

Abstract
An efficient, highly stereoselective asymmetric synthesis of fully functionalized cyclopentanes bearing an oxindole moiety and several other functional groups in one pot has been developed. Key step is an organocatalytic triple Michael domino reaction forming three C–C bonds and six stereocenters, including a quaternary one. Starting from equimolar amounts of simple substrates, a high molecular complexity can be reached after a Wittig olefination in one pot. The new protocol can easily be scaled up to gram amounts.
Keywords: asymmetric synthesis, cyclopentanes, domino reactions, organocatalysis, oxindoles
Significant effort has been devoted to the development of synthetic strategies that rapidly construct diverse and complex molecular structures, not only for the total synthesis of natural products but also to provide practical and sustainable methods for the preparation of bioactive compounds, such as pharmaceuticals and agrochemicals.[1] In this regard, asymmetric organocatalytic domino/cascade reactions have been intensively investigated in recent years owing to their well-known intrinsic advantages over classical approaches.[2] In 2006, our group reported the first multicomponent organocatalytic triple domino reaction, which opened an efficient access to functionalized cyclohexene derivatives.[3] While the majority of newly developed organocatalytic cascade reactions focuses on six-membered ring systems,[4] polysubstituted cyclopentanes[5] as common motifs in natural products and pharmaceuticals (Figure 1) are of equal importance. Thus, several efficient organocatalytic procedures for the asymmetric synthesis of cyclopentane derivatives have recently been reported.[6] We envisaged to develop a new organocatalytic approach to fully substituted cyclopentanes bearing multiple stereocenters by employing a triple domino reaction.
Figure 1.
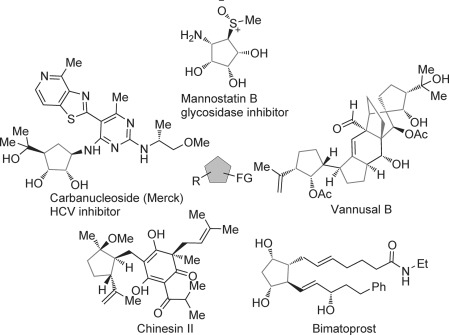
Representative natural products and drugs with highly functionalized cyclopentane cores.
Oxindoles[7] have been intensively investigated owing to their broad biological and pharmacological activities.[8] In 2010, Björkling and co-workers reported that cyclopentane-containing oxindoles possess antitumor activity.[9] The racemic compounds were synthesized by a traditional stepwise procedure, and it was shown that the absolute configuration plays an important role for the antitumor activity.[10] Therefore, it is highly desirable to develop enantioselective catalytic procedures for the synthesis of such core structures. We envisaged that a triple domino reaction involving a Michael addition of an oxindole to a strong electron-withdrawing unsaturated conjugated diene and subsequent double Michael additions with cinnamaldehyde could provide an efficient and straightforward route for the asymmetric synthesis of fully substituted cyclopentane core structures bearing an oxindole unit.
Herein, we report such a novel asymmetric Michael/Michael/Michael sequence to construct a variety of potentially bioactive products containing a cyclopentane ring through the formation of three C–C bonds and six stereocenters (Scheme 1).
Scheme 1.
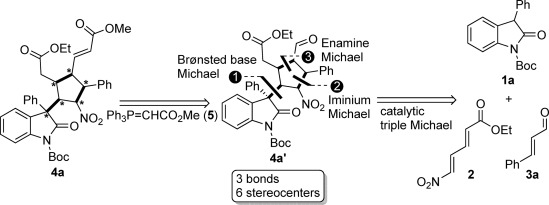
Asymmetric synthesis of fully substituted cyclopentanes bearing an oxindole moiety through a triple Michael addition (retrosynthetic analysis).
