

In this study, Sun et al. generate genomic and proteomic data sets to discover an essential new role for calmodulin family member CALML5 in epidermal differentiation. The upstream regulators of CALML5—a ZNF750 transcription factor and a lncRNA—were identified in addition to a 14-3-3 protein, SFN, required for CALML5 function, thus providing insight into a new ZNF750–TINCR–CALML5–SFN network needed for epidermal differentiation.
Keywords: differentiation, epidermis, transcription
Abstract
Outward migration of epidermal progenitors occurs with induction of hundreds of differentiation genes, but the identities of all regulators required for this process are unknown. We used laser capture microdissection followed by RNA sequencing to identify calmodulin-like 5 (CALML5) as the most enriched gene in differentiating outer epidermis. CALML5 mRNA was up-regulated by the ZNF750 transcription factor and then stabilized by the long noncoding RNA TINCR. CALML5 knockout impaired differentiation, abolished keratohyalin granules, and disrupted epidermal barrier function. Mass spectrometry identified SFN (stratifin/14-3-3σ) as a CALML5-binding protein. CALML5 interacts with SFN in suprabasal epidermis, cocontrols 13% of late differentiation genes, and modulates interaction of SFN to some of its binding partners. A ZNF750–TINCR–CALML5–SFN network is thus essential for epidermal differentiation.
The epidermis is a renewable somatic tissue that consists of distinct cellular strata. Keratinocyte progenitors reside in the basal stratum and are capable of self-renewal as well as generation of daughter cells that form the overlying differentiated layers. As keratinocytes differentiate, they activate transcription of genes necessary for tissue function, including proteins essential for barrier function, antimicrobial defense, and desquamation. Epidermal homeostasis depends on balanced loss and repletion of keratinocyte strata. Disruption of homeostasis characterizes skin diseases affecting >20% of the population (Lopez-Pajares et al. 2013), underscoring the importance of epidermal homeostasis to human health.
Epidermal differentiation is coordinated by diverse regulators that include receptors, transcription factors, chromatin modifiers, signaling molecules, structural proteins, long noncoding RNAs (lncRNAs), and enzymes (Blanpain and Fuchs 2009; Klein and Andersen 2015). However, the complete set of regulators that generate epidermal gene expression programs and how they interact with one another are still incompletely understood.
Here, we performed laser capture microdissection (LCM) and transcriptome sequencing in progenitor and differentiated strata of human epidermis to discover uncharacterized genes that are highly enriched in each state. We identified calmodulin-like 5 (CALML5) as a new regulator of terminal epidermal differentiation genes and barrier function proteins and defined its role within a pathway of established epidermal regulators, including ZNF750, the TINCR lncRNA, and SFN (stratifin/14-3-3σ).
Results and Discussion
LCM/RNA sequencing (RNA-seq) identifies CALML5 as highly expressed in differentiated epidermis
To identify genes enriched in progenitor and differentiated skin layers, we performed LCM to separate basal and suprabasal layers of human epidermis (Fig. 1A). Microdissected layers from two independent samples were subjected to RNA-seq. Expression of protein-coding genes with an average RPKM (reads per kilobase per million mapped reads) of >1 was scored as a ratio of differentiated to progenitor levels and then rank-ordered by enrichment score. We observed canonical basal layer genes (TP63, KRT5, and KRT14) enriched in the progenitor strata and late differentiation genes (FLG, LOR, and SPRR1B) in the differentiated strata, as expected (Fig. 1B).
Figure 1.
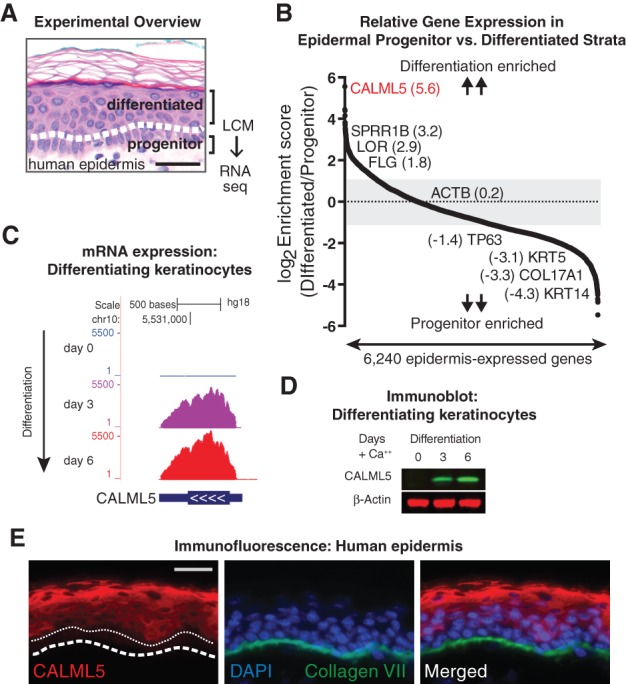
LCM of progenitor and differentiated layers of human epidermis identifies CALML5 as highly expressed in epidermal differentiation. (A) LCM was used to separate progenitor and differentiated layers of human epidermis, which were then subjected to RNA-seq. Bar, 50 µm. (B) Expression ratio scores of each gene in differentiated/progenitor strata. Genes with an average RPKM of >1 were rank-ordered. Representative genes are shown with enrichment scores. (C) RNA expression of CALML5 during in vitro keratinocyte differentiation. (D) Immunoblot of keratinocyte lysates at days 0, 3, and 6 of differentiation in vitro. (E) Immunofluorescence of CALML5 in human epidermis. The white dashed line indicates the basement membrane. The white dotted line indicates the top of the progenitor layer. Bar, 50 µm.
