

Abstract
A new low-energy pathway is reported for the electrochemical reduction of CO2 to formate and syngas at low overpotentials, utilizing a reactive ionic liquid as the solvent. The superbasic tetraalkyl phosphonium ionic liquid [P66614][124Triz] is able to chemisorb CO2 through equimolar binding of CO2 with the 1,2,4-triazole anion. This chemisorbed CO2 can be reduced at silver electrodes at overpotentials as low as 0.17 V, forming formate. In contrast, physically absorbed CO2 within the same ionic liquid or in ionic liquids where chemisorption is impossible (such as [P66614][NTf2]) undergoes reduction at significantly increased overpotentials, producing only CO as the product.
Keywords: CO2 reduction, electrolysis, formate, ionic liquids, superbases
Although CO2 is a greenhouse gas thought to be involved in climate change,[1] it can also be considered as an abundant carbon building block for carbon neutral fuels and chemicals.[2] Electrochemical reduction is one route to achieve this goal. Indeed, the reduction of CO2 at low applied overpotentials with high efficiencies is a significant current challenge owing to its thermodynamic stability and kinetic inertness.[3] The high overpotential for CO2 reduction is related to the large reorganization energy associated with reduction of linear CO2 to the bent [.CO2]− radical anion. Thus, a very negative reduction potential is required for the first electron reduction, that is, −1.9 V vs. NHE,[4] rendering reduction highly energy inefficient. Materials that form complexes with CO2 in a non-linear geometry can decrease this reorganization energy and catalyze the electrochemical CO2 reduction.[5]
Recently, promising results have been reported utilizing room-temperature ionic liquids (RTILs) for the electrochemical reduction of CO2. High CO2 solubility, intrinsic ionic conductivity, and wide potential windows of RTILs make them attractive solvents for CO2 electroreduction.[6] Initial reports on CO2 reduction in RTILs formed dialkyl carbonates through generation of .CO2 radicals, which were reacted with alcohols using 1-alkyl-3-methylimidazolium ([Cnmim]+) based RTILs with a range of non-coordinating anions.[7]
Further RTIL studies focused on CO2 reduction to products other than dialkyl carbonates. Rosen et al. reported the use of Ag electrodes in [C2mim][BF4], which was found to decrease the energy of formation of the [.CO2]− radical anion through the complexation of CO2 with the [C2mim]+ cation.[8] This significantly reduced the overpotential for CO2 reduction to CO to <0.2 V. Furthermore, the cation was shown to suppress the competing H2 production reaction by forming a monolayer on the electrode.[9] Further decreases of the applied potential have been achieved by substitution of the Ag working electrode with MoS2, giving overpotentials as low as 0.054 V.[10] Brennecke and co-workers also showed that the anion influenced the product selectivity, with oxalate formation favored over CO in [C2mim][NTf2].[11] This change in product selectivity was also shown by Watkins and Bocarsly, where formate was produced in [C2mim][TFA] using Pb and Sn working electrodes.[12] Therein, no evidence was found for a [C2mim]+-CO2 complex;[13] however, the RTIL was thought to stabilize intermediates in formate production.
Although interesting, CO2 reduction in non-coordinating [Cnmim]+ based RTILs is of limited applicability, owing to low CO2 solubility. For example, CO2 solubility in [C4mim][NTf2] is <0.04 CO2 mole fraction at 10 °C and 0.1 MPa.[14] The only coordinating IL used to date for CO2 reduction studies is [C4mim][OAc],[15] which has a CO2 solubility of 0.274 CO2 mole fraction at 25 °C and 0.1 MPa.[16] The low solubility affects the rate of product formation and limits the industrial significance of these systems.
These low solubilities have been overcome by the use of superbasic ionic liquids. In these systems, [P66614][124Triz], for example, has been shown to absorb equimolar quantities of CO2 through the chemical interaction of CO2 with the anion and physical absorption of CO2 in the solution free space (Scheme 1).[6b, 17] This set of ILs have been studied extensively for CO2 capture but, to date, no reports of their use in CO2 conversion have been published. A key feature of the anion–CO2 interaction is that the CO2 chemically binds without prior reduction to [.CO2]−. Notably, since CO2 is transformed from a linear to bent geometry on binding to the anion, this can significantly lower the CO2 reduction potential.