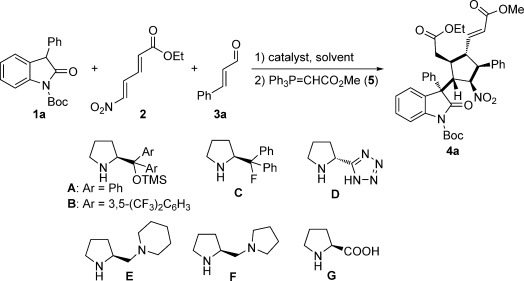
For the initial screening, we performed the reaction of oxindole 1 a, unsaturated conjugated diene 2, and (E)-cinnamaldehyde (3 a) by using diphenyl prolinol trimethylsilyl ether A as catalyst in CHCl3 at room temperature, followed by a one-pot Wittig reaction (Table 1). To our delight, the reaction using 0.3 equivalents of catalyst A proceeded well, affording product 4 a in a good domino yield (40 %) with very good diastereo- and enantioselectivity (15:1 d.r. and 89 % ee; entry 1). When lowering the catalyst loading to 0.1 equivalents, the yield remained the same and the diastereoselectivity decreased to 5.5:1; however, the enantioselectivity increased to 94 % (entry 2). The addition of benzoic acid as additive increased the yield and diastereoselectivity slightly, but had a negative effect on the enantioselectivity (entry 3). Furthermore, no improvement was obtained when the reaction was performed at a lower temperature (entry 4). Increasing the catalyst loading to 0.5 equivalents led to product 4 a in a good yield of 52 % with excellent diastereo- and enantioselectivity (15:1 d.r. and 98 % ee; entry 5). When the starting materials 1 a, 2, and 3 a were added in portions, a better result was obtained (63 % yield, 15:1 d.r., and 99 % ee; entry 6). Using CH2Cl2 instead of CHCl3 as solvent resulted in an improved yield of 70 % and excellent enantioselectivity (98 %), albeit with a lower diastereoselectivity (7:1 d.r.; entry 7). A subsequent solvent screening revealed that CHCl3 was the best choice (entries 7–10). Several structurally similar proline-derived catalysts B, C, D, E, and F were tested, but no better results were obtained (entries 11–15). In addition, (S)-proline (G) was ineffective for this reaction (entry 16).
Table 1.
Screening of the reaction conditions[a]
 | |||||
|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | Yield [%][b] | d.r.[c] | ee [%][d] |
| 1[e] | A | CHCl3 | 40 | 15:1 | 89 |
| 2[f] | A | CHCl3 | 40 | 5:1 | 94 |
| 3[f, g] | A | CHCl3 | 49 | 8:1 | 86 |
| 4[f, g, h] | A | CHCl3 | 48 | 3:1 | 87 |
| 5 | A | CHCl3 | 52 | 15:1 | 98 |
| 6[i, j] | A | CHCl3 | 63 | 15:1 | 99 |
| 7 | A | CH2Cl2 | 70 | 7:1 | 98 |
| 8 | A | toluene | 52 | 6:1 | 96 |
| 9 | A | ether | 13 | 15:1 | 98 |
| 10 | A | dioxane | 21 | –[k] | 98 |
| 11 | B | CHCl3 | 40 | 10:1 | 98 |
| 12 | C | CHCl3 | 31 | 7:1 | 95 |
| 13 | D | CHCl3 | 25 | 8:1 | n.d.[l] |
| 14 | E | CHCl3 | 32 | n.d. | n.d. |
| 15 | F | CHCl3 | 26 | n.d. | n.d. |
| 16 | G | CHCl3 | 0 | – | – |
[a] Reaction conditions (method A): all reactions were performed by using 1 a (0.25 mmol, 77.3 mg), 2 (0.25 mmol, 43.0 mg), 3 a (0.25 mmol, 33 mg), and catalyst (0.5 equiv) in CHCl3 (2 mL) for 22 h at room temperature followed by a one-pot olefination reaction for 5 h (Wittig reagent 5: 0.375 mmol, 125 mg), unless otherwise stated. [b] Yield of isolated product 4 a as a mixture of diastereomers after column chromatography. [c] Determined by 1H NMR spectroscopy. [d] Determined by chiral HPLC analysis for the major diastereomer. [e] Using 0.3 equiv catalyst. [f] Using 0.1 equiv catalyst. [g] Addition of 0.2 equiv PhCO2H. [h] Performed at −20 °C. [i] Method B: 1 a, 2, and 3 a were added in three portions at 0, 3, and 6 h. Wittig reagent 5 (0.5 mmol, 167.0 mg) was added in two portions at 22 and 25 h (stirring for 6 h in total). [j] 0.5 mmol scale. [k] Mixture of diastereomers. [l] Not determined.