With the goal of identifying novel regulators of late differentiation, we examined genes enriched in differentiated strata. The most enriched gene was CALML5, which encodes the 146-amino-acid CALML5 protein, also previously described as calmodulin-like skin protein (CLSP) (Méhul et al. 2000, 2001). CALML5 shares 49% amino acid identity with calmodulin but displays significant residue changes that indicate a distinct protein interaction profile (Babini et al. 2006). Unlike ubiquitously expressed calmodulin, CALML5 expression is restricted to the epidermis and other stratified epithelial tissues (Méhul et al. 2001; Hwang and Morasso 2003; Hwang et al. 2007). CALML5 is up-regulated after tape stripping (Hwang et al. 2007) and in atopic dermatitis (Donovan et al. 2013), suggesting a potential role in responding to skin barrier disruption. We confirmed that CALML5 induction is recapitulated in differentiation of primary keratinocytes in vitro (Kretz et al. 2013) at the mRNA and protein levels (Fig. 1C,D). In vivo, the CALML5 protein is absent in the progenitor-containing basal layer and is present most strongly in the outer differentiated epidermal strata (Fig. 1E).
CALML5 is required for late epidermal differentiation
To determine whether CALML5 plays a functional role in epidermal differentiation, CALML5 was depleted in organotypic human epidermal tissue (Fig. 2A,B). Expression of late differentiation genes was inhibited with CALML5 loss (Fig. 2A). CALML5-depleted keratinocytes could stratify into tissue but showed a deficiency in expression of filaggrin and loricrin (Fig. 2C), two proteins essential to barrier function. Nile Red staining revealed reduced lipid deposition (Supplemental Fig. 1A), consistent with impaired lipid barrier formation. CALML5 depletion did not affect proliferation of basal progenitors (Supplemental Fig. 1B,C) and did not significantly impact apoptosis (Supplemental Fig. 1B–D), suggesting that its impact on skin homeostasis is predominantly in the regulation of differentiation. In addition, some differentiation genes were unaffected by CALML5 depletion (Supplemental Fig. 1E), indicating that CALML5 impacts specific aspects of differentiation and does not cause global differentiation arrest.
Figure 2.
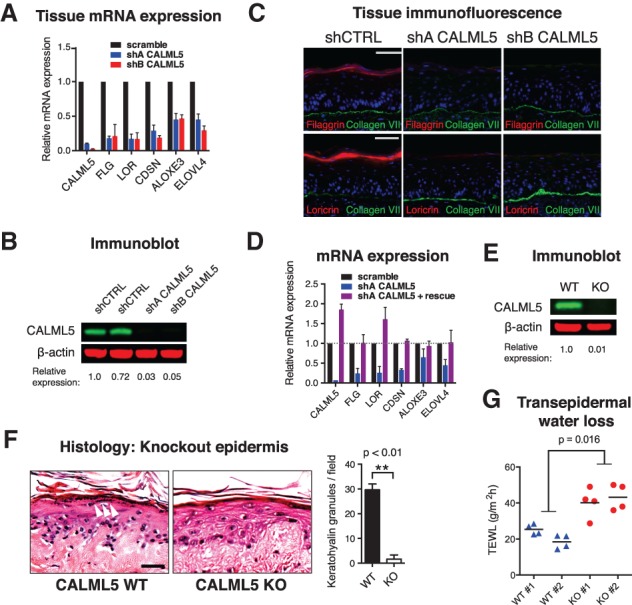
CALML5 is required for epidermal differentiation. (A) Quantitative RT–PCR (qRT–PCR) of mRNA from human organotypic epidermal tissue. n = 3. Error bars indicate SEM. (B) Immunoblot of CALML5-depleted cells. Quantitation is relative to scramble control. (C) Immunofluorescence of control (shCTRL) and CALML5-depleted (shA and shB) human organotypic epidermis. Bar, 50 µm. (D) qRT–PCR of in vitro differentiated keratinocytes after CALML5 rescue. The dotted line indicates expression of control. n = 2. Error bars indicate SEM. (E) Immunoblot of CALML5 knockout cells generated by CRISPR/Cas9 editing of clone 103 human keratinocytes. Scrambled guide RNA was used to produce wild-type control. (F) Hematoxylin and eosin stain of CALML5 wild-type and knockout xenografted human epidermal tissue. White arrowheads indicate keratohyalin granules. Bar, 50 µm. Quantitation of keratohyalin granules is shown at right. (G) Transepidermal water loss of CALML5 wild-type and knockout xenografts on live mice. n = 2 per condition. Four measurements were performed per graft; horizontal lines denote the average.
