

Abstract
PTEN is a dual-specificity protein tyrosine phosphatase. As one of the central tumor suppressors, a thorough regulation of its activity is essential for proper cellular homeostasis. The precise implications of PTEN inhibition by reactive oxygen species (e.g. H2O2) and the subsequent structural consequences remain elusive. To study the effects of PTEN inhibition, bisperoxidovanadium (bpV) complexes serve as important tools with the potential for the treatment of nerve injury or cardiac ischemia. However, their mode of action is unknown, hampering further optimization and preventing therapeutic applications. Based on protein crystallography, mass spectrometry, and NMR spectroscopy, we elucidate the molecular basis of PTEN inhibition by H2O2 and bpV complexes. We show that both molecules inhibit PTEN via oxidative mechanisms resulting in the formation of the same intramolecular disulfide, therefore enabling the reactivation of PTEN under reductive conditions.
Keywords: bpV-phen, disulfides, inhibitors, protein tyrosine phosphatase, tumor suppressors
PTEN (phosphatase and tensin homologue deleted on chromosome 10) is a member of the protein tyrosine phosphatase (PTP) superfamily and one of the most important tumor suppressors.[1] In addition, it constitutes a critical factor in regenerative processes.[2] These implications are mainly associated with the inhibitory effect on AKT signaling which is conveyed by its lipid phosphatase activity.[1] PTEN function is tightly regulated by posttranslational modifications such as phosphorylation, ubiquitination, and oxidation.[1, 3] PTEN and a number of other PTPs harbor an active site with a characteristic phosphate-binding loop (P-loop: [I/V]HCXXGXXR[S/T]) involving a deprotonated catalytic cysteine,[4, 5] which is particularly susceptible to oxidation.[6] In a cellular context, reactive oxygen species (e.g. H2O2)[7] can trigger the oxidation of thiols to sulfenic acid, which can either be irreversibly oxidized to sulfinic and sulfonic acid[6] or react with properly aligned nucleophiles (e.g. thiols and activated amides) to form reversible modifications such as disulfides and sulfenyl-amides (Figure 1 a).[6] Importantly, these reversible modifications constitute a protective mechanism preventing irreversible impairment of phosphatase activity.[6] In the case of PTEN, H2O2 inhibits phosphatase activity by triggering the formation of a disulfide bond between catalytic cysteine C124 and closely aligned cysteine C71.[8–10] So far, the structural consequences of this oxidation remain elusive.
Figure 1.

a) H2O2 triggers oxidation of thiols to sulfenic acid, which can react with nucleophiles to form reversible modifications, or it reacts irreversibly to yield sulfinic as well as sulfonic acid (PTP: protein tyrosine phosphatase). b) Chemical structure of bpV-phen (potassium oxidobis(η2-peroxido)phenanthrolinevanadate).
PTEN inhibition and subsequent activation of prosurvival signaling stimulates cellular regeneration thereby promoting axonal regrowth and neural survival.[11] Therefore, PTEN inhibition is considered a therapeutic approach in response to nerve injury and cardiac ischemia. Bisperoxidovanadium (bpV, also: bisperoxovanadium) complexes inhibit PTEN activity and serve as model compounds to study these implications.[12–16] The precise mode of action of bpV inhibitors is under debate,[13, 16] and its elucidation can support further optimization campaigns. To obtain structural insight into PTEN inhibition by H2O2 and bpV complexes, we applied protein crystallography, mass spectrometry (MS), and 51V NMR spectroscopy. Strikingly, both molecules inhibit PTEN by oxidative mechanisms that result in the formation of the same disulfide-bridged PTEN species. In both cases, disulfide formation is reversible under reductive conditions.