With the optimized conditions in hand (Table 1, entry 6), the substrate scope and limitations were explored. As shown in Scheme 2, substrates with various substituents at the 3- or 5-position of the oxindole were tolerated. Oxindoles 1 a–c bearing a phenyl group at the 3-position reacted well with cinnamaldehyde 3 a, providing the corresponding products 4 a–c in yields ranging from 63–72 % with good diastereo- (7:1–15:1) and excellent enantioselectivities (97–99 %). Under the conditions of method A, product 4 d was also formed in good results as well (57 % yield, 11:1 d.r., and 99 % ee). Reactions with oxindoles containing a benzyl or methyl group at the 3-position afforded products 4 e or 4 f with excellent enantioselectivities, albeit with diminished yields and diastereoselectivities (4 e: 30 % yield, 3:1 d.r.; 4 f: 44 % yield, 2:1 d.r.).
Scheme 2.

The substrate scope of oxindoles 1. Yield refers to isolated product 4 as a mixture of diastereomers after column chromatography. The d.r. value was determined by 1H NMR spectroscopy and the ee value by HPLC analysis for the major diastereomer. [a] Method A: all the reactions were performed by using 1 (0.5 mmol), 2 (0.5 mmol), 3 a (0.5 mmol), and catalyst A (0.25 mmol) in CHCl3 (4 mL) for 22 h at room temperature followed by a one-pot Wittig reaction for 5 h (reagent 5: 0.75 mmol), unless otherwise stated. [b] Method B (see Table 1).
Next, we applied this protocol to a broad range of aldehydes 3 b–f bearing various electron-rich or electron-deficient aromatic groups (Scheme 3). For example, methoxy, chloro, and nitro groups were well tolerated in the reaction of cinnamaldehydes 3 b–d with oxindole 2 a, providing the corresponding products 4 g–i in yields ranging from 61–65 % with excellent diastereo- and enantioselectivities (9:1–15:1 d.r. and 97–99 % ee). No significant difference in the outcome for product 4 h was observed when employing either method A or B. The domino reaction also worked well when using a lower catalyst loading of 0.3 equivalents to obtain the desired product 4 h (49 % yield, 7:1 d.r., and 98 % ee). For substrate 3 e with a nitro group at the ortho-position, the reaction also proceeded well, affording product 4 j in 61 % yield with 15:1 d.r. and 97 % ee. To further extend the substrate scope, a heterocyclic 2-furyl α,β-unstaturated aldehyde 3 f was tested under the conditions and product 4 k was obtained in a good yield of 47 % with excellent enantioselectivity (99 % ee), albeit with a low diastereoselectivity (3:1 d.r.). To our delight, substrate 1 e with a benzyl group at the 3-position reacted well with cinnamaldehydes 3 b and 3 g, providing products 4 l and 4 m in good yields with good diastereoselectivities (47 % yield, 8:1 d.r. and 63 % yield, 10:1 d.r., respectively), as well as excellent enantioselectivities (99 % ee).
Scheme 3.