Scheme 1.

Addition of CO2 to [P66614][124Triz], showing binding of CO2 to the triazolide anion.
In this study, we report the first electrochemical reduction of CO2 captured within the superbasic RTIL [P66614][124Triz], providing a new low-energy pathway for CO2 reduction in RTILs. Full product characterization of solution and gas phase products is reported using 1H NMR spectroscopy and online GC analysis, respectively.
The influence of the anion identity on CO2 reduction was examined by recording cyclic voltammograms (CVs) at a Ag working electrode in 0.1 mol L−1 [P66614][NTf2] and [P66614][124Triz] in acetonitrile (MeCN). The reference electrode was 0.01 mol L−1 Ag+/Ag formed by dissolving AgNO3 in [C4mim][NO3] and separated from the bulk solution by a glass frit, as reported previously.[18] As well as the reactive IL, [P66614][NTf2] was examined as a comparison because CO2 is unable to chemically bind to this anion. However, by keeping the [P66614]+ cation consistent, the physically absorbed CO2 should be stabilized to the same extent once reduced to the radical anion ([.CO2]−), allowing reduction processes for the chemically and physically absorbed CO2 to be distinguished. The CVs recorded at a scan rate of 100 mV s−1 at 22 °C in [P66614][NTf2] and [P66614][124Triz] are shown in Figure 1 a and b, respectively. First, the CVs were taken in RTIL solutions saturated with argon (Figure 1). Consistent with previous reports of CVs using metal electrodes in dialkylimidazolium based RTILs, only small capacitive currents are observed in a wide potential window, with no additional features other than the solvent or RTIL reduction in the cathodic range.
Figure 1.

Cyclic voltammograms recorded with a silver working electrode in acetonitrile solutions with 0.1 mol L−1 of the ionic liquids (IL) a) [P66614][NTf2] and b) [P66614][124Triz]. Inset: magnified sections.
The CVs taken for solutions of both ionic liquids purged with CO2 for 30 min (Figure 1) exhibited cathodic features that can be associated with CO2 reduction. As the onset potential is often difficult to define, in agreement with previous reports, the potentials that result in a current density of 6 A m−2 were selected as the onset potential for CO2 reduction.[11, 19] For the [NTf2]−-based RTIL, upon the addition of CO2, the current starts to increase at −0.9 V followed by a rapid increase at −1.9 V. The rapid increase in current at −1.9 V can be attributed to reduction of physically bound CO2, forming [.CO2]− stabilized by the [P66614]+ cation. The small current increases from −0.9 V are attributed to trace water contamination (see below). For the [124Triz]−-based RTIL, upon the addition of CO2, a small current increase is observed at circa −0.9 V and a further small increase is observed at circa −1.5 V; these features are likely to be due to the reduction of CO2 bound to the [124Triz]− anion (see below). This is followed by a rapid increase in current at −1.9 V owing to the reduction of physically bound CO2 to [.CO2]− stabilized by the [P66614]+ cation. This potential is identical to the [NTf2]−-based RTIL system and is consistent with the [P66614]+ cation providing the same stabilization to [.CO2]− in both systems. Owing to the highly hygroscopic nature of RTILs[20] and the high solubility of O2 within MeCN, the small features at less negative potential (−0.9 V) are attributed to trace amounts of moisture or oxygen within the system. Prior to this analysis, the RTILs were rigorously dried under high vacuum and purged with Ar for 1 h to limit the influence of adventitious water; however, complete removal is very difficult.