Initially, phosphatase activity of full-length PTEN was investigated using the malachite green assay with phosphatidylinositol trisphosphate (PI(3,4,5)P3) as substrate. Half-maximal inhibitory concentrations (IC50) of H2O2 and bpV-phen (Figure 1 b) were determined providing values in agreement with previous reports (H2O2: (60±23) μm, bpV-phen: (0.18±0.02) μm, Figure S2 in the Supporting Information).[8, 12] To verify the reversibility of PTEN inhibition under reductive conditions, PTEN was pretreated with H2O2 (3.5 mm) and then diluted with buffer either lacking reducing agent (mock), or containing dithiothreitol (DTT, 4 mm) and glutathione (GSH, 4 mm), respectively (Figure 2 a). In the absence of reducing agent, efficient PTEN inhibition is observed (residual phosphatase activity: (8±4) %) while addition of DTT or GSH resulted in reactivation (phosphatase activity: (99±3) % and (73±4) %, respectively). Given the presence of peroxido ligands in bpV-phen and the so far unknown inhibitory mechanism, we tested whether PTEN inhibition by bpV-phen can also be reversed under reductive conditions. Again, PTEN inhibition (residual phosphatase activity: (4±4) %) was abolished after DTT or GSH treatment (phosphatase activity: (95±7) % or (69±5) %) indicating oxidative inhibition by bpV-phen (Figure 2 a).
Figure 2.
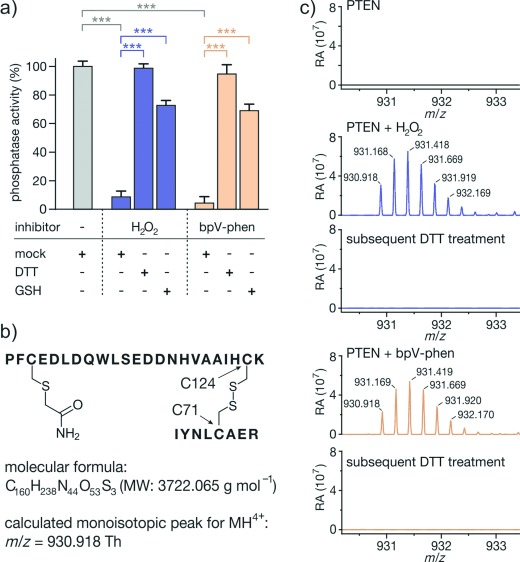
a) Inhibition of PTEN (100 μm) by H2O2 (3.5 mm, blue) or bpV-phen (400 μm, orange) is reversed by thiols (T=25 °C, 4 mm DTT or GSH, triplicates, errors account for 1σ; *** P<0.001). b) Tryptic fragment with disulfide between C71 and C124 (Th: Thomson=Da/e). c) High-resolution MS spectra of tryptic fragment (Figure 2 b) after H2O2 (1 mm, blue) or bpV-phen (1 mm, orange) treatment (T=25 °C, 100 μm PTEN, t=10 min; upper panels). The fragment is absent in the untreated control (black) and after incubation with 10 mm DTT (lower panels, for details see Figure S6).
H2O2 triggers disulfide formation between PTEN's active site cysteines C124 and C71.[8] To investigate whether bpV-phen treatment results in the same disulfide, we performed HPLC-coupled high-resolution MS. After incubation of PTEN with H2O2 or bpV-phen, samples were treated with iodoacetamide to protect free cysteine thiols. This was followed by tryptic digestion in the absence of reducing agent to preserve existing disulfide bridges. Resulting peptides were analyzed by MS. H2O2 and bpV-phen treatment resulted in the formation of a tryptic fragment with a monoisotopic mass of 930.918 Th (MH4+) which was absent in the untreated sample. MS/MS sequencing of this peak (Figure S7) identified a fragment consisting of two peptides linked by a disulfide between C124 and C71 (Figure 2 b). Subsequent treatment of the tryptic mixture with DTT led to a complete loss of the corresponding isotope pattern (Figure 2 c), verifying disulfide formation. This confirms oxidative inhibition of PTEN by bpV-phen.
