

Abstract
Retinitis pigmentosa (RP) is a rare genetic disease that causes gradual blindness through retinal degeneration. Intriguingly, seven of the 24 genes identified as responsible for the autosomal-dominant form (adRP) are ubiquitous spliceosome components whose impairment causes disease only in the retina. The fact that these proteins are essential in all organisms hampers genetic, genomic, and physiological studies, but we addressed these difficulties by using RNAi in Caenorhabditis elegans. Our study of worm phenotypes produced by RNAi of splicing-related adRP (s-adRP) genes functionally distinguishes between components of U4 and U5 snRNP complexes, because knockdown of U5 proteins produces a stronger phenotype. RNA-seq analyses of worms where s-adRP genes were partially inactivated by RNAi, revealed mild intron retention in developing animals but not in adults, suggesting a positive correlation between intron retention and transcriptional activity. Interestingly, RNAi of s-adRP genes produces an increase in the expression of atl-1 (homolog of human ATR), which is normally activated in response to replicative stress and certain DNA-damaging agents. The up-regulation of atl-1 correlates with the ectopic expression of the pro-apoptotic gene egl-1 and apoptosis in hypodermal cells, which produce the cuticle, but not in other cell types. Our model in C. elegans resembles s-adRP in two aspects: The phenotype caused by global knockdown of s-adRP genes is cell type-specific and associated with high transcriptional activity. Finally, along with a reduced production of mature transcripts, we propose a model in which the retina-specific cell death in s-adRP patients can be induced through genomic instability.
Keywords: C. elegans, retinitis pigmentosa, RNA-seq, spliceosome, intron retention
INTRODUCTION
Retinitis pigmentosa (RP) is a rare disease that affects approximately one out of 4000 individuals (more than 1.5 million people worldwide). RP causes a progressive loss of vision and ultimately blindness. Patients affected by this disease present defects in photoreceptors (rods and cones) and RPE (retinal pigment epithelium) cells that trigger cellular apoptosis and degeneration of the retina (Hartong et al. 2006; Ferrari et al. 2011). Although sporadic RP is not rare, most RP patients present inherited mutations, through diverse modes of genetic transmission, in any of the 50 genes that have been related to the disease (Daiger et al. 2013). Of all these RP familial cases, 30%–40% show autosomal-dominant genetic inheritance (Hartong et al. 2006). To date, mutations in 24 genes have been linked to autosomal-dominant RP (adRP) (Rossmiller et al. 2012). Among these genes, seven (PRPF3, PRPF4, PRPF6, PRPF8, PRPF31, SNRNP200/BRR2, and RP9) are coding for ubiquitous components of the spliceosome machinery and, when defective, cause disease only in the retina (Mordes et al. 2006; Zhao et al. 2009; Tanackovic et al. 2011b; Chen et al. 2014). All of them but RP9/PAP-1, which interacts at the protein level with PRPF3, are evolutionarily conserved (Supplemental Fig. S1; Maita et al. 2005).
A better understanding of the mechanisms of action of these genes will provide a new insight into the disease that could help in the search of potential therapies for adRP patients. However, the essential function of s-adRP genes limits genetic and cellular studies. As a consequence, most of the knowledge that we have about how the spliceosome works comes from biochemical or in vitro studies. The multicellular organism Caenorhabditis elegans, thanks to the extraordinary conservation of these splicing-related proteins through evolution, offers a shortcut to understand why a mild reduction in the activity of genes required in all cells is critical only for a specific tissue.
The spliceosome is probably the most complex molecular machine of the cell and most of its components are evolutionarily conserved (Fabrizio et al. 2009). Moreover, splicing mechanisms are also similar from yeast to humans (Wahl et al. 2009; Will and Lührmann 2011). More than 200 proteins form this macromolecular complex that removes introns from eukaryotic pre-mRNAs. This is a highly dynamic process, with proteins going in and out of the complex depending on the splicing step that is taking place. The core of the spliceosome is composed of small nuclear ribonucleoproteins (snRNPs), which are complexes of proteins and uridine-rich small nuclear RNAs (named U1, U2, U4, U5, and U6 snRNAs). Particularly, the splicing-related genes linked with adRP encode proteins present in the U4/U6·U5 tri-snRNP complex, which is a main actor in the process of splicing. Among these proteins, PRP3, PRP4, and PRP31 are U4 snRNP components, whereas SNRNP200/BRR2, PRP6, and PRP8 are part of the U5 snRNP (Maita et al. 2005; Liu et al. 2006). For the sake of simplicity, we can place the s-adRP proteins within the process of splicing as follows: An intron flanked by two exons is recognized by the pre-spliceosome (U1 and U2 snRNPs complexes). Next, the U4/U6·U5 tri-snRNP remodels the spliceosome to later exclude U1 and U4 snRNPs, and keep U2, U5, and U6 snRNPs to form the activated spliceosome. Then, two catalytic steps occur to remove the intron and join the flanking exons.
In the present study, we use genetic and genomic tools to investigate in a multicellular organism how the alteration of a common and essential process such as pre-mRNA maturation can affect specific cells only.
RESULTS
A C. elegans toolkit to study splicing-related autosomal-dominant retinitis pigmentosa (s-adRP): RNAi, mutants, and transgenic animals
Despite the genetic nature of s-adRP, development of animal models to study this disorder has been hampered by the essential functions of the genes whose mutations are responsible of the disease. In multicellular organisms, complete depletion of s-adRP genes leads to lethality and, in consequence, described mutations in splicing-related genes causing adRP are either partial-loss-of-function mutations or deletion alleles that cause haploinsufficiency. To mimic and study the effect of this mild impairment of gene functions in C. elegans we took advantage of RNA interference (RNAi).
RNAi was first discovered and developed as a research tool in C. elegans (Fire et al. 1998). RNAi in this nematode is systemic, which implies that the effect of a specific dsRNA spreads through the whole organism. Because C. elegans feeds on bacteria, the feeding of worms with a clone producing a gene-specific RNAi results in the delivery of the specific dsRNA into the animal cells. As a complementary RNAi technique, dsRNA can also be administered by microinjection into the worm germline, which normally produces a stronger phenotype. RNAi clones corresponding to s-adRP genes were obtained, and validated by sequencing, from the two existing C. elegans RNAi libraries (Kamath et al. 2003; Rual et al. 2004), with the exception of the RNAi clone for snrp-200, which was generated in our laboratory by Gateway cloning (Fig. 1A). By quantitative PCR we investigated the effectiveness of the RNAi for some s-adRP genes and found that >50% of the targeted transcripts were depleted in our RNAi conditions (Fig. 1B).
FIGURE 1.

C. elegans toolkit to study s-adRP. (A) Scheme of s-adRP genes in C. elegans including the regions that are targeted by RNAi clones (black bars), the deleted fragment in the prp-31(gk1094) and prp-8(gk3511) alleles (red bars), and the elements of the prp-8 and prp-31 reporters generated in this study (green rectangles). The prp-31(gk1094) allele consists of a 5-bp insertion/1953-bp deletion. prp-8(gk3511) is a 1823-bp deletion. (B) Quantification of prp genes’ expression levels after their respective inactivation by RNAi. mRNA levels of prp genes are represented relative to the expression in gfp(RNAi) control animals (arbitrary value of 1, indicated with a red line). Transcript levels are normalized against tbb-2 levels in each case. RNA for analysis was obtained from up to four biological replicates (n). RNA from samples used for RNA-seq analyses were included. Error bars represent standard error of the mean. (C) Representative confocal image showing a transgenic worm expressing the transgene sEx12486, which consists of the GFP under the control of a promoter region for prp-8. (D) Representative confocal image showing a transgenic worm expressing the transgene cerEx79, which consists of the fusion protein GFP::H2B under the control of prp-31 promoter and 3′UTR.
Haploinsufficiency in s-adRP has been reported for human PRPF31 but not for PRPF8, which does not allow the full loss of one copy of the gene (Abu-Safieh et al. 2006). We investigated haploinsufficiency in C. elegans by using deletion alleles for prp-31 and prp-8. One copy of prp-31(gk1094) or prp-8(gk3511) alleles (The C. elegans Deletion Mutant Consortium 2012), balanced with the translocation hT2 or with classic genetic markers, did not produce haploinsufficiency in terms of fertility or embryonic lethality (Supplemental Fig. S2A).