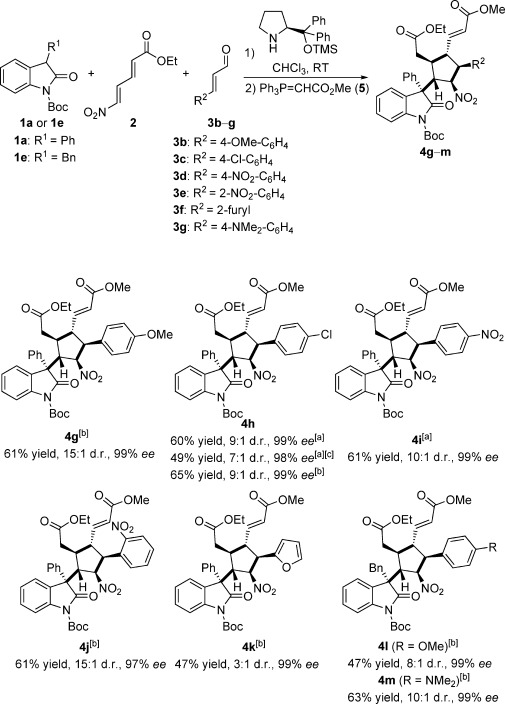
The substrate scope of cinnamaldehydes 3. Yield refers to isolated product 4 as a mixture of diastereomers after column chromatography. The d.r. value was determined by 1H NMR spectroscopy and the ee value by HPLC analysis for the major diastereomer. [a] Method A. [b] Method B. [c] Using 0.3 equiv catalyst A.
To test the scalability of the new protocol, we performed the reaction on a gram scale under the optimized conditions (method B); product 4 a was obtained in a good yield of 68 % (2.3 g) with good diastereo- and excellent enantioselectivity (8:1 d.r. and 99 % ee). Furthermore, the tert-butoxycarbonyl (Boc) protecting group could be easily removed in the presence of trifluoroacetic acid (TFA) at room temperature from oxindole 4 a to get the deprotected compound 6 in 94 % yield with 95 % ee (Scheme 4). The absolute configuration of the stereocenters in compound 4 a was unambiguously determined by X-ray crystallography (Figure 2).[11]
Scheme 4.
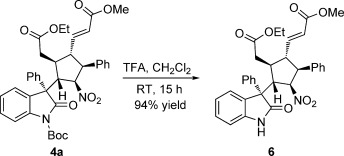
Removal of the N-Boc group from oxindole 4 a.
Figure 2.
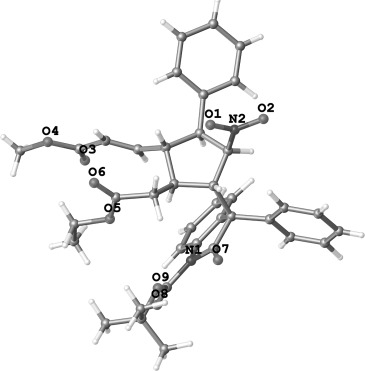
X-ray crystal structure of product 4a.
A possible mechanism is proposed in Scheme 5. The first Michael addition of oxindole 1 a to (E,E)-5-nitro-2,4-pentadienoic acid ethyl ester (2) is initiated by the diarylprolinol silyl ether catalyst, which acts as a Brønsted base in the process, providing an active intermediate I that was observed by mass spectroscopy. The active intermediate I reacts quickly with cinnamylaldehyde 3 a through a second Michael reaction via iminium activation, providing an intermediate II, which is further transformed to the product aldehyde 4 a′ by a third Michael reaction via enamine activation. The final product 4 a is then obtained by olefination of 4 a′ using the Wittig reagent 5.
Scheme 5.