To elucidate the structural consequences of disulfide formation, we applied protein crystallography. The so far only available crystal structure of PTEN shows the active site in its reduced state (PDB: 1D5R) and was generated using a truncated version of PTEN that involves its central phosphatase and C2 domain (tPTEN, 7–353 aa with a sequence deletion from 286 to 309).[17] To ensure applicability of tPTEN for our studies, we confirmed that treatment with H2O2 as well as bpV-phen results in the same disulfide-bridged tryptic fragment observed for full-length PTEN (Figures S8 and S9). We identified crystallization conditions similar to that previously reported,[17] which provided crystals diffracting with a resolution of up to 2.2 Å. tPTEN crystallizes in a space group distinct from the original structure (C2221 vs. I4) with a significantly larger unit cell harboring four PTEN molecules per asymmetric unit (Figure S10). The four protomers align closely with each other (maximal RMSD=1.46 Å for 314 Cα atoms) and with the previous PTEN structure (maximal RMSD=1.29 Å, Figures S11 and S12). As reported before, the active site is bound to one molecule of tartrate originating from the crystallization buffer (Figure S13).
Having obtained suitable tPTEN crystals, we proceeded with soaking experiments to study structural changes upon H2O2 treatment. Crystals were incubated with 1 mm H2O2 for 1 h providing a crystal structure with 2.4 Å resolution that appears to be very similar to the reduced form of tPTEN (maximal RMSD=1.57 Å for 314 Cα atoms, Figure 3 a and S14). While the electron density between catalytic cysteine C124 and adjacent C71 is clearly separated before H2O2 treatment (Figure 3 b), it is evidently connected after exposure to H2O2 (PTEN(ox), Figure 3 c). The oxidized active site still harbors one molecule of tartrate.[18] Disulfide formation promotes a rotation of catalytic cysteine C124 toward C71 without changing the position of their Cα atoms significantly (Cα–Cα distance reduced: 5.2 Å vs. oxidized: 5.0 Å). Also, the entire active site including the P-loop (IHCKAGKGRT) displayed only minimal conformational changes (RMSD=0.37 Å for 10 Cα atoms, Figure 3 d). This is in analogy to human lymphoid tyrosine phosphatase (LYP) which also exhibits small conformational changes in the P-loop upon disulfide formation (RMSD=0.39 Å, Figure S15).[19] The reversibly oxidized human PTPs, CDC25B (disulfide, RMSD=2.48 Å),[20] and PTP1B (sulfenyl-amide, RMSD=2.38 Å)[21] exhibit substantial structural changes upon oxidation (Figure S15). These PTPs also experience irreversible oxidation of the active site cysteine (sulfinic or sulfonic acid) upon long incubation times with H2O2 (t>20 min).[20, 21] To investigate the reversibility of PTEN oxidation, we performed time-dependent activity measurements with increased H2O2 concentrations (Figure S4). Notably, reactivation upon DTT treatment occurred up to 20 mm H2O2 and 80 min incubation time, verifying the absence of irreversible modifications and indicating very efficient protection of PTEN from constitutive inactivation by oxidative agents.
Figure 3.
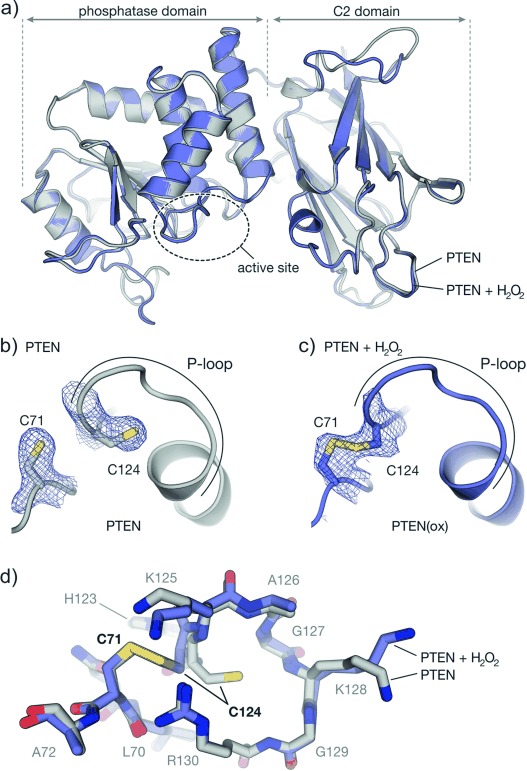
a) Superimposed structures of PTEN before (gray, PDB: 5BZZ, chain A) and after oxidation with H2O2 (blue, PDB: 5BUG, chain A). b, c) Crystal structure of the active site in reduced and H2O2-treated PTEN, respectively (both chain A). Side chains of C71 and C124 are shown explicably (mesh: 2 Fo−Fc electron density omit map around C71 and C124 contoured at 1σ). d) Superimposed crystal structures of active site residues of reduced (gray) and H2O2-treated (blue) PTEN.