We further explored other evidences of haploinsufficiency in worms carrying the prp-31 deletion by investigating different penetrance among individuals. We monitored the progeny of prp-31(gk1094) heterozygous individuals and observed no inter-individual variability in terms of brood size (Supplemental Fig. S2B). Moreover, the Mendelian proportions in the offspring of heterozygous animals were normal, with approximately one-quarter of arrested larvae (prp-31(gk1094) homozygous) (Supplemental Fig. S2C).
In conclusion, prp-31(gk1094) and prp-8(gk3511) are haplosufficient in terms of somatic and germline development, although we cannot discard haploinsufficiency in any other specific cellular process. Strains carrying these mutations could be useful to screen for enhancers and therefore for potential modifiers of the disease.
Finally, we studied the expression of s-adRP genes using transgenic reporter strains. We analyzed the expression pattern of a prp-8 transcriptional reporter (Pprp-8::GFP) and observed an ubiquitous expression (Fig. 1C; Hunt-Newbury et al. 2007). This transgene, although informative, did not contain all the regulatory regions of the gene and moreover was not integrated in the worm genome, resulting in mosaicism in its expression. Thus, we made several attempts to express s-adRP proteins tagged with green fluorescent protein (GFP), but we were not able to obtain transgenic animals by these means, suggesting that ectopic expression of these proteins could be toxic for animal development. Alternatively, we generated a transcriptional reporter containing the promoter region of prp-31 in frame with the nuclear histone 2B, the GFP gene, and the 3′ UTR of prp-31. Animals were transformed by gene bombardment or microinjection, and we obtained several strains carrying this reporter either integrated in the genome or as an extrachromosomal array. All these strains broadly expressed the transgenes along the animal providing a useful genetic background to screen for other genes or small molecules capable of regulating the expression levels of prp-31 (Fig. 1D).
In summary, RNAi clones for s-adRP genes, strains carrying deletion alleles for prp-31 and prp-8 in heterozygosity, and several GFP reporter strains for prp-31 constitute the first version of a toolkit to investigate s-adRP genes in C. elegans.
RNAi phenotypes of s-adRP genes encoding U5 snRNP components are more dramatic than those corresponding to U4 snRNP proteins
To study the RNAi phenotypes of prp-3, prp-4, prp-6, prp-8, prp-31, and snrp-200, we performed RNAi by feeding in a synchronized population of worms at the first larval stage (L1) (Porta-de-la-Riva et al. 2012). We clearly distinguished two phenotypic groups: Whereas prp-6, prp-8 and snrp-200 RNAi produced larval arrest, animals treated with RNAi for prp-3, prp-4, and prp-31 produced adult animals that were sterile (Table 1; Fig. 2A).
TABLE 1.
Phenotypes observed after RNAi of s-adRP genes by two different methods

FIGURE 2.
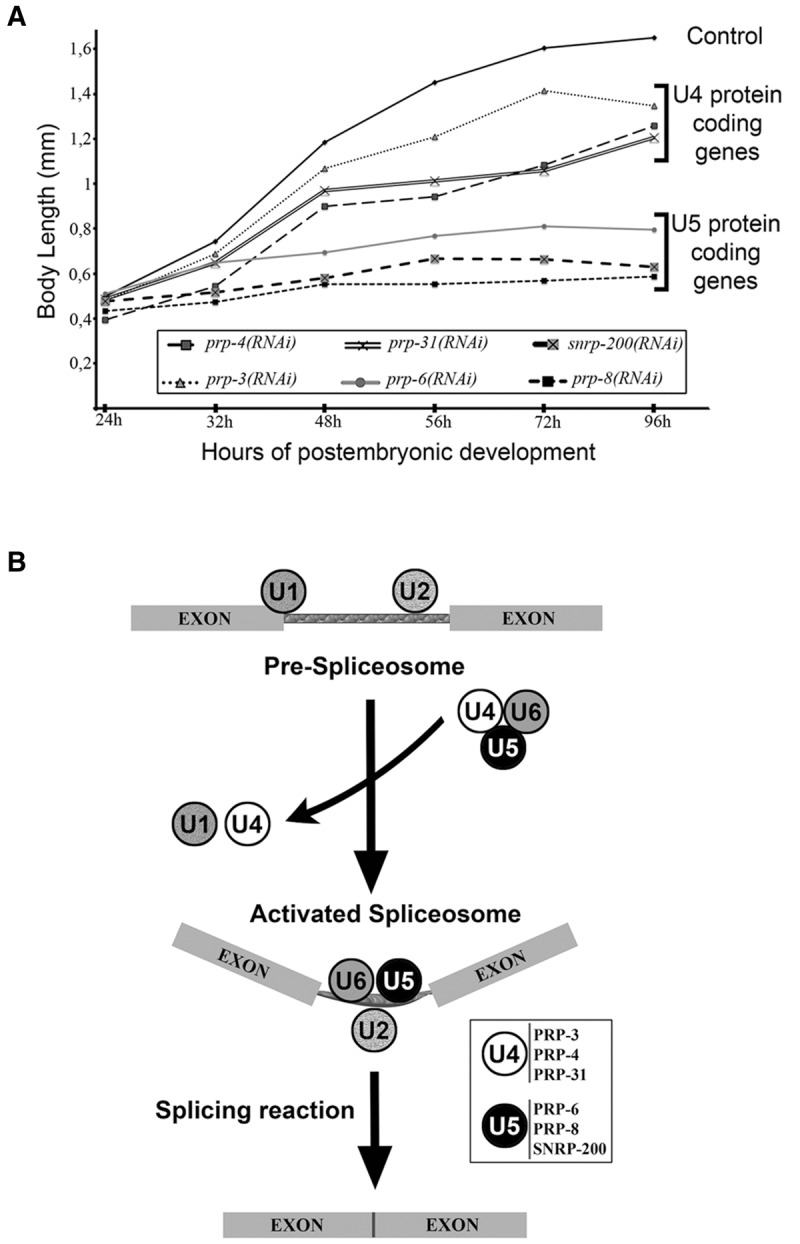
RNAi experiments classify s-adRP genes in two phenoclusters. (A) Developmental growth in animals treated with RNAi for s-adRP genes. Body length of wild-type worms with the indicated RNAi clones, starting from synchronized L1 and grown at 20°C. Mean body length of 50 worms per RNAi condition was scored at the indicated time points. Animal length was measured using the ImageJ software. Control worms were fed with the L4440 plasmid (empty vector). (B) Simplified scheme of the role of s-adRP genes in the splicing process. The six C. elegans s-adRP proteins are part of the tri-snRNP U4/U6·U5 complex. After the formation of the activated spliceosome, U5, but not U4, s-adRP proteins are required for subsequent splicing steps involving transesterification reactions. Exons are linked by thinner rectangles that represent an intron. White box: PRP-3, PRP-4, and PRP-31 are U4-specific, whereas PRP-6, PRP-8, and SNRNP-200 are U5-specific.
As mentioned in the introduction, all six proteins are components of the U4/U6·U5 tri-snRNP, but only PRP-6, PRP-8 and SNRP-200 are present in the U5 snRNP and participate in the activated spliceosome (Fig. 2B). This functional difference could explain the stronger phenotype caused by their corresponding RNAi feeding clones.
To increase the efficiency of the RNAi, we synthesized and microinjected dsRNA for prp-3, prp-4, and prp-31. These RNAi by injection caused embryonic lethality for all three, confirming the phenotype observed for all the s-adRP genes in a previous large-scale RNAi by injection study (Sönnichsen et al. 2005).
Therefore, a strong RNAi inactivation of s-adRP genes produces a nonviable phenotype. However, RNAi by feeding of s-adRP genes produces milder defects, with prp-6, prp-8, and snrp-200 displaying more dramatic RNAi phenotypes probably because of their functions in the activated or catalytic spliceosome.