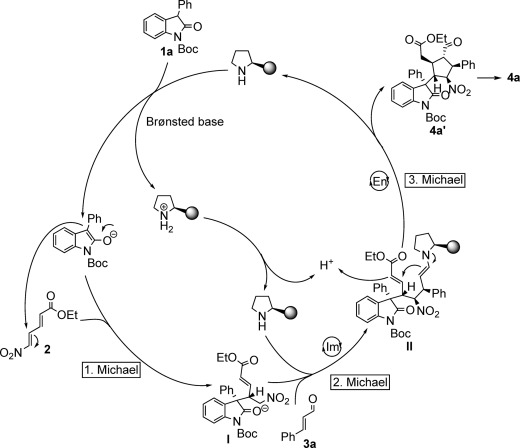
A proposed pathway for the asymmetric synthesis of fully substituted cyclopentanes. Im=iminium activation; En=enamine activation.
In conclusion, a novel organocatalytic domino protocol was developed to construct a series of potentially bioactive, fully substituted cyclopentanes bearing different functional groups including an oxindole moiety in good yields, very good diastereo-, and excellent enantioselectivities. It is worth noting that the one-pot triple Michael sequence creates three bonds and six stereocenters, including one quaternary center, by using equimolar amounts of reactants under mild reaction conditions. Although a high catalyst loading (0.5 equiv) was required for most substrates to obtain high diastereoselectivities, the protocol was also successfully applied to some substrates using a lower amount of catalyst (0.3 equiv). The scalability of the new protocol to grams was demonstrated without loss in efficiency.
Experimental Section
General procedure for the organocatalytic domino reaction (described for the synthesis of 4 a as a typical example)
Method A: A glass vial (10 mL) equipped with a magnetic stirring bar was charged with 1 a (154.6 mg, 0.5 mmol), 2 (86 mg, 0.5 mmol), 3 a (66 mg, 0.5 mmol), and catalyst A (81.5 mg, 0.25 mmol). After adding CHCl3 (4 mL), the reaction tube was purged by using an argon flow for 1 min, then covered with a Teflon-coated screw cap. The reaction mixture was stirred at room temperature for 22 h. After addition of the Wittig reagent 5 (250 mg, 0.75 mmol) and purging by using an argon flow for 1 min, the mixture was stirred for another 5 h. The resulting solution was then directly applied to flash chromatography (at first using pentane/ethyl acetate=10:1, then pentane/ethyl acetate=5:1 as eluent) to afford the desired product 4 a as a yellow solid (180 mg, 54 % yield, 15:1 d.r., 98 % ee).
Method B: A glass vial (10 mL) equipped with a magnetic stirring bar was charged with 1 a (51.5 mg, 0.17 mmol), 2 (28.7 mg, 0.17 mmol), 3 a (22 mg, 0.17 mmol), and catalyst A (81.5 mg, 0.25 mmol; starting materials 1 a, 2, and 3 a were divided into three portions). The reaction tube was purged by using an argon flow for 1 min, then covered with a Teflon-coated screw cap after adding CHCl3 (4 mL). After stirring at room temperature for 3 h, the second portion of 1 a (51.5 mg, 0.17 mmol), 2 (28.7 mg, 0.17 mmol), and 3 a (22 mg, 0.17 mmol) was added to the reaction tube. After purging by using an argon flow for 1 min, the mixture was stirred for another 3 h. After that, the third portion of 1 a (51.5 mg, 0.17 mmol), 2 (28.7 mg, 0.17 mmol), and 3 a (22 mg, 0.17 mmol) was added to the reaction mixture, which was stirred for another 16 h after purging by using an argon flow for 1 min. Then, the first portion of Wittig reagent 5 (83.5 mg, 0.25 mmol) was added to the reaction tube. The mixture was stirred for 3 h after purging by using an argon flow for 1 min. After that, the second portion of Wittig reagent 5 (83.5 mg, 0.25 mmol) was added to the reaction mixture. The mixture was stirred for another 3 h after purging by using an argon flow for 1 min. The resulting solution was then directly applied to flash chromatography (at first using pentane/ethyl acetate=10:1, then pentane/ethyl acetate=5:1 as eluent) to afford product 4 a as a yellow solid (209 mg, 0.31 mmol, 63 % yield, 15:1 d.r., 99 % ee).