These studies were compared with the response following the addition of H2O (0.7 mol L−1), the presence of which is required to form protonated reduction products and CO. The presence of H2O+CO2 within the basic IL should enable the formation of carbonate, which may itself be reduced. CVs of Ar and CO2 saturated RTILs in the presence of water are shown in Figure 1. For the [NTf2]−-based RTIL, the addition of H2O to the Ar saturated sample (Figure 1 a) shows a small current increase at circa −0.9 V. This is in the same position that current increases were observed in the anhydrous CO2 saturated sample and may suggest the presence of small amounts of moisture in the anhydrous CO2-saturated sample. A rapid increase in current is then observed at circa −2.3 V, suggesting the reduction of H2O to H2 upon the Ag electrode. The addition of H2O (0.7 mol L−1) to the CO2 saturated solution shows a small reduction feature at −0.7 V and a strong cathodic current from −1.9 V. Although a small anodic shift is observed, the responses are similar. The small reduction feature at −0.7 V can be associated with those observed at −0.9 V in the anhydrous CO2 saturated sample and in the Ar saturated sample containing H2O. The large reduction feature at −1.9 V is identical to that seen in the solely CO2 saturated sample and is attributed to the reduction of CO2 to [.CO2]− (Scheme 2, Equation (4)).
Scheme 2.

Proposed reduction processes taking place at polarized Ag electrode in ionic liquids saturated with CO2.
The addition of H2O to the argon-saturated [124Triz]−-based RTIL shows two small reduction peaks at circa −1.1 V and −1.5 V followed by a rapid current increase at −1.9 V. The superbasic RTIL, [P66614][124Triz] is able to react with H2O to form [P66614][OH]+[124Triz]−H, the latter of which provides an additional route to H+. The first reduction peak at −1.1 V is at a more negative potential than that observed in the anhydrous CO2 saturated sample (−0.9 V); however, the second current reduction is in an identical position (−1.5 V). The rapid reduction of current is due to reduction of H2O to H2. This is at a less negative potential than that observed for the [NTf2]− based RTIL (−2.3 V) suggesting the choice of anion has an effect on the H2O reduction potential. The most significant changes are observed in the addition of H2O (0.7 mol L−1) to the CO2 saturated [124Triz]− based RTIL, which shows an anodic shift and large enhancement of the two low potential reduction features at −0.7 V and −1.3 V (Figure 1 b) followed by a large increase in current at −1.9 V. The first reduction feature is attributed to the reduction of CO2 bound to the [124Triz]− anion to form formate (Scheme 2, Equation (2)). The second reduction feature is also associated with formate product formation via a different mechanism (see below). The third reduction feature, at −1.9 V in Figure 1 b, is assigned to both the reduction of the physically bound CO2 stabilized by the [P66614]+ cation, consistent with the CV observed in [P66614][NTf2] (Figure 1 a) and the reduction of H2O to H2. To examine the origin of these features in more detail, an electrolysis study has been performed.
In Scheme 2, Equation (2) indicates that the anion is regenerated and, therefore, should be catalytic. However, any formic acid produced may protonate this anion and the system would not be recyclable. To examine whether the feature at low potential is still present in the presence of the products, one mole equivalent of formic acid to [P66614][124Triz] was added to the system (AcN+IL+CO2+H2O) and the voltammetry compared with the system without formic acid addition (Supporting Information, Figure S1). No reduction in the low potential feature was observed, suggesting no inhibition of the anion binding site. Furthermore, 1H NMR spectrum of this sample revealed no evidence for the neutral 124-triazole. It should also be noted that the addition of formic acid to the same system in the absence of CO2 does not result in the appearance of peaks below −1 V (not shown).
Electrolysis was performed using a Ag working electrode on the hydrated CO2-saturated RTILs within a gas-tight electrochemical cell. For each potential, 10 C of charge was passed before analysis of the products b< 1H NMR spectroscopy and online gas chromatography. The variation in product selectivity with applied potential using the RTIL [P66614][124Triz] is shown in Figure 2 and the Supporting Information, Table S1. It should be noted that no degradation of the IL was observed in the 1H NMR throughout the electrolysis studies.
Figure 2.
