

Abstract
In the canonical (G-X-Y)n sequence of the fibrillar collagen triple helix, stabilizing direct inter-chain hydrogen bonding connects neighboring chains. Mutations at G can disrupt these interactions and are linked to connective tissue diseases. Here we integrate computational approaches with NMR to obtain a dynamic view of hydrogen bonding distributions in the (POG)4-(POA)-(POG)5 peptide, showing that the solution conformation, dynamics and hydrogen bonding deviate from the reported x-ray crystal structure in many aspects. The simulations and NMR data provide clear evidence for inequivalent environments in the three chains. MD simulations indicate direct inter-chain hydrogen bonds in the leading chain, water-bridges in the middle chain, and non-bridging waters in the trailing chain at the G→A substitution site. Theoretical calculations of NMR chemical shifts using a quantum fragmentation procedure can account for the unusual downfield NMR chemical shifts at the substitution sites and are used to assign the resonances to the individual chains. The NMR and MD data highlight the sensitivity of amide shifts to changes in the acceptor group from peptide carbonyls to water. The results are used to interpret solution NMR data for a variety of glycine substitutions and other sequence triplet interruptions, to provide new connections between collagen sequences, their associated structures, dynamical behavior and ability to recognize collagen receptors.
Keywords: Collagen, Interruption, Osteogenesis Imperfecta Diseases, Hydrogen Bonding, Chemical Shifts, Molecular Dynamics Simulations, NMR
Graphical Abstract
INTRODUCTION
Collagen is the most abundant protein in the human body and is remarkable as it plays a dual role as a structural protein that provides tensile strength to skin, bone, cartilage and blood vessel walls, and as a protein that is highly biologically active and involved in many extracellular matrix interactions1–3. The linear, (G-X-Y)n repetitive triple helical collagen motif presents a very different picture from a typical folded globular protein, and its interactions have different features or specificity. The triple helix is a rod-like folded form in which the glycine residues are all buried near a central axis, while the residues in the X and Y positions are largely exposed to solvent4,5. The collagen triple helix has a one-residue stagger and the hydrogen bonding (H-bond) of the triple helix is between the three chains with the backbone amide protons of the Gly residues forming inter-chain hydrogen bonds (H-bonds) with the carbonyl oxygen of the X residue in the adjacent chain. All polar residues and hydrophobic side chains are on the exterior, and there is no hydrophobic core. The X and Y positions are frequently occupied by Pro and hydroxyproline (abbreviated by the three letter code Hyp or one letter code O), respectively, which are important to triple helix stability, and Gly-Pro-Hyp is the most common and most stabilizing tripeptide sequence.
Defects in the triple helix domain of collagen have been associated with a number of human collagen diseases1,6–8. Osteogenesis Imperfecta (OI) or brittle bone disease affects roughly one in 10,000 individuals and results from mutations in type I collagen, a heterotrimer comprised of two α1(I) chains and one α2(I) chain6–8. The most common mutation is a single base change that leads to the replacement of a single Gly by another residue anywhere along the (G-X-Y)n sequence, breaking the repeating tripeptide pattern. The severity of the diseases varies widely, ranging from mild cases with multiple fractures to perinatal lethal cases. The molecular basis of the disease is still not understood but it has been shown that mutations result in perturbation of the local conformation and dynamics of the triple helix structure around the mutation site6,9,10.
Collagen model peptides (CMPs) have been used to model the conformation, dynamics, folding and stability of the collagen triple-helix11–15. The simplest model of an OI mutation, a Gly to Ala substitution in the center of the repeating (POG)10 sequence (referred to as G→A peptide hereafter), has been shown to form a very stable triple helix but with a significant decrease in thermal stability relative to the peptide with repeating (POG)10 sequence16. Studies on a heterotrimer model of a Gly to Ala substitution show that the thermal stability varies as a function of the number of chains that contain an Ala substitution17. The X-ray crystal structure18,19 reveals a highly localized disrupted screw symmetry at the mutation site and shows a loss of direct inter-chain H-bonds. For each chain, the direct H-bonds are replaced with water mediated hydrogen bonds in which water creates a bridge between the backbone group of the Ala NH to the O=C group of the Pro in the neighboring chain.
Because the unique rod-like structure and repeating sequence of the triple helix make assignments difficult, solution NMR structural studies are challenging20. However, NMR data on G→A peptides suggest that the structural and dynamic perturbations are very sensitive to the local sequence context surrounding the mutation 21–24, and that perturbations induced by the Gly to Ala mutation in the (POG) context deviate from those in the crystal environment. The NMR data indicates that the Gly to Ala substitutions lead to nonequivalence of the Ala residues in the three strands with one of the strands likely to form a good backbone hydrogen bond while the other two do not, and one strand having a very significant down field-shifted 1H resonance relative to other two resonances in the HSQC spectra. The NMR23 data suggest that the nature of the H-bonding in the G→A peptide is more asymmetric and dynamic than that of the crystal structure. However, because of the lack of chain-specific NMR assignments and in light of the complex H-bonding configurations of the triple helix 18,19 which include inter-chain H-bonding, an extensive hydration network, and in certain instances water-mediated H-bonds, a full description of the H-bonding structure and dynamics is impossible using NMR data alone.
