

Abstract
Mutations in isocitrate dehydrogenase 1 (IDH1) have been found in the vast majority of low grade and progressive infiltrating gliomas and are characterized by the production of 2-hydroxyglutarate from α-ketoglutarate. Recent investigations of malignant gliomas have identified additional genetic and chromosomal abnormalities which cluster with IDH1 mutations into two distinct subgroups. The astrocytic subgroup was found to have frequent mutations in ATRX, TP53 and displays alternative lengthening of telomeres. The second subgroup with oligodendrocytic morphology has frequent mutations in CIC or FUBP1, and is linked to co-deletion of the 1p/19q arms. These mutations reflect the development of two distinct molecular pathways representing the majority of IDH1 mutant gliomas. Unfortunately, due to the scarcity of endogenously derived IDH1 mutant models, there is a lack of accurate models to study mechanism and develop new therapy. Here we report the generation of an endogenous IDH1 anaplastic astrocytoma in vivo model with concurrent mutations in TP53, CDKN2A and ATRX. The model has a similar phenotype and histopathology as the original patient tumor, expresses the IDH1 (R132H) mutant protein and exhibits an alternative lengthening of telomeres phenotype. The JHH-273 model is characteristic of anaplastic astrocytoma and represents a valuable tool for investigating the pathogenesis of this distinct molecular subset of gliomas and for preclinical testing of compounds targeting IDH1 mutations or alternative lengthening of telomeres.
Keywords: IDH1, ATRX, alternative lengthening of telomeres, ALT, glioma model, xenograft, astrocytoma
Introduction
Glioblastoma (GBM), a grade IV astrocytoma, is a highly aggressive tumor which can occur either de novo (primary GBM), or can progress from a WHO grade II or III glioma (secondary or progressive GBM). Since its identification as an oncogene in 2008, mutations in isocitrate dehydrogenase 1 have been found in the majority of grade II–III gliomas and secondary glioblastoma. Driver mutations in IDH1 are restricted to a single residue, R132, which normally encodes an arginine residue located in the substrate binding pocket. Mutations in this residue impart a novel enzymatic reaction: the conversion of α-ketoglutarate (α-KG) to D-2-hydroxyglutarate (2-HG). Though normally present at very low levels in the cell, intracellular 2-HG concentrations can be increased up to 10–30 mM in IDH1 mutant tumors [1–3]. Owing to the close structural similarity between the metabolites, 2-HG is believed to promote tumorigenesis by competitively inhibiting α-KG dependent dioxygenases including the Jumonji C-domain containing histone demethylases and the TET family of DNA methylcytosine dioxygenases, believed to function in DNA demethylation [4]. Ultimately, continued exposure to 2-HG results in widespread cellular changes, including characteristic hypermethylation of genomic DNA, suppression of cellular differentiation and metabolic deficits [5–8].
Recent sequencing efforts in grade II–III gliomas have identified additional genetic and chromosomal abnormalities, many of which cluster with IDH1 mutations in two distinct subgroups. One subgroup of IDH1 mutant tumors was found to have frequent mutations in ATRX, TP53 and displayed alternative lengthening of telomeres (ALT). The second subgroup of IDH1 mutant tumors was found to have frequent mutations in CIC or FUBP1 and was linked to co-deletion of the 1p/19q arms where these genes reside. Low grade gliomas without IDH1 mutations were classified as a separate molecular subgroup [9–12]. Remarkably, these genetic signatures corresponded tightly with clinical outcome to a much greater degree than histopathological stratification and suggest that in addition to the IDH1 mutation there are two separate molecular pathways that can be used to contribute to transformation [9]. The IDH1/TP53/ATRX and IDH1/CIC/FUBP1 mutated tumors are distinct molecular classes of glioma that are useful to consider separately, both for prognosis and molecular targeting.
Although our understanding of IDH1 mutated gliomas grows, the development of relevant models remains a challenge. Patient derived IDH1 mutant tumors have been difficult to culture and published xenografts are restricted to oligodendroglioma and oligoastrocytoma backgrounds [13–15]. The development and molecular characterization of additional endogenous IDH1 mutant astrocytoma models is important for preclinical testing of molecular based therapies which target progressive gliomas.