For both cases, the major diastereoisomer could be isolated as a colorless solid by using a preparative TLC plate (hexane/isopropanol=15:1 as eluent).
Acknowledgments
We thank the European Research Council (ERC Advanced Grant 320493 “DOMINOCAT”) for financial support. Dr. Zou is grateful to the Chinese Scholarship Council (CSC) for supporting him to study abroad.
Supporting Information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information
References
- 1a.Davies HML, Sorensen EJ. Chem. Soc. Rev. 2009;38:2981–3272. For a themed issue on the rapid formation of complexity in organic synthesis, see. [Google Scholar]
- 1b.Cossy J, Arseniyadis S. Modern Tools for the Synthesis of Complex Bioactive Molecules. Weinheim: Wiley-VCH; 2012. [Google Scholar]
- 2a.Enders D, Grondal C, Hüttl MRM. Angew. Chem. Int. Ed. 2007;46:1570–1581. doi: 10.1002/anie.200603129. For selected reviews on organocatalytic domino reactions, see. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. 2007;119 [Google Scholar]
- 2b.Grondal C, Jeanty M, Enders D. Nat. Chem. 2010;2:167–178. doi: 10.1038/nchem.539. [DOI] [PubMed] [Google Scholar]
- 2c.Pellissier H. Adv. Synth. Catal. 2012;354:237–294. [Google Scholar]
- 2d.Grossmann A, Enders D. Angew. Chem. Int. Ed. 2012;51:314–325. doi: 10.1002/anie.201105415. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. 2012;124 [Google Scholar]
- 2e.Lu L-Q, Chen J-R, Xiao X-J. Acc. Chem. Res. 2012;45:1278–1293. doi: 10.1021/ar200338s. [DOI] [PubMed] [Google Scholar]
- 2f.Goudedranche S, Raimondi W, Bugaut X, Constantieux T, Bonne D, Rodriguez J. Synthesis. 2013;45:1909–1930. [Google Scholar]
- 2g.Volla CMR, Atodiresei I, Rueping M. Chem. Rev. 2014;114:2390–2431. doi: 10.1021/cr400215u. [DOI] [PubMed] [Google Scholar]
- 3.Enders D, Hüttl MRM, Grondal C, Raabe G. Nature. 2006;441:861–863. doi: 10.1038/nature04820. [DOI] [PubMed] [Google Scholar]
- 4a.Dalko PI. Comprehensive Enantioselective Organocatalysis. Weinheim: Wiley-VCH; 2013. [Google Scholar]
- 4b.Rios Torres R. Stereoselective Organocatalysis. Weinheim: Wiley-VCH; 2013. [Google Scholar]
- 5a.Trost BM. Angew. Chem. Int. Ed. Engl. 1986;25:1–20. For reviews on the synthesis of cyclopentanes, see. [Google Scholar]
- Angew. Chem. 1986;98 [Google Scholar]
- 5b.Silva LF. Tetrahedron. 2002;58:9137–9161. [Google Scholar]
- 5c.Heasley B. Eur. J. Org. Chem. 2009:1477–1489. [Google Scholar]
- 5d.Heasley B. Curr. Org. Chem. 2014;18:641–686. [Google Scholar]
- 6a.Vignola N, List B. J. Am. Chem. Soc. 2004;126:450–451. doi: 10.1021/ja0392566. [DOI] [PubMed] [Google Scholar]
- 6b.Zu L, Li H, Xie H, Wang J, Jiang W, Tang Y, Wang W. Angew. Chem. Int. Ed. 2007;46:3732–3734. doi: 10.1002/anie.200700485. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. 2007;119 [Google Scholar]
