

Abstract
HnRNP L is a ubiquitous splicing-regulatory protein that is critical for the development and function of mammalian T cells. Previous work has identified a few targets of hnRNP L-dependent alternative splicing in T cells and has described transcriptome-wide association of hnRNP L with RNA. However, a comprehensive analysis of the impact of hnRNP L on mRNA expression remains lacking. Here we use next-generation sequencing to identify transcriptome changes upon depletion of hnRNP L in a model T-cell line. We demonstrate that hnRNP L primarily regulates cassette-type alternative splicing, with minimal impact of hnRNP L depletion on transcript abundance, intron retention, or other modes of alternative splicing. Strikingly, we find that binding of hnRNP L within or flanking an exon largely correlates with exon repression by hnRNP L. In contrast, exons that are enhanced by hnRNP L generally lack proximal hnRNP L binding. Notably, these hnRNP L-enhanced exons share sequence and context features that correlate with poor nucleosome positioning, suggesting that hnRNP may enhance inclusion of a subset of exons via a cotranscriptional or epigenetic mechanism. Our data demonstrate that hnRNP L controls inclusion of a broad spectrum of alternative cassette exons in T cells and suggest both direct RNA regulation as well as indirect mechanisms sensitive to the epigenetic landscape.
Keywords: alternative splicing, functional genomics, hnRNP L, RNA-seq, RASL-seq
INTRODUCTION
Alternative pre-mRNA splicing is a nearly ubiquitous mechanism by which eukaryotic cells generate multiple protein-coding mRNAs from a single genetic locus (Nilsen and Graveley 2010). As many as 95% of human multiexon genes generate more than one processed product by alternative pre-mRNA splicing (Pan et al. 2008; Wang et al. 2008). Alternative mRNA isoforms may differ in open reading frames, thus impacting the function of the encoded protein, or may alter the presence of regulatory sequences in untranslated regions (UTRs) thereby influencing RNA stability, transport, or translational control (Nilsen and Graveley 2010). Importantly, tissue-specific differences in alternative splicing (AS) have been suggested to shape cell-fate decisions (Gehman et al. 2012; Licatalosi et al. 2012; Xue et al. 2013; Raj et al. 2014; Quesnel-Vallieres et al. 2015), while signal-induced changes in AS have been observed in response to metabolic, neuronal, or immune cues (Patel et al. 2001; Xie and Black 2001; Shin and Manley 2004; An and Grabowski 2007; Lynch 2007; Heyd and Lynch 2011; Martinez et al. 2012; Fu and Ares 2014). Moreover, mutations that alter splicing have been causally linked to cancer, neurodegenerative diseases, and autoimmune disorders, among others (Jacobsen et al. 2000; Cartegni and Krainer 2002; Cooper et al. 2009; David and Manley 2010; Xiong et al. 2014).
Given the prevalence and importance of alternative splicing, it is critical to understand the factors and mechanisms that dictate alternative splicing patterns. The process of splicing itself requires multiple RNA–RNA, RNA–protein, and protein–protein interactions to identify splice sites and join them within the catalytic spliceosome complex (Wahl et al. 2009; Fu and Ares 2014). This complex assembly process provides many opportunities for regulation, both through altering the affinity of these numerous protein and RNA interactions and through control of the kinetics of spliceosome assembly at a particular site relative to competing processes (Wahl et al. 2009; Fu and Ares 2014). Typically, the study of alternative splicing has focused on the regulatory influence of trans-acting RNA-binding proteins that associate with the substrate pre-mRNA near the sites of alternative splicing. However, it is increasingly apparent that other RNA processing events such as 5′ and 3′ end formation as well as epigenetic marks and transcription elongation rate also can influence splicing patterns (Braunschweig et al. 2013; Fu and Ares 2014).
One family of RNA-binding proteins that has been strongly implicated in alternative splicing regulation is the hnRNP proteins. The hnRNP proteins were originally identified by their coassociation with pre-mRNA (Pinol-Roma et al. 1988), although these proteins share little sequence homology with one another other than containing one or more RRM or KH-type RNA-binding domains. Most hnRNP family members have been shown to regulate alternative splicing as well as other RNA processing steps such as 3′ end processing, mRNA translation, and mRNA stability (Bomsztyk et al. 2004; Martinez-Contreras et al. 2007; Licatalosi et al. 2008). Moreover, global comparisons of transcriptome-wide binding of individual hnRNPs with their effect on transcriptome expression have led to the general model that the functional consequence of a particular hnRNP on a given transcript is highly dependent on where it binds within an RNA (Licatalosi and Darnell 2009; Fu and Ares 2014; Shi and Manley 2015).
One hnRNP for which global comparison of binding and function is still lacking, however, is hnRNP L. HnRNP L is a highly abundant nuclear protein comprised of four RRM domains, a glycine-rich N-terminal domain and a proline-rich sequence separating RRMs 2 and 3 (Shankarling and Lynch 2013). Although ubiquitously expressed, hnRNP L is critical for the proper development and function of the mammalian immune system. The first connection between hnRNP L and the immune system was the discovery that hnRNP L regulates the splicing of the gene encoding the hematopoietic-specific protein tyrosine phosphatase CD45 (Rothrock et al. 2005). HnRNP L binds to, and represses, the three variable exons of the CD45 pre-mRNA, leading to the expression of the smallest isoform of CD45 essential for maintaining T-cell homeostasis (Rothrock et al. 2005; Tong et al. 2005; Motta-Mena et al. 2010; Preussner et al. 2012). Furthermore, lymphoid-specific knockout of hnRNP L in mice causes significant defects in thymic development and reduced migration of hnRNP L−/− T cells to the periphery (Gaudreau et al. 2012). Importantly, hnRNP L is expressed at consistent levels throughout T-cell development and activation and CD45 misregulation is insufficient to account for all of the physiological defects observed in the hnRNP L−/− T cells (Gaudreau et al. 2012). Together these data indicate that hnRNP L plays a critical role throughout T-cell maturation and function via more than one target or mechanism.
A handful of additional targets of hnRNP L splicing-regulatory activity have been identified in both T cells and other cell types (Hung et al. 2008; Gaudreau et al. 2012; Shankarling et al. 2014); however, validated targets of hnRNP L splicing activity remain relatively few. Moreover, a transcriptome-wide comparison of the binding and function of hnRNP L has not been performed. A comparative study of the physical and functional targets of hnRNP L is of particular interest as studies with model substrates suggest that hnRNP L binding can either enhance or repress exon inclusion, in a manner dependent not on location but rather on splice site strength and associated proteins (Motta-Mena et al. 2010; Shankarling et al. 2014). The fact that the location of hnRNP L-RNA interaction is not sufficient to predict splicing-regulatory outcome differs from the position-dependent effects of many other hnRNPs. Thus, further investigation of the correlation, or lack thereof, between hnRNP L binding and splicing activity is predicted to reveal novel rules governing the regulation of alternative splicing.
In previous studies, we have carried out UV crosslinking and immunoprecipitation (CLIP-seq) in a cultured T-cell line (Jurkat) to identify the transcriptome-wide association of hnRNP L with RNA (Shankarling et al. 2014). Here we report the genome-wide analysis of alternative splicing and mRNA expression upon depletion of hnRNP L in this same cellular system. We identify ∼1300 targets of hnRNP L-dependent alternative splicing; however, we detect few if any mRNAs that differ in overall abundance upon hnRNP L depletion. Thus, at least in Jurkat T cells, hnRNP L appears to act as a dedicated splicing regulator, with little impact on total mRNA expression. Of further interest, upon comparing isoform expression with hnRNP L CLIP signals, we made the unanticipated discovery that while we observe both exon enhancement and repression by hnRNP L, only those exons repressed by hnRNP L exhibit significant binding of hnRNP L within or flanking the regulated exon. In contrast, exons whose inclusion is enhanced in an hnRNP L-dependent manner share common sequence features, most notably GC-rich motifs in and around the alternative exons. A class of exons flanked by short GC-rich introns have been described previously and shown to lack the clear nucleosome occupancy typically observed over exons and correlates with efficient exon inclusion (Amit et al. 2012). Indeed, our data indicate that nucleosome occupancy over hnRNP L-enhanced exons is generally less than hnRNP L-repressed or unresponsive exons. Moreover, we observe at least several instances in which nucleosome binding at hnRNP L-enhanced exons is decreased upon depletion of hnRNP L. Therefore, we propose a model in which hnRNP L regulates splicing in at least two ways; either through direct repression of exons when bound to an RNA substrate, or indirectly through exploiting the chromatin landscape.