To confirm that the phenotype was not due to off-target effects, CALML5 expression was restored with a rescue construct (Fig. 2D). CALML5 repletion rescued late epidermal gene expression, demonstrating that the phenotype was specific to CALML5 loss. To evaluate the phenotype in vivo, CALML5 knockout keratinocytes were created with CRISPR/Cas9 genomic editing. Edited keratinocyte pools showed a milder phenotype (Supplemental Fig. 1F), but sequencing revealed incomplete locus deletion (Supplemental Fig. 1G). To achieve complete knockout in a defined cell population, we generated immortalized keratinocytes by HPV E6/E7 infection, isolated clones, and tested early-passage clones for their ability to differentiate. We isolated a clonal line with preserved differentiation capacity (Supplemental Fig. 1H) and used CRISPR/Cas9 editing to generate a CALML5 knockout clone, verifying protein loss by immunoblot (Fig. 2E). To examine the impact of CALML5 loss in regenerated tissue in vivo, we xenografted wild-type and CALML5 knockout keratinocytes onto immune-deficient mice. CALML5 knockout keratinocytes regenerated stratified tissue that lacked keratohyalin granules, structures that are a hallmark of the stratum granulosum (Fig. 2F). Terminal differentiation gene expression was inhibited in CALML5 knockout epidermis, mirroring the phenotype observed in organotypic tissue (Supplemental Fig. 1I,J). To assess physiologic barrier function in vivo, we measured transepidermal water loss on live mouse xenografts (Fig. 2G), which showed increased evaporative loss in CALML5 knockout epidermis, consistent with impaired epidermal barrier function due to CALML5 deletion.
We next examined whether CALML5 was itself sufficient to induce differentiation. CALML5 was overexpressed in primary keratinocytes in low (0.15 mM) and high (1.2 mM) calcium conditions. We did not observe significant induction of differentiation genes (Supplemental Fig. 1K), suggesting that CALML5 function likely requires other factors that are only produced upon differentiation. We also tested whether calcium binding was required for CALML5 function. We generated alanine substitution mutants of the acidic amino acids that form the calcium-binding pocket of the N-terminal and C-terminal pair of EF hands. Wild-type or EF mutant CALML5 was repleted into CALML5 knockout keratinocytes. Only wild-type CALML5 was capable of transcriptional rescue, indicating that intact calcium-binding motifs are required for CALML5 effects on differentiation (Supplemental Fig. 1L).
Taken together, experiments in primary keratinocytes, organotypic tissue, and in vivo xenografted epidermis demonstrated that CALML5 is required for epidermal differentiation and establishment of the barrier function of the skin.
CALML5 is up-regulated by ZNF750
To gain insight into the genes controlled by CALML5, RNA-seq was performed with CALML5-depleted tissue versus normal control. CALML5 depletion resulted in greater than twofold expression change of 547 protein-coding genes compared with control (Fig. 3A). Gene ontology (GO) analysis revealed enrichment for genes involved in epidermal development and differentiation (Fig. 3B) and phenotypic terms related to palmoplantar keratoderma and congenital erythroderma (Supplemental Fig. 2A). Gene set overlap against progenitor, early, and late differentiation gene signatures (Lopez-Pajares et al. 2015) showed the strongest association to the late differentiation signature (Supplemental Fig. 2B). Viewed together, RNA-seq analysis demonstrated that CALML5 affects the expression of genes directly relevant to skin differentiation and disease.
Figure 3.
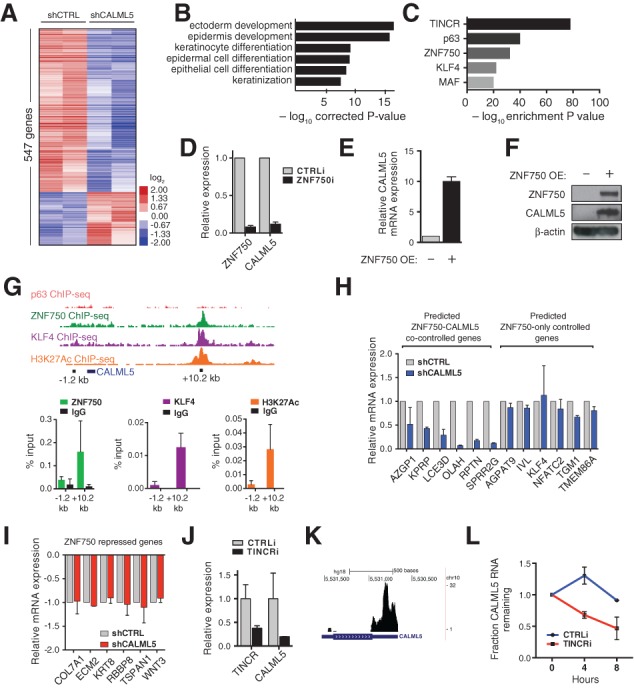
CALML5 is induced by ZNF750 and TINCR. (A) Heat map representing RNA-seq of CALML5-depleted epidermal tissue versus control. (B) GO enrichment analysis of CALML5-controlled genes. (C) Gene set overlap analysis of CALML5 gene set against known epidermal regulators. (D) qRT–PCR of ZNF750-depleted keratinocytes. n = 2. Error bars indicate SEM. (E) qRT–PCR of CALML5 mRNA with ZNF750 overexpression. (F) Immunoblot of primary progenitor keratinocytes with ZNF750 overexpression. (G) ChIP-seq (chromatin immunoprecipitation [ChIP] combined with deep sequencing) of ZNF750, KLF4 (Boxer et al. 2014), p63 (Zarnegar et al. 2012), and histone 3 Lys27 acetylation (H3K27ac; ENCODE) at the CALML5 genomic locus. (Below) Targeted ChIP of a candidate enhancer downstream from CALML5. n = 2. Error bars indicate SEM. (H) qRT–PCR of ZNF750 target differentiation genes in wild-type and CALML5-depleted keratinocytes with ZNF750 overexpression. n = 2. Error bars indicate SEM. (I) qRT–PCR of ZNF750 target progenitor genes in wild-type and CALML5-depleted keratinocytes with ZNF750 overexpression. n = 2. Error bars indicate SEM. (J) qRT–PCR of TINCR-depleted keratinocytes. n = 2. Error bars indicate SEM. (K) TINCR RNA interactome sequencing track shows potential RNA:RNA interaction in the CALML5 3′ untranslated region (UTR). (L) Actinomycin D mRNA half-life assay in wild-type and TINCR-depleted keratinocytes. n = 3. Error bars indicate SEM.