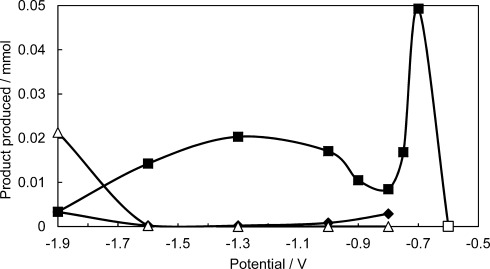
Variation of product yield as a function of applied potential vs Ag/AgNO3 after 10 C of charge is passed using a CO2 saturated hydrated (0.7 mol L−1) 0.1 mol L−1 [P66614][124Triz] in MeCN electrolyte at a Ag electrode. (▪ formate, ♦ CO, ▵ H2). Note the formate generated at −0.6 V (□) was obtained following 24 h of electrolysis during which only 1.5 C of charge had passed.
In [P66614][124Triz], formate was the only detectable product in the solution phase. At −0.7 V, 0.05 mmol of formate is produced at a Faradaic efficiency of 95 %. The open circuit potential of the system is −0.53 V; therefore, holding at −0.7 V corresponds to an applied overpotential of 0.17 V. Production of formate at −0.7 V relates to the first reduction peak seen in the CV of the [P66614][124Triz]+H2O+CO2 system. No formate production was observed at less negative applied potentials. A second, albeit smaller, formate maxima is seen at −1.3 V with a Faradaic efficiency of 39 %. This potential corresponds to the second reduction feature seen in the CVs. The formation of formate at −1.3 V is plausibly formed via an alternative mechanism to that seen at −0.7 V, for example by the decomposition of formaldehyde to formate or a radical reaction with superoxide. Further experiments are ongoing to ascertain the origin of the second reduction feature. Finally, at −1.9 V formate production is dramatically reduced and syngas is detected. This corresponds to the rapid increase in reduction current noted on the CV. A summary of the reductive process can be found in Scheme 2.
Formate production is greatly decreased on the onset of CO production, which suggests that reduction of physically absorbed CO2 competes with the reduction of chemically bound CO2 in this potential range. This may be due to blocking of the electrode surface by adsorbed CO, CO2 or other reaction intermediates.
In contrast with the reactive RTIL, electrolysis at −0.7 V in a hydrated CO2 saturated [P66614][NTf2]/MeCN electrolyte showed no formate production. This is consistent with the postulation that no lower energy pathway for CO2 reduction is available owing to a lack of CO2 binding site on the [NTf2]− anion. Furthermore, there is a significant decrease in the production of CO with only 0.2 μmol formed at −1.9 V; that is, over an order of magnitude lower than the amount formed in [P66614][124Triz] at 3.3 μmol. This is plausibly due to the higher CO2 capacity of [P66614][124Triz] over [P66614][NTf2]. Hydrogen was also detected at −1.9 V with 21.3 μmol detected in [P66614][124Triz] and only 6.8 μmol in [P66614][NTf2] at Faradaic efficiencies of 41.1 and 13.2 %, respectively. The difference can be related to the reduction peak for H2 production being more negative for [P66614][NTf2] (−2.3 V) than [P66614][124Triz] (−1.9 V). An alternative explanation for the increased formate production at less negative potentials for the reactive anion system is that the cation, [P66614]+, is catalytic and, when the anion, [NTf2]−, is used it acts as a poison to the Ag electrode. To address this issue, [P66614][BF4] was also examined. Performing electrolysis at −0.7 V resulted in a small amount of formate (0.010 mmol) being produced with a low Faradaic efficiency (19 %). Furthermore, to examine the effect of the cation [N4444][124Triz] was also synthesized (see the Supporting Information) and compared with the [P66614][124Triz]. Performing electrolysis using this ionic liquid at −0.7 V for 10 C of charge resulted in the production of formate (0.021 mmol) as well as formaldehyde (0.006 mmol) with a combined Faradaic efficiency of 63 %. The contrast of high versus low Faradaic efficiencies and product formation at low potentials when reactive and non-reactive anions are employed, respectively, provides support for the proposal that the reactive anions can provide a new low energy pathway for CO2 electroreduction.