RESULTS
HnRNP L has a widespread impact on the inclusion of cassette exons, but not intron retention or transcript abundance
HnRNP L is critical for proper T cell development and function (Gaudreau et al. 2012); however, only a relatively few confirmed targets of hnRNP L-dependent splicing regulation have been identified. To determine the full breadth of hnRNP L regulated splicing in T cells, we used complementary knockdown and sequencing approaches to generate high depth- and breadth-analysis of hnRNP L-dependent mRNA expression in Jurkat T cells. To obtain a global view of the effect of hnRNP L on the transcriptome, we analyzed poly(A)-selected mRNA from wild-type and hnRNPL-depleted Jurkat cells by paired-end mRNA sequencing (RNA-seq). For these experiments, hnRNP L was depleted by transfection with a translation-blocking antisense morpholino oligonucleotide (AMO) (Fig. 1A,B). In addition, to increase sequencing depth at known alternative splice junctions and leverage an orthogonal method for knockdown, we also performed RASL-seq of RNA from Jurkat cells depleted of hnRNP L by inducible expression of an shRNA (Fig. 1A,B). RASL-seq uses a pool of junction-specific primer pairs to interrogate and quantify changes in ∼5600 known alternative splicing events (Li et al. 2012; Zhou et al. 2012).
FIGURE 1.

Complementary high-throughput sequencing approaches identify hnRNP L-dependent alternative splicing events in Jurkat T cells. (A) Experimental design in which Jurkat T cells were depleted of hnRNP L by transfection of antisense oligonucleotides (AMO) or transduction with a lentiviral-based shRNA, then grown under unstimulated conditions or stimulated with PMA for 48 h prior to RNA and protein extraction. (B) Western blot of hnRNP L depletion by AMO or lentiviral shRNA. HnRNP A1 is used as a loading control. (C) Regression analysis comparing the ΔPSI (hnRNP L KD-WT) determined by RASL-seq versus rMATS for cassette exons queried by both methods. Only ΔPSI with P < 0.05 are shown. In C–E, the gray lines in graph indicate an absolute value of ΔPSI = 10. (D) Scatterplot of significant (P < 0.05) inclusion level changes in unstimulated and stimulated conditions, as calculated by either sequencing methods. (E) Regression analysis comparing RASL/rMATS inferred values of ΔPSI versus RT-PCR-determined values for 48 cassette exons exhibiting either stringent non-responsiveness (ΔPSI < 3, P > 0.05) or significant responsiveness (ΔPSI > 10, P > 0.05) upon hnRNP L depletion (see also Supplemental Table S4). In cases in which both rMATS and RASL-seq provide evidence of a significant change, the ΔPSI value from the RASL-seq analysis is used. (F) GO categories for genes housing cassette exons that are significantly enhanced or repressed by hnRNP L (as defined in E). Top five categories that meet the threshold of P < 0.05, fold enrichment >1.5 are shown (see also Supplemental Table S5). (G) Regression analysis comparing RPKM for introns in wild-type versus hnRNP L-depleted cells. Slope of best-fit line is 0.98 (R2 = 0.92) for unstimulated conditions and 1.08 (R2 = 0.86) for stimulated conditions. Shown are only values for ∼800 introns identified in Braunschweig et al. (2014) to be retained in 20%–80% of messages. See Supplemental Figure S1B for regression analysis of all ∼200,000 introns (slope is 1.1 [R2 = 0.99] for both cell conditions).
Both the AMO and shRNA reduced the hnRNP L protein level by over 50% (Fig. 1B), with little to no effect on the expression of the paralog hnRNP LL (Supplemental Fig. S1A). The use of two depletion methods allows us to control for technique-specific biases or off-target effects. Furthermore, in each case we analyzed hnRNP L depletion in both unstimulated (unstim) and phorbol myristal acetate (PMA)-stimulated (stim) cells, providing independent physiological conditions for the identification of hnRNP L-responsive pre-mRNA processing events (Fig. 1A). PMA-stimulation mimics T cell receptor signaling and is known to induce widespread changes in transcript abundance and alternative splicing (Ip et al. 2007; Martinez et al. 2012); however, the expression and activity of hnRNP L is not altered by PMA (Rothrock et al. 2005; Topp et al. 2008). Thus, we predict hnRNP L to have a similar impact on splicing in both unstimulated and PMA-stimulated cells, as we observed previously in our studies of CD45 (Rothrock et al. 2005; Topp et al. 2008); although the targets of hnRNP L activity may differ in these two cell states due to differences in what genes are expressed.
We first focused on the impact of hnRNP L on the splicing of cassette exons, the most common class of alternative splicing. Using the rMATS algorithm (Shen et al. 2012) to analyze the RNA-seq data, we observe 826 cassette exons that differ significantly (change in Percent Spliced In [ΔPSI] > 10, P < 0.05) upon hnRNP L depletion in unstimulated cells, while 635 cassette exons meet this threshold upon hnRNP L depletion in PMA-stimulated cells (Table 1; Supplemental Tables S1–S3). This represents ∼1%–2% of the ∼50,000 exons for which we obtained sufficient read depth to quantify inclusion (Table 1; Supplemental Tables S1, S2). Similarly, of the ∼3000 cassette exons for which we obtained >10 RASL-seq reads, 113 and 86 cassette exons exhibit a significant (P < 0.05) hnRNP L-dependent change in inclusion of at least 10% PSI in unstimulated and stimulated Jurkat cells, respectively (Table 1; Supplemental Tables S1–S3). Importantly, the statistically significant alternative splicing predictions from both experiments were well correlated in both unstimulated (P = 7.45 × 10−16) and stimulated (P = 7.41 × 10−13) conditions (Fig. 1C; Supplemental Fig. S1B), confirming that our assays have identified bona fide targets of hnRNP L-regulated splicing. The slightly dampened ΔPSI calculated by rMATS versus RASL-seq could be due either to methodology differences or to the reduced efficiency of knockdown by the AMO as compared to the shRNA. For genes expressed in both cell states we also observed a high degree of correlation between the effect of hnRNP L on a given exon (Fig. 1D), consistent with our prediction that hnRNP L-regulation of alternative cassette exon splicing is largely shared between conditions. Finally, 48 of 54 events (∼90%) tested by low-cycle RT-PCR yielded ΔPSI measurements that are highly consistent with the sequencing results (Fig. 1E; Supplemental Table S4), further demonstrating the robustness of both the RASL-seq and rMATS platforms.
TABLE 1.
Summary of transcriptome analysis
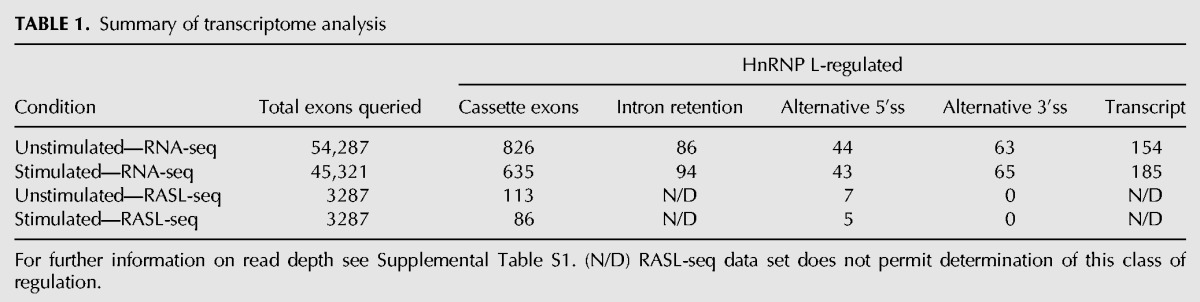
To gain initial insight into the functional impact of hnRNP L-regulated alternative splicing in Jurkat cells, we used GO analysis to identify functional categories enriched within the set of genes that contains repressed exons and the set of genes within enhanced exons. HnRNP L-enhanced exons showed little bias toward any functional category, with only weak significance observed for genes encoding proteins with RNA-binding activity (Fig. 1F; Supplemental Table S5). This is consistent with a well-reported tendency of RNA-binding proteins to be targets of splicing regulation (Lareau et al. 2007; Huelga et al. 2012). In contrast, genes containing hnRNP L-repressed exons are largely enriched in functions related to chromatin structure and transcription (Fig. 1F; Supplemental Table S5). The potential implication of this enrichment is discussed below.