Genetic regulators of epidermal differentiation are becoming increasingly well characterized. To determine how CALML5 relates to other known regulators, we performed gene set overlap analysis between the CALML5-regulated gene set and compiled data of 44 epidermal regulators (Fig. 3C; Supplemental Fig. 2C,D; Supplemental Table 4; Lopez-Pajares et al. 2015). This revealed the most significant overlap between CALML5 and three regulators: p63, a master transcription factor for stratified epithelia (Mills et al. 1999; Yang et al. 1999); ZNF750, a transcription factor downstream from p63 (Boxer et al. 2014); and TINCR, a lncRNA that stabilizes differentiation gene mRNAs (Kretz et al. 2013).
To generate a working model of how these regulators interact, we first performed quantitative RT–PCR (qRT–PCR) of these candidates in CALML5-depleted keratinocytes and found that the expression of ZNF750, p63, and TINCR was not significantly affected by CALML5 loss (Supplemental Fig. 2E), suggesting that CALML5 is not likely a proximal upstream regulator. However, reanalysis of published data (Kretz et al. 2013; Boxer et al. 2014) indicated that ZNF750 and TINCR depletion leads to reduced CALML5 expression, suggesting that they may be upstream regulators of CALML5. In addition, ZNF750 is a direct target of p63 (Sen et al. 2012). Taken together, these findings suggested a model in which p63-activated ZNF750 induces CALML5 expression, with CALML5 mRNA then post-transcriptionally stabilized by TINCR.
To test this working model, we used RNAi to deplete ZNF750 in differentiated keratinocytes. ZNF750 depletion inhibited CALML5 up-regulation, confirming a requirement for ZNF750 to induce CALML5 (Fig. 3D). We next overexpressed ZNF750 and found that its overexpression was sufficient to induce CALML5 mRNA and protein expression (Fig. 3E,F). To determine whether this regulatory relationship was direct, we examined ChIP-seq (chromatin immunoprecipitation [ChIP] combined with deep sequencing) of ZNF750, p63, and KLF4 in differentiated keratinocytes, which revealed candidate genomic binding for ZNF750 and KLF4, but not p63, at a site ∼10 kb downstream from the CALML5 locus (Zarnegar et al. 2012; Boxer et al. 2014). ENCODE ChIP-seq for histone 3 Lys27 acetylation (H3K27ac) demonstrated enrichment at the same region, consistent with features of an active enhancer (Fig. 3G). Site-directed ChIP confirmed ChIP-seq results (Fig. 3G). Taken together, these data indicate that ZNF750 binds an enhancer ∼10 kb downstream from CALML5 and is necessary and sufficient for CALML5 expression during skin differentiation.
CALML5 functions downstream from ZNF750 and TINCR to regulate differentiation
We next wished to characterize how ZNF750 and CALML5 interact to regulate differentiation. ZNF750 acts dually to induce differentiation genes and repress progenitor genes by associating with distinct protein complexes at target loci (Boxer et al. 2014). To determine whether CALML5 affects ZNF750 gene target expression, we assayed the capability of ZNF750 to regulate target genes in the context of CALML5 depletion. We found that a subset of ZNF750 target differentiation genes was not fully induced with CALML5 depletion, whereas a separate subset of ZNF750-controlled differentiation genes was unaffected by CALML5 loss (Fig. 3H). In contrast, progenitor genes repressed by ZNF750 were unaffected by CALML5 depletion (Fig. 3I). These results indicate that CALML5 functions downstream from ZNF750 to mediate up-regulation of a subset of ZNF750 differentiation gene targets but does not contribute to repression of ZNF750 targeted progenitor genes. Finally, we evaluated whether CALML5 could rescue ZNF750 loss by overexpression in ZNF750-depleted keratinocytes (Supplemental Fig. 2F). Differentiation gene expression was not restored by CALML5, indicating that other ZNF750-induced factors are required for CALML5 function.
We next examined whether the lncRNA TINCR controls CALML5 mRNA levels. First, we depleted TINCR in differentiated keratinocytes using RNAi. CALML5 mRNA and protein levels were significantly reduced with TINCR loss (Fig. 3J; Supplemental Fig. 2G). TINCR RNA interactome analysis sequencing (RIA-seq) (Kretz et al. 2013) identified a peak in the CALML5 3′ untranslated region (UTR), indicating that CALML5 mRNA could be a TINCR stabilization target (Fig. 3K). To test this, control versus TINCR-depleted keratinocytes were treated with actinomycin D to inhibit transcription, and mRNA levels were measured by qRT–PCR at time points after treatment. CALML5 mRNA, but not the HBP1 control differentiation gene, was significantly less stable with TINCR depletion (Fig. 3L; Supplemental Fig. 2H), confirming that TINCR promotes CALML5 mRNA levels through stabilization.
