

Abstract
The Mitsunobu reaction is renowned for its mild reaction conditions and broad substrate tolerance, but has limited utility in process chemistry and industrial applications due to poor atom economy and the generation of stoichiometric phosphine oxide and hydrazine by-products that complicate purification. A catalytic Mitsunobu reaction using innocuous reagents to recycle these by-products would overcome both of these shortcomings. Herein we report a protocol that is catalytic in phosphine (1-phenylphospholane) employing phenylsilane to recycle the catalyst. Integration of this phosphine catalytic cycle with Taniguchi’s azocarboxylate catalytic system provided the first fully catalytic Mitsunobu reaction.
Keywords: organocatalysis, phosphorus heterocycles, reaction kinetics, silanes, synthetic methods
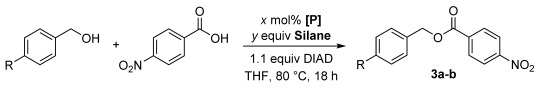
The Mitsunobu reaction is the displacement of an alcohol with a pronucleophile (Nu–H) mediated by phosphine and azocarboxylate reagents, which work in concert to activate the pronucleophile through deprotonation and convert the alcohol to a reactive alkoxyphosphonium species.[1] Renowned for its mild reaction conditions and broad substrate tolerance, the Mitsunobu reaction is capable of forming C–O, C–N, C–S, C–X, and C–C bonds.[2] However, the Mitsunobu reaction is highly underutilized in process chemistry and manufacturing due to arduous purification from by-products and poor atom economy.[3] Although several innovative reagents have been developed that can be removed by liquid–liquid or solid–liquid extractions to facilitate purification,[4] the ideal Mitsunobu reaction would be catalytic in phosphine and azocarboxylate, and use innocuous reagents to recycle these catalysts.[5] Toward this goal, Toy and co-workers rendered the Mitsunobu catalytic in the azocarboxylate using PhI(OAc)2 to oxidize the hydrazine by-product[6] whereas Taniguchi and co-workers developed an iron(II) phthalocyanine catalytic system employing oxygen as the terminal oxidant.[7] O’Brien and co-workers disclosed the first example of a Mitsunobu reaction that is catalytic in phosphine in the patent literature,[8] and optimization of this reaction is heavily desired.[4, 9] Herein we report the development and optimization of a Mitsunobu reaction catalytic in phosphorus utilizing dibenzophosphole and phospholane precatalysts 1 and 2 (Scheme 1 B), inspired by the development of the catalytic Appel,[10a] Staudinger,[10b] and Wittig[11] reactions. We then combined this catalytic cycle with the Taniguchi iron–phthalocyanine catalytic system to generate the first fully catalytic Mitsunobu reaction (Scheme 1 A).
Scheme 1.
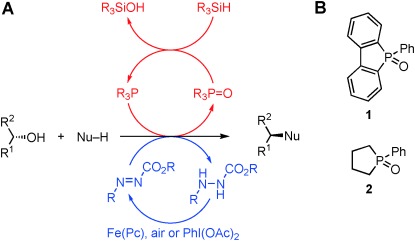
Proposed catalytic Mitsunobu reaction.