Recent reports have suggested that a second class of alternative splicing, intron retention, is more abundant than previously recognized (Braunschweig et al. 2014). Furthermore, hnRNP LL, a paralog of hnRNP L, has been shown to regulate at least some intron retention events in primary mouse T cells (Cho et al. 2014). However, our rMATS analysis only predicted ∼100 intron retention events impacted by hnRNP L, many fewer than the number of cassette exons that are regulated (Table 1). Since rMATS requires a minimal number of reads to quantify splicing changes, we considered that this algorithm might miss hnRNP L-induced intron retention if the intron is never retained in wild-type cells. Therefore, we also counted the raw number of reads in wild-type and hnRNP L-depleted cells that map to a set of ∼200,000 introns defined in a recent study of intron retention (Braunschweig et al. 2014). Consistent with the rMATS data, we observe a close correlation in the number of reads that map to introns (RPKM) in wild-type and hnRNP L-depleted cells, whether we look at all introns (Supplemental Fig. S1C) or a set of ∼27,000 introns shown previously to be susceptible to retention (Fig. 1G; Braunschweig et al. 2014). Thus, while there may be isolated instances of intron removal regulated by hnRNP L, we conclude that intron retention is not broadly altered by the loss of hnRNP L. Similarly, we observe relatively few alternative 5′ and 3′ splice site-switching events that are dependent on hnRNP L (Table 1).
Finally, since many splicing factors have been shown to also influence other steps in RNA biogenesis such as transcription or mRNA stability (Braunschweig et al. 2013; Yarosh et al. 2015), we leveraged the transcriptome-wide coverage of RNA-seq data to ask if hnRNP L controls transcript abundance in addition to, or coupled with, isoform choice. We first confirmed our ability to accurately detect transcript changes in our RNA-seq data by comparing our RNA-seq data from unstimulated and PMA-stimulated wild-type cells with the limma package (Ritchie et al. 2015). Consistent with published data on gene expression during T-cell signaling (Ip et al. 2007), we observe a widespread increase of transcript abundance upon PMA-stimulation of Jurkat cells (Fig. 2A). Furthermore, the extent of PMA-induced change in gene-expression inferred by limma was strongly corroborated by qRT-PCR (Fig. 2B). In contrast, the same pipeline identified very few transcripts that differ >1.5-fold between wild-type and hnRNP L depleted cells, regardless of cell growth condition (Fig. 2C,D). Moreover, most of the few predicted hnRNP L-dependent changes in gene expression were deemed to be false positives, as they were not substantiated by qPCR analysis of independent knockdown samples (Supplemental Fig. S2A). Importantly, we also observe no correlation between the expression level of a given transcript and the extent of regulation of exon splicing within the transcript by hnRNP L (Supplemental Fig. S2B). Taken together, our data indicate that the primary impact of hnRNP L on the transcriptome of T cells is through controlling inclusion of cassette exons, and that hnRNP L plays a limited role in regulating other forms of alternative splicing or transcript abundance.
FIGURE 2.
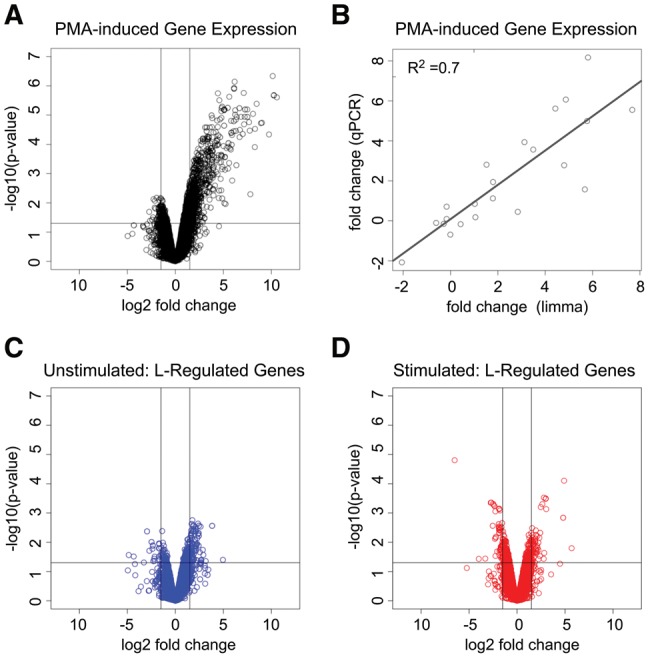
Gene-expression analysis of hnRNP L-responsive differential gene expression. (A) Volcano plot of differential mRNA expression following PMA stimulation of Jurkat cells as determined by limma analysis of RNA-seq data. Vertical lines represent a 1.5-fold log2 change in expression. (B) Regression analysis of qPCR validation of PMA-induced differential transcript levels compared with limma inferred values from the RNA-seq data. (C) Volcano plot of hnRNP L depletion-induced differential mRNA expression in unstimulated Jurkat cells. (D) Volcano plot of hnRNP L depletion-induced mRNA gene expression in PMA-stimulated cells. Vertical lines in A,C,D represent an absolute log2 change in mRNA expression of 1.5-fold.
HnRNP L uses distinct modes of enhancement and repression of exon splicing
Given the pervasive role of hnRNP L in regulating cassette exon inclusion in T cells, we wanted to understand the determinants of hnRNP L function. Specifically, we are interested in features that may correlate with hnRNP L-dependent exon inclusion versus skipping. We first defined sets of exons that met a rigorous definition as an exon that is hnRNP L-repressed, hnRNP L-enhanced or hnRNP L-unresponsive. Stringent hnRNP L-repressed (n = 352) or enhanced (n = 177) exons were defined as exhibiting a significant change (|ΔPSI| ≥ 10, P < 0.05) in the same direction upon knockdown in both unstimulated and stimulated conditions; while hnRNP L-unresponsive exons (n = 250) were defined as those that did not respond to hnRNP L knockdown in either condition (|ΔPSI| < 3) and were queried by both the RASL-seq and RNA-seq techniques (Supplemental Table S2D–F). Previous studies from our group have suggested that splice site strength plays a role in determining the directionality of hnRNP L-regulated alternative splicing, with extremely weak splice sites prone to enhancement while splice sites of moderate strength prone to repression by hnRNP L (Motta-Mena et al. 2010; Chiou et al. 2013; Shankarling et al. 2014). To compare splice site strengths among responsive and unresponsive cassettes, we extracted splice site scores for each of the four splice sites involved in the inclusion of a cassette exon (Fig. 3A). Consistent with our previous studies as well as published analyses of general alternative cassette exons, we observed that both hnRNP L-repressed and hnRNP L-enhanced exons have weaker 3′ splice sites (ss) than those found in unresponsive cassettes (repressed P = 0.0293, enhanced P = 6.51 × 10−6, t-test), and that enhanced exons have even weaker 3′ss than repressed exons (P = 0.013). In contrast, we observe no general bias in the strength of either the 5′ss of L-regulated cassette exons or their flanking splice sites (Fig. 3A).
FIGURE 3.

Exon repression by hnRNP L correlates with direct binding, while exon enhancement does not. (A) Box plots of splice site scores calculated by MaxEnt (Yeo and Burge 2004) for the 5′ and 3′ splice sites of hnRNP L-responsive cassette exons and their flanking exons, compared with hnRNP L-unresponsive cassette exons. hnRNP L-responsive and unresponsive categories are as defined in the text. Median values for each category are given above the plots. (B) Fraction of cassettes containing hnRNP L binding sites defined by CLIP-seq at single-nucleotide resolution within and around hnRNP L-repressed (red) and unresponsive (gray) cassette exons (top) or hnRNP L-enhanced (green) or unresponsive (gray) cassette exons (bottom). (C) Total percent of stringently defined (see text) hnRNP L-responsive and unresponsive cassette exons that contain a CLIP-seq signal for hnRNP L binding anywhere within the exon, or within a 300-nt region of flanking intron. (D) Representative RT-PCR gels of splicing changes observed in wild-type (WT) and hnRNP L-depleted conditions (KD) under unstimulated (U) or stimulated (S) conditions. Mean PSI and standard deviation (SD) are given below gels. In all cases (*) P < 0.05, (**) P < 0.01, (***) P < 0.001.
We next investigated the possibility of positional dependence of hnRNP L occupancy on splicing outcomes. Previously, we published CLIP analysis of hnRNP L binding to mRNA transcripts in Jurkat cells (Shankarling et al. 2014). We therefore generated an RNA map by computing the fraction of instances in which each nucleotide around a cassette exon is encompassed within a CLIP-defined peak of hnRNP L-binding (Fig. 3B). Notably, we observed a general increase in hnRNP L occupancy surrounding hnRNP L-repressed exons relative to exons that are unresponsive to hnRNP L expression, with similar enrichment around the 3′ss and 5′ss of hnRNP L-repressed exons (Fig. 3B,C). In contrast, we observe no significant enrichment of hnRNP L binding around exons it enhances (Fig. 3B,C), even when the entirety of the flanking introns was considered (Supplemental Fig. S3). Importantly, we independently confirmed by RT-PCR several examples of hnRNP L-dependent enhancement of exons that lack evidence of surrounding hnRNP L binding, as well as repression of exons with surrounding hnRNP L binding (Fig. 3D; Supplemental Table S4). It is common for splicing-regulatory proteins to function as both enhancers and repressors of exon inclusion; however, in most instances in which it has been studied, this differential activity has been attributed to location of binding of the splicing factor relative to the exon (Fu and Ares 2014). Strikingly, the above data reveal no evidence for such position-dependent activity of hnRNP L. Instead, our data suggest that, at least in T cells, hnRNP L functions primarily as a repressor of exon inclusion when bound to a transcript and that enhancement of exon inclusion may largely occur in an indirect manner that does not require close association of hnRNP L with the RNA target.