The imprint of s-adRP genes on splicing catalysis
To study the molecular consequences of a partial inactivation of s-adRP genes, we analyzed the transcriptomes of animals treated with RNAi against prp-6, prp-8, and prp-31. L1 animals were fed with the corresponding RNAi clone and harvested after 24 h at 20°C, before developmental alterations due to RNAi became perceptible. In order to estimate the global levels of intron retention, we quantified the proportion of reads (read length 73 bp) mapping in intronic sequences in experimental and control samples. To ensure that our analysis was detecting intron retention and not stabilization of excised introns, we scored the proportion of reads mapping within introns and reads mapping in exon–intron junctions (Fig. 3A). Mild intron retention levels were observed in animals treated with RNAi against these three s-adRP genes. The strongest levels of intron retention were detected in prp-8(RNAi) samples. More precisely, 6.49% and 4.07% of the reads mapped in exon–intron junctions in prp-8(RNAi) and gfp(RNAi) animals, respectively. These results indicate a basal level of unspliced transcripts in control animals, suggesting that the splicing machinery is not 100% efficient in the excision of introns in normal conditions. We corroborated the increase of intron retention events in prp-8(RNAi) animals by semiquantitative RT-PCR (Supplemental Fig. S3A) and by detecting reads crossing the exon–intron border of some pre-mRNAs (Supplemental Fig. S3B).
FIGURE 3.

Intron retention caused by RNAi of s-adRP genes in different backgrounds and stages. Proportion of reads (73 bp) mapping within intron or in the intron–exon border in L3 N2 and smg-1 mutants or in adult glp-4 mutants, upon the indicated RNAis.
By considering only RNA-seq reads that mapped in annotated introns, we identified 69 statistically significant (P-value ≤ 0.01) common intron retention events in the whole genome of L3 animals treated with RNAi against s-adRP genes (Supplemental Table S1). We did not detect any common characteristic of the retained introns in terms of length (ranging from 114 to 4506 bp) or position within the gene (Supplemental Table S1).
The nonsense-mediated-decay (NMD) machinery prevents the expression of truncated proteins by degrading transcripts with premature termination codons (PTCs) (Hodgkin et al. 1989; Chang et al. 2007). Previous transcriptomic studies estimated that 20% of C. elegans genes produce transcripts that are degraded by the NMD pathway and many of those transcripts arise from splicing errors as retention of introns or wrong splice site selection (Ramani et al. 2009). Therefore, in our RNAi experiments the NMD system could be masking a stronger effect on intron retention. To address this concern we performed the same transcriptomic studies in the NMD defective strain smg-1(r861). Although the proportion of reads on introns was slightly higher in all smg-1(r861) samples, results were comparable with those obtained in wild-type animals and we did not observe a more dramatic effect on intron retention in NMD mutants upon RNAi of s-adRP genes (Fig. 3A).
Interestingly, in adult animals without a germline (5-d-old glp-4(bn2) mutants) we did not observe the mild intron retention associated with prp-8(RNAi), probably because of the much more reduced levels of transcriptional activity in adult animals compared with animals during development (Fig. 3B).
RNAi of s-adRP genes induces the expression of the pro-apoptotic factor egl-1 and apoptosis in a cell type-specific manner
The transcriptome of prp-6(RNAi), prp-8(RNAi), and prp-31(RNAi) animals compared with those exposed to control RNAi brought to light a list of genes misregulated with distinct P-values derived from our bioinformatic analysis (Supplemental Table S2). There are different strategies to take benefit of this transcriptomic data but we followed a candidate-gene approach because among the genes with the lowest P-value (≤0.0001) we found the effector of apoptosis egl-1, which encodes a conserved BH3-only domain protein (Conradt and Horvitz 1998; Nehme and Conradt 2008). We validated this up-regulation by qPCR (Fig. 4A,B).
FIGURE 4.

Up-regulation of atl-1 and egl-1 after RNAi of s-adRP genes follows a gradient from prp-8(RNAi) to prp-31(RNAi). (A) RNA-seq data of atl-1 and egl-1 after RNAi in wild-type worms. FPKM represents the fragments per kilobase of transcript per million mapped reads. Bars indicate the average “confidence_high” and “confidence_low” values provided by Cufflinks (Trapnell et al. 2012) for each gene. (B) Validation of the RNA-seq data by qPCR. mRNA levels of atl-1 and egl-1 upon RNAi of some s-adRP genes are represented relative to their expression in gfp(RNAi) control animals (arbitrary value of 1, indicated with a gray line). qPCR expression data were normalized to transcript levels of tbb-2. Three separate experiments were analyzed. Error bars represent the standard deviation. Student's t-test for independent samples was used to analyze the statistical significance: One, two, and three asterisks indicate P < 0.05, P < 0.01, and P < 0.001, respectively. (C) Both DNA insults, UV and hydroxyurea, produce an increase in atl-1 and egl-1 expression. Quantification of expression levels of atl-1 and egl-1 in wild-type animals treated either with UV (100 J/m2) or hydroxyurea (25 mM). Expression levels of these genes are represented relative to the ones in untreated worms.
Because we analyzed the transcriptome of a multicellular organism, we wondered whether the over-expression of egl-1 was global or cell type-specific. Hence, we treated EGL-1::GFP transgenic animals with prp-8(RNAi) and observed GFP expression in somatic cells that are not expected to suffer apoptosis in normal development (Sulston and Horvitz 1977). In wild-type worms, programmed cell death occurs in 18 cells at the L2 stage (Fig. 5A; Lettre and Hengartner 2006). However, prp-8(RNAi) animals displayed ectopic EGL-1::GFP expression that was particularly evident in hypodermal seam cells, which form a row from the head to the tail of the worm (Fig. 5B).
FIGURE 5.

egl-1::GFP expression is ectopically induced in prp-8(RNAi) animals. Expression of the transgene opIs56 [Pegl-1::2xNLS::GFP] after 24 h at 20°C in (A) control RNAi (empty vector) and (B) prp-8(RNAi). Size of the animals and the germline development stage indicate that in our experimental conditions prp-8(RNAi) worms develop to a similar stage as N2 during the first 24 h. prp-8(RNAi) worms ectopically express GFP in additional cells, including hypodermal seam cells (magnified area). Blue fluorescence is shown to label areas with autofluorescence. Images displayed are representative of three different experimental replicates. (C,D) Animals treated with prp-8(RNAi) display additional apoptotic cell corpses. The ced-6 mutant larvae treated with (C) control RNAi and (D) prp-8(RNAi). Black arrows indicate apoptotic cells (button-like refractile corpses) that were found in prp-8(RNAi) worms but not in control animals. The right panel shows a magnified image of the area highlighted with a white box.
To confirm that ectopic egl-1 expression results in apoptosis, we looked for apoptotic cell-corpses in larval cells using the mutant strain ced-6(n2095), which is defective in the clearance of apoptotic cells. As expected, we detected apoptotic corpses in somatic hypodermal cells of animals treated with prp-8(RNAi) that were not present in wild-type worms (Fig. 5C,D). Moreover, using a GFP reporter for hypodermal seam cells, we observed a reduced number of seam cells in prp-8(RNAi) worms (Supplemental Fig. S4).
We further investigated this cell-specific phenotype by using strains where the RNAi effect was tissue-specific. These strains contain a mutation in the rde-1 gene, which is essential for RNAi, and ectopically express rde-1 either in muscle or hypodermal cells only. Strikingly, we observed the characteristic growth arrest of prp-8(RNAi) when the RNAi machinery was functional in hypodermal cells, but we did not detect such phenotype when the prp-8(RNAi) was effective in muscle cells (Fig. 6A,B). Interestingly, these hypodermal cells synthesize the proteins that form the cuticle and, similarly to retinal cells, require high transcriptional activity (Page and Johnstone 2007).
FIGURE 6.