Next, we employed 51V NMR spectroscopy to investigate the reaction of bpV-phen with PTEN. bpV-Phen provided the expected chemical shift (δ(51V)=−744 ppm)[22] and was titrated (50–200 μm) to full-length PTEN (100 μm). At low inhibitor concentration (50 μm), the bpV-phen signal rapidly diminishes (complete conversion: <15 min). This coincides with the occurrence of the product peak (δ(51V)=−553 ppm) which was assigned to dihydrogen orthovanadate (H2VO4−, Figure 4 a). At increased inhibitor concentrations (100 and 200 μm), we observed a residual bpV-phen signal indicating quantitative oxidation of PTEN. To explore the structural impact of PTEN oxidation by bpV-phen, tPTEN crystals were soaked with 1 mm bpV-phen for 4 h. Since we were interested in the precise localization of potential vanadium species, crystallographic data collection was performed at a longer wavelength (λ=1.77 Å) allowing the detection of X-ray diffraction as well as anomalous scattering originating only from certain atoms such as vanadium and sulfur. We obtained a crystal structure with 2.5 Å resolution that appears to be very similar to the structure after H2O2 treatment (maximal RMSD=1.37 Å for 314 Cα atoms, Figure S16). The 2 Fo−Fc omit map verifies disulfide formation with connected electron densities between C124 and C71 in all four protomers (Figure S17). Based on the anomalous scattering signal, an additional electron density map was generated applying the Bijvoet difference Fourier method.[23] Under our conditions, the resulting density is indicative for vanadium and sulfur atoms. This map verifies disulfide formation in all protomers and it shows additional anomalous density in the active site of protomer A. This indicates the presence of a vanadium species (Figure 4 b) which together with the 2 Fo−Fc electron density confirms the presence of orthovanadate (Figure S18). Interestingly in chains B and D, the active site harbors one molecule of tartrate analogous to the H2O2-treated crystals, which is also reflected by a lack of the corresponding anomalous signal. Chain C exhibits an intermediate state with partial population by orthovanadate and tartrate (about 30:70). Despite the varying ligand occupancy, all four active sites align closely (Figure S16). An overlay of reduced PTEN (gray) with the H2O2 (blue) as well as bpV-phen (orange) oxidized forms reveals minimal structural variations in the active site indicating a low dependency of P-loop conformation on the oxidation state and the nature of the bound ligand (Figures 4 e and S17).
Figure 4.
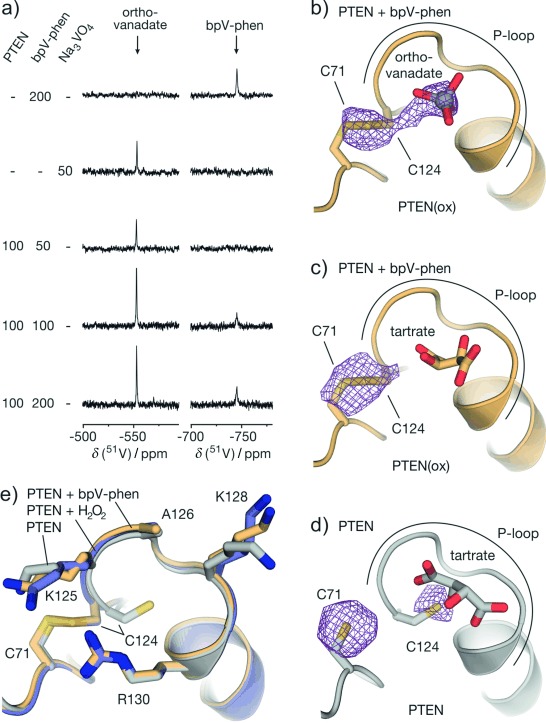
a) 51V NMR titration measurements of bpV-phen (50–200 μm) in the presence of full-length PTEN (100 μm). For complete spectra see Figure S19. b–d) Crystal structures of the active site in bpV-phen-treated (orange, PDB: 5BZX, b: chain A, c: chain: B) and reduced PTEN (gray, PDB: 5BZZ, d: chain A). Orthovanadate and side chains of C71 and C124 are shown explicably. Anomalous scattering density map (mesh, contoured at 3.4σ) was generated using the Bijvoet difference Fourier method at a resolution of 5.0 Å (vanadium and sulfur contribute to electron density). e) Superimposed structures of active site (all chain A) in reduced (gray), H2O2- (blue), and bpV-phen-treated (orange) PTEN.