HnRNP L enhanced exons share common sequence and context features
To better understand the apparent indirect enhancement of a subset of exons by hnRNP L, we next looked for enriched sequence motifs within and around hnRNP L-regulated exons (Fig. 4A). Consistent with the binding data in Figure 3, we observe an enrichment of CA-rich hexamers, the optimal binding site for hnRNP L, within or flanking hnRNP L-repressed exons but not hnRNP L-enhanced exons (Fig. 4A, left). We next partitioned all regulated exons based on the absence (defined as “unbound”) of any hnRNP L CLIP-seq binding site within the cassette exon or flanking introns and exons (n = 180 enhanced, n = 239 repressed). We identified hexamers enriched in regions upstream of, downstream from, or within the “unbound” enhanced or repressed exons as compared with cognate sequences extracted from all RefSeq internal exons. No sequence features were found to be significantly enriched upstream of, within, or downstream from “unbound” hnRNP L-repressed exons (Fig. 4B). In contrast, GC-rich sequence features were found to be significantly enriched upstream of and within enhanced unbound exons (Fig. 4B, logo of all significant hexamers is displayed, see Supplemental Table S6 for all enriched hexamers). A further comparison of all hnRNP L-repressed, -enhanced, and -unresponsive exons demonstrates that GC-rich motifs are uniquely enriched within or flanking hnRNP L-enhanced exons, but depleted around hnRNP L-repressed exons (Fig. 4A, right).
FIGURE 4.

HnRNP L-enhanced exons are preferentially flanked by short GC-rich introns. (A) Fraction of repressed, enhanced, and unresponsive cassette exons that have an CA-repeat hexamer (left) or one of the enriched GC-rich hexamers (right) (see Supplemental Table S6) within the exon or the flanking 300 nt. (B) De novo motif enrichment analysis of sequence features enriched in exonic and exon-proximal regions for repressed and enhanced exons within cassettes that do not contain hnRNP L CLIP-seq peaks. Logo of enriched hexamers is shown. See Supplemental Table S6 for all enriched hexamers. (C) Box plots of exon and intron lengths in hnRNP L-repressed, hnRNP L-enhanced, and unresponsive cassettes. (D) Length of upstream intron (I1, left) and downstream intron (I2, right) of repressed, enhanced, and unresponsive cassettes partitioned by presence or absence of the GC-rich motif in the upstream 300-nt interval or within the exon. “Total” represents all exons in the class irrespective of GC content. In all cases (*) P < 0.05, (**) P < 0.01, (***) P < 0.001.
The enrichment of a GC motif around hnRNP L-enhanced exons suggested that perhaps a GC-binding protein is itself regulated by hnRNP L that, in turn, directly mediates the splicing changes observed upon knockdown of hnRNP L. We considered this less likely as we detect little impact of hnRNP L depletion on gene expression. Nevertheless, we tested the expression of three RNA-binding proteins for which there is some evidence of GC-binding, namely RBM4, Y14, and SRSF2 (Ray et al. 2013). In no case did we observe any change in the expression of these proteins upon depletion of hnRNP L (Supplemental Fig. S4). Moreover, using publicly available CLIP and RNA-seq data for RBM4 (Uniacke et al. 2012; Wang et al. 2014), we find no evidence for significant binding or regulation of hnRNP L-enhanced exons by this protein. Therefore, while we cannot fully rule out the possibility that hnRNP L enhances exon inclusion through regulation of a secondary splicing factor, we find no evidence to support this model.
We thus considered other possible mechanisms for indirect exon enhancement by hnRNP L. In particular, we note that exons flanked by short GC-rich introns have previously been described as a distinct class of exons with unique properties (Amit et al. 2012; Braunschweig et al. 2014). To investigate if hnRNP L-enhanced exons might correlate with this exon population, we analyzed the length of the genomic features surrounding hnRNP L-regulated exons. Strikingly, we find that the introns upstream (I1) and downstream (I2) of enhanced exons are significantly shorter than those flanking repressed exons (median I1 length for repressed = 3.8 kb versus enhanced = 1.6 kb; P = 0.00077, median I2 length for repressed = 2.8 kb versus enhanced = 1.3 kb; P = 0.03924) (Fig. 4C). In contrast, exon lengths are only marginally different between L-responsive and unresponsive populations (median exon lengths for unresponsive = 99 nt, repressed = 116 nt, enhanced = 117 nt). We also asked whether the short flanking introns that characterize enhanced exons co-occur with the presence of the GC-rich motif. We used the presence (with GC motif) or absence (no GC motif) of the enriched hexamers from Figure 4B within or upstream of the exon to segregate enhanced, repressed, and unresponsive cassettes and compared flanking intron lengths. Consistent with other reports, in all cases the presence of GC-rich motifs correlated strongly with shorter introns (Fig. 4D), including the particularly short introns flanking hnRNP L-enhanced exons. Taken together, these findings provide evidence that hnRNP L preferentially enhances a subset of exons flanked by short GC-rich introns. Indeed, we find significant mutual enrichment of hnRNP L-enhanced exons with a genome-wide set of exons flanked by short GC-rich introns (Supplemental Table S7).
HnRNP L impacts nucleosome occupancy of enhanced exons
Typically, exons contain a high GC-content relative to their flanking introns. This “GC-differential” has been suggested to promote nucleosome positioning over the exons (Tillo and Hughes 2009; Brown et al. 2012). In contrast, exons flanked by short GC-rich introns have been shown to lack such a GC-differential and correspondingly, exhibit reduced nucleosome occupancy (Amit et al. 2012). Strikingly, we observe a similar lack of GC-differential (Fig. 5A) and reduced nucleosome occupancy (Fig. 5B) for the hnRNP L-enhanced exons relative to exons that are repressed by, or unresponsive to, hnRNP L.
FIGURE 5.
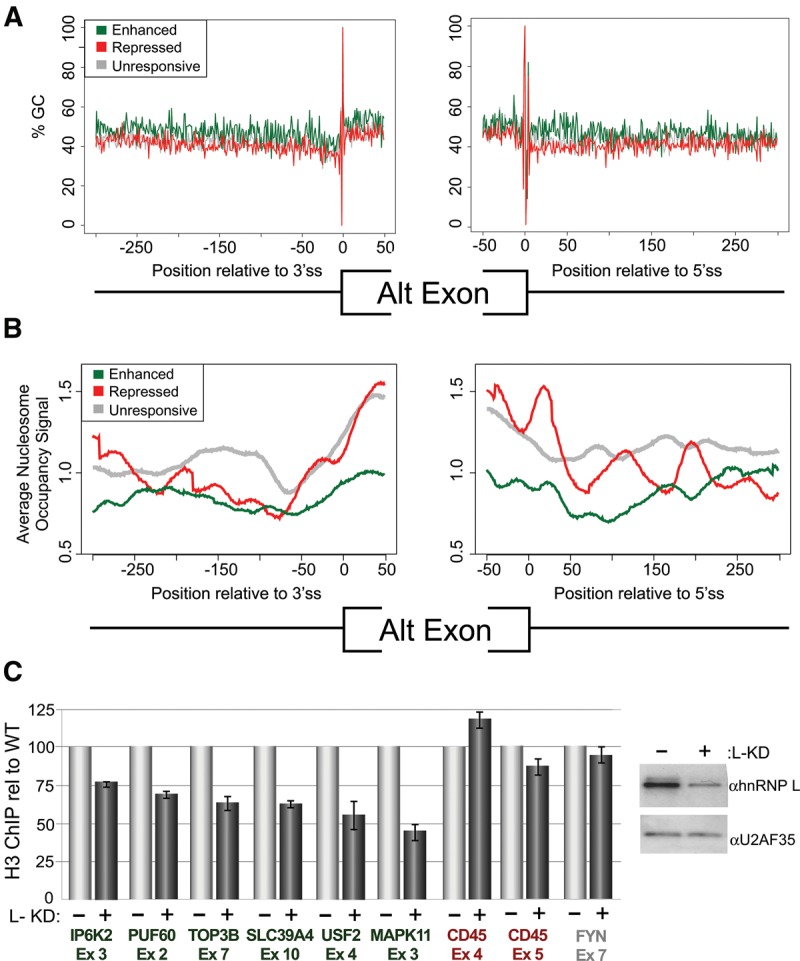
HnRNP L-enhanced exons share epigenetic features that are altered upon hnRNP L depletion. (A) Percentage of GC dinucleotides as a function of distance away from the 3′ss (left) or 5′ss (right) of hnRNP L repressed, enhanced, or unresponsive alternative exons. (B) Similar to A but plotting nucleosome occupancy derived from ENCODE data on K562 cells. (C) Relative qPCR signal of DNA corresponding to indicated hnRNP L-enhanced (green), repressed (red) or unresponsive (gray) exons following ChIP with an antibody that recognizes H3.3 and H3.1. Signal from H3 ChIP in hnRNP L depleted cells (+L-KD) is calculated relative to bead-alone control and then normalized by the H3/bead signal in wild-type Jurkat cells (−L-KD), which is set to 100. On right is a Western blot showing hnRNP L knockdown relative to U2AF35 loading controls.