prp-8(RNAi) animals display tissue-specific phenotype. (A) Larval arrest phenotype was observed in wild-type- and hypodermis-specific RNAi animals (rde-1 (ne219) V; KzIs9), while no obvious phenotype was observed in muscle-specific RNAi worms (rde-1 (ne219) V; KzIs20). Representative images were taken under the stereoscope after 48 h at 20°C. Scale bar, 1 mm. (B) Body length measure of wild-type, and hypodermis- and muscle-specific RNAi animals fed with prp-8(RNAi) and control (empty vector) clones. Animals synchronized at L1 stage were treated with the corresponding RNAi clone and grown at 20°C. More than 25 worms were measured after 48 h of treatment in each condition. Animal length is showed in millimeters and was scored using the ImageJ software. Statistical significance was calculated using Student's t-test for independent samples. Three asterisks indicate statistical significance with P < 0.001. Whiskers were plotted by Tukey's test. (C) Up-regulation of atl-1 and egl-1 after prp-8(RNAi) is tissue specific. qPCR results for atl-1 and egl-1 expression in wild-type and hypodermis- and muscle-specific RNAi animals represented in a bar graph. mRNA levels of these genes after prp-8(RNAi) are relative to their expression in control animals. Results obtained from three independent biological replicates. mRNA transcript levels of atl-1 and egl-1 are normalized against tbb-2 levels and represented in a log10 scale in both experiments. Error bars represent standard deviation.
atl-1, the homolog of human ATR, is up-regulated after RNAi of s-adRP genes
Following the candidate gene approach, among the top up-regulated genes atl-1/ATR also called our attention (Fig. 4A,B). ATM and ATR are the primary sensors of DNA damage in humans. Whereas ATM mainly senses DNA double-strand breaks (DSBs), ATR responds to other DNA lesions such as those produced by replication fork stalling or UV damage, and it is recruited together with Replication protein A (RPA) to single-strand DNA regions originated by DNA damage (Kastan and Bartek 2004; Falck et al. 2005; Garcia-Muse and Boulton 2005).
Thus, we were persuaded to investigate whether a slight defect in the spliceosome activity could produce DNA lesions. It has been described that inactivation of various RNA processing factors induces the accumulation of DNA damage foci (Montecucco and Biamonti 2013). Therefore, we used the RPA-1::YFP reporter to study the effect of prp-8(RNAi) on the foci formation of the DNA damage sensing protein RPA-1. RPA-1::YFP is expressed in all worm cells, but the distribution of this tagged protein is diffuse and does not form visible foci (Vermezovic et al. 2012). We did not observe RPA-1::GFP foci after prp-8(RNAi) in normal growth conditions but the number of foci was higher in prp-8(RNAi) worms than in control animals upon UV exposure (Fig. 7). This result suggests that a partial depletion of prp-8 activity may cause an alteration in the DNA that is not reported by our RPA-1::YFP transgenic in normal conditions, but it is uncovered upon UV exposure.
FIGURE 7.

Higher sensitivity to UV damage observed in prp-8(RNAi)-treated animals. After UV exposure, foci formation evidenced by the transgene opIs263 [Prpa-1::rpa-1::YFP+unc-119(+)] is more abundant in animals with partial inactivation of prp-8 compared with control worms (fed with empty vector clone). (A) Representative confocal images of transgenic worms carrying the RPA-1::YFP transgene under control and prp-8(RNAi) conditions. RNAi treatment started from L1 stage at 20°C. After 24 h, animals were exposed to a 100 J/m2 dose of UV-C radiation, and 24 h later foci formation was assessed through confocal microscopy. Foci formation was observed in somatic and germline cells in both conditions. White squares show magnified images of germline cells with RPA-1::YFP foci. (B) Quantification of foci formation per cell represented in a dispersion graph. Each square (control) and dot (prp-8(RNAi)) represents the number of cells displaying the corresponding amount of foci. Only somatic and germline cells that displayed one or more foci were scored.
Another DNA insult that can increase the activity of atl-1 is replicative stress. Replication forks can be collapsed by the presence of R-loops (DNA–RNA hybrids), whose formation can be promoted by alterations in the spliceosome (Aguilera and García-Muse 2012; Hamperl and Cimprich 2014). Such impact of R-loops on the replication machinery may induce replicative stress (Mazouzi et al. 2014). To induce replicative stress in C. elegans larvae, we exposed developing worms to hydroxyurea and observed, similarly to the effect of prp-8(RNAi), ectopic expression of egl-1::GFP and arrested development (Supplemental Fig. S5).
To confirm the impact of DNA damage or replicative stress in the expression of egl-1 and atl-1, larvae were exposed to UV or treated with HU, respectively, and expression levels of these two genes were studied by qPCR 24 h after L1 (the same stage at which animals were harvested for RNA-seq experiments). These two DNA insults were capable of increasing the expression levels of atl-1 and egl-1 (Fig. 4C). Interestingly, we observed that the tissue-specific up-regulation of atl-1 and egl-1 occurred when prp-8(RNAi) was efficient in the hypodermis but did not happen when the RNAi worked in muscle cells only (Fig. 6C).
All together, these results suggest that larval arrest, cell type-specific apoptosis, and up-regulation of egl-1 and atl-1 could be consequences of DNA alterations driven by impaired functions of the spliceosome in a particular cell type.
DISCUSSION
Despite the publication of more than two hundred reports about the implication of spliceosome components in RP, the molecular mechanisms behind the role of ubiquitous proteins in a retinal-specific disease are still unclear. Similarly, SMN1 and SMN2 are widely expressed in human tissues but their mutations only produce defects in motor neurons. Such neuronal defects cause spinal muscular atrophy, a human disease that has also been investigated in C. elegans through the worm ortholog smn-1 (Briese et al. 2009; Sleigh et al. 2011). This is just one example of the multiple human diseases (others are cancer or metabolic disorders) that have been studied and modeled in the multicellular organism C. elegans (Markaki and Tavernarakis 2010). Conveniently, s-adRP genes and the mechanisms of splicing in which they are implicated are very well conserved, which justifies the study of splicing-associated diseases in worms.
Alterations in U4 or U5 snRNP coding genes have a different impact on the organism
Based on the analysis of RNAi phenotypes, our study distinguishes between s-adRP genes coding for U4 or U5 components. All s-adRP proteins are required to activate the spliceosome, but U5 proteins remain in the activated complex whereas U4 proteins do not. This wider role of U5 snRNPs in splicing could explain why human genes coding for U5 snRNPs allow less harmful mutations than U4 snRNP coding genes. For example, most PRP8 adRP mutations consist of a single amino acid change, caused by a point mutation in the last exon (exon 42) (Grainger and Beggs 2005; Towns et al. 2010), whereas genes coding U4 snRNP components such as PRPF31 include null alleles that are able to be maintained in heterozygosis in adRP patients (Mordes et al. 2006; Rio Frio et al. 2008), and probably somatic loss of heterozygosity (Abu-Safieh et al. 2006). Accordingly, it is likely that human nonretinal cells can only handle the loss of a functional copy of the gene in s-adRP genes coding for U4 snRNPs components such as PRP31. Because there are still adRP patients whose disease-causing mutation has not been identified, other genes encoding proteins required for the tri-snRNP U4/U6·U5 formation, such as SNU114 (U5 snRNP) or SNU13 (U4 snRNP), are candidates to be screened for mutations responsible of adRP (de Sousa Dias et al. 2013).
Mutations in the U4 component PRP31 cause adRP with a variable penetrance that allows some carriers to stay asymptomatic. Recent studies focused on factors influencing the regulation of PRPF31 expression and therefore the pathogenic effect of these mutations (Venturini et al. 2012; Rose et al. 2014). Our reporter strains carrying both the promoter and the 3′ UTR of prp-31 can be used as a system to identify genes or conditions that could influence the expression of prp-31. Although we did not observe haploinsufficiency in worms heterozygous for a deletion in prp-31, these animals can serve as a sensitive background to search for factors capable of influencing prp-31 functions.
Because all s-adRP proteins function in early splicing steps, future studies should focus on spliceosome assembly rather than on intron removal (catalysis) to unmask common mechanisms of action fueling the pathology.