Chemoselective reduction of the phosphine oxide product back to the phosphine in the presence of a reactive azo compound is required in order to complete the phosphine catalytic cycle. We initially investigated dibenzophosphole oxide 1 as a precatalyst due to its facile reduction by silanes and its ability to tune the catalyst through modification of the aryl rings.[10] The coupling of 4-nitrobenzoic acid and benzyl alcohol or 4-trifluoromethylbenzyl alcohol was studied with a model system employing stoichiometric DIAD, 1 (10 mol %), and various silanes (Table 1). To benchmark these results, the yield obtained with stoichiometric triphenylphosphine (TPP) was determined as 84 % (Table 1, entry 1). Polymethylhydrosiloxane and triphenylsilane did not provide catalytic turnover (Table 1, entries 3 and 4). Diphenylsilane (entry 5) provided the first encouraging yield of 42 %, whereas phenylsilane (entry 6) furnished an improved yield of 63 %. We next explored the impact of PhSiH3 stoichiometry (entries 8–11; see also Table S2) on the outcome of the reaction. We observed that excess silane was detrimental, although it led to faster conversion. Conversely, a lower amount of silane led to substantially slower conversion, but did result in slightly improved yields. As a control, we also measured the background reaction in the absence of PhSiH3 confirming its vital role (entry 8). The reaction of PhSiH3 with alcohols in the presence of Lewis acids (i.e., 4-nitrobenzoic acid) at elevated temperatures is precedented[12] and contributed to the lower yields (Figure S1). Thus, even the yield of the stoichiometric TPP reaction was reduced from 84 % to 77 % when PhSiH3 was included in the reaction (entries 1 and 2). Based on these considerations, we settled on 1.1 equivalents of PhSiH3 as optimal, to minimize both reaction time and undesired reactivity. Finally, we evaluated phospholane precatalyst 2 using our optimized conditions and were elated to obtain 77 % yield (entry 12), which is identical to the stoichiometric TPP version under the same conditions. The catalyst loading could be lowered to 5 mol % without affecting the yield, with 1 mol % of catalyst still providing a respectable 54 % yield (entries 12–15).
Table 1.
Development of conditions for the Mitsunobu reaction catalytic in phosphine[a]
| Entry | R | [P] | x | Silane | y | Product | Yield [%][b] |
|---|---|---|---|---|---|---|---|
| 1 | H | TPP | 110 | none | 3 a | 84 | |
| 2 | H | TPP | 110 | PhSiH3 | 1.1 | 3 a | 77 |
| 3 | H | 1 | 10 | PHMS | 1.5 | 3 a | 0 |
| 4 | H | 1 | 10 | Ph3SiH | 2.0 | 3 a | 0 |
| 5 | H | 1 | 10 | Ph2SiH2 | 1.1 | 3 a | 42 |
| 6 | H | 1 | 10 | PhSiH3 | 1.1 | 3 a | 63 |
| 7 | H | 1 | 10 | none | – | 3 a | 0[c] |
| 8 | CF3 | 1 | 10 | PhSiH3 | 0.5 | 3 b | 66[d] |
| 9 | CF3 | 1 | 10 | PhSiH3 | 1.5 | 3 b | 63 |
| 10 | CF3 | 1 | 10 | PhSiH3 | 3.0 | 3 b | 52 |
| 11 | CF3 | 1 | 10 | PhSiH3 | 6.0 | 3 b | 43 |
| 12 | H | 2 | 10 | PhSiH3 | 1.1 | 3 a | 77 |
| 13 | H | 2 | 5 | PhSiH3 | 1.1 | 3 a | 77[e] |
| 14 | H | 2 | 2 | PhSiH3 | 1.1 | 3 a | 58[f] |
| 15 | H | 2 | 1 | PhSiH3 | 1.1 | 3 a | 54[g] |
[a] Reactions performed on 1 mmol scale at 0.25 m. [b] Isolated average of two reactions. [c] Reactions with the reduced form of 1 without silane added produced 7 % of product. [d] 46 h. [e] Reaction at 10 mmol scale was performed with 78 % yield. [f] 38 h. [g] 69 h.
We speculated that the greater reactivity of catalyst 2 was due to its more facile reduction by PhSiH3, which is the rate-limiting step in the phosphine catalytic cycle. The group of van Delft had indicated that 1 and 2 were nearly equivalent in reactivity as measured by reduction with Ph2SiH2 at 100 °C in 1,4-dioxane.[10] To study the relative reactivity of 1 and 2 more rigorously, we measured their rates of reduction by 31P NMR spectroscopy under pseudo first-order conditions (30, 15, 7.5 equiv PhSiH3) at various temperatures ranging from 25 to 80 °C.[12] The activation energies (ΔG≠) were then calculated from the temperature dependence of the second-order rate constants using Arrhenius (Figure 1) and Eyring plots. Reduction of 2 was facile, even at 25 °C, as evident of its low ΔG≠ of 14.1±0.4 kcal mol−1. Whereas 1 was significantly less reactive with a ΔG≠ of 21.3±3.6 kcal mol−1 and was not reduced at 25 °C even after 120 h. These data would indicate that the catalytic Mitsunobu reaction should readily occur at room temperature with 2. Unfortunately, this is not true in practice and the catalytic Mitsunobu reaction requires elevated temperatures to achieve complete conversion. The activation energies were determined in the absence of other reagents (pronucleophile, alcohol, and DIAD), which attenuate the rate of phosphine reduction (Table S7).