TINCR stabilizes its targets through a STAU1-dependent, UPF1/2-independent mechanism that is not yet fully characterized (Kretz et al. 2013). Since polyA tail length is a major determinant of mRNA stability, we examined CALML5 mRNA polyA length in control versus TINCR-depleted tissue (Supplemental Fig. 2I,J). TINCR depletion resulted in significant reduction of CALML5 polyA length, suggesting that TINCR interaction may preserve polyA length. Together, these results demonstrate that CALML5 is transcriptionally induced by ZNF750, is stabilized post-transcriptionally by pairing with the lncRNA TINCR, and functions to control induction of late epidermal differentiation genes.
CALML5 interacts with SFN
We next sought to identify factors that cooperate with the CALML5 protein to regulate skin differentiation. Biochemical fractionation of differentiated keratinocytes did not identify stable CALML5-containing complexes (Supplemental Fig. 3A), so we hypothesized that functional CALML5 interactions may be transient or unstable. To identify transient interacting proteins, we used a proximity-mediated biotin ligation assay/mass spectrometry (BioID/MS), which identifies vicinal proteins in live cells (Roux et al. 2012).
BioID/MS for CALML5 in differentiated keratinocytes identified specific interaction candidates (Fig. 4A,B). Interactome likelihood analysis (SAINT score) was performed (Choi et al. 2010), and only candidates with a SAINT score of 1.0 were examined further (Fig. 4B). These candidates had known roles in protein modification, protein transport, and signal transduction. Because our primary objective was to identify interactors that cooperate to regulate epidermal differentiation, candidates that had mouse knockouts with no skin phenotype (CLIC1, FHL1, PCMT1, CASP3, and PTPRA) were not evaluated further. The remaining candidates were screened by RNAi-mediated gene depletion and expression analysis of the 15 top representative CALML5-controlled genes. This phenocopy assay found that depletion of SFN inhibited expression of the largest subset of CALML5 target genes (Fig. 4C).
Figure 4.
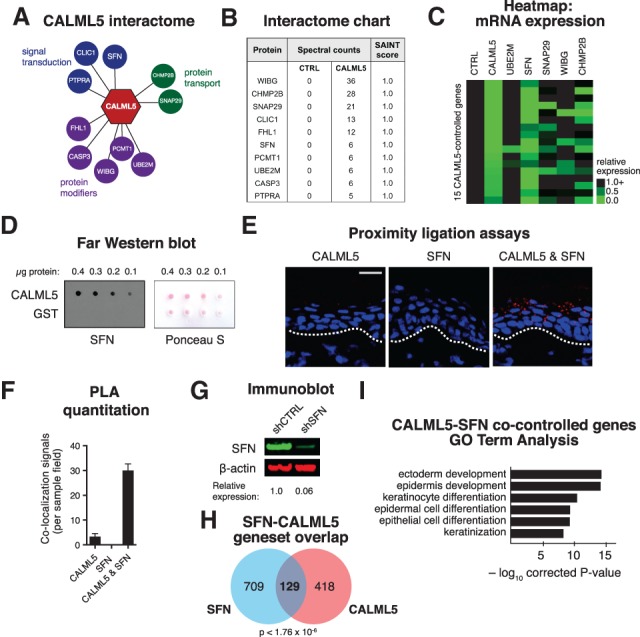
CALML5 interacts with SFN to promote epidermal differentiation. (A) The CALML5 protein interactome as defined by proximity-dependent BioID/MS. (B) Table of spectral counts and SAINT scores of interacting proteins. (C) Heat map of 15 CALML5 target gene expression levels (rows), as measured by qRT–PCR, after RNAi depletion of candidate interacting genes (columns). (D) Far Western blot with recombinant CALML5 or glutathione S-transferase (GST) control, probed with recombinant SFN protein. At right, Ponceau S loading control. (E) Proximity ligation assays (PLAs) in human epidermis with antibodies to CALML5, SFN, or both. Bar, 50 µm. (F) Quantitation of PLA colocalization. (G) Immunoblot of control and SFN-depleted keratinocytes. Quantitation is relative to scramble control. (H) Venn diagram showing overlap of SFN- and CALML5-controlled gene sets. (I) GO analysis of the CALML5–SFN overlap gene set.
SFN is a 14-3-3 protein subtype, a family of molecules that can bind and modulate phosphoserine/threonine-containing proteins (Morrison 2009). Unique to 14-3-3 proteins, SFN expression is largely restricted to skin and other epithelia. A SFN mutation causes the repeated epilation (Er) mouse phenotype, characterized by a hyperproliferative epidermis with impaired differentiation (Herron et al. 2005; Li et al. 2005). In keratinocytes, SFN is involved with diverse and far-ranging effects, including protein kinase C signaling (Dellambra et al. 2000), regulation of epidermal cell growth through mTOR/AKT (Kim et al. 2006), and extracellular signaling to fibroblasts (Ghahary et al. 2005). Recent reports also demonstrate that SFN controls keratinocyte differentiation through regulation of YAP1 localization (Sambandam et al. 2015). In keratinocytes, CALML5 resides predominantly in the cytoplasm, while SFN is present in the nucleus, cytoplasm, and extracellular space (Fig. 1E; Supplemental Fig. 4A; Ghahary et al. 2005; Kim et al. 2006). This raised the possibility that CALML5 could affect gene transcription by regulating interacting partners, such as SFN, that can translocate to the nucleus and affect gene expression.