- 6c.Enders D, Wang C, Bats JW. Angew. Chem. Int. Ed. 2008;47:7539–7542. doi: 10.1002/anie.200802532. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. 2008;120 [Google Scholar]
- 6d.Tan B, Shi Z, Chua PJ, Zhong G. Org. Lett. 2008;10:3425–3428. doi: 10.1021/ol801246m. [DOI] [PubMed] [Google Scholar]
- 6e.Tan B, Chua PJ, Zeng X, Lu M, Zhong G. Org. Lett. 2008;10:3489–3492. doi: 10.1021/ol801273x. [DOI] [PubMed] [Google Scholar]
- 6f.Hong B-C, Nimje RY, Lin C-W, Liao J-H. Org. Lett. 2011;13:1278–1281. doi: 10.1021/ol1030487. [DOI] [PubMed] [Google Scholar]
- 6g.Hong B-C, Dange NS, Hsu C-S, Liao J-H, Lee G-H. Org. Lett. 2011;13:1338–1341. doi: 10.1021/ol200006e. [DOI] [PubMed] [Google Scholar]
- 6h.Tan B, Candeias NR, Barbas CF. Nat. Chem. 2011;3:473–477. doi: 10.1038/nchem.1039. [DOI] [PubMed] [Google Scholar]
- 6i.Albertshofer K, Tan B, Barbas CF., III Org. Lett. 2012;14:1834–1837. doi: 10.1021/ol300441z. [DOI] [PubMed] [Google Scholar]
- 6j.Zhao G-L, Ibrahem I, Dziedzic P, Sun J, Bonneau C, Córdova A. Chem. Eur. J. 2008;14:10007–10011. doi: 10.1002/chem.200801082. [DOI] [PubMed] [Google Scholar]
- 6k.Sun W, Zhu G, Wu C, Hong L, Wang R. Chem. Eur. J. 2012;18:6737–6741. doi: 10.1002/chem.201200478. [DOI] [PubMed] [Google Scholar]
- 6l.Remeš M, Vesely J. Eur. J. Org. Chem. 2012:3747–3752. [Google Scholar]
- 6m.Liu G, Shirley ME, Van KN, McFarlin RL, Romo D. Nat. Chem. 2013;5:1050–1058. doi: 10.1038/nchem.1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7a.Bui T, Syed S, Barbas CF. J. Am. Chem. Soc. 2009;131:8758–8759. doi: 10.1021/ja903520c. For selected asymmetric syntheses of oxindoles, see. [DOI] [PubMed] [Google Scholar]
- 7b.Wang C, Yang X, Loh CCJ, Raabe G, Enders D. Chem. Eur. J. 2012;18:11531–11535. doi: 10.1002/chem.201201262. [DOI] [PubMed] [Google Scholar]
- 7c.Zhong F, Dou X, Han X, Yao W, Zhu Q, Meng Y, Lu Y. Angew. Chem. Int. Ed. 2013;52:943–947. doi: 10.1002/anie.201208285. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. 2013;125 [Google Scholar]
- 7d.Huang J-Z, Wu X, Gong L-Z. Adv. Synth. Catal. 2013;355:2531–2537. [Google Scholar]
- 7e.Cui B-D, Han W-Y, Wu Z-J, Zhang X-M, Yuan W-C. J. Org. Chem. 2013;78:8833–8839. doi: 10.1021/jo401154b. [DOI] [PubMed] [Google Scholar]
- 7f.Gao J, Chen J-R, Duan S-W, Li T-R, Lu L-Q, Xiao W-J. Asian J. Org. Chem. 2014;3:530–535. [Google Scholar]
- 7g.Ošeka M, Noole A, žari S, Öeren M, Järving I, Lopp M, Kanger T. Eur. J. Org. Chem. 2014:3599–3606. [Google Scholar]
- 7h.Li T-Z, Wang X-B, Sha F, Wu X-Y. J. Org. Chem. 2014;79:4332–4339. doi: 10.1021/jo500145w. [DOI] [PubMed] [Google Scholar]