Orthovanadate binds to the active sites of numerous enzymes including phosphatases and kinases.[24] To estimate the affinity of orthovanadate for PTEN(ox), we performed 51V NMR experiments with preoxidized PTEN. In these measurements, the orthovanadate peak neither exhibits line broadening nor chemical shift changes, suggesting a very low affinity (Kd>1 mm, Figure S20). Taken together with the low PTEN affinity of tartrate, this explains the presence of orthovanadate, tartrate, and mixed states in the active site during X-ray crystallography. Notably, orthovanadate inhibits certain human phosphatases at sub-micromolar concentrations (e.g. PTP1B (Ki=0.38 μm),[25] and alkaline phosphatase (Ki=0.6 μm)[26]). For this reason, we were interested whether it also contributes to PTEN inhibition by bpV-phen. In activity assays using full-length PTEN and Na3VO4, we did not detect phosphatase inhibition up to 0.1 mm orthovanadate (Figure S5) and even at 1 mm Na3VO4 only 20 % inhibition was observed. This indicates that inhibition of PTEN by bpV-phen is primarily induced by disulfide formation.
Herein, we show that H2O2 and bpV-phen inhibit PTEN via reversible disulfide formation between cysteines C124 and C71. In both cases, oxidation of PTEN results in only small structural changes implying minimal impact on phosphatase-independent functions. Reversible oxidation of active site cysteines is a crucial feature for the regulation of many phosphatases, including LYP,[19] PTP1B,[27] VHR,[27] CDC25,[28] SHP-1/2,[29, 30] LMW-PTP,[31] and PTEN.[32] For reversible oxidation involving disulfide formation, two distinct cysteine arrangements are discussed based on the relative distance of both cysteines within the primary sequence:[19, 32] A) Cysteines are separated by only a few amino acids (e.g. i,i+5 for LMW-PTP) and B) disulfide formation occurs between two residues that are separated in terms of sequence but located in close proximity. With respect to the second case, CDC25B, LYP, and now PTEN have been structurally characterized.[19, 20] Compared with PTEN, CDC25B exhibits severe structural changes upon reversible oxidation with H2O2, whereas disulfide formation in LYP also results in only minor structural changes. It remains to be seen whether these differences in the conformation of the oxidized state convey certain biological properties. For PTEN, it is important to note that oxidative inhibition of phosphatase activity appears to be highly reversible even after stringent H2O2 treatment. This distinguishes PTEN from other studied phosphatases and could indicate more efficient protection from irreversible inactivation by reactive oxygen species.
The inhibition of PTEN by bpV-phen and H2O2 results in the same oxidized protein species. However, the underlying mechanisms differ since thiol oxidation by bpV complexes involves an electron-transfer step rather than intermediate formation of sulfenic acid.[33] The fact that inhibition of PTEN by bpV-phen yields orthovanadate is an important finding raising the question whether orthovanadate contributes to the neuro- and cardioprotective effects of bpV complexes. In addition, it remains to be seen whether the oxidative mode of action observed for PTEN also applies for other phosphatases thereby providing an explanation for the inhibitory effect of bpV complexes on other PTPs.[13, 16] Finally, it is important to note that the reversible inhibition of PTEN by bpV-phen may represent a highly desirable feature for therapeutic applications since irreversible inhibition of this central tumor suppressor holds the potential to induce systemic disturbance.