Previous studies have also shown that exons with a strong GC-differential are more highly included as compared to exons that do not differ in GC-content from their surrounding sequences (Amit et al. 2012), presumably due to the lack in the latter population of nucleosome positioning and/or other epigenetic marks that promote splicing (Kornblihtt et al. 2009; Schwartz and Ast 2010; Brown et al. 2012). Therefore, it is possible that hnRNP L enhances inclusion of low GC-differential exons through promoting their marking by nucleosomes and/or other epigenetic features. Consistent with this model, we find that depletion of hnRNP L reduces signal for bulk H3, a marker for nucleosomes (Grimaldi et al. 2013), over hnRNP L-enhanced exons, with less effect observed for hnRNP L-repressed or unresponsive exons (Fig. 5C). This local alteration in H3 occupancy is unlikely to result from, or induce, changes in transcription, as we observe no correlation between hnRNP L-mediated exon inclusion and transcript expression (Supplemental Fig. S2B). Notably, we also find no evidence in Jurkat cells for the regulation of H3K36 trimethylation by hnRNP L (Supplemental Fig. S5), as has been reported in other cell types (Yuan et al. 2009). However, as discussed above, we do observe hnRNP L-dependent alternative splicing of several genes encoding enzymes involved in histone and/or DNA modifications (Fig. 1F) that could potentially impact nucleosome positioning (Chodavarapu et al. 2010; Gelfman and Ast 2013; Portela et al. 2013; Venkatesh and Workman 2013).
In sum, our data indicate that binding of hnRNP L to a pre-mRNA generally leads to exon repression, while the ability of hnRNP L to enhance exon inclusion is likely not through direct binding to the RNA. Although the exact mechanism by which hnRNP L indirectly enhances exon inclusion remains to be determined, the data we present here demonstrate that hnRNP L enhances a set of exons that share unique sequence and context features, suggesting that hnRNP L may either manipulate or exploit the epigenetic landscape to achieve splicing regulation.
DISCUSSION
Previous studies have demonstrated pervasive association of hnRNP L with RNA in human T cells as well as a critical role for hnRNP L in proper T-cell development and function (Gaudreau et al. 2012; Shankarling et al. 2014). Here we expand the understanding of the biologic function of hnRNP L by identifying changes in transcriptome expression upon knockdown of hnRNP L in a human T-cell line. We find that hnRNP L primarily regulates cassette-type alternative splicing, with minimal impact on transcript abundance, intron retention, or other modes of alternative splicing. Specifically, we identify ∼1300 exons that are regulated by hnRNP L, with about two-thirds of these exons exhibiting hnRNP L-dependent repression. These hnRNP L-regulated exons are enriched in genes involved in controlling gene expression and other core cellular functions. Therefore, our work indicates a primary role of hnRNP L in repressing the splicing of alternative cassette exons in T cells and highlights numerous targets of hnRNP L-dependent splicing that are likely to have broad relevance in T cell biology.
In comparing the pattern of hnRNP L binding to RNA with the functional impact of hnRNP L on mRNA processing, we made the unexpected discovery that, atypically, the location of hnRNP L binding relative to an exon does not predict function. Location-dependent activity has been observed for many splicing-regulatory proteins, such as NOVA and ESRP2, in which different effects on exon inclusion are observed when a protein binds intronic sequences upstream versus downstream from the regulated exon (Ule et al. 2006; Warzecha et al. 2010). In contrast, we observe that binding of hnRNP L to any location within or flanking an exon primarily correlates with exon repression, suggesting that hnRNP L binding may interfere with the assembly of various splicing complexes on an alternative exon. Indeed, studies of individual genes have already revealed several mechanisms through which hnRNP L can repress splicing, including competition with enhancers, competition with core splicing machinery and trapping of catalytically inactive spliceosome intermediates (Hui et al. 2005; House and Lynch 2006; Hung et al. 2008; Motta-Mena et al. 2010; Chiou et al. 2013).
Importantly, however, we do observe that about a third of exons are up-regulated upon depletion of hnRNP L, specifying these as hnRNP L-enhanced exons. Strikingly, these hnRNP L-enhanced exons generally lack any direct association of hnRNP L with the pre-mRNA in their vicinity; indicating that exon enhancement by hnRNP L most likely occurs through a distinct mechanism(s). Notably, the hnRNP L-enhanced exons share sequence and context features, particularly the presence of GC-rich exonic motifs and being flanked by short GC-rich introns.
Exons flanked by short GC-rich introns have been identified as a distinct functional group in previous studies (Amit et al. 2012; Gelfman et al. 2013; Braunschweig et al. 2014). One attribute ascribed to these exons is the potential tendency to be spliced through intron-definition mechanisms, as opposed to the exon-definition pathway more typical of mammalian splicing (Amit et al. 2012). Since hnRNP L has been proposed to increase intron-definition (Hui et al. 2005; House and Lynch 2008), an appealing model is that hnRNP L-enhanced exons are regulated through hnRNP L-dependent intron definition. However, we consider a model of hnRNP L-regulated intron definition to be insufficient for two reasons. First, the median length of introns flanking the hnRNP L-enhanced exons is much greater than the length maximum for intron-definition (>1.2 kb versus 250 nt) (Fox-Walsh et al. 2005). Second, if hnRNP L regulated exons through intron-definition, loss of hnRNP L would be predicted to lead to intron retention rather than exon skipping. Indeed, one study has shown that short GC-rich introns are overrepresented among retained introns (Braunschweig et al. 2014). However, we observe little intron retention relative to exon skipping upon depletion of hnRNP L (Fig. 1). Therefore, we predict that the features of exons flanked by short, GC-rich introns that are most relevant to enhancement by hnRNP L are separate from their propensity toward intron-definition.
The initial study to highlight a class of exons flanked by short GC-rich introns demonstrated that the primary determinant of exon inclusion is the differential in GC content between an exon and its flanking introns (“GC-differential”) (Amit et al. 2012). Specifically, a greater GC-differential was shown to result in greater exon inclusion (Amit et al. 2012). This study and others have further shown that reduced GC-differential between an exon and flanking introns results in less optimal positioning of nucleosomes over exon-encoding DNA (Amit et al. 2012; Brown et al. 2012; Gelfman et al. 2013). Importantly, nucleosome positioning at exons is well established to correlate with increased exon inclusion during splicing, through mechanisms including local slowing of transcription elongation and/or increased local recruitment of splicing factors or other regulatory proteins via interaction with histones (Brown et al. 2012; Braunschweig et al. 2013). Thus, while other mechanisms may be at play, reduced nucleosome occupancy may account for the weaker inclusion observed for exons with reduced GC-differential.
We propose that hnRNP L enhances inclusion of exons with reduced GC-differential by promoting the recognition of these poorly marked exons. Given that hnRNP L does not associate with the RNA in the vicinity of the enhanced exons, we suggest that hnRNP L may impact these sensitized exons through direct or indirect interaction with the transcription machinery. For example, in other cases of alternative splicing, changes to the DNA polymerase complex or alterations in histone or DNA modifications have been shown to facilitate the recognition of poorly defined exons (Zhang et al. 2006; Luco et al. 2010; Pradeepa et al. 2012; Gelfman and Ast 2013). Alternatively, or in addition, hnRNP L may promote nucleosome occupancy on otherwise poorly nucleosome-associated exons. Interestingly, previous studies have implicated hnRNP L as a component of the mediator complex (Huang et al. 2012). Furthermore, we show here that the most enriched class of genes containing hnRNP L-repressed exons are those with functions in chromatin regulation, DNA modification and transcription, raising the possibility that hnRNP L-repressed splicing events may alter the epigenetic landscape to manifest broader splicing regulation. In sum, there are many potential mechanisms by which hnRNP L may link transcription, epigenetics, and alternative splicing. The results we present here lay the foundation and motivation for future studies to unravel these important connections.