Figure 1.

The Arrhenius plots of 1 and 2.
With the optimal catalytic conditions established, we then investigated the substrate scope with a range of alcohols and pronucleophiles (Table 2). For comparison, the yields of the stoichiometric reaction conducted at room temperature are also presented. Primary benzylic, allylic, and alkyl alcohols (Table 2, entries 1–5) were reacted with 4-nitrobenzoic acid to afford the corresponding esters in moderate to good yields. The lower yield of the benzylic substrates, relative to the stoichiometric reaction, is due to competitive silane reactivity of the alcohols (Figure S1). Notably, the 82 % yield obtained with the simple alkyl alcohol, 3-(4-fluorophenyl)propanol (entry 5) was identical to the stoichiometric version. Propargyl alcohol was also esterified with 84 % yield (entry 6), similar to its stoichiometric counterpart. Secondary alcohols were also competent substrates (entries 7 and 8) providing respectable yields with high enantiomeric purities (e.r.>94:6). We next examined the coupling of 2-phenylethanol with benzoic acid, aminoacid, phenol, and sulfamide pronucleophiles (entries 9–12). These substrates all afforded the corresponding products in good to excellent yields compared to the stoichiometric reaction. Intramolecular reaction of Boc-protected homoserine furnished the γ-lactone in an impressive 87 % yield (entry 13). The background reaction without the phosphine catalyst was less than 2 %. Reaction of 2′,3′-O-isopropylideneinosine with Boc-protected sulfamide provided the coupled product in 70 % yield (entry 14) highlighting the utility of the catalytic Mitsunobu reaction with more challenging substrates.[14]
Table 2.
The substrate scope of the catalytic Mitsunobu reaction[a]
| Entry | Product | Catalytic yield [%][b] | Stoichiometric yield [%][b,c] | |
|---|---|---|---|---|
| 1 |  |
3 a | 77 | 94 |
| 2 | 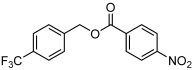 |
3 b | 76 | 92 |
| 3 | 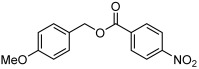 |
3 c | 61 | 80 |
| 4 | 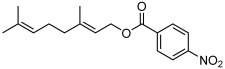 |
3 d | 50 | 50 |
| 5 | 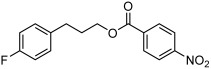 |
3 e | 82 | 82 |
| 6 | 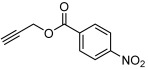 |
3 f | 84 | 90 |
| 7 | 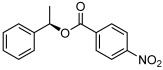 |
3 g | 69[d] | 77[d] |
| 8 |  |
3 h | 68[e] | 78[e] |
| 9 | 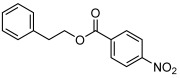 |
3 i | 76 | 96 |
| 10 |  |
3 j | 63 | 83 |
| 11 | 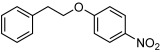 |
3 k | 51[f] | 85[f] |
| 12 | 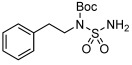 |
3 l | 72 | 90 |
| 13 | 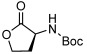 |
3 m | 87[g,h] | 98[g] |
| 14 |  |
3 n | 70[i] | 93[i] |
[a] Reactions performed on 0.5–1.0 mmol scale at 0.25 m employing 1.5 equiv of pronucleophile, 10 mol % loading of catalyst 2, and 1.1 equivalents of both DIAD and PhSiH3. Reactions were all run at 80 °C. [b] Isolated average of two reactions. [c] Reactions performed at 23 °C with 1.5 equivalents of pronucleophile, TPP, and DIAD without phenylsilane for 18 h. [d] e.r. 94:6. [e] e.r.>99.5:0.5. [f] 48 h. [g] Concentration was 0.04 m. [h] Background reaction with only Boc-homoserine-OH, DIAD, and PhSiH3 only produced traces. [i] N-Boc sulfamide (3 equiv) was used as the pronucleophile.