Worms as a model for s-adRP
This study proposes the usage of C. elegans as a model to investigate the cellular mechanisms causing a subtype of the rare disease RP. Such a proposal is strongly supported by the following arguments: (i) Through RNAi we can mimic the partial loss of function of s-adRP genes occurring in RP patients (Longman et al. 2000). (ii) RNAi of s-adRP genes causes mild intron retention similar to what has been observed in other s-adRP models (Gamundi et al. 2008; Tanackovic et al. 2011b). (iii) Some retina cells of s-adRP patients suffer apoptotic cell death, whereas cells in other tissues are unaffected, and our model induces the ectopic expression of the pro-apoptotic gene egl-1 and the onset of apoptotic cell corpses in developing hypodermal cells. (iv) RNAi by feeding of s-adRP genes supposes a constraint to developmental processes requiring high transcriptional activity (larval development for U5 genes and germline development for U4 genes), and s-adRP patients present disease only in the retina, a human tissue with very high transcriptional requirements (Derlig et al. 2015), whereas other tissues are asymptomatic.
Why do mutations in s-adRP genes have the ability to cause retina-specific diseases in humans?
Several hypotheses have been formulated to address this question and probably most of them are valid considering the complexity of the retina and the diversity of mutations leading to s-adRP. Taking into consideration the published data and our study, we find three compatible possibilities, which are defects in splicing reactions, lower transcriptional efficiency, and genome instability through the formation of R-loops (Fig. 8).
FIGURE 8.

Model for the tissue-specific apoptosis in s-adRP. (A) Similarly to humans, s-adRP genes are expressed in all C. elegans cell types. Partial inactivation of s-adRP genes causes tissue-specific defects both in humans and worms. (B) In normal condition, splicing occurs cotranscriptionally. (C) When the functioning of the splicesome is altered, the activity of the RNAPII is affected and R-loops (DNA–RNA hybrids) are produced, creating single-strand DNA regions that are more sensitive to DNA insults. Moreover, R-loops may cause collisions between the transcriptional and the replicative machineries.
The most straightforward explanation is that splicing is inefficient in the retina, where cells present a high transcriptional activity, causing a reduction in the mRNA production and consequently a deficit in the amount of proteins. However, these defects in constitutive splicing are not general because studies in lymphoblastoid cell lines from s-adRP patients demonstrates that only five out of 57 introns tested presented unspliced products (Tanackovic et al. 2011b). Moreover, only three of these five intron retention events were common for all the PRPF mutations tested (Tanackovic et al. 2011a,b). Nevertheless, although a limited/mild intron retention has been observed in s-adRP models (Wilkie et al. 2008; Tanackovic et al. 2011b), a correlation between slight intron retention in certain genes and retinal degeneration has not been clearly established yet (Mordes et al. 2006).
In C. elegans, by RT-PCR and by observing intron retention events in a genome browser (i.e., a fmo-5 intron [Supplemental Fig. S3]), we conclude that the likelihood of a partial depletion of s-adRP genes keeping transcripts unspliced is low, underscoring a high buffering capacity of spliceosomal proteins to maintain their essential functions despite an important reduction of their activity.
The alteration of these adRP splicing components may affect alternative splicing (AS) and thus the balance among isoforms. In fact, it has been shown that AS is affected in cell lines derived from patients carrying s-adRP mutations (Tanackovic et al. 2011b). The alterations in the levels of alternative transcripts do not seem to be similar in cell lines with different s-adRP mutations (Tanackovic et al. 2011b). On the contrary, a recent report based on siRNA in cell lines has uncovered a link between patterns of AS alterations and functionally related spliceosome components (Papasaikas et al. 2014). To explore the effect of our RNAi assays on AS we checked a panel of eight AS events occurring at L2 and L3 larval stages (Ramani et al. 2011) and did not find common AS alterations in s-adRP RNAi samples (Supplemental Table S3).
In any case, it is difficult to justify C. elegans’ phenotypes, as well as the retinal degeneration in humans, just by slight intron retention or by a modest unbalance of alternative transcripts, which argues in favor of a more complex mechanism.
The second possible answer relies on co-transcriptional splicing. It is well established that splicing and transcription are coordinated processes and that alterations in the spliceosome can affect the transcriptional machinery (Das et al. 2007; Fong et al. 2013; Fontrodona et al. 2013; Montecucco and Biamonti 2013). As an example of such co-transcriptional splicing in a developing multicellular animal, our laboratory demonstrated that RSR-2 interacts with PRP-8 and the RNA polymerase II (RNAPII) in C. elegans, and that the impairment of RSR-2 functions results in a lower transcriptional efficiency (Fontrodona et al. 2013). Consistent with this second hypothesis, the mutant allele prp-8(rr40), which reduces the levels of wild-type PRP-8, diminishes the production of highly expressed germline transcripts, but it does not affect the splicing reaction (Hebeisen et al. 2008).
Finally, our results support a third hypothesis that can coexist with the previous ones: A reduction in the activity of s-adRP proteins produces genomic instability that, in cells with high transcriptional activity, can contribute to programmed cell death. The impairment of the splicing machinery can form R-loops (RNA/DNA duplex) that expose the single-strand DNA (ssDNA) to DNA-damaging agents such as UV light at active transcriptional regions (Aguilera and García-Muse 2012, 2013). This would explain the sensitivity of prp-8(RNAi) animals to UV exposure. Moreover, R-loops are also a source of transcriptional associated mutagenesis (TAM), which is elevated under high transcriptional activity (Kim et al. 2007) and can induce apoptosis (Hendriks et al. 2010).
In addition to the formation of these DNA damage-prone sites, R-loops can contribute to genome instability because replication and transcriptional machineries could meet and collide at these R-loop sites leading to replicative stress (Brambati et al. 2015). In C. elegans the collision with the replication machinery is feasible because hypodermal cells are actively dividing during larval stages and also suffer a round of endoreplication (Sulston and Horvitz 1977; Hedgecock and White 1985). In mammalian cells, the existence of a proliferative capacity in adult retinal cells has been reported (Tropepe et al. 2000; Kiilgaard et al. 2007; Al-Hussaini et al. 2008), providing a possible scenario for replicative stress in dividing cells.
However, in the process of understanding the pathogenesis of the disease, we should take into consideration that the retinal degeneration of adRP patients is a gradual process that may take many years. Here we present C. elegans as a promising model to investigate mechanisms of the disease and potential therapies in a short time frame.
Future prospects
In this study, we present a robust model to study s-adRP in C. elegans. We focused our attention in atl-1 and egl-1, but we have identified other interesting genes in our data set (which is available at the Gene Expression Omnibus [GEO] repository) that are up-regulated in animals with reduced levels of spliceosomal proteins and that may have a role in compensating or repairing such deficiency. Among them, we identified genes coding for spliceosome components such as mog-4, mog-5, prpf-4, or ddx-23 (Supplemental Table S1; Kerins et al. 2010).
Through an increased sensitivity to DNA insults and replicative stress we have opened a third way to explain the retina-specific apoptosis in s-adRP. This alternative way should be explored in terms of therapies that could alleviate the progressive loss of vision in s-adRP patients. To such aim, C. elegans can play a key role thanks to its amenability for high-throughput screening.
Our approach takes advantage of the RNAi but the new technologies for genome editing and the functional conservation of spliceosomal genes are making feasible the introduction of human s-adRP mutations in worms (Waaijers and Boxem 2014). This approach could be an ideal platform to develop personalized medicine and drug discovery.
MATERIALS AND METHODS
Strains
Standard methods were used to culture and manipulate worms (Stiernagle 2006; Porta-de-la-Riva et al. 2012). In this study, we used the wild-type Bristol N2 and the strains TR1331: smg-1(r861) I, SS104: glp-4(bn2) I, CER87: unc-119(ed3) III, cerEx79[Pprp-31::GFP::H2B::prp-31 3′ UTR + pGH8 + unc-119(+)], CER148: unc-119(ed3) III; cerIs05[Pprp-31::GFP::H2B::prp-31 3′ UTR +unc-119(+)], CER149: unc-119(ed3) III; cerIs06[Pprp-31::GFP::H2B::prp-31 3′ UTR +unc-119(+)], VC2709: prp-31(gk1094) I/hT2[bli-4(e937) let-?(q782) qIs48](I;III), CER100: prp-31(gk1094)/unc-57(ad592) dpy-5(e61) I, CER101: +/unc-57(ad592) dpy-5(e61) I, VC3460: prp-8(gk3511) III/hT2 [bli-4(e937) let-?(q782) qIs48] (I;III), BC12486: dpy-5(e907) I; sEx12486[Pprp-8::GFP + pCeh361], WS1973: unc-119(ed3) III; opIs56[Pegl-1::2xNLS::GFP + unc-119(+)], MT4970: ced-6(n2095) III, NR222: rde-1(ne219) V; kzIs9[pKK1260(lin-26p::nls::GFP) + pKK1253(lin-26p::rde-1) + pRF6(rol-6(su1006)], NR350: rde-1(ne219) V;kzIs20[pDM#715(hlh-1p::rde-1) + pTG95(sur-5p::NLS::GFP)], WS4581: unc-119(ed3) III; opIs263[rpa-1p::rpa-1::YFP + unc-119(+)].