MATERIALS AND METHODS
Cell culture, cell stimulation, and hnRNP L depletion
The clonal Jurkat T-cell line, JSL1, has been described previously (Lynch and Weiss 2000). JSL1 Jurkat cells were cultured in RPMI medium with 5% heat-inactivated fetal bovine serum (FBS). Stimulation of JSL1 Jurkat cells was achieved by supplementing culture medium with the phorbol ester PMA (Sigma-Aldrich) at a final concentration of 20 ng/mL. Depletion of hnRNP L by antisense morpholino oligonucleotide (AMO) was achieved by electrotransfection of 20 million cells with 10 µL of 1 nmol/µL translation-blocking AMO. Electroporated cells were allowed to recover in RPMI medium supplemented with fetal bovine serum for 24 h before stimulation. For shRNA depletion of hnRNP L, JSL1 Jurkat cells were transduced with a lentivirus that expresses an shRNA against nucleotides 1499–1509 of the hnRNP L ORF, then grown under neomycin selection for 3 wk to establish clonal lines in which the shRNA expression cassette is stably integrated. For depletion of hnRNP L, expression of the shRNA was induced with doxycycline (1 mg/mL) for 48 h prior to cell harvest.
RNA-seq library preparation
Illumina TRU-seq v2 paired-end high-throughput polyA mRNA sequencing libraries were prepared according to the manufacturer's protocol. Briefly, 1 μg of total RNA was diluted to 50 µL with water, mixed with 50 µL of RNA purification beads [poly(dT) beads provided with kit], and incubated for 5 min at 65°. Beads magnetically separated and washed, then mRNA was eluted from the beads with 50 µL of elute-preime-fragment buffer in a 2-min 80° incubation followed by bead extraction with provided bead-binding buffer. Purified mRNAs were then fragmented to a median size of 160 nt as per the manufacturer's protocol and then subjected to first-strand and second-strand synthesis. Products were then purified with Ampure beads, repaired, and eluted. Adapters were individually added to each sample according to a unique barcoding strategy in which each sample received its own barcode. Barcoded products were then amplified by 13 cycles of amplification with the manufacturer's provided PCR cycling program, then repurified by a single round of Ampure bead purification. The resulting libraries were submitted to the Next-Generation Sequencing core at the University of Pennsylvania for normalization, pooling, and high-throughput sequencing on the Illumina HiSeq 2000 apparatus.
Splicing analysis from RNA-seq data
Alternative splicing was inferred from the RNA-seq data using the Multivariate Analysis of Transcript Splicing (rMATS) software for replicates, which takes raw sequence reads in FASTQ format as the input (Shen et al. 2012; Park et al. 2013). Parameter values for rMATS were set by optimizing the accuracy of the MATS estimation of splicing difference of known standards (genes previously analyzed by RT-PCR). The final rMATS analysis of hnRNP L-responsive alternative splicing was done using replicate support, –c 0.001, -analysis P, and the ReadsOnTargetAndJunctionCounts scoring method, comparing duplicate hnRNP L-depleted to mock-depleted sample groups in unstimulated and stimulated conditions separately. The output from rMATS was then filtered for the same criteria as were applied to RASL-seq output: P-value <0.05 and absolute value of ΔPSI ≥ 10%. Intron retention was analyzed as described in Braunschweig et al. (2014). Briefly, RNA-seq reads were aligned against an index generated from a set of 202,972 human introns and separately against a set of introns that are commonly included (20% ≤ retention ≤ 80%) in at least 20 cell types (n = 815). Differential intron retention was analyzed by the limma package as described above for gene-expression analysis
RASL-seq
RASL-seq was performed as previously described using a set of probes that interrogate ∼5600 specific splicing events (Li et al. 2012). In brief, total RNA was harvested from biologic triplicate samples of wild-type and hnRNP L-depleted JSL1 Jurkat cells grown under unstimulated or PMA-stimulated conditions. These RNA samples were individually hybridized to the probe set and selected by oligo(dT). Juxtaposed probes annealed to selected RNAs were then ligated and amplified and barcoded by PCR for subsequent multiplexed sequencing on a HiSeq2000. On average, 2 million reads were obtained for each RNA sample. Splicing events were filtered for a minimum of 10 reads average across all biologic replicates and conditions and then isoform ratios were calculated by comparing number of reads representing the longest isoform to the number of total reads for that splicing event (PSI = percent spliced in of variable exon). The change in PSI (ΔPSI) was then calculated as the difference between the average PSI across the three biologic replicates of RNA from wild-type versus hnRNP L-depleted cells grown under the same condition. HnRNP L-dependent splicing events were identified as splicing events for which the absolute value of ΔPSI comparing wild-type or hnRNP L-depleted cells was ≥10, with P < 0.05 (unpaired Student's t-test).
Differential gene-expression analysis
To analyze gene-expression changes from RNA-seq alignments, aligned reads were analyzed by the limma package (Ritchie et al. 2015). Transcripts were discarded if they did not have at least 1 read per million in at least half of the replicates under comparison.
RNA map generation
To examine CLIP-seq binding patterns within responsive and unresponsive cassettes, coordinates for 350-nt intervals containing 50 nt of exonic sequence and 300 nt of exon-proximal intronic sequence were generated for the C1 exon 5′ss (the exon upstream of the alternative exon), both splice sites of the alternative exon, and the 3′ss of the C2 exon. Next, for each nucleotide in each of these intervals, the fraction of cassettes containing a CLIP-seq peak at that position was computed. This analysis normalizes variable peak size by allowing a given position to only be either considered occupied (value of 1) or unoccupied (value of 0).
Binomial motif enrichment analysis of exonic and periexonic intervals
To investigate potentially enriched sequence features within and around exons of interest, we compared the fraction of intervals of interest containing each kmer to the fraction of analogous intervals containing that kmer of the same type from all internal RefSeq exons. Sequences were extracted from intervals −300 to −20 nt upstream of the exons' 3′ splice sites, from the entire exon excluding the three outermost nucleotides, and from +6 to +300 nt downstream from the 5′ splice sites. Binomial test P-values were computed using hexamer frequencies obtained from analogous intervals from all RefSeq internal exons. To control for multiple hypothesis testing a Bonferroni α level was computed as 0.05/4**k, or 1.22 × 10−5 for k = 6. All kmers with P-values below this corrected α level were then aligned together with ClustalW2 and the multiple sequence alignments were used to generate sequence logos with WebLogo version 2.8.2.
RNA extraction, RT-PCR splicing assay, and qRT-PCR gene-expression assay
RNA was isolated with the RNA-bee (Tel-Test) reagent and protocol. Low-cycle radiolabeled RT-PCR assay was carried out as described previously (Lynch and Weiss 2000) as an orthogonal method to quantify splicing changes. Gene-expression changes inferred by limma were independently tested by qRT-PCR. Total RNA isolated as described above was reverse transcribed with a cDNA reverse transcription kit (Applied Biosystems) using random 9-mer primers. cDNA was then mixed in optical plates (Applied Biosystems) with SYBR green PCR master mix (Applied Biosystems) and primers that span exon–exon junctions to minimize the possibility of genomic DNA amplification. After amplification on a SDS7000 qRT-PCR thermal cycler (ABI), standard curves were inspected for linearity and PCR products were analyzed by 1.5% agarose gel electrophoresis to confirm expected amplicon size. Quantification was by ABI Prism software and normalized to ACTB quantification achieved by qRT-PCR from the same RT-PCR reactions. Gene-expression changes were computed as the average of the log2 (knockdown/mock-depleted) for hnRNP L depletion and as log2 (stimulated/unstimulated) for stimulation-responsive differential gene-expression analyses. Primer sequences for all RT-PCR and RT-qPCR primers are listed in Supplemental Table S8.
Chromatin immunoprecipitation
Briefly, 5 × 106 cells were fixed with 1% formaldehyde at room temperature for 10 min. Cross-linked cells were sonicated at 40% amplitude (30 sec on, 90 sec off, for a total of 24 min) using a Branson 101-135-126 Sonifier. ChIP was performed using 2 µL of a polyclonal anti-Histone H3 antibody (Abcam, ab1791) and 5 µL protein A agarose (Roche, 11719408001). ChIPed samples were then reverse cross-linked at 65°C for 16 h. ChIPed DNA was purified using SpinSmart columns (Denville Scientific, CM-500-50) and eluted twice with 50 µL water. qPCR analysis of DNA from H3 ChIP was done with primers in Supplemental Table S7 and normalized to signal obtained from ChIP with protein A beads alone.
DATA DEPOSITION
The raw sequence reads supporting the results of this article are available in the NCBI SRA repository under accession number SRP059357 (alias PRJNA285906) at http://www.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?study=SRP059357.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank the University of Pennsylvania NGS core facility for assistance with sequencing of the Tru-Seq libraries, Y. Xing and members of the Xing laboratory for guidance with the rMATS analysis, and Matthew Gazzara, Yoseph Barash, and Shalini Oberdoerffer for helpful discussions and/or critical reading of the manuscript. This work was funded by R01GM067719 and R01GM084034 to K.W.L. and R01HG004659 to X.-D.F.