Here we report the generation of an endogenous patient derived IDH1 anaplastic astrocytoma in vivo model with driver mutations in TP53, CDKN2A and ATRX. Although this IDH1 mutant line does not proliferate in vitro, the model faithfully resembles the patient tumor and robustly expresses the IDH1 (R132H) mutant protein when grown in both the flank and orthotopically. Additionally, the model exhibits a phenotype characteristic of an anaplastic astrocytoma characteristic including robust production of 2-HG, genome hypermethylation and alternative lengthening of telomeres (ALT).
Material and Methods
Xenograft establishment
Flank and orthotopic xenografts were established as previously described [16]. Briefly, tissue was obtained during the resection of an anaplastic astrocytoma (WHO grade III) from an adult male patient. The tissue was mechanically disassociated, mixed with an equal volume of growth factor–reduced Matrigel (BD Biosciences, CA), and injected subcutaneously into the flanks of athymic nude mice (0.2cc/flank). All animal protocols and procedures were performed in accordance with the Johns Hopkins Animal Care and Use Committee guidelines. Animals were monitored frequently for signs of tumor growth. Xenografts were passaged in a similar fashion. Cross-sectional samples were obtained at each passage and either snap frozen or fixed in formalin. The samples were then embedded in paraffin and stained by H&E or used for immunohistochemistry. The IDH1 (R132H) mutation was validated by direct sequencing at every passage.
For orthotopic xenografts, flank xenografts were resected and enzymatically disassociated using a 2:1 ratio of collagenase (10mg/mL, Invitrogen, NY) and hyaluronidase (1000 units/mL, Sigma, MO). Cell number and viability was assessed using Trypan blue exclusion. For intracranial implantations, 500,000 cells were stereotactically implanted into the right frontal cortex of 4–6 week old female athymic nude mice (NCI-Frederick) as previously described [17]. Mice were sacrificed upon showing symptoms of distress and the brains removed and formalin fixed for subsequent gross pathological examination of tumor formation and immunohistochemistry.
Sequencing of IDH1
Genomic DNA was isolated from patient tissue and flank xenografts using the DNeasy Blood and Tissue Kit (Qiagen, CA) according to manufacturer’s instructions. PCR and sequencing was conducted as previously described [18]. Briefly, 60 ng of genomic DNA was added to a standard PCR reaction to amplify a portion of exon 4 of IDH1 (forward 5′-GTAAAACGACGGCCAGTTGAGCTCTATATGCCATCACTGC 3′, reverse 5′-CAATTTCATACCTTGCTTAATGGG-3′). The PCR product was purified using the QIAquick Gel Extraction Kit (Qiagen, CA) and submitted for sequencing (Genewiz, NJ) using targeted primers (forward 5′-CGGTCTTCAGAGAAGCCATT-3′, and reverse 5′-GCAAAATCACATTATTGCCAAC-3′).
Histology and Immunohistochemistry
All histopathological and immunohistochemical analyses were performed using tissue fixed in 10% formalin and embedded in paraffin. Tissue was obtained from patient samples after appropriate approval from the Johns Hopkins University Institutional Review Board. Paraffin-embedded sections were cut at 5 microns, deparaffinized, and stained with either hematoxylin and eosin (H&E) or immunohistochemical stains as specified. Heat-induced epiotope retrieval was performed for 36 minutes at 98°C in EDTA buffer (pH 9.0). Immunohistochemical staining was performed using antibodies specific for IDH1 (R132H) (dilution 1:50, Dianova, clone H09) and visualized using the ultraView DAB detection system (Ventana Medical Systems).
Whole Exome Sequencing
Genomic DNA was isolated from normal human whole blood and flank xenograft tissue as described above. gDNA fragmentation was performed with the Bioruptor (Diagenode, NJ), and size selection at 200 bp – 300 bp was carried out. The exomes of gDNA were captured using the SureSelect All Exon 50Mb Target Enrichment kit (Agilent, CA) according to the manufacturer’s instructions. DNA captured was run on the HiSeq2000 platform (Illumina, CA) according to the manufacturer’s instructions, to generate 100-base paired-end reads.