RNA-mediated interference
To induce RNA-mediated interference (RNAi) by feeding, nematode growth medium (NGM) plates were supplemented with 50 µg/mL ampicillin, 12.5 µg/mL tetracycline, and 1 mM IPTG. NGM-supplemented plates were seeded with the corresponding RNAi clone. All RNAi clones but the one corresponding to snrp-200 were obtained either from the ORFeome library (Rual et al. 2004) or the Ahringer library (Kamath et al. 2003). From cDNA we generated an RNAi clone (pCUC24) for snrp-200 by Gateway cloning (amplicon size 1091 bp), which includes the spliced sequence from 106 to 1197 bp (primers available under request). Each clone was validated by PCR and/or sequencing. To study the RNAi phenotypes of s-adRP genes, a synchronized population of N2 worms at L1 stage was fed with each clone and grown at 20°C. To induce RNAi by microinjection, dsRNA was synthesized from the RNAi feeding clone by using MEGAscript T7 kit (Ambion). Wild-type young adults were injected with 1 μg/μL of dsRNA.
RNA sequencing
RNA-seq analyses were performed using RNA of the following population sets: synchronized N2 and smg-1(r861) L1 larvae fed for 24 h at 20°C with the RNAi clones of prp-6, prp-8, prp-31, and gfp; and 5-d adult glp-4(bn2) worms grown at 25°C and fed first, for 72 h with OP50 and next, for 48 more hours with the RNAi clones of prp-8, prp-31, and gfp. RNA was isolated with TRI Reagent (MRC, Inc.) and purified by using the Purelink RNA Mini kit (Ambion) and Purelink DNAse (Ambion). RNA samples were subjected to quality and yield controls on the Agilent 2010 Bioanalyzer and Nanodrop Spectrophotometer, respectively. Poly(A)-enriched samples were multiplexed in libraries for paired-end RNA sequencing on Illumina Hiseq 2000 platform, through the CNAG (Centro Nacional de Análisis Genómico) sequencing facility. To generate BAM files, more than 50 million reads (length of 73 bp) for sample were mapped against the C. elegans worm version WS236 following the GEMTools pipeline (http://gemtools.github.io/). These BAM files were analyzed using the SeqSolve software, which use Cufflinks/Cuffdiff for differential gene/transcript expression analyses (Trapnell et al. 2012). A full 98.7% of the reads from the control gfp(RNAi) sample mapped in exons. The sequence data for the 11 transcriptomes analyzed in this study are available at the NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE72952.
Real-time PCR
For gene expression analyses, synchronized worms were harvested, washed, and frozen in TRI Reagent to perform total RNA isolation. cDNA was synthesized with oligo(dT) primers using the RevertAid H Minus First Strand cDNA synthesis kit (ThermoScientific) following the manufacturer's instructions. To determine gene expression, the LightCycler 480 SYBR Green I Master kit (Roche) was used.
Generation of transgenic reporter strains
The molecular construct to study the expression and localization of prp-31 (pCUC32) was made by the Gateway recombination cloning system (Invitrogen). The 5′ entry clone includes 664 bp of the promoter region, and 3′ entry clone contains the 77 bp of the 3′ UTR sequence (primer sequences available upon request). Plasmid pCM1.35 was used as the middle entry clone and pCFJ150 was the destination vector.
Microinjection
Transgenic animals were generated by microinjecting 2 ng/µL of pCUC32 [Pprp-31::GFP::H2B::prp-31 3′ UTR +unc-119(+)] together with 20 ng/µL of the red neuronal marker pGH8 [Prab-3::mCherry::unc-54utr] into DP38 [unc-119(ed3)] young adult worms. 78 ng/µL of 1 Kb Plus DNA ladder (Invitrogen) were used as carrier. Wild-type animals were selected over uncoordinated (Unc) worms, and mCherry expression was checked under the dissecting microscope. GFP expression was observed at high magnification using an inverted fluorescence Nikon ECLIPSE Ti-S microscope.
Bombardment
One and a half milligrams of gold powder (Chempur, gold powder 0.3–3.0 micron 99.9%) was transferred to a low protein binding 1.5-mL tube, resuspended in 150 µL of 50 mM spermidine, and sonicated for 10 sec. Twenty-four micrograms of pCUC32 DNA was added to the mixture, deionized water was added up to 540 µL, and the mixture was incubated for 10 min. Next, we added 150 µL of 1 M CaCl2. The mixture was incubated for another 10 min and centrifuged at maximum speed for 30 sec. After three washes with 96% ethanol, the pellet was resuspended in 300 µL PVP 0.1 mg/mL in EtOH. Shots with 20 µL of the suspension were performed using a Caenotec gene gun on pre-chilled 35-mm plates with 20–30 µL of DP38 [unc-119(ed3)] young adults. Then the NGM agar of each plate was sliced into six pieces, and each slice was placed on a 90-mm plate. These plates were incubated at 20°C for 2 wk and then screened for transgenic worms that rescued the unc-119(ed3) phenotype.
Replicative stress induction
To induce replicative stress we used hydroxyurea (HU) (Sigma-Aldrich, Cat# H8627). In all cases 25 mM HU plates were used and prepared adding 500 µL of 0.5 M HU to plates containing 10 mL of NGM medium and seeded with E. coli OP50. To observe egl-1 expression, synchronized Pegl-1::GFP (WS1973) L1 worms were grown in either HU or standard plates for 24 and 48 h at 20°C. After each time point, worms were recovered with M9 buffer, washed, and mounted using 0.3 mM of levamisole for in vivo observation through an inverted fluorescence microscope. To quantify the expression levels of atl-1 and egl-1 in a replicative stress context, synchronized N2 L1 worms were grown at 20°C for 24 h in HU plates and then recovered with M9 buffer for total RNA isolation.
DNA damage induction through UV-C exposure
Synchronized worms were exposed to UV-C light (254 nm) at different time points depending on the experiment. A UV crosslinker (model 2400, Stratagene) was used to apply 100 J/m2 in all the cases. Before the exposure to UV-C, worms were washed several times to get rid of bacteria and placed in a bacteria-free NGM plate.
To quantify the expression levels of atl-1 and egl-1 upon DNA damage induced by UV-C exposure, synchronized L1 N2 worms were grown in standard conditions at 20°C. UV-C exposure was performed 18 h post L1 and total RNA isolation 6 h later.
Foci quantification
For RPA-1 foci observation synchronized Prpa::YFP L1 worms were grown at 20°C for 24 h. Then, they were exposed to UV-C and later placed again in control or prp-8(RNAi) plates for an additional 24 h. Worms were then mounted using 0.3 mM of levamisole and observed using a confocal fluorescence microscope. RPA-1::YFP foci formation was quantified by counting the number of bright foci present in germline and somatic cells.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Juan Valcarcel and Dr. Ana Méndez for critical reading of the manuscript. Some strains were provided by the CGC (Caenorhabditis Genetics Center), which is funded by the National Institutes of Health Office of Research Infrastructure Programs (P40 OD010440). J.C. is a Miguel Servet Researcher (ISCIII). K.R.-P. has a FINCyT PhD fellowship from the government of Peru. This study was supported by La Marató de TV3 foundation (Ref. 100910) and by a grant from the Instituto de Salud Carlos III (ISCIII) (Exp. PI12/01554), which is cofunded by FEDER funds/European Regional Development Fund (ERDF)—a way to build Europe.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.053397.115.