Integration of the phosphine and azocarboxylate catalytic cycles would provide the first fully catalytic Mitsunobu reaction. Given the opposing requirements for catalyst turnover (phosphine oxide reduction versus hydrazine oxidation), it was unclear whether these two cycles would be compatible. Among the two described azocarboxylate catalytic systems,[6, 7] we chose the Taniguchi iron(II) phthalocyanine [Fe(pc)] protocol that utilizes catalytic hydrazine 4, because it employs oxygen as the terminal oxidant. As a proof of concept, we studied the coupling of 4-methoxybenzyl alcohol[15] with 4-nitrobenzoic acid. Our first attempt of employing our optimized protocol with Taniguchi’s conditions (10 mol % [Fe(pc)], 10 mol % 4) furnished a 15 % yield (Table 3, entry 1). While not impressive, this result indicated that both catalytic cycles could be combined as at least one turnover was noted. Performing the reaction under an oxygen-enriched atmosphere quickly improved the yield from 15 to 35 % (Table 3, entries 1 and 3). Switching to 5 Å molecular sieves slightly improved yields; however, combining an oxygen-enriched atmosphere with 5 Å molecular sieves proved to be crucial and increased the yield to 63 % (entry 5), which is comparable to the yield obtained with our catalytic phosphine conditions alone (Table 2, entry 3). As noted before, catalyst 2 was superior to catalyst 1 in the fully catalytic Mitsunobu reaction (Table 3, entries 3 and 5).
Table 3.
The initial optimization of the fully catalytic Mitsunobu reaction[a]
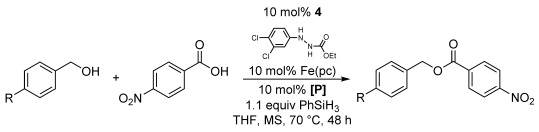
| Entry | R | [P] | MS [Å][b] | Atmosphere | Yield[c] |
|---|---|---|---|---|---|
| 1 | OMe | 1 | 4 | air | 15 |
| 2 | OMe | 1 | 5 | air | 19 |
| 3 | OMe | 1 | 5 | O2 enriched[d] | 35 |
| 4 | OMe | 2 | 5 | air | 35 |
| 5 | OMe | 2 | 5 | O2 enriched[d] | 63[e] |
| 6 | H | 2 | 5 | O2 enriched[d] | 68 |
[a] Reactions performed on 0.5 mmol scale at 0.17 m employing 1.5 equivalents of 4-nitrobenzoic acid. [b] All MS are powdered. [c] Average yield of the isolated product of two reactions. [d] Oxygen prepared by the reaction of NaOCl and H2O2. [e] Average yield of the isolated product of three reactions ranging from 60–68 % yield.
We were able to successfully combine Taniguchi’s approach with our novel catalytic phosphorus methodology. Both the phosphorous catalyst and the catalytic hydrazine can also be recovered easily in greater than 95 % quantities during chromatography. Fe(pc) is nontoxic and inexpensive, and 2 is synthesized in only two steps without the need for halogenated solvents or carcinogenic butadiene.[11b, 16] While there is still room for improvement, the concept of a fully catalytic Mitsunobu reaction has finally been realized. Improvements in yield and reaction setup will turn this concept into a more practical application, but this reaction still fulfills the requirements of a sustainable, safe, and economic Mitsunobu reaction for process chemistry and manufacturing. We are currently engaged in seeking improvements to our methodology with new catalysts.[17] We are also seeking a fully catalytic Mitsunobu reaction that is feasible at room temperature[18] to ensure the widest possible application of this powerful transformation.