Reads in fastq format were initially processed with GATK to remove Illumina adaptor sequences and Phred-scaled base qualities of ≤ 10 (-QT 10). After GATK trimming step, reads were mapped using the Burrows-Wheeler Aligner (version 0.6.1) with a –q 20 setting for read trimming, which removes the 3′ portion of reads from an alignment if it is below the quality threshold specified. The alignments were used to generate Sequence Alignment/Map (SAM) paired-end read files. All SAM files were converted to BAM files then sorted BAM files with Samtools (version 0.1.18). PCR and optical duplicates and multiple reads likely to have been read from a single cluster on the flow-cell image were marked with Picard tools. Regions that needed to be realigned were identified using the GATK Realigner Target Creator. The reads covering localized indels were realigned, and quality values were recalibrated using GATK. The GATK was also used to locate, filter and annotate variants. gDNA derived from the whole blood of two unrelated individuals as well as dbSNP135 to remove common SNPs. All predicted deleterious mutations not existing in dbSNP were counted.
Mean exonic coverage was calculated for all exonic baits in xenograft and control samples by GATK. The mean exonic coverage was subsequently normalized by average whole exome coverage of the sample. Individual case vs. control log2 ratios were then calculated for all the exons in the data set and plotted. The presence of copy-number alterations was detected using a combined approach involving a set of statistical Wilcoxon signed-rank tests performed on a 500,000 bp sliding windows along the genome. Amplifications were defined as greater than 4 copies of the gene (case vs. control log2 ratio greater than 8).
Telomere-specific FISH and microscopy
Telomere-specific FISH was conducted as previously described [19–20] Briefly, deparaffinized slides were hydrated, steamed for 20 minutes in citrate buffer (Vector Laboratories, GA), dehydrated, and hybridized with a Cy3-labeled peptide nucleic acid (PNA) probe complementary to the mammalian telomere repeat sequence ([N-terminus to C-terminus] CCCTAACCCTAACCCTAA). As a positive control for hybridization efficiency, a FITC-labeled PNA probe having specificity for human centromeric DNA repeats (ATTCGTTGGAAACGGGA; CENP-B binding sequence) was also included in the hybridization solution [21]. Slides were imaged with a Nikon 50i epifluorescence microscope equipped with X-Cite series 120 illuminator (EXFO Photonics Solutions Inc., Ontario, CA) and appropriate fluorescence excitation/emission filters. Grayscale images were captured for using Nikon NIS-Elements software and an attached PhotometricsCoolsnapEZ digital camera, pseudo-colored and merged. The telomerase-independent alternative lengthening of telomeres (ALT) phenotype was confirmed by the presence of abnormally large and intense intra-nuclear telomere FISH signals; a hallmark of cells utilizing the ALT pathway. Such foci are not observed in normal cells, nor are they observed in ALT-negative cancer cells, and thus serve as specific biomarkers of ALT.
Results and Discussion
The flank xenograft was established from an adult male patient with a recurrent WHO grade III anaplastic astrocytoma bearing a heterozygous IDH1 mutation. The patient originally presented with a WHO grade II glioma and underwent a full resection. The tumor recurred as a WHO grade III two years later and was found to have areas of hypercellularity, and anaplasia but lacked necrosis or vascular proliferation. IDH1 (R132H) staining revealed robust expression of the mutant protein (Fig. 1A). The patient underwent an additional surgical resection upon which time tissue was harvested. Prior to this point the patient had not received any radiation or chemotherapy. The fresh tissue was implanted subcutaneously into the flanks of athymic nude mice and neurosphere culture was attempted. A first generation tumor grew approximately one month after implantation as a large, localized mass. The resulting IDH1 mutant tumor was designated as JHH-273 and maintained by serial passage in athymic nude mice. Sequencing of original patient tumor revealed a heterozygous IDH1 (G395A) mutation. The first generation xenograft maintained the IDH1 mutation but with a loss of the wild type copy (Figure 1B). Attempts to expand freshly disassociated tumor in vitro have been unsuccessful, despite relatively high viability following disassociation.
Fig. 1. IDH1 mutation, mutant protein expression and in vivo growth JHH-273.

(A) H&E staining of resected patient tumor tissue showed areas of hypercellularity and mitotic figures (left) as well as strong IDH1 (R132H) protein expression (right) leading to a diagnosis of IDH1 mutant anaplastic astrocytoma (WHO grade III) (B) Sequencing of exon 4 of IDH1 shows the original heterozygous G395A (R132H) mutation (left) in the original patient tumor, with loss of the wild type copy in the first passage xenograft (right) (C) Orthotopically implanted xenografts have a median survival time of 63 days and are uniformly lethal (n=15).