In conclusion, the superbasic RTIL [P66614][124Triz] has been shown to provide an alternative low-energy pathway for CO2 conversion to formate. This is the first time that chemical binding of the neutral CO2 molecule to the anion of a RTIL has been shown to decrease the activation energy required for electrochemical reduction. This subsequently leads to the lowest reported applied potentials for CO2 reduction to formate on Ag electrodes to date with high Faradaic efficiencies.
Acknowledgments
This work was carried out as part of the “4CU” programme grant, aimed at sustainable conversion of carbon dioxide into fuels, led by the University of Sheffield and carried out in collaboration with the University of Manchester, Queen’s University Belfast, and University College London. The authors acknowledge gratefully the Engineering and Physical Sciences Research Council (EPSRC) for supporting this work financially (Grant No EP/K001329/1). S.F.R.T. and N.H. wish to thank the ELSOL workshop, Researcher Links, FAPESP, and Newton fund for funding. M.T.G. thanks the RSC and CNPq for funding.
Supporting Information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information
References
- 1.Karl TR, Trenberth KE. Science. 2003;302:1719–1723. doi: 10.1126/science.1090228. [DOI] [PubMed] [Google Scholar]
- 2.Aresta M. Carbon Dioxide as Chemical Feedstock. Weinheim: Wiley-VCH; 2010. pp. 1–13. [Google Scholar]
- 3.Jhong H-RM, Ma S, Kenis PJA. Curr. Opin. Chem. Eng. 2013;2:191–199. [Google Scholar]
- 4.Schneider J, Jia H, Muckerman JT, Fujita E. Chem. Soc. Rev. 2012;41:2036–2051. doi: 10.1039/c1cs15278e. [DOI] [PubMed] [Google Scholar]
- 5.Roldan A, Hollingsworth N, Roffey A, Islam HU, Goodall JBM, Catlow CRA, Darr JA, Bras W, Sankar G, Holt KB, Hogarth G, de Leeuw NH. Chem. Commun. 2015;51:7501–7504. doi: 10.1039/c5cc02078f. [DOI] [PubMed] [Google Scholar]
- 6a.Welton T. Chem. Rev. 1999;99:2071–2084. doi: 10.1021/cr980032t. [DOI] [PubMed] [Google Scholar]
- 6b.Wang C, Luo X, Luo H, Jiang D-e, Li H, Dai S. Angew. Chem. Int. Ed. 2011;50:4918–4922. doi: 10.1002/anie.201008151. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. 2011;123 [Google Scholar]
- 6c.Brennecke JF, Gurkan BE. J. Phys. Chem. Lett. 2010;1:3459–3464. [Google Scholar]
- 7a.Feng Q, Liu S, Wang X, Jin G. Appl. Surf. Sci. 2012;258:5005–5009. [Google Scholar]
- 7b.Liu F, Liu S, Feng Q, Zhuang S, Zhang J, Bu P. Int. J. Electrochem. Sci. 2012;7:4381–4387. [Google Scholar]
- 7c.Yuan X, Lu B, Liu J, You X, Zhao J, Cai Q. J. Electrochem. Soc. 2012;159:E183–E186. [Google Scholar]
- 8.Rosen BA, Salehi-Khojin A, Thorson MR, Zhu W, Whipple DT, Kenis PJA, Masel RI. Science. 2011;334:643–644. doi: 10.1126/science.1209786. [DOI] [PubMed] [Google Scholar]
- 9.Rosen BA, Haan JL, Mukherjee P, Braunschweig B, Zhu W, Salehi-Khojin A, Dlott DD, Masel RI. J. Phys. Chem. C. 2012;116:15307–15312. [Google Scholar]