REFERENCES
- Abu-Safieh L, Vithana EN, Mantel I, Holder GE, Pelosini L, Bird AC, Bhattacharya SS. 2006. A large deletion in the adRP gene PRPF31: evidence that haploinsufficiency is the cause of disease. Mol Vis 12: 384–388. [PubMed] [Google Scholar]
- Aguilera A, García-Muse T. 2012. R loops: from transcription byproducts to threats to genome stability. Mol Cell 46: 115–124. [DOI] [PubMed] [Google Scholar]
- Aguilera A, García-Muse T. 2013. Causes of genome instability. Annu Rev Genet 47: 1–32. [DOI] [PubMed] [Google Scholar]
- Al-Hussaini H, Kam JH, Vugler A, Semo M, Jeffery G. 2008. Mature retinal pigment epithelium cells are retained in the cell cycle and proliferate in vivo. Mol Vis 14: 1784–1791. [PMC free article] [PubMed] [Google Scholar]
- Brambati A, Colosio A, Zardoni L, Galanti L, Liberi G. 2015. Replication and transcription on a collision course: eukaryotic regulation mechanisms and implications for DNA stability. Front Genet 6: 166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briese M, Esmaeili B, Fraboulet S, Burt EC, Christodoulou S, Towers PR, Davies KE, Sattelle DB. 2009. Deletion of smn-1, the Caenorhabditis elegans ortholog of the spinal muscular atrophy gene, results in locomotor dysfunction and reduced lifespan. Hum Mol Genet 18: 97–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y-F, Imam JS, Wilkinson MF. 2007. The nonsense-mediated decay RNA surveillance pathway. Annu Rev Biochem 76: 51–74. [DOI] [PubMed] [Google Scholar]
- Chen X, Liu Y, Sheng X, Tam PO, Zhao K, Chen X, Rong W, Liu Y, Liu X, Pan X, et al. 2014. PRPF4 mutations cause autosomal dominant retinitis pigmentosa. Hum Mol Genet 23: 2926–2939. [DOI] [PubMed] [Google Scholar]
- Conradt B, Horvitz HR. 1998. The C. elegans protein EGL-1 is required for programmed cell death and interacts with the Bcl-2-like protein CED-9. Cell 93: 519–529. [DOI] [PubMed] [Google Scholar]
- Daiger SP, Sullivan LS, Bowne SJ. 2013. Genes and mutations causing retinitis pigmentosa. Clin Genet 84: 132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das R, Yu J, Zhang Z, Gygi MP, Krainer AR, Gygi SP, Reed R. 2007. SR proteins function in coupling RNAP II transcription to pre-mRNA splicing. Mol Cell 26: 867–881. [DOI] [PubMed] [Google Scholar]
- Derlig K, Gießl A, Brandstätter JH, Enz R, Dahlhaus R. 2015. Special characteristics of the transcription and splicing machinery in photoreceptor cells of the mammalian retina. Cell Tissue Res 10.1007/s00441-015-2204-x. [DOI] [PubMed] [Google Scholar]
- De Sousa Dias M, Hernan I, Pascual B, Borràs E, Mañé B, Gamundi MJ, Carballo M. 2013. Detection of novel mutations that cause autosomal dominant retinitis pigmentosa in candidate genes by long-range PCR amplification and next-generation sequencing. Mol Vis 19: 654–664. [PMC free article] [PubMed] [Google Scholar]
- Fabrizio P, Dannenberg J, Dube P, Kastner B, Stark H, Urlaub H, Lührmann R. 2009. The evolutionarily conserved core design of the catalytic activation step of the yeast spliceosome. Mol Cell 36: 593–608. [DOI] [PubMed] [Google Scholar]
- Falck J, Coates J, Jackson SP. 2005. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 434: 605–611. [DOI] [PubMed] [Google Scholar]
- Ferrari S, Di Iorio E, Barbaro V, Ponzin D, Sorrentino FS, Parmeggiani F. 2011. Retinitis pigmentosa: genes and disease mechanisms. Curr Genomics 12: 238–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. 1998. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391: 806–811. [DOI] [PubMed] [Google Scholar]
- Fong YW, Cattoglio C, Tjian R. 2013. The intertwined roles of transcription and repair proteins. Mol Cell 52: 291–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontrodona L, Porta-de-la-Riva M, Morán T, Niu W, Díaz M, Aristizábal-Corrales D, Villanueva A, Schwartz S, Reinke V, Cerón J. 2013. RSR-2, the Caenorhabditis elegans ortholog of human spliceosomal component SRm300/SRRM2, regulates development by influencing the transcriptional machinery. ed. N. Maizels. PLoS Genet 9: e1003543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamundi MJ, Hernan I, Muntanyola M, Maseras M, López-Romero P, Alvarez R, Dopazo A, Borrego S, Carballo M. 2008. Transcriptional expression of cis-acting and trans-acting splicing mutations cause autosomal dominant retinitis pigmentosa. Hum Mutat 29: 869–878. [DOI] [PubMed] [Google Scholar]
- Garcia-Muse T, Boulton SJ. 2005. Distinct modes of ATR activation after replication stress and DNA double-strand breaks in Caenorhabditis elegans. EMBO J 24: 4345–4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grainger RJ, Beggs JD. 2005. Prp8 protein: at the heart of the spliceosome. RNA 11: 533–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamperl S, Cimprich KA. 2014. The contribution of co-transcriptional RNA:DNA hybrid structures to DNA damage and genome instability. DNA Repair (Amst) 19: 84–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartong DT, Berson EL, Dryja TP. 2006. Retinitis pigmentosa. Lancet 368: 1795–1809. [DOI] [PubMed] [Google Scholar]
- Hebeisen M, Drysdale J, Roy R. 2008. Suppressors of the cdc-25.1(gf)-associated intestinal hyperplasia reveal important maternal roles for prp-8 and a subset of splicing factors in C. elegans. RNA 14: 2618–2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedgecock EM, White JG. 1985. Polyploid tissues in the nematode Caenorhabditis elegans. Dev Biol 107: 128–133. [DOI] [PubMed] [Google Scholar]
- Hendriks G, Jansen JG, Mullenders LHF, de Wind N. 2010. Transcription-coupled repair and apoptosis provide specific protection against transcription-associated mutagenesis by ultraviolet light. Transcription 1: 95–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin J, Papp A, Pulak R, Ambros V, Anderson P. 1989. A new kind of informational suppression in the nematode Caenorhabditis elegans. Genetics 123: 301–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt-Newbury R, Viveiros R, Johnsen R, Mah A, Anastas D, Fang L, Halfnight E, Lee D, Lin J, Lorch A, et al. 2007. High-throughput in vivo analysis of gene expression in Caenorhabditis elegans. PLoS Biol 5: e237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath RS, Fraser AG, Dong Y, Poulin G, Durbin R, Gotta M, Kanapin A, Le Bot N, Moreno S, Sohrmann M, et al. 2003. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature 421: 231–237. [DOI] [PubMed] [Google Scholar]
- Kastan MB, Bartek J. 2004. Cell cycle checkpoints and cancer. Nature 432: 316–323. [DOI] [PubMed] [Google Scholar]
- Kerins JA, Hanazawa M, Dorsett M, Schedl T. 2010. PRP-17 and the pre-mRNA splicing pathway are preferentially required for the proliferation versus meiotic development decision and germline sex determination in Caenorhabditis elegans. Dev Dyn 239: 1555–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiilgaard JF, Prause JU, Prause M, Scherfig E, Nissen MH, la Cour M. 2007. Subretinal posterior pole injury induces selective proliferation of RPE cells in the periphery in in vivo studies in pigs. Invest Ophthalmol Vis Sci 48: 355–360. [DOI] [PubMed] [Google Scholar]
- Kim N, Abdulovic AL, Gealy R, Lippert MJ, Jinks-Robertson S. 2007. Transcription-associated mutagenesis in yeast is directly proportional to the level of gene expression and influenced by the direction of DNA replication. DNA Repair (Amst) 6: 1285–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lettre G, Hengartner MO. 2006. Developmental apoptosis in C. elegans: a complex CEDnario. Nat Rev Mol Cell Biol 7: 97–108. [DOI] [PubMed] [Google Scholar]