Since in vitro culture was not successful, orthotopic implantation required enzymatic disassociation of flank tumor, in the absence of serum or growth factors. Mice bearing intracranial JHH-273 tumors begin to lose weight and show neurological deficits approximately 7 weeks following implantation and median survival time for mice bearing intracranial tumors is 63 days following implantation (Fig. 1C). The model is uniformly lethal and the time to death has remained consistent between passages.
Orthotopically implanted tumors exhibit robust IDH1 (R132H) protein expression and a diffuse growth pattern, characteristic of anaplastic astrocytomas (Fig. 2). The tumor cells showed infiltrative growth throughout the cortex and deep grey matter structures. In some xenografts tumor cells infiltrated through the corpus collosum to the contralateral hemisphere, a characteristic finding of infiltrating gliomas (Fig. 2). In addition, some xenografts showed growth within the leptomeninges and ventricles. As in the primary tumors, the growth demonstrated increased cellularity with scattered mitotic figures and scattered tumor giant cells, consistent with a diagnosis of anaplastic astrocytoma (Fig. 2C, inset). While some gemistocytic features were noted within the intracranial xenograft, these were less prominent in the primary tumor.
Fig. 2. Infiltrative growth of orthotopically implanted JHH-273.
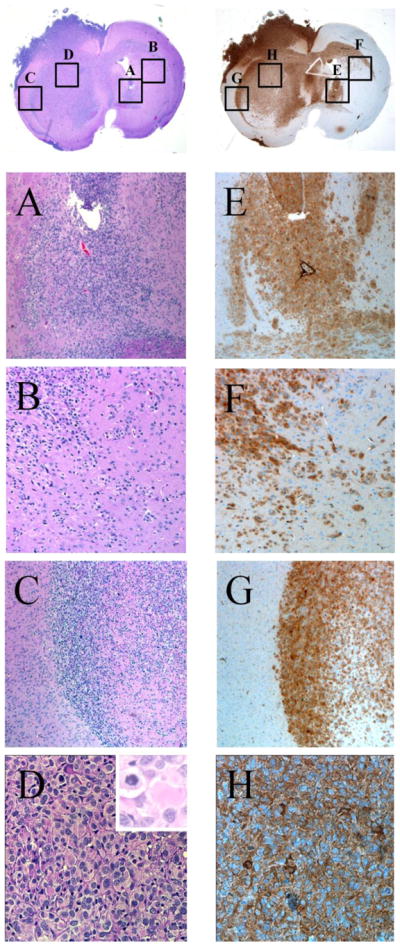
Sections of orthotopically implanted tumor stained with H&E (left) or by immunohistochemistry using an IDH1 (R132H) specific antibody (right) to show an infiltrative growth pattern at the site of implantation (A,E) through the corpus callosum (B,F) and diffusion to the contralateral hemisphere (C, G). Additionally, high magnification images show typical astrocytic morphology with gemistocytic cells and mitotic figures (D, H).
To identify additional molecular abnormalities in our IDH1 mutant model, we performed whole exome capture and next-generation sequencing on gDNA obtained from flank xenograft tissue. We achieved a mean coverage of 124× across the exome, with 94% covered by at least 10×. Copy number variation analysis revealed deletion of genetic content at multiple points throughout the genome, resulting in a complete loss of 39 known genes and including a 1.3 Mb deletion in the p arm of chromosome 9 containing CDKN2A. No significant genetic amplifications were observed. Although matched control DNA was not available for analysis, we utilized gDNA derived from the whole blood of two unrelated individuals as well as dbSNP135 to remove common SNPs. After removal of common SNPs, our analysis revealed 231 candidate somatic mutations in 170 genes. These mutations consisted of missense (83.1%), frameshift (7.4%), splice site (3.0%), nonsense (0.9%), and insertions/deletions (5.6%).
Promising candidate mutations were selected based on a comprehensive list of known driver gene mutations [22]. Of the 125 published Mut-driver genes, five were present in the JHH-273 model. Exome sequencing confirmed the known IDH1 mutation, and additionally revealed missense mutations in TP53 and a frameshift mutation in ATRX (Table 1). The frameshift mutation in ATRX is predicted to produce an early stop codon, resulting in the loss of 81% of the protein. Concurrent mutations in TP53, IDH1, CDKN2A and ATRX have been well reported in multiple anaplastic astrocytoma samples and have been used as genetic signatures in the classification of glioma. Whole exome sequencing demonstrates that the JHH-273 model bears a mutant profile consistent with IDH1 mutant anaplastic astrocytoma.