SFN binds CALML5 in differentiated epidermis to control late differentiation
We first corroborated the BioID/MS interaction result with a Far Western blot, which showed a specific interaction between nondenatured CALML5 and SFN recombinant proteins (Fig. 4D). This interaction was unaffected by the absence of free calcium (Supplemental Fig. 4B). To visualize their temporal–spatial interaction in vivo, proximity ligation assays (PLAs) for CALML5 and SFN were performed on human epidermis. CALML5–SFN interactions were observed abundantly in suprabasal strata and peaked in late differentiated layers (Fig. 4E,F). To visualize their intracellular interaction, we performed PLA in keratinocytes in vitro, finding that their interaction occurred in the cytoplasm (Supplemental Fig. 4C). To assess whether their interaction was dependent on intact calcium-binding EF hand motifs, we performed PLA using the EF hand mutants, which showed preserved CALML5–SFN colocalization compared with wild type (Supplemental Fig 4C,D).
To delineate the global genetic relationship between CALML5 and SFN, SFN-depleted organotypic tissue was generated, and mRNA expression was quantitated by RNA-seq (Fig. 4G,H; Supplemental Fig. 4E). SFN depletion altered expression of 838 genes compared with control tissue. These genes included both progenitor and late differentiation genes (Supplemental Fig. 4F). Gene set overlap between SFN and CALML5 identified 129 genes (P < 1.76 × 10−6) present in both gene sets (Fig. 4H), which was enriched with late differentiation genes (Fig. 4I; Supplemental Fig. 4G). Intersecting these 129 genes with the late epidermal differentiation gene signature (Lopez-Pajares et al. 2015) demonstrated that CALML5 and SFN cocontrol 50 late differentiation signature genes (13% of total; P < 1.94 × 10−24) (Supplemental Fig. 4H). Together, these results supported the model of CALML5 and SFN functioning together to control genes required for late differentiation.
CALML5 affects YAP1 localization and binding of SFN to interacting partners
To investigate the impact of CALML5–SFN interaction, we evaluated whether CALML5 depletion affected known SFN functions in keratinocytes. SFN sequesters the transcriptional regulator YAP1 to the cytoplasm in differentiating keratinocytes (Sambandam et al. 2015). The translocation of YAP1 from the nucleus to the cytoplasm mediates a critical transcriptional switch from the progenitor to the differentiated state (Schlegelmilch et al. 2011). We investigated whether CALML5 impacts this process. In low-density conditions, YAP1 is predominantly nuclear (Supplemental Fig. 5A). Upon plating to high-density conditions that promote differentiation, YAP1 translocates to the cytoplasm within several hours. However, in CALML5 knockout cells, we found that the kinetics of YAP1 cytoplasmic translocation was significantly impaired (Supplemental Fig. 5B), indicating that the SFN–YAP1 signaling switch was inhibited with CALML5 loss.
We next sought to identify whether CALML5 may function more generally to modulate the binding of its interactors with their downstream effectors and proteins. To test this, we performed SFN BioID/MS in wild-type and CALML5-depleted differentiated keratinocytes. This revealed known SFN interactors, such as PKP3 (Roberts et al. 2013), as well as previously unreported proteins (Supplemental Fig. 5C). Interestingly, some of these proteins displayed strongly altered spectral counts in the CALML5-depleted state, suggesting that their interactions with SFN may be modulated by CALML5. A number of these proteins have known roles in skin disease, such as EXPH5 (McGrath et al. 2012). We verified these results by targeted BioID IP of EXPH5, which confirmed an interaction with SFN that was diminished by 44% in CALML5-depleted cells (Supplemental Fig. 5D). On an interactome level, these data support a role for CALML5 in modulating the binding of SFN with its downstream proteins, demonstrating a potential mechanism that may govern its function with other interactors as well.
A new regulator of late epidermal differentiation
Epidermal differentiation requires coordinated induction and repression of gene expression programs. A significant number of essential regulators of this process have been characterized, but the integrated relationship between these regulators and cross-talk between downstream pathways is still being defined. In addition, the ability of the epidermis to restratify faithfully after injury suggests that epidermal gene expression must be a dynamic process capable of restoring homeostasis after disruption.
Here, we used expression-based analysis of human epidermis to identify CALML5 as an essential gene required for late epidermal differentiation and provisionally placed CALML5 in the context of known epidermal regulators to define a molecular pathway that establishes expression of functional barrier proteins (Fig. 5). Calmodulins and 14-3-3 proteins are both highly conserved, broadly expressed protein families that perform diverse functions in many tissues. Interestingly, CALML5 and SFN both appear to have arisen from their ubiquitous parent proteins to have tissue-specific expression in stratified epithelia, raising the possibility that their functions and interaction may have evolved to regulate epithelial stratification. These findings contribute to the understanding of the interplay of molecular regulators required to generate and maintain dynamic and complex epithelial tissues such as the skin.