In this study, we integrate NMR and molecular dynamics (MD) simulations with explicit solvent to elucidate the complex H-bonding dynamics of a triple helix that contains a Gly mutation site that models a disease state. The MD simulations capture structural perturbations consistent with the NMR studies, supporting the notion that the solution conformation of CMPs deviates from the x-ray crystal structure in many aspects. MD simulations using explicit waters on the simplest model peptide of OI, the G→A peptide, show that the rod like molecule has significant bending motions, on a time scale of hundreds of picoseconds and that the three triple helical chains display distinctly different H-bonding dynamics. The H-bonding occupancies at the Ala mutation site are different in the three chains and have complex dynamics; water is moving in and out to allow the formation of direct H-bonds or to form dynamic interstitial water-mediated H-bonds. Theoretical calculations of NMR chemical shifts on the MD snapshots that contain interstitial waters can account for the unusual downfield NMR chemical shifts observed at Gly substitution sites and highlight the sensitivity of the 1H chemical shift to the complex dynamics of the H-bonding occupancies. We can explore in this unique system the differences between direct (interchain) hydrogen bonding and water-mediated interactions in ways that are generally not possible with globular proteins. These results, combined with NMR data for other peptides, suggest a general model for the structural and dynamic consequences of glycine interruptions in collagen.
MATERIALS AND METHODS
MD System Description and Setup
The initial coordinates of the collagen-like peptides, (POG)4-(POA)-(POG)5 [referred as G→A peptide] and (POG)10 were taken from the published crystal structures in PDB code 1CAG18 and 1V7H25, respectively. MD simulations were performed with the Amber 99SB58 protein force field using the AMBER10 programs59. In the simulation, the protein is placed in a truncated octahedral periodic box of the explicit SPC water model with about 27,700 water molecules. The distance from the surface of the box to the closet atom of the solute is set to 15 Å. The system is heated from 0 to 293 K over 1ns and followed by 3 ns simulation with restraints then 1 ns without restraints in the NPT ensemble. Finally, MD production simulation is performed in the NVE ensemble. In MD simulations, SHAKE algorithm is used to constrain all the bonds involving hydrogen atoms and the time step is 1 fs. The potential energy function represents the contribution from bond, dihedral angles, van der Waals, and electrostatic terms. The particle-mesh Ewald method was used to calculate the electrostatics interaction and a non-bonded cutoff of 8 Å was used during the MD simulation. The simulations were run for 170 ns for G→A peptide, and 100 ns for (POG)10 peptides. A separate 1 μs simulation of the G→A peptide in solution (see Fig. S3 in Supporting Information) confirmed that structure is stable on this longer time scale, and that no significant changes were seen compared to the 170 ns simulation analyzed here. All trajectories were saved every 2ps for future analysis. Post processing and analysis were carried out using the cpptraj analysis tool60.
Crystal Simulations
Simulations of the G→A peptide used the same force field parameters described above. We created a supercell consisting of 6 unit cells: 1 copy along the a (triple-helix) direction, and 3 and 2 copies along the b and c directions, respectively. Acetate ions modeled in the 1CGD pdb entry19 were modeled, and neutralized by an equal number of sodium ions. The number of water molecules needed to maintain the observed unit cell dimensions was determined by trial and error to be 3384. The simulation temperature was maintained at 263 K (the temperature of data collection) by a Langevin thermostat with a collision frequency of 5 ps−1. Constant pressure was maintained with a weak-coupling (Berendsen) barostat with a time constant of 3 ps. After 280 ns of equilibration (during which time restraints on the peptide to keep it near its crystallographic position were gradually relaxed), 300 ns of production simulation were carried out. The final supercell dimensions were 173.36, 42.15, and 50.58 Å, compared to 173.46, 42.17, and 50.62 Å in the experiment. (The β angle of 95.82° was not allowed to vary.) In a typical snapshot, the backbone atoms of the triple helix differed by 1.2 Å (root-mean-square distance) from the deposited model in the 1CGD pdb entry.
Theoretical Chemical Shift Calculation
Proton and nitrogen chemical shifts for the NH atoms of the Gly and Ala residues were estimated using the automated fragmentation model originally introduced by He et al.31. In this model, a quantum region is defined around the residue whose shift is being calculated, that includes that residue and its neighbors out to a cutoff of about 3.5 Å. Water molecules within a similar distance are also included. Protein residues beyond the cutoff are included as MM (molecular mechanics) atoms, and the effects of waters beyond the cutoff are treated with a continuum dielectric (Poisson) model. Details are given elsewhere32,35,61. For these calculations, we used the OLYP density functional with a TZVP basis set (as recommended elsewhere61), using the demon-2k (version 3) program. For each calculation, we averaged over 400 equally spaced snapshots with an interval of 50 ps from the last 20 ns of the MD simulation. The computed shielding was compared to those for amide nitrogen and hydrogen in ubiquitin (using the same computational model), to obtain the reported chemical shifts. The same snapshots were analyzed by the SHIFTX2 program33 using default parameters, with no homology information being used.
NMR Experiments
Peptide (POG)4POA(POG)5 [designed as G→A peptide] was synthesized by the Tufts University Core Facility and purified using a Waters XTerra Prep C18 column on an Amersham Biosciences fast protein liquid chromatography system23. The G→A peptide was made with selectively 15N labeled residues at Ala15 and Gly24 positions; labeling was limited by the repetitive sequence. NMR samples for the G→A peptide were prepared in 10% D2O/90% H2O at pH 2 with concentrations of 3 mm.
NMR experiments for the G→A peptide were performed on a Varian INOVA 500-MHz spectrometer. The HSQC experiment was performed at 15 °C. The 3JHNHA coupling constants were measured and corrected. For the measurements of amide proton temperature gradients, 1H-15N HSQC spectra were obtained at 0–25 °C with an interval of 5 °C. The sample was equilibrated at