Experimental Section
Typical procedure for the Mitsunobu reaction catalytic in phosphine: To a 15 mL pressure tube equipped with a stir bar was added catalyst 2 (18 mg, 0.10 equiv, 0.10 mmol) and 4-nitrobenzoic acid (250 mg, 1.5 equiv, 1.5 mmol). Then, THF (4 mL) was added followed by benzyl alcohol (103 μL, 1.0 equiv, 1.0 mmol), DIAD (216 μL, 1.1 equiv, 1.1 mmol), and phenylsilane (135 μL, 1.1 equiv, 1.1 mmol). The reaction vessel was sealed with a #15 O-ring and heated to 80 °C for 18 h. The reaction was cooled to 23 °C and concentrated under reduced pressure. The residue was purified by column chromatography eluting with hexane (to recover reduced 2) followed by a step-wise gradient from 0 to 10 % EtOAc in hexane. Benzyl 4-nitrobenzoate was isolated as an off-white solid (197 mg, 0.77 mmol, 77 %).
Typical procedure for the fully catalytic Mitsunobu reaction: To a 35 mL pressure tube equipped with a stir bar was added catalyst 2 (9.0 mg, 0.10 equiv, 0.05 mmol), catalyst 4 (12.5 mg, 0.10 equiv, 0.05 mmol), Fe(pc) (28.5 mg, 0.10 equiv, 0.05 mmol), 4-nitrobenzoic acid (125 mg, 1.5 equiv, 0.75 mmol), and 5 Å powdered molecular sieves (500 mg). THF (3 mL) was added followed by 4-methoxybenzyl alcohol (62 μL, 1.0 equiv, 0.50 mmol), and phenylsilane (68 μL, 1.1 equiv, 0.55 mmol). The vessel was purged with oxygen gas and sealed with a #15 O-ring. The reaction was heated at 70 °C for 48 h. The reaction was cooled to 23 °C, filtered to remove the sieves and the filtrate was partitioned between EtOAc (30 mL) and saturated aqueous NaHCO3 (30 mL). The organic layer was separated and washed with saturated aqueous NaCl (30 mL), dried (MgSO4) and concentrated under reduced pressure. The residue was purified by column chromatography eluting with hexane (to recover reduced 2 and oxidized 4) followed by a step-wise gradient from 0 to 10 % EtOAc in hexane. 4-Methoxybenzyl 4-nitrobenzoate was isolated as a yellow solid (90.5 mg, 0.32 mmol, 63 %).
Acknowledgments
We thank the National Science Foundation for primary support of this work (pre-doctoral fellowship to Joe Buonomo, GRFP-ID: 00039202). We thank Christopher D. Brown for acting as a “checker” to confirm reproducibility of reaction results.
Supporting Information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information
References
- 1a.Mitsunobu O, Yamada M, Mukaiyama T. Bull. Chem. Soc. Jpn. 1967;40:935–939. [Google Scholar]
- 1b.Mitsunobu O, Yamada MT. Bull. Chem. Soc. Jpn. 1967;40:2380–2382. [Google Scholar]
- 2a.Mitsunobu O. Synlett. 1981:1–28. For reviews, see. [Google Scholar]
- 2b.Hughes DL. Org. React. 1992;42:335–656. [Google Scholar]
- 2c.Kumara Swamy KC, Bhuvan Kumar NN, Balaraman E, Pavan Kumar KVP. Chem. Rev. 2009;109:2551–2651. doi: 10.1021/cr800278z. [DOI] [PubMed] [Google Scholar]
- 3.Carey JS, Laffan D, Thomson C, Willams MT. Org. Biomol. Chem. 2006;4:2337–2347. doi: 10.1039/b602413k. [DOI] [PubMed] [Google Scholar]
- 4a.But TYS, Toy PH. Chem. Asian J. 2007;2:1340–1355. doi: 10.1002/asia.200700182. For a brief review, see. [DOI] [PubMed] [Google Scholar]
- 4b.Fletcher S. Org. Chem. Front. 2015;2:739–752. for a brief review on catalytic nucleophilic substitution reactions, see. [Google Scholar]
- 4c.An J, Denton RM, Lambert TH, Nacsa ED. Org. Biomol. Chem. 2014;12:2993–3003. doi: 10.1039/c4ob00032c. [DOI] [PubMed] [Google Scholar]
- 5.Constable DJC, Dunn PJ, Hayler JD, Humphrey GR, Leazer JL, Jr, Linderman RJ, Lorenz K, Manley J, Pearlman BA, Wells A, Zaks A, Zhang TY. Green Chem. 2007;9:411–420. [Google Scholar]
- 6.But TYS, Toy PH. J. Am. Chem. Soc. 2006;128:9636–9637. doi: 10.1021/ja063141v. [DOI] [PubMed] [Google Scholar]
- 7.Hirose D, Taniguchi T, Ishibashi H. Angew. Chem. Int. Ed. 2013;52:4613–4617. doi: 10.1002/anie.201300153. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. 2013;125 [Google Scholar]
- 8a.O’Brien CJ. 2014. (Univ. Texas, USA), US Patent 8901365.