Author contributions: B.S.C. performed the AMO-based depletion of hnRNP L, prepared the RNA for RNA-seq, performed qPCR of RNA samples, and carried out all of the computational analysis in this study. I.T. planned, carried out, and analyzed the RT-PCR examination of splicing; S.J.A. performed and analyzed the qPCR of ChIP samples; M.J.M. generated the hnRNP L-depleted cell lines and prepared RNA for RASL-seq; R.J.L. and H-Y.F. performed the ChIP analysis and provided expertise on the nucleosome studies; J.Q. and X-D.F. performed the RASL-seq analysis; K.W.L. conceived and oversaw the project, was involved in analyzing and interpreting the data, and wrote the paper with the assistance of the other authors.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.052969.115.
REFERENCES
- Amit M, Donyo M, Hollander D, Goren A, Kim E, Gelfman S, Lev-Maor G, Burstein D, Schwartz S, Postolsky B, et al. 2012. Differential GC content between exons and introns establishes distinct strategies of splice-site recognition. Cell Rep 1: 543–556. [DOI] [PubMed] [Google Scholar]
- An P, Grabowski PJ. 2007. Exon silencing by UAGG motifs in response to neuronal excitation. PLoS Biol 5: e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bomsztyk K, Denisenko O, Ostrowski J. 2004. hnRNP K: one protein multiple processes. Bioessays 26: 629–638. [DOI] [PubMed] [Google Scholar]
- Braunschweig U, Gueroussov S, Plocik AM, Graveley BR, Blencowe BJ. 2013. Dynamic integration of splicing within gene regulatory pathways. Cell 152: 1252–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braunschweig U, Barbosa-Morais NL, Pan Q, Nachman EN, Alipanahi B, Gonatopoulos-Pournatzis T, Frey B, Irimia M, Blencowe BJ. 2014. Widespread intron retention in mammals functionally tunes transcriptomes. Genome Res 24: 1774–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown SJ, Stoilov P, Xing Y. 2012. Chromatin and epigenetic regulation of pre-mRNA processing. Hum Mol Genet 21: R90–R96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartegni L, Krainer AR. 2002. Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat Genet 30: 377–384. [DOI] [PubMed] [Google Scholar]
- Chiou NT, Shankarling G, Lynch KW. 2013. hnRNP L and hnRNP A1 induce extended U1 snRNA interactions with an exon to repress spliceosome assembly. Mol Cell 49: 972–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho V, Mei Y, Sanny A, Chan S, Enders A, Bertram EM, Tan A, Goodnow CC, Andrews TD. 2014. The RNA-binding protein hnRNPLL induces a T cell alternative splicing program delineated by differential intron retention in polyadenylated RNA. Genome Biol 15: R26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chodavarapu RK, Feng S, Bernatavichute YV, Chen PY, Stroud H, Yu Y, Hetzel JA, Kuo F, Kim J, Cokus SJ, et al. 2010. Relationship between nucleosome positioning and DNA methylation. Nature 466: 388–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper TA, Wan L, Dreyfuss G. 2009. RNA and disease. Cell 136: 777–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David CJ, Manley JL. 2010. Alternative pre-mRNA splicing regulation in cancer: pathways and programs unhinged. Genes Dev 24: 2343–2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox-Walsh KL, Dou Y, Lam BJ, Hung SP, Baldi PF, Hertel KJ. 2005. The architecture of pre-mRNAs affects mechanisms of splice-site pairing. Proc Natl Acad Sci 102: 16176–16181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu XD, Ares M Jr. 2014. Context-dependent control of alternative splicing by RNA-binding proteins. Nat Rev Genet 15: 689–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudreau MC, Heyd F, Bastien R, Wilhelm B, Moroy T. 2012. Alternative splicing controlled by heterogeneous nuclear ribonucleoprotein L regulates development, proliferation, and migration of thymic pre-T cells. J Immunol 188: 5377–5388. [DOI] [PubMed] [Google Scholar]
- Gehman LT, Meera P, Stoilov P, Shiue L, O'Brien JE, Meisler MH, Ares M Jr, Otis TS, Black DL. 2012. The splicing regulator Rbfox2 is required for both cerebellar development and mature motor function. Genes Dev 26: 445–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelfman S, Ast G. 2013. When epigenetics meets alternative splicing: the roles of DNA methylation and GC architecture. Epigenomics 5: 351–353. [DOI] [PubMed] [Google Scholar]
- Gelfman S, Cohen N, Yearim A, Ast G. 2013. DNA-methylation effect on cotranscriptional splicing is dependent on GC architecture of the exon-intron structure. Genome Res 23: 789–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimaldi Y, Ferrari P, Strubin M. 2013. Independent RNA polymerase II preinitiation complex dynamics and nucleosome turnover at promoter sites in vivo. Genome Res 24: 117–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyd F, Lynch KW. 2011. DEGRADE, MOVE, REGROUP: signaling control of splicing proteins. Trends Biochem Sci 36: 397–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- House AE, Lynch KW. 2006. An exonic splicing silencer represses spliceosome assembly after ATP-dependent exon recognition. Nat Struct Mol Biol 13: 937–944. [DOI] [PubMed] [Google Scholar]
- House AE, Lynch KW. 2008. Regulation of alternative splicing: more than just the ABCs. J Biol Chem 283: 1217–1221. [DOI] [PubMed] [Google Scholar]
- Huang Y, Li W, Yao X, Lin QJ, Yin JW, Liang Y, Heiner M, Tian B, Hui J, Wang G. 2012. Mediator complex regulates alternative mRNA processing via the MED23 subunit. Mol Cell 45: 459–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huelga SC, Vu AQ, Arnold JD, Liang TY, Liu PP, Yan BY, Donohue JP, Shiue L, Hoon S, Brenner S, et al. 2012. Integrative genome-wide analysis reveals cooperative regulation of alternative splicing by hnRNP proteins. Cell Rep 1: 167–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui J, Hung LH, Heiner M, Schreiner S, Neumuller N, Reither G, Haas SA, Bindereif A. 2005. Intronic CA-repeat and CA-rich elements: a new class of regulators of mammalian alternative splicing. EMBO J 24: 1988–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung LH, Heiner M, Hui J, Schreiner S, Benes V, Bindereif A. 2008. Diverse roles of hnRNP L in mammalian mRNA processing: a combined microarray and RNAi analysis. RNA 14: 284–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ip JY, Tong A, Pan Q, Topp JD, Blencowe BJ, Lynch KW. 2007. Global analysis of alternative splicing during T-cell activation. RNA 13: 563–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen M, Schweer D, Ziegler A, Gaber R, Schock S, Schwinzer R, Wonigeit K, Lindert RB, Kantarci O, Hemmer B, et al. 2000. A point mutation in PTPRC is associated with the development of multiple sclerosis. Nat Genet 26: 495–499. [DOI] [PubMed] [Google Scholar]
- Kornblihtt AR, Schor IE, Allo M, Blencowe BJ. 2009. When chromatin meets splicing. Nat Struct Mol Biol 16: 902–903. [DOI] [PubMed] [Google Scholar]
- Lareau LF, Inada M, Green RE, Wengrod JC, Brenner SE. 2007. Unproductive splicing of SR genes associated with highly conserved and ultraconserved DNA elements. Nature 446: 926–929. [DOI] [PubMed] [Google Scholar]
- Li H, Qiu J, Fu XD. 2012. RASL-seq for massively parallel and quantitative analysis of gene expression. Curr Protoc Mol Biol 98: 4.13.1–4.13.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licatalosi DD, Darnell RB. 2009. RNA processing and its regulation: global insights into biological networks. Nat Rev Genet 11: 75–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licatalosi DD, Mele A, Fak JJ, Ule J, Kayikci M, Chi SW, Clark TA, Schweitzer AC, Blume JE, Wang X, et al. 2008. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature 456: 464–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licatalosi DD, Yano M, Fak JJ, Mele A, Grabinski SE, Zhang C, Darnell RB. 2012. Ptbp2 represses adult-specific splicing to regulate the generation of neuronal precursors in the embryonic brain. Genes Dev 26: 1626–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luco RF, Pan Q, Tominaga K, Blencowe BJ, Pereira-Smith OM, Misteli T. 2010. Regulation of alternative splicing by histone modifications. Science 327: 996–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch KW. 2007. Regulation of alternative splicing by signal transduction pathways. Adv Exp Med Biol 623: 161–174. [DOI] [PubMed] [Google Scholar]