Figure 5.
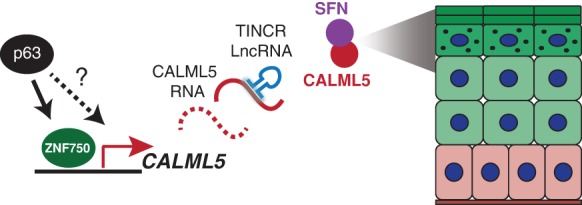
Provisional model. During epidermal differentiation, CALML5 is induced by ZNF750, is post-transcriptionally stabilized by TINCR, and functions together with SFN to promote the expression of late differentiation genes.
Materials and methods
LCM/RNA-seq
Clinically normal human epidermis of sun-protected truncal sites was collected from elective surgeries. Duplicate samples from unrelated adults were placed into RNALater (Ambion) and embedded into O.C.T. (Sakura), and 7-µM sections were mounted on PEN slides (Leica). Slides were stained using an LCM staining kit (Ambion), and sections were harvested with a Leica AS LMD laser capture microscope. Samples were processed with the SMART-Seq version 3 ultralow-input RNA kit (Clontech), and sequencing libraries were prepared with the Nextera XT kit (Illumina). Sequencing was performed on an Illumina HiSeq 2500 with 50-base-pair single-end reads. Sequence data have been deposited at National Institutes of Health Sequence Read Archive (SRA 064531, 064538, and 064547). Please see Supplemental Figure 6 for discussion of RNA-seq analysis methods.
Proximity-mediated BioID
A hemagluttinin (HA) tag and BirA* protein (Roux et al. 2012) were fused to the CALML5 or SFN protein in the pLEX vector (Thermo Scientific). Primary human keratinocytes were infected, selected with 1 µg/mL puromycin for 48 h, and then plated to confluence with addition of 1.2 mM CaCl2 to induce differentiation. After 96 h of differentiation, biotin was supplemented at 10 µM. Cells were harvested 24 h after biotin induction. Prior to affinity purification, cell lysates were centrifuged through Microsep columns (Pall) at 2000g for 1 h to deplete free biotin. The protocol was otherwise performed as described (Roux et al. 2013). Proteins were sequenced by microcapillary reverse-phase high-performance liquid chromatography (HPLC) nanoelectrospray tandem MS (µLC/MS/MS) on a Thermo LTQ-Orbitrap mass spectrometer.
Plasmids, primers, antibodies, and additional methods
Plasmids and additional methods are detailed in Supplemental Figure 6. Primer sequences are provided in Supplemental Table 5. Antibodies are listed in Supplemental Table 6.
Supplementary Material
Acknowledgments
We thank M. Snyder, A. Oro, J. Crabtree, H. Chang, K. Wang, A. Bhaduri, C. Lee, and X. Bao for presubmission review. We thank K. Reiger and S. Tao for technical support, and L. Morcom and P. Bernstein for administrative assistance. This work was supported by the U.S. Department of Veterans Affairs Office of Research and Development and National Institute of Arthritis and Musculoskeletal and Skin Diseases National Institutes of Health R01 AR45192 to P.A.K., and National Institute of Arthritis and Musculoskeletal and Skin Diseases National Institutes of Health F32 AR063508 to B.K.S.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.267708.115.
References
- Babini E, Bertini I, Capozzi F, Chirivino E, Luchinat C. 2006. A structural and dynamic characterization of the EF-hand protein CLSP. Structure 14: 1029–1038. [DOI] [PubMed] [Google Scholar]
- Blanpain C, Fuchs E. 2009. Epidermal homeostasis: a balancing act of stem cells in the skin. Nat Rev Mol Cell Biol 10: 207–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boxer LD, Barajas B, Tao S, Zhang J, Khavari PA. 2014. ZNF750 interacts with KLF4 and RCOR1, KDM1A, and CTBP1/2 chromatin regulators to repress epidermal progenitor genes and induce differentiation genes. Genes Dev 28: 2013–2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi H, Larsen B, Lin Z-Y, Breitkreutz A, Mellacheruvu D, Fermin D, Qin ZS, Tyers M, Gingras A-C, Nesvizhskii AI. 2010. SAINT: probabilistic scoring of affinity purification-mass spectrometry data. Nat Methods 8: 70–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dellambra E, Golisano O, Bondanza S, Siviero E, Lacal P, Molinari M, D'Atri S, De Luca M. 2000. Downregulation of 14-3-3σ prevents clonal evolution and leads to immortalization of primary human keratinocytes. J Cell Biol 149: 1117–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan M, Ambach A, Thomas-Collignon A, Prado C, Bernard D, Jammayrac O, Gollnick H, de Lacharriere O. 2013. Calmodulin-like skin protein level increases in the differentiated epidermal layers in atopic dermatitis. Exp Dermatol 22: 836–837. [DOI] [PubMed] [Google Scholar]
- Ghahary A, Marcoux Y, Karimi-Busheri F, Li Y, Tredget EE, Kilani RT, Lam E, Weinfeld M. 2005. Differentiated keratinocyte-releasable stratifin (14-3-3σ) stimulates MMP-1 expression in dermal fibroblasts. J Invest Dermatol 124: 170–177. [DOI] [PubMed] [Google Scholar]