- 10.Asadi M, Kumar B, Behranginia A, Rosen BA, Baskin A, Repnin N, Pisasale D, Phillips P, Zhu W, Haasch R, Klie RF, Král P, Abiade J, Salehi-Khojin A. Nat. Commun. 2014;5:4470. doi: 10.1038/ncomms5470. [DOI] [PubMed] [Google Scholar]
- 11.Sun L, Ramesha GK, Kamat PV, Brennecke JF. Langmuir. 2014;30:6302–6308. doi: 10.1021/la5009076. [DOI] [PubMed] [Google Scholar]
- 12.Watkins JD, Bocarsly AB. ChemSusChem. 2014;7:284–290. doi: 10.1002/cssc.201300659. [DOI] [PubMed] [Google Scholar]
- 13.Shiflett MB, Yokozeki A. J. Chem. Eng. Data. 2009;54:108–114. [Google Scholar]
- 14.Anthony JL, Anderson JL, Maginn EJ, Brennecke JF. J. Phys. Chem. B. 2005;109:6366–6374. doi: 10.1021/jp046404l. [DOI] [PubMed] [Google Scholar]
- 15.Barrosse-Antle LE, Compton RG. Chem. Commun. 2009:3744–3746. doi: 10.1039/b906320j. [DOI] [PubMed] [Google Scholar]
- 16.Shiflett MB, Kasprzak DJ, Junk CP, Yokozeki A. J. Chem. Thermodyn. 2008;40:25–31. [Google Scholar]
- 17a.Taylor SFR, McCrellis C, McStay C, Jacquemin J, Hardacre C, Mercy M, Bell R, de Leeuw N. J. Solution Chem. 2015;44:511–527. [Google Scholar]
- 17b.Wang C, Luo H, Jiang D-E, Li H, Dai S. Angew. Chem. Int. Ed. 2010;49:5978–5981. doi: 10.1002/anie.201002641. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. 2010;122 [Google Scholar]
- 17c.Seo S, Quiroz-Guzman M, DeSilva MA, Lee TB, Huang Y, Goodrich BF, Schneider WF, Brennecke JF. J. Phys. Chem. B. 2014;118:5740–5751. doi: 10.1021/jp502279w. [DOI] [PubMed] [Google Scholar]
- 17d.Gurkan B, Goodrich BF, Mindrup EM, Ficke LE, Massel M, Seo S, Senftle TP, Wu H, Glaser MF, Shah JK, Maginn EJ, Brennecke JF, Schneider WF. J. Phys. Chem. Lett. 2010;1:3494–3499. [Google Scholar]
- 17e.Luo X, Guo Y, Ding F, Zhao H, Cui G, Li H, Wang C. Angew. Chem. Int. Ed. 2014;53:7053–7057. doi: 10.1002/anie.201400957. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. 2014;126 [Google Scholar]
- 17f.Wang C, Luo H, Li H, Zhu X, Yu B, Dai S. Chem. Eur. J. 2012;18:2153–2160. doi: 10.1002/chem.201103092. [DOI] [PubMed] [Google Scholar]
- 17g.Seo S, DeSilva MA, Brennecke JF. J. Phys. Chem. B. 2014;118:14870–14879. doi: 10.1021/jp509583c. [DOI] [PubMed] [Google Scholar]
- 18a.Aldous L, Silvester DS, Villagran C, Pitner WR, Compton RG, Lagunas MC, Hardacre C. New J. Chem. 2006;30:1576–1583. [Google Scholar]
- 18b. M. Alvarez-Guerra, J. Albo, E. Alvarez-Guerra, A. Irabien, Energy Env. Sci, in press.
- 18c.Manan NSA, Aldous L, Alias Y, Murray P, Yellowlees LJ, Lagunas MC, Hardacre C. J. Phys. Chem. B. 2011;115:13873–13879. doi: 10.1021/jp208159v. [DOI] [PubMed] [Google Scholar]
- 19.Nakagawa T, Beasley CA, Murray RW. J. Phys. Chem. C. 2009;113:12958–12961. [Google Scholar]
- 20.Köddermann T, Wertz C, Heintz A, Ludwig R. Angew. Chem. Int. Ed. 2006;45:3697–3702. doi: 10.1002/anie.200504471. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. 2006;118 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
miscellaneous_information