Table 1.
Genetic mutations in the IDH1 mutant anaplastic astrocytoma model, JHH-273
| Gene Symbol | Mutation | Genetic Mutation | Protein Mutation |
|---|---|---|---|
| ATRX | T1635 | Frameshift | Nonsense |
| CDKN2A | N/A | Deletion | N/A |
| IDH1 | G395A | Missense | R132H |
| TP53 | C736T | Missense | E246L |
| TP53 | C619T | Missense | M207V |
Due to the association of ATRX mutations with the alternative lengthening of telomeres phenotype, telomere length was assessed by telomere specific fluorescent in situ hybridization (FISH) in primary tumor, flank and orthotopic xenografts. An ALT positive phenotype was identified by the presence of large ultra-bright telomeric FISH foci in a subset of tumor cells [23]. The primary patient tumor exhibited ALT-associated foci (Fig. 3A), which was robustly maintained in the flank and orthotopic xenografts (Fig. 3B and C, respectively). Interestingly, although the original patient sample showed ALT-associated foci only in a subset of the tumor cells, the flank and orthotopic xenograft had a more homogenous pattern, with nearly every cell being positive for ALT-associated foci. In contrast, the ALT phenotype was not present in an IDH1 wild type glioma patient sample (Fig. 3D).
Fig. 3. ALT characterization in JHH-273.

(A) Telomere-specific FISH analysis in the original patient tumor, (B) the flank xenograft tumor (C) and the orthotopically implanted tumor show a strongly ALT positive phenotype, as indicated by large, ultrabright telomere FISH signals. D. IDH1 wild type GBM is ALT negative.
Although the original patient tumor displayed heterogeneity with regards to IDH1 (R132H) tissue distribution and ALT staining, the JHH-273 tumor has been remarkably homogenous, with nearly every cell expressing mutant protein and the ALT phenotype. Additionally, the morphology and growth kinetics of the xenograft have remained consistent across passage, suggesting that the JHH-273 model represents a relatively homogenous population of IDH1 mutant anaplastic astrocytoma.
Conclusions
In this work, we characterize JHH-273, an in vivo model of a patient derived IDH1 mutant anaplastic astrocytoma which was originally reported in a recent preclinical drug study [16]. Prior work from our laboratory showed that the JHH-273 model maintains the IDH1 mutation across passage, produces 2-HG in vivo, and bears a hypermethylated phenotype characteristic of IDH1 mutant gliomas [16]. Further genetic sequencing revealed that the JHH-273 model bears mutations in TP53, CDKN2A and ATRX, and exhibits the ALT telomere maintenance phenotype. Additionally we report in this study that JHH-273 maintains infiltrative intracranial growth with a reasonable time to death, making it the only practically usable animal model for IDH1 mutant anaplastic astrocytoma. This model represents the only documented IDH1 mutant anaplastic astrocytoma model with ALT and only the third documented glioma model with ALT, making this model useful for preclinical studies targeting the ALT pathway [24, 25]. Collectively this data suggests that the patient derived JHH-273 model is characteristic of an IDH1 mutant anaplastic astrocytoma and represents a valuable tool for preclinical testing of compounds targeting IDH1 mutations or ALT and for investigating this distinct molecular subset of gliomas.
The JHH-273 xenograft was established after many attempts, underscoring the obstacles that many other groups have faced in developing glioma models bearing endogenous IDH1 mutations. Attempts to expand freshly disassociated xenograft in vitro have been unsuccessful, despite relatively high viability following disassociation. Efforts to expand the population included culture in several types of adherent and neurosphere medium, as well as serum-free medium containing FGF, EGF and PDGF. Since growth was observed in vivo but not in vitro, it was assumed that there may exist a synergy between host environment and tumor cell culture. For this reason, co-culture was attempted using lethally irradiated mouse embryonic feeder cell in serum-free medium however this was also unsuccessful.