- Lynch KW, Weiss A. 2000. A model system for the activation-induced alternative-splicing of CD45 implicates protein kinase C and Ras. Mol Cell Biol 20: 70–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez NM, Pan Q, Cole BS, Yarosh CA, Babcock GA, Heyd F, Zhu W, Ajith S, Blencowe BJ, Lynch KW. 2012. Alternative splicing networks regulated by signaling in human T cells. RNA 18: 1029–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Contreras R, Cloutier P, Shkreta L, Fisette J-F, Revil T, Chabot B. 2007. hnRNP proteins and splicing Control. Adv Exp Med Biol 623: 123–147. [DOI] [PubMed] [Google Scholar]
- Motta-Mena LB, Heyd F, Lynch KW. 2010. Context-dependent regulatory mechanism of the splicing factor hnRNP L. Mol Cell 29: 223–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsen TW, Graveley BR. 2010. Expansion of the eukaryotic proteome by alternative splicing. Nature 463: 457–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. 2008. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet 40: 1413–1415. [DOI] [PubMed] [Google Scholar]
- Park JW, Tokheim C, Shen S, Xing Y. 2013. Identifying differential alternative splicing events from RNA sequencing data using RNASeq-MATS. Methods Mol Biol 1038: 171–179. [DOI] [PubMed] [Google Scholar]
- Patel NA, Chalfant CE, Watson JE, Wyatt JR, Dean NM, Eichler DC, Cooper DR. 2001. Insulin regulates alternative splicing of protein kinase C beta II through a phosphatidylinositol 3-kinase-dependent pathway involving the nuclear serine/arginine-rich splicing factor, SRp40, in skeletal muscle cells. J Biol Chem 276: 22648–22654. [DOI] [PubMed] [Google Scholar]
- Pinol-Roma S, Choi YD, Matunis MJ, Dreyfuss G. 1988. Immunopurification of heterogeneous nuclear ribonucleoprotein particles reveals an assortment of RNA-binding proteins. Genes Dev 2: 215–227. [DOI] [PubMed] [Google Scholar]
- Portela A, Liz J, Nogales V, Setien F, Villanueva A, Esteller M. 2013. DNA methylation determines nucleosome occupancy in the 5′-CpG islands of tumor suppressor genes. Oncogene 32: 5421–5428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradeepa MM, Sutherland HG, Ule J, Grimes GR, Bickmore WA. 2012. Psip1/Ledgf p52 binds methylated histone H3K36 and splicing factors and contributes to the regulation of alternative splicing. PLoS Genet 8: e1002717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preussner M, Schreiner S, Hung LH, Porstner M, Jack HM, Benes V, Ratsch G, Bindereif A. 2012. HnRNP L and L-like cooperate in multiple-exon regulation of CD45 alternative splicing. Nucleic Acids Res 40: 5666–5678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quesnel-Vallieres M, Irimia M, Cordes SP, Blencowe BJ. 2015. Essential roles for the splicing regulator nSR100/SRRM4 during nervous system development. Genes Dev 29: 746–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj B, Irimia M, Braunschweig U, Sterne-Weiler T, O'Hanlon D, Lin ZY, Chen GI, Easton LE, Ule J, Gingras AC, et al. 2014. A global regulatory mechanism for activating an exon network required for neurogenesis. Mol Cell 56: 90–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray D, Kazan H, Cook KB, Weirauch MT, Najafabadi HS, Li X, Gueroussov S, Albu M, Zheng H, Yang A, et al. 2013. A compendium of RNA-binding motifs for decoding gene regulation. Nature 499: 172–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. 2015. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43: e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothrock CR, House AE, Lynch KW. 2005. HnRNP L represses exon splicing via a regulated exonic splicing silencer. EMBO J 24: 2792–2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S, Ast G. 2010. Chromatin density and splicing destiny: on the cross-talk between chromatin structure and splicing. EMBO J 29: 1629–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankarling G, Lynch KW. 2013. Minimal functional domains of paralogues hnRNP L and hnRNP LL exhibit mechanistic differences in exonic splicing repression. Biochem J 453: 271–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankarling G, Cole BS, Mallory MJ, Lynch KW. 2014. Transcriptome-wide RNA interaction profiling reveals physical and functional targets of hnRNP L in human T cells. Mol Cell Biol 34: 71–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S, Park JW, Huang J, Dittmar KA, Lu ZX, Zhou Q, Carstens RP, Xing Y. 2012. MATS: a Bayesian framework for flexible detection of differential alternative splicing from RNA-seq data. Nucleic Acids Res 40: e61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Manley JL. 2015. The end of the message: multiple protein-RNA interactions define the mRNA polyadenylation site. Genes Dev 29: 889–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin C, Manley JL. 2004. Cell signalling and the control of pre-mRNA splicing. Nat Rev 5: 727–738. [DOI] [PubMed] [Google Scholar]
- Tillo D, Hughes TR. 2009. G+C content dominates intrinsic nucleosome occupancy. BMC Bioinformatics 10: 442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong A, Nguyen J, Lynch KW. 2005. Differential expression of CD45 isoforms is controlled by the combined activity of basal and inducible splicing-regulatory elements in each of the variable exons. J Biol Chem 280: 38297–38304. [DOI] [PubMed] [Google Scholar]
- Topp JD, Jackson J, Melton AA, Lynch KW. 2008. A cell-based screen for splicing regulators identifies hnRNP LL as a distinct signal-induced repressor of CD45 variable exon 4. RNA 14: 2038–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ule J, Stefani G, Mele A, Ruggiu M, Wang X, Taneri B, Gaasterland T, Blencowe BJ, Darnell RB. 2006. An RNA map predicting Nova-dependent splicing regulation. Nature 444: 580–586. [DOI] [PubMed] [Google Scholar]
- Uniacke J, Holterman CE, Lachance G, Franovic A, Jacob MD, Fabian MR, Payette J, Holcik M, Pause A, Lee S. 2012. An oxygen-regulated switch in the protein synthesis machinery. Nature 486: 126–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesh S, Workman JL. 2013. Set2 mediated H3 lysine 36 methylation: regulation of transcription elongation and implications in organismal development. Wiley Interdiscip Rev Dev Biol 2: 685–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl MC, Will CL, Luhrmann R. 2009. The spliceosome: design principles of a dynamic RNP machine. Cell 136: 701–718. [DOI] [PubMed] [Google Scholar]
- Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. 2008. Alternative isoform regulation in human tissue transcriptomes. Nature 456: 470–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Chen D, Qian H, Tsai YS, Shao S, Liu Q, Dominguez D, Wang Z. 2014. The splicing factor RBM4 controls apoptosis, proliferation, and migration to suppress tumor progression. Cancer Cell 26: 374–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warzecha CC, Jiang P, Amirikian K, Dittmar KA, Lu H, Shen S, Guo W, Xing Y, Carstens RP. 2010. An ESRP-regulated splicing programme is abrogated during the epithelial-mesenchymal transition. EMBO J 29: 3286–3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J, Black DL. 2001. A CaMK IV responsive RNA element mediates depolarization-induced alternative splicing of ion channels. Nature 410: 936–939. [DOI] [PubMed] [Google Scholar]
- Xiong HY, Alipanahi B, Lee LJ, Bretschneider H, Merico D, Yuen RK, Hua Y, Gueroussov S, Najafabadi HS, Hughes TR, et al. 2014. RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science 347: 1254806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Ouyang K, Huang J, Zhou Y, Ouyang H, Li H, Wang G, Wu Q, Wei C, Bi Y, et al. 2013. Direct conversion of fibroblasts to neurons by reprogramming PTB-regulated microRNA circuits. Cell 152: 82–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarosh CA, Iacona JR, Lutz CS, Lynch KW. 2015. PSF: nuclear busy-body or nuclear facilitator? Wiley Interdiscip Rev RNA 6: 351–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo G, Burge CB. 2004. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol 11: 377–394. [DOI] [PubMed] [Google Scholar]
- Yuan W, Xie J, Long C, Erdjument-Bromage H, Ding X, Zheng Y, Tempst P, Chen S, Zhu B, Reinberg D. 2009. Heterogeneous nuclear ribonucleoprotein L Is a subunit of human KMT3a/Set2 complex required for H3 Lys-36 trimethylation activity in vivo. J Biol Chem 284: 15701–15707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Du J, Sun B, Dong X, Xu G, Zhou J, Huang Q, Liu Q, Hao Q, Ding J. 2006. Structure of human MRG15 chromo domain and its binding to Lys36-methylated histone H3. Nucleic Acids Res 34: 6621–6628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Qiu J, Liu W, Zhou Y, Plocinik RM, Li H, Hu Q, Ghosh G, Adams JA, Rosenfeld MG, et al. 2012. The Akt-SRPK-SR axis constitutes a major pathway in transducing EGF signaling to regulate alternative splicing in the nucleus. Mol Cell 47: 422–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.