- Liu S, Rauhut R, Vornlocher H-P, Lührmann R. 2006. The network of protein–protein interactions within the human U4/U6.U5 tri-snRNP. RNA 12: 1418–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longman D, Johnstone IL, Cáceres JF. 2000. Functional characterization of SR and SR-related genes in Caenorhabditis elegans. EMBO J 19: 1625–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maita H, Kitaura H, Ariga H, Iguchi-Ariga SMM. 2005. Association of PAP-1 and Prp3p, the products of causative genes of dominant retinitis pigmentosa, in the tri-snRNP complex. Exp Cell Res 302: 61–68. [DOI] [PubMed] [Google Scholar]
- Markaki M, Tavernarakis N. 2010. Modeling human diseases in Caenorhabditis elegans. Biotechnol J 5: 1261–1276. [DOI] [PubMed] [Google Scholar]
- Mazouzi A, Velimezi G, Loizou JI. 2014. DNA replication stress: causes, resolution and disease. Exp Cell Res 329: 85–93. [DOI] [PubMed] [Google Scholar]
- Montecucco A, Biamonti G. 2013. Pre-mRNA processing factors meet the DNA damage response. Front Genet 4: 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mordes D, Luo X, Kar A, Kuo D, Xu L, Fushimi K, Yu G, Sternberg P Jr, Wu JY. 2006. Pre-mRNA splicing and retinitis pigmentosa. Mol Vis 12: 1259–1271. [PMC free article] [PubMed] [Google Scholar]
- Nehme R, Conradt B. 2008. egl-1: a key activator of apoptotic cell death in C. elegans. Oncogene 27Suppl 1: S30–S40. [DOI] [PubMed] [Google Scholar]
- Page AP, Johnstone IL. 2007. The cuticle. wormbook. www.wormbook.org. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papasaikas P, Tejedor JR, Vigevani L, Valcárcel J. 2014. Functional splicing network reveals extensive regulatory potential of the core spliceosomal machinery. Mol Cell 57: 7–22. [DOI] [PubMed] [Google Scholar]
- Porta-de-la-Riva M, Fontrodona L, Villanueva A, Cerón J. 2012. Basic Caenorhabditis elegans methods: synchronization and observation. J Vis Exp e4019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramani AK, Nelson AC, Kapranov P, Bell I, Gingeras TR, Fraser AG. 2009. High resolution transcriptome maps for wild-type and nonsense-mediated decay-defective Caenorhabditis elegans. Genome Biol 10: R101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramani AK, Calarco JA, Pan Q, Mavandadi S, Wang Y, Nelson AC, Lee LJ, Morris Q, Blencowe BJ, Zhen M, et al. 2011. Genome-wide analysis of alternative splicing in Caenorhabditis elegans. Genome Res 21: 342–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rio Frio T, Wade NM, Ransijn A, Berson EL, Beckmann JS, Rivolta C. 2008. Premature termination codons in PRPF31 cause retinitis pigmentosa via haploinsufficiency due to nonsense-mediated mRNA decay. J Clin Invest 118: 1519–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose AM, Shah AZ, Venturini G, Rivolta C, Rose GE, Bhattacharya SS. 2014. Dominant PRPF31 mutations are hypostatic to a recessive CNOT3 polymorphism in retinitis pigmentosa: a novel phenomenon of “linked trans-acting epistasis”. Ann Hum Genet 78: 62–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossmiller B, Mao H, Lewin AS. 2012. Gene therapy in animal models of autosomal dominant retinitis pigmentosa. Mol Vis 18: 2479–2496. [PMC free article] [PubMed] [Google Scholar]
- Rual J-F, Ceron J, Koreth J, Hao T, Nicot A-S, Hirozane-Kishikawa T, Vandenhaute J, Orkin SH, Hill DE, van den Heuvel S, et al. 2004. Toward improving Caenorhabditis elegans phenome mapping with an ORFeome-based RNAi library. Genome Res 14: 2162–2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleigh JN, Buckingham SD, Esmaeili B, Viswanathan M, Cuppen E, Westlund BM, Sattelle DB. 2011. A novel Caenorhabditis elegans allele, smn-1(cb131), mimicking a mild form of spinal muscular atrophy, provides a convenient drug screening platform highlighting new and pre-approved compounds. Hum Mol Genet 20: 245–260. [DOI] [PubMed] [Google Scholar]
- Sönnichsen B, Koski LB, Walsh A, Marschall P, Neumann B, Brehm M, Alleaume A-M, Artelt J, Bettencourt P, Cassin E, et al. 2005. Full-genome RNAi profiling of early embryogenesis in Caenorhabditis elegans. Nature 434: 462–469. [DOI] [PubMed] [Google Scholar]
- Stiernagle T. 2006. Maintenance of C. elegans. www.wormbook.org. [DOI] [PMC free article] [PubMed]
- Sulston JE, Horvitz HR. 1977. Post-embryonic cell lineages of the nematode, Caenorhabditis elegans. Dev Biol 56: 110–156. [DOI] [PubMed] [Google Scholar]
- Tanackovic G, Ransijn A, Ayuso C, Harper S, Berson EL, Rivolta C. 2011a. A missense mutation in PRPF6 causes impairment of pre-mRNA splicing and autosomal-dominant retinitis pigmentosa. A.m. J Hum Genet 88: 643–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanackovic G, Ransijn A, Thibault P, Abou Elela S, Klinck R, Berson EL, Chabot B, Rivolta C. 2011b. PRPF mutations are associated with generalized defects in spliceosome formation and pre-mRNA splicing in patients with retinitis pigmentosa. Hum Mol Genet 20: 2116–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The C. elegans Deletion Mutant Consortium. 2012. large-scale screening for targeted knockouts in the Caenorhabditis elegans genome. G3 (Bethesda) 2: 1415–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towns KV, Kipioti A, Long V, McKibbin M, Maubaret C, Vaclavik V, Ehsani P, Springell K, Kamal M, Ramesar RS, et al. 2010. Prognosis for splicing factor PRPF8 retinitis pigmentosa, novel mutations and correlation between human and yeast phenotypes. Hum Mutat 31: E1361–E1376. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. 2012. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7: 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tropepe V, Coles BL, Chiasson BJ, Horsford DJ, Elia AJ, McInnes RR, van der Kooy D. 2000. Retinal stem cells in the adult mammalian eye. Science 287: 2032–2036. [DOI] [PubMed] [Google Scholar]
- Venturini G, Rose AM, Shah AZ, Bhattacharya SS, Rivolta C. 2012. CNOT3 is a modifier of PRPF31 mutations in retinitis pigmentosa with incomplete penetrance. PLoS Genet 8: e1003040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermezovic J, Stergiou L, Hengartner MO, d'Adda di Fagagna F. 2012. Differential regulation of DNA damage response activation between somatic and germline cells in Caenorhabditis elegans. Cell Death Differ 19: 1847–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waaijers S, Boxem M. 2014. Engineering the Caenorhabditis elegans genome with CRISPR/Cas9. Methods 68: 381–388. [DOI] [PubMed] [Google Scholar]
- Wahl MC, Will CL, Lührmann R. 2009. The spliceosome: design principles of a dynamic RNP machine. Cell 136: 701–718. [DOI] [PubMed] [Google Scholar]
- Wilkie SE, Vaclavik V, Wu H, Bujakowska K, Chakarova CF, Bhattacharya SS, Warren MJ, Hunt DM. 2008. Disease mechanism for retinitis pigmentosa (RP11) caused by missense mutations in the splicing factor gene PRPF31. Mol Vis 14: 683–690. [PMC free article] [PubMed] [Google Scholar]
- Will CL, Lührmann R. 2011. Spliceosome structure and function. Cold Spring Harb Perspect Biol 3: a003707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Bellur DL, Lu S, Zhao F, Grassi MA, Bowne SJ, Sullivan LS, Daiger SP, Chen LJ, Pang CP, et al. 2009. Autosomal-dominant retinitis pigmentosa caused by a mutation in SNRNP200, a gene required for unwinding of U4/U6 snRNAs. Am J Hum Genet 85: 617–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.