Acknowledgments
We thank Dr. Stefan Heinrichs for providing the PTEN gene construct and Dr. Ingrid Vetter for support in the structure determination. We thank Georg Holtermann and Franziska Müller for technical support in protein crystallography and mass spectrometry, respectively. We thank Prof. Dr. Dieter Lenz for initial support with 51V NMR spectroscopy, and Inke Siewert for useful discussions. We appreciate the beamline staff at Swiss Light Source PXII-X10SA. J.H. thanks the Fonds der Chemischen Industrie for a PhD stipend. This work was supported by the German Research Foundation (DFG, Emmy Noether program RA1944/2-1 and GR3592/2-1), AstraZeneca, Bayer CropScience, Bayer HealthCare, Boehringer Ingelheim, Merck KGaA, and the Max Planck Society.
Supporting Information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information
References
- 1.Song MS, Salmena L, Pandolfi PP. Nat. Rev. Cancer. 2012;13:283–296. doi: 10.1038/nrm3330. [DOI] [PubMed] [Google Scholar]
- 2.Sun F, Park KK, Belin S, Wang D, Lu T, Chen G, Zhang K, Yeung C, Feng G, Yankner BA, et al. Nature. 2011;480:372–375. doi: 10.1038/nature10594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Worby CA, Dixon JE. Annu. Rev. Biochem. 2014;83:641–669. doi: 10.1146/annurev-biochem-082411-113907. [DOI] [PubMed] [Google Scholar]
- 4.Zhang ZY, Dixon JE. Biochemistry. 1993;32:9340–9345. doi: 10.1021/bi00087a012. [DOI] [PubMed] [Google Scholar]
- 5.Peters GH, Frimurer TM, Olsen OH. Biochemistry. 1998;37:5383–5393. doi: 10.1021/bi971187i. [DOI] [PubMed] [Google Scholar]
- 6.Tonks NK. Nat. Rev. Mol. Cell Biol. 2006;7:833–846. doi: 10.1038/nrm2039. [DOI] [PubMed] [Google Scholar]
- 7.Winterbourn CC. Nat. Chem. Biol. 2008;4:278–286. doi: 10.1038/nchembio.85. [DOI] [PubMed] [Google Scholar]
- 8.Lee SR, Yang KS, Kwon J, Lee C, Jeong W, Rhee SG. J. Biol. Chem. 2002;277:20336–20342. doi: 10.1074/jbc.M111899200. [DOI] [PubMed] [Google Scholar]
- 9.Leslie NR, Bennett D, Lindsay YE, Stewart H, Gray A, Downes CP. EMBO J. 2003;22:5501–5510. doi: 10.1093/emboj/cdg513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schwertassek U, Haque A, Krishnan N, Greiner R, Weingarten L, Dick TP, Tonks NK. FEBS J. 2014;281:3545–3558. doi: 10.1111/febs.12898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brunet A, Datta SR, Greenberg ME. Curr. Opin. Neurobiol. 2001;11:297–305. doi: 10.1016/s0959-4388(00)00211-7. [DOI] [PubMed] [Google Scholar]
- 12.Schmid AC, Byrne RD, Vilar R, Woscholski R. FEBS Lett. 2004;566:35–38. doi: 10.1016/j.febslet.2004.03.102. [DOI] [PubMed] [Google Scholar]
- 13.Rosivatz E, Matthews JG, McDonald NQ, Mulet X, Ho KK, Lossi N, Schmid AC, Mirabelli M, Pomeranz KM, Erneux C, et al. ACS Chem. Biol. 2006;1:780–790. doi: 10.1021/cb600352f. [DOI] [PubMed] [Google Scholar]