It has been well documented that the generation and maintenance of endogenous IDH1 mutant tumors has proven challenging [26]. We speculate that the levels of 2-HG produced by heterozygous mutants inhibits many cellular processes necessary for propagation in a laboratory setting. In some cases, disruption of the wild type allele appears to decreases intracellular 2-HG to levels which are amenable to laboratory domestication such as in our JHH-273 model and the anaplastic oligoastrocytoma model BT142 mut/− line (ATCC ACS-1018).
This model was developed in order to enhance translational studies with more accurate in vivo models. In addition to JHH-273, there also exist several IDH1 mutant xenograft models including a panel of patient-derived IDH1 mutant xenografts, two of which are able stable in culture [13, 14, 27, 28] and the BT142 mut/− line (ATCC ACS-1018) which retains the ability to grow both in vitro and in vivo and is currently available from the ATCC. In addition to the tumor type from which the model was derived, the BT142 mut/− line differs from the JHH-273 model by the presence of a codeletion of the 1p/19q arms, a lack of mutation in TP53 and slower growth rate in vivo. Since IDH1 mutant tumors likely develop by distinct molecular pathways, each model represents a valuable tool for investigating the pathogenesis of these tumors and for preclinical testing of molecular based therapies which target progressive gliomas.
Acknowledgments
Funding: This work was supported by the Conrad N. Hilton Foundation, the Virginia and D.K. Ludwig Fund for Cancer Research, and the Irving J. Sherman Professorship in Neurosurgery Research to GJR.
We thank Meghan J. Seltzer, Vafi Salmasi, Gilson S. Baia, Zev A. Binder, Kelli M. Wilson and Andrew R. Larsen for technical assistance and constructive support.
List of abbreviations
- IDH1
isocitrate dehydrogenase 1
- α-KG
α-ketoglutarate
- 2-HG
2-hydroxyglutarate
- GBM
glioblastoma
- CIC
capicua transcriptional repressor
- FUBP1
far-upstream binding protein 1
- ATRX
alpha thalassemia/mental retardation syndrome X-linked
- TP53
tumor protein p53
- CDKN2A
cyclin-dependent kinase inhibitor 2A
- ALT
alternative lengthening of telomeres
Footnotes
Competing interests
GJR is co-inventor on intellectual property pertaining to the IDH1 mutation discover and receives royalties under licensing agreements managed by Johns Hopkins University in accordance with its conflict of interest policy.
References
- 1.Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, Marks KM, Prins RM, Ward PS, Yen KE, Liau LM, Rabinowitz JD, Cantley LC, Thompson CB, Vander Heiden MG, Su SM. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–744. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gross S, Cairns RA, Minden MD, Driggers EM, Bittinger MA, Jang HG, Sasaki M, Jin S, Schenkein DP, Su SM, Dang L, Fantin VR, Mak TW. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J Exp Med. 2010;207:339–344. doi: 10.1084/jem.20092506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Choi C, Ganji SK, DeBerardinis RJ, Hatanpaa KJ, Rakheja D, Kovacs Z, Yang XL, Mashimo T, Raisanen JM, Marin-Valencia I, Pascual JM, Madden CJ, Mickey BE, Malloy CR, Bachoo RM, Maher EA. 2-hydroxyglutarate detection by magnetic resonance spectroscopy in IDH-mutated patients with gliomas. Nat Med. 2012;18:624–629. doi: 10.1038/nm.2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Xiao MT, Liu LX, Jiang WQ, Liu J, Zhang JY, Wang B, Frye S, Zhang Y, Xu YH, Lei QY, Guan KL, Zhao SM, Xiong Y. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19:17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP, Verhaak RG, Hoadley KA, Hayes DN, Perou CM, Schmidt HK, Ding L, Wilson RK, Van Den Berg D, Shen H, Bengtsson H, Neuvial P, Cope LM, Buckley J, Herman JG, Baylin SB, Laird PW, Aldape K. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17:510–522. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, Campos C, Fabius AW, Lu C, Ward PS, Thompson CB, Kaufman A, Guryanova O, Levine R, Heguy A, Viale A, Morris LG, Huse JT, Mellinghoff IK, Chan TA. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012;483:479–483. doi: 10.1038/nature10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, Edwards CR, Khanin R, Figueroa ME, Melnick A, Wellen KE, O’Rourke DM, Berger SL, Chan TA, Levine RL, Mellinghoff IK, Thompson CB. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483:474–478. doi: 10.1038/nature10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seltzer MJ, Bennett BD, Joshi AD, Gao P, Thomas AG, Ferraris DV, Tsukamoto T, Rojas CJ, Slusher BS, Rabinowitz JD, Dang CV, Riggins GJ. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res. 2010;70:8981–8987. doi: 10.1158/0008-5472.CAN-10-1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiao Y, Killela PJ, Reitman ZJ, Rasheed AB, Heaphy CM, de Wilde RF, Rodriguez FJ, Rosemberg S, Oba-Shinjo SM, Nagahashi Marie SK, Bettegowda C, Agrawal N, Lipp E, Pirozzi C, Lopez G, He Y, Friedman H, Friedman AH, Riggins GJ, Holdhoff M, Burger P, McLendon R, Bigner DD, Vogelstein B, Meeker AK, Kinzler KW, Papadopoulos N, Diaz LA, Yan H. Frequent ATRX, CIC, FUBP1 and IDH1 mutations refine the classification of malignant gliomas. Oncotarget. 2012;3:709–722. doi: 10.18632/oncotarget.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sahm F, Koelsche C, Meyer J, Pusch S, Lindenberg K, Mueller W, Herold-Mende C, von Deimling A, Hartmann C. CIC and FUBP1 mutations in oligodendrogliomas, oligoastrocytomas and astrocytomas. Acta Neuropathol. 2012;123:853–860. doi: 10.1007/s00401-012-0993-5. [DOI] [PubMed] [Google Scholar]
- 11.Killela PJ, Pirozzi CJ, Reitman ZJ, Jones S, Rasheed BA, Lipp E, Friedman H, Friedman AH, He Y, McLendon RE, Bigner DD, Yan H. The genetic landscape of anaplastic astrocytoma. Oncotarget. 2013 doi: 10.18632/oncotarget.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu XY, Gerges N, Korshunov A, Sabha N, Khuong-Quang DA, Fontebasso AM, Fleming A, Hadjadj D, Schwartzentruber J, Majewski J, Dong Z, Siegel P, Albrecht S, Croul S, Jones DT, Kool M, Tonjes M, Reifenberger G, Faury D, Zadeh G, Pfister S, Jabado N. Frequent ATRX mutations and loss of expression in adult diffuse astrocytic tumors carrying IDH1/IDH2 and TP53 mutations. Acta Neuropathol. 2012;124:615–625. doi: 10.1007/s00401-012-1031-3. [DOI] [PubMed] [Google Scholar]
- 13.Luchman HA, Stechishin OD, Dang NH, Blough MD, Chesnelong C, Kelly JJ, Nguyen SA, Chan JA, Weljie AM, Cairncross JG, Weiss S. An in vivo patient-derived model of endogenous IDH1-mutant glioma. Neuro Oncol. 2012;14:184–191. doi: 10.1093/neuonc/nor207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klink B, Miletic H, Stieber D, Huszthy PC, Valenzuela JA, Balss J, Wang J, Schubert M, Sakariassen PO, Sundstrom T, Torsvik A, Aarhus M, Mahesparan R, von Deimling A, Kaderali L, Niclou SP, Schrock E, Bjerkvig R, Nigro JM. A novel, diffusely infiltrative xenograft model of human anaplastic oligodendroglioma with mutations in FUBP1, CIC, and IDH1. PLoS One. 2013;8:e59773. doi: 10.1371/journal.pone.0059773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Navis AC, Niclou SP, Fack F, Stieber D, van Lith S, Verrijp K, Wright A, Stauber J, Tops B, Otte-Holler I, Wevers RA, van Rooij A, Pusch S, von Deimling A, Tigchelaar W, van Noorden CJ, Wesseling P, Leenders WP. Increased mitochondrial activity in a novel IDH1-R132H mutant human oligodendroglioma xenograft model: in situ detection of 2-HG and alpha-KG. Acta Neuropathol Commun. 2013;1:18. doi: 10.1186/2051-5960-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Borodovsky A, Salmasi V, Turcan S, Fabius AW, Baia GS, Eberhart CG, Weingart JD, Gallia GL, Baylin SB, Chan TA, Riggins GJ. 5-azacytidine reduces methylation, promotes differentiation and induces tumor regression in a patient-derived IDH1 mutant glioma xenograft. Oncotarget. 2013;4:1737–1747. doi: 10.18632/oncotarget.1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Siu IM, Tyler BM, Chen JX, Eberhart CG, Thomale UW, Olivi A, Jallo GI, Riggins GJ, Gallia GL. Establishment of a human glioblastoma stemlike brainstem rodent tumor model. J Neurosurg Pediatr. 6:92–97. doi: 10.3171/2010.3.PEDS09366. [DOI] [PubMed] [Google Scholar]