- Herron BJ, Liddell RA, Parker A, Grant S, Kinne J, Fisher JK, Siracusa LD. 2005. A mutation in stratifin is responsible for the repeated epilation (Er) phenotype in mice. Nat Genet 37: 1210–1212. [DOI] [PubMed] [Google Scholar]
- Hwang M, Morasso MI. 2003. The novel murine Ca2+-binding protein, Scarf, is differentially expressed during epidermal differentiation. J Biol Chem 278: 47827–47833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang J, Kalinin A, Hwang M, Anderson DE, Kim MJ, Stojadinovic O, Tomic-Canic M, Lee SH, Morasso MI. 2007. Role of Scarf and its binding target proteins in epidermal calcium homeostasis. J Biol Chem 282: 18645–18653. [DOI] [PubMed] [Google Scholar]
- Kim S, Wong P, Coulombe PA. 2006. A keratin cytoskeletal protein regulates protein synthesis and epithelial cell growth. Nature 441: 362–365. [DOI] [PubMed] [Google Scholar]
- Klein RH, Andersen B. 2015. Dynamic networking for epidermal differentiation. Dev Cell 32: 661–662. [DOI] [PubMed] [Google Scholar]
- Kretz M, Siprashvili Z, Chu C, Webster DE, Zehnder A, Qu K, Lee CS, Flockhart RJ, Groff AF, Chow J, et al. 2013. Control of somatic tissue differentiation by the long non-coding RNA TINCR. Nature 493: 231–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Lu Q, Estepa G, Verma IM. 2005. Identification of 14-3-3σ mutation causing cutaneous abnormality in repeated-epilation mutant mouse. Proc Natl Acad Sci 102: 15977–15982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Pajares V, Yan K, Zarnegar BJ, Jameson KL, Khavari PA. 2013. Genetic pathways in disorders of epidermal differentiation. Trends Genet 29: 31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Pajares V, Qu K, Zhang J, Webster DE, Barajas BC, Siprashvili Z, Zarnegar BJ, Boxer LD, Rios EJ, Tao S, et al. 2015. A lncRNA–MAF:MAFB transcription factor network regulates epidermal differentiation. Dev Cell 32: 693–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JA, Stone KL, Begum R, Simpson MA, Dopping-Hepenstal PJ, Liu L, McMillan JR, South AP, Pourreyron C, McLean WH, et al. 2012. Germline mutation in EXPH5 implicates the Rab27b effector protein Slac2-b in inherited skin fragility. Am J Hum Genet 91: 1115–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Méhul B, Bernard D, Simonetti L, Bernard MA, Schmidt R. 2000. Identification and cloning of a new calmodulin-like protein from human epidermis. J Biol Chem 275: 12841–12847. [DOI] [PubMed] [Google Scholar]
- Méhul B, Bernard D, Schmidt R. 2001. Calmodulin-like skin protein: a new marker of keratinocyte differentiation. J Invest Dermatol 116: 905–909. [DOI] [PubMed] [Google Scholar]
- Mills AA, Zheng B, Wang XJ, Vogel H, Roop DR, Bradley A. 1999. p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature 398: 708–713. [DOI] [PubMed] [Google Scholar]
- Morrison DK. 2009. The 14-3-3 proteins: integrators of diverse signaling cues that impact cell fate and cancer development. Trends Cell Biol 19: 16–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts BJ, Reddy R, Wahl K. 2013. Stratifin limits plakophilin-3 exchange with the desmosomal plaque. PLoS One 8: e77012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux KJ, Kim DI, Raida M, Burke B. 2012. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J Cell Biol 196: 801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux KJ, Kim DI, Burke B. 2013. BioID: a screen for protein–protein interactions. Curr Protoc Protein Sci 74: 19.23.1–19.23.14. [DOI] [PubMed] [Google Scholar]
- Sambandam SAT, Kasetti RB, Xue L, Dean DC, Lu Q, Li Q. 2015. 14-3-3σ regulates keratinocyte proliferation and differentiation by modulating Yap1 cellular localization. J Invest Dermatol 135: 1621–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlegelmilch K, Mohseni M, Kirak O, Pruszak J, Rodriguez JR, Zhou D, Kreger BT, Vasioukhin V, Avruch J, Brummelkamp TR, et al. 2011. Yap1 act downstream of α-catenin to control epidermal proliferation. Cell 144: 782–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen GL, Boxer LD, Webster DE, Bussat RT, Qu K, Zarnegar BJ, Johnston D, Siprashvili Z, Khavari PA. 2012. ZNF750 is a p63 target gene that induces KLF4 to drive terminal epidermal differentiation. Dev Cell 22: 669–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang A, Schweitzer R, Sun D, Kaghad M, Walker N, Bronson RT, Tabin C, Sharpe A, Caput D, Crum C, et al. 1999. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature 398: 714–718. [DOI] [PubMed] [Google Scholar]
- Zarnegar BJ, Webster DE, Lopez-Pajares V, Hunt BVS, Qu K, Yan KJ, Berk DR, Sen GL, Khavari PA. 2012. Genomic profiling of a human organotypic model of AEC syndrome reveals ZNF750 as an essential downstream target of mutant TP63. Am J Hum Genet 91: 435–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.