- 14.Christie KJ, Webber CA, Martinez JA, Singh B, Zochodne DW. J. Neurosci. 2010;30:9306–9315. doi: 10.1523/JNEUROSCI.6271-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Walker CL, Walker MJ, Liu N-K, Risberg EC, Gao X, Chen J, Xu X-M. PLoS ONE. 2012;7:e30012. doi: 10.1371/journal.pone.0030012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Spinelli L, Lindsay YE, Leslie NR. Adv. Biol. Regul. 2015;57:102–111. doi: 10.1016/j.jbior.2014.09.012. [DOI] [PubMed] [Google Scholar]
- 17.Lee JO, Yang H, Georgescu MM, Di Cristofano A, Maehama T, Shi Y, Dixon JE, Pandolfi P, Pavletich NP. Cell. 1999;99:323–334. doi: 10.1016/s0092-8674(00)81663-3. [DOI] [PubMed] [Google Scholar]
- 18. Given the fact that the active site harbors one molecule of tartrate (Figure S13), we verified that tartrate does not affect PTEN oxidation and reactivation under our assay conditions (for details see Figure S3)
- 19.Tsai SJ, Sen U, Zhao L, Greenleaf WB, Dasgupta J, Fiorillo E, Orrú V, Bottini N, Chen XS. Biochemistry. 2009;48:4838–4845. doi: 10.1021/bi900166y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Buhrman G, Parker B, Sohn J, Rudolph J, Mattos C. Biochemistry. 2005;44:5307–5316. doi: 10.1021/bi047449f. [DOI] [PubMed] [Google Scholar]
- 21.Salmeen A, Andersen JN, Myers MP, Meng T-C, Hinks JA, Tonks NK, Barford D. Nature. 2003;423:769–773. doi: 10.1038/nature01680. [DOI] [PubMed] [Google Scholar]
- 22.Cerovac Z, Ban J, Morinville A, Yaccato K, Shaver A, Maysinger D. Neurochem. Int. 1999;34:337–344. doi: 10.1016/s0197-0186(99)00018-2. [DOI] [PubMed] [Google Scholar]
- 23.Strahs G, Kraut J. Annu. Rev. Biochem. 1968;35:503–512. [Google Scholar]
- 24.Pessoa JCosta, Garribba E, Santos MFA, Santos-Silva T. Coord. Chem. Rev. 2015;301–302:49–86. [Google Scholar]
- 25.Huyer G, Liu S, Kelly J, Moffat J, Payette P, Kennedy B, Tsaprailis G, Gresser MJ, Ramachandran C. J. Biol. Chem. 1997;272:843–851. doi: 10.1074/jbc.272.2.843. [DOI] [PubMed] [Google Scholar]
- 26.Seargeant LE, Stinson RA. Biochem. J. 1979;181:247–250. doi: 10.1042/bj1810247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Denu JM, Tanner KG. Biochemistry. 1998;37:5633–5642. doi: 10.1021/bi973035t. [DOI] [PubMed] [Google Scholar]
- 28.Sohn J, Rudolph J. Biochemistry. 2003;42:10060–10070. doi: 10.1021/bi0345081. [DOI] [PubMed] [Google Scholar]
- 29.Cunnick JM, Dorsey JF, Mei L, Wu J. Biochem. Mol. Biol. Int. 1998;45:887–894. doi: 10.1002/iub.7510450506. [DOI] [PubMed] [Google Scholar]
- 30.Meng T-C, Fukada T, Tonks NK. Mol. Cell. 2002;9:387–399. doi: 10.1016/s1097-2765(02)00445-8. [DOI] [PubMed] [Google Scholar]
- 31.Caselli A, Marzocchini R, Camici G, Manao G, Moneti G, Pieraccini G, Ramponi G. J. Biol. Chem. 1998;273:32554–32560. doi: 10.1074/jbc.273.49.32554. [DOI] [PubMed] [Google Scholar]
- 32.Cho SH, Lee CH, Ahn Y, Kim H, Kim H, Ahn CY, Yang KS, Lee SR. FEBS Lett. 2004;560:7–13. doi: 10.1016/s0014-5793(04)00112-7. [DOI] [PubMed] [Google Scholar]
- 33.Ballistreri FP, Barbuzzi EG, Tomaselli GA, Toscano RM. J. Inorg. Biochem. 2000;80:173–176. doi: 10.1016/s0162-0134(00)00027-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
miscellaneous_information