- 7i.Zhu X-L, Xu J-H, Cheng D-J, Zhao L-J, Liu X-Y, Tan B. Org. Lett. 2014;16:2192–2195. doi: 10.1021/ol5006888. [DOI] [PubMed] [Google Scholar]
- 7j.Yang H-B, Zhao Y-Z, Sang R, Shi M. J. Org. Chem. 2014;79:3519–3528. doi: 10.1021/jo5003246. [DOI] [PubMed] [Google Scholar]
- 7k.Wu H, Wang YM. Chem. Eur. J. 2014;20:5899–5904. doi: 10.1002/chem.201402002. [DOI] [PubMed] [Google Scholar]
- 7l.Wu C, Li G, Sun W, Zhang M, Hong L, Wang R. Org. Lett. 2014;16:1960–1963. doi: 10.1021/ol500517d. [DOI] [PubMed] [Google Scholar]
- 7m.Liu Z-M, Li N-K, Huang X-F, Wu B, Li N, Kwok C-Y, Wang Y, Wang X-W. Tetrahedron. 2014;70:2406–2415. [Google Scholar]
- 7n.Rueping M, Liu X, Bootwicha T, Pluta R, Merkens C. Chem. Commun. 2014;50:2508–2511. doi: 10.1039/c3cc49877h. [DOI] [PubMed] [Google Scholar]
- 7o.Ramachary DB, Prasad MS, Laxmi SV, Madhavachary R. Org. Biomol. Chem. 2014;12:574–580. doi: 10.1039/c3ob42100g. [DOI] [PubMed] [Google Scholar]
- 8a.Hewawasam P, Erway M, Moon SL, Knipe J, Weiner H, Boissard CG, Post-Munson DJ, Gao Q, Huang S, Gribkoff VK, Meanwell NA. J. Med. Chem. 2002;45:1487–1499. doi: 10.1021/jm0101850. [DOI] [PubMed] [Google Scholar]
- 8b.Neel DA, Brown ML, Lander PA, Grese TA, Defauw JM, Doti RA, Fields T, Kelley SA, Smith S, Zimmerman KM, Steinberg MI, Jadhav PK. Bioorg. Med. Chem. Lett. 2005;15:2553–2557. doi: 10.1016/j.bmcl.2005.03.086. [DOI] [PubMed] [Google Scholar]
- 8c.Ding K, Lu YP, Nikolovska-Coleska Z, Wang G, Qiu S, Shangary S, Gao W, Qin D, Stuckey J, Krajewski K, Roller PP, Wang S. J. Med. Chem. 2006;49:3432–3435. doi: 10.1021/jm051122a. [DOI] [PubMed] [Google Scholar]
- 8d.Jiang T, Kuhen KL, Wolff K, Yin H, Bieza K, Caldwell J, Bursulaya B, Wu TY-H, He Y. Bioorg. Med. Chem. Lett. 2006;16:2105–2108. doi: 10.1016/j.bmcl.2006.01.073. [DOI] [PubMed] [Google Scholar]
- 8e.Uddin MK, Reignier SG, Coulter T, Montalbetti C, Grånäs C, Butcher S, Krog-Jensen C, Felding J. Bioorg. Med. Chem. Lett. 2007;17:2854–2857. doi: 10.1016/j.bmcl.2007.02.060. [DOI] [PubMed] [Google Scholar]
- 9.Christensen MK, Erichsen KD, Trojel-Hansen C, Tjørnelund J, Nielsen SJ, Frydenvang K, Johansen TN, Nielsen B, Sehested M, Jensen PB, Ikaunieks M, Zaichenko A, Loza E, Kalvinsh I, Björkling F. J. Med. Chem. 2010;53:7140–7145. doi: 10.1021/jm100763j. [DOI] [PubMed] [Google Scholar]
- 10. According to the Björkling group, some S-configured oxindole derivatives possess potential antitumor activity, while the racemic compounds show complete tumor regression. The enantiomers were separated by chiral stationary-phase chromatography.
- 11. The absolute configuration of 4 a was determined by X-ray crystallographic analysis. CCDC 1028110 (4 a) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
miscellaneous_information