- 8b.O’Brien CJ. 2012. (Univ. Texas, USA), Eur. Pat. Appl. EP2417082.
- 9.Davey S. Nat. Chem. 2013;5:358. [Google Scholar]
- 10a.van Kalkeren HA, Leenders SHAM, Hommersom CRA, Rutjes FPJT, van Delft FL. Chem. Eur. J. 2011;17:11290–11295. doi: 10.1002/chem.201101563. [DOI] [PubMed] [Google Scholar]
- 10b.van Kalkeren HA, te Grotenhuis C, Haasjes FS, Hommersom CRA, Rutjes FPJT, van Delft FL. Eur. J. Org. Chem. 2013:7059–7066. [Google Scholar]
- 11a.O’Brien CJ, Tellez JL, Nixon ZS, Kang LJ, Carter AL, Kunkel SR, Przeworski KC, Chass GA. Angew. Chem. Int. Ed. 2009;48:6836–6839. doi: 10.1002/anie.200902525. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. 2009;121 [Google Scholar]
- 11b.O’Brien CJ, Nixon ZS, Holohan AJ, Kunkel SR, Tellez JL, Doonan BJ, Coyle EE, Lavigne F, Kang LJ, Przeworski KC. Chem. Eur. J. 2013;19:15281–15289. doi: 10.1002/chem.201301444. [DOI] [PubMed] [Google Scholar]
- 12a.Adlington MG, Orfanopoulos M, Fry JL. Tetrahedron Lett. 1976;17:2955–2958. [Google Scholar]
- 12b.Gevorgyan V, Rubin M, Benson S, Liu J-X, Yamamoto Y. J. Org. Chem. 2000;65:6179–6186. doi: 10.1021/jo000726d. [DOI] [PubMed] [Google Scholar]
- 13. See the Supporting Information for details.
- 14. Competitive intramolecular cyclization through attack of the purine N-3 nitrogen on the activated 5′-alcohol results in cyclonucleoside formation, a well-known side reaction that can plague Mitsunobu couplings, which we thought could be exacerbated at the elevated reaction temperatures required for the catalytic Mitsunobu reaction.
- 15. p11-Methoxybenzyl alcohol was selected for our model studies because in H NMR spectroscopy the methoxy group at 3.76 undergoes a diagnostic chemical shift to 3.91 in the ester product that allows easy monitoring by H NMR spectroscopy.
- 16.Hecht SS. J. Natl. Cancer Inst. 1999;91:1194–1210. doi: 10.1093/jnci/91.14.1194. [DOI] [PubMed] [Google Scholar]
- 17a.Zhao W, Yan PK, Radosevich AT. J. Am. Chem. Soc. 2015;137:616–619. doi: 10.1021/ja511889y. [DOI] [PubMed] [Google Scholar]
- 17b.Reichl KD, Dunn NL, Fastuca NJ, Radosevich AT. J. Am. Chem. Soc. 2015;137:5292–5295. doi: 10.1021/jacs.5b01899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O’Brien CJ, Lavigne F, Coyle EE, Holohan AJ, Doonan BJ. Chem. Eur. J. 2013;19:5854–5858. doi: 10.1002/chem.201300546. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
miscellaneous_information