- 18.Seltzer MJ, Bennett BD, Joshi AD, Gao P, Thomas AG, Ferraris DV, Tsukamoto T, Rojas CJ, Slusher BS, Rabinowitz JD, Dang CV, Riggins GJ. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res. 70:8981–8987. doi: 10.1158/0008-5472.CAN-10-1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meeker AK, Gage WR, Hicks JL, Simon I, Coffman JR, Platz EA, March GE, De Marzo AM. Telomere length assessment in human archival tissues: combined telomere fluorescence in situ hybridization and immunostaining. Am J Pathol. 2002;160:1259–1268. doi: 10.1016/S0002-9440(10)62553-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Montgomery E, Argani P, Hicks JL, DeMarzo AM, Meeker AK. Telomere lengths of translocation-associated and nontranslocation-associated sarcomas differ dramatically. Am J Pathol. 2004;164:1523–1529. doi: 10.1016/S0002-9440(10)63710-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen C, Hong YK, Ontiveros SD, Egholm M, Strauss WM. Single base discrimination of CENP-B repeats on mouse and human Chromosomes with PNA-FISH. Mamm Genome. 1999;10:13–18. doi: 10.1007/s003359900934. [DOI] [PubMed] [Google Scholar]
- 22.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heaphy CM, de Wilde RF, Jiao Y, Klein AP, Edil BH, Shi C, Bettegowda C, Rodriguez FJ, Eberhart CG, Hebbar S, Offerhaus GJ, McLendon R, Rasheed BA, He Y, Yan H, Bigner DD, Oba-Shinjo SM, Marie SK, Riggins GJ, Kinzler KW, Vogelstein B, Hruban RH, Maitra A, Papadopoulos N, Meeker AK. Altered telomeres in tumors with ATRX and DAXX mutations. Science. 2011;333:425. doi: 10.1126/science.1207313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heaphy CM, Schreck KC, Raabe E, Mao XG, An P, Chu Q, Poh W, Jiao Y, Rodriguez FJ, Odia Y, Meeker AK, Eberhart CG. A glioblastoma neurosphere line with alternative lengthening of telomeres. Acta Neuropathol. 2013;126:607–608. doi: 10.1007/s00401-013-1174-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Silvestre DC, Pineda JR, Hoffschir F, Studler JM, Mouthon MA, Pflumio F, Junier MP, Chneiweiss H, Boussin FD. Alternative lengthening of telomeres in human glioma stem cells. Stem Cells. 2011;29:440–451. doi: 10.1002/stem.600. [DOI] [PubMed] [Google Scholar]
- 26.Piaskowski S, Bienkowski M, Stoczynska-Fidelus E, Stawski R, Sieruta M, Szybka M, Papierz W, Wolanczyk M, Jaskolski DJ, Liberski PP, Rieske P. Glioma cells showing IDH1 mutation cannot be propagated in standard cell culture conditions. Br J Cancer. 2011;104:968–970. doi: 10.1038/bjc.2011.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kelly JJ, Blough MD, Stechishin OD, Chan JA, Beauchamp D, Perizzolo M, Demetrick DJ, Steele L, Auer RN, Hader WJ, Westgate M, Parney IF, Jenkins R, Cairncross JG, Weiss S. Oligodendroglioma cell lines containing t(1;19)(q10;p10) Neuro Oncol. 2010;12:745–755. doi: 10.1093/neuonc/noq031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wakimoto H, Tanaka S, Curry WT, Loebel F, Zhao D, Tateishi K, Chen J, Klofas LK, Lelic N, Kim JC, Dias-Santagata D, Ellisen LW, Borger DR, Fendt SM, Vander Heiden MG, Batchelor TT, Iafrate AJ, Cahill DP, Chi AS. Targetable signaling pathway mutations are associated with malignant phenotype in IDH-mutant gliomas. Clin Cancer Res. 2014;20:2898–2909. doi: 10.1158/1078-0432.CCR-13-3052. [DOI] [PMC free article] [PubMed] [Google Scholar]