and facilitate RISC assembly. RNA 19: 1583–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazumder A, Bose M, Chakraborty A, Chakrabarti S, Bhattacharyya SN. 2013. A transient reversal of miRNA-mediated repression controls macrophage activation. EMBO Rep 14: 1008–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi T, Takeuchi A, Siomi H, Siomi MC. 2010. A direct role for Hsp90 in pre-RISC formation in Drosophila. Nat Struct Mol Biol 17: 1024–1026. [DOI] [PubMed] [Google Scholar]
- Pare JM, Tahbaz N, Lopez-Orozco J, LaPointe P, Lasko P, Hobman TC. 2009. Hsp90 regulates the function of Argonaute 2 and its recruitment to stress granules and P-bodies. Mol Biol Cell 20: 3273–3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pare JM, Lopez-Orozco J, Hobman TC. 2011. MicroRNA-binding is required for recruitment of human Argonaute 2 to stress granules and P-bodies. Biochem Biophys Res Commun 414: 259–264. [DOI] [PubMed] [Google Scholar]
- Pare JM, LaPointe P, Hobman TC. 2013. Hsp90 cochaperones p23 and FKBP4 physically interact with hAgo2 and activate RNA interference-mediated silencing in mammalian cells. Mol Biol Cell 24: 2303–2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi HH, Ongusaha PP, Myllyharju J, Cheng D, Pakkanen O, Shi Y, Lee SW, Peng J, Shi Y. 2008. Prolyl 4-hydroxylation regulates Argonaute 2 stability. Nature 455: 421–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudel S, Wang Y, Lenobel R, Korner R, Hsiao HH, Urlaub H, Patel D, Meister G. 2011. Phosphorylation of human Argonaute proteins affects small RNA binding. Nucleic Acids Res 39: 2330–2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybak A, Fuchs H, Hadian K, Smirnova L, Wulczyn EA, Michel G, Nitsch R, Krappmann D, Wulczyn FG. 2009. The let-7 target gene mouse lin-41 is a stem cell specific E3 ubiquitin ligase for the miRNA pathway protein Ago2. Nat Cell Biol 11: 1411–1420. [DOI] [PubMed] [Google Scholar]
- Schirle NT, MacRae IJ. 2012. The crystal structure of human Argonaute2. Science 336: 1037–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo GJ, Kincaid RP, Phanaksri T, Burke JM, Pare JM, Cox JE, Hsiang TY, Krug RM, Sullivan CS. 2013. Reciprocal inhibition between intracellular antiviral signaling and the RNAi machinery in mammalian cells. Cell Host Microbe 14: 435–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Xia W, Khotskaya YB, Huo L, Nakanishi K, Lim SO, Du Y, Wang Y, Chang WC, Chen CH, et al. 2013. EGFR modulates microRNA maturation in response to hypoxia through phosphorylation of AGO2. Nature 497: 383–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan CS, Ganem D. 2005. A virus-encoded inhibitor that blocks RNA interference in mammalian cells. J Virol 79: 7371–7379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahbaz N, Carmichael JB, Hobman TC. 2001. GERp95 belongs to a family of signal-transducing proteins and requires Hsp90 activity for stability and Golgi localization. J Biol Chem 276: 43294–43299. [DOI] [PubMed] [Google Scholar]
- Tahbaz N, Kolb FA, Zhang H, Jaronczyk K, Filipowicz W, Hobman TC. 2004. Characterization of the interactions between mammalian PAZ PIWI domain proteins and Dicer. EMBO Rep 5: 189–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dijk E, Cougot N, Meyer S, Babajko S, Wahle E, Seraphin B. 2002. Human Dcp2: a catalytically active mRNA decapping enzyme located in specific cytoplasmic structures. EMBO J 21: 6915–6924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Mercier R, Hobman TC, LaPointe P. 2013. Regulation of RNA interference by Hsp90 is an evolutionarily conserved process. Biochim Biophys Acta 1833: 2673–2681. [DOI] [PubMed] [Google Scholar]
- Wu C, So J, Davis-Dusenbery BN, Qi HH, Bloch DB, Shi Y, Lagna G, Hata A. 2011. Hypoxia potentiates microRNA-mediated gene silencing through post-translational modification of Argonaute2. Mol Cell Biol 31: 4760–4774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Y, Sankala H, Zhang X, Graves PR. 2008. Phosphorylation of Argonaute 2 at serine-387 facilitates its localization to processing bodies. Biochem J 413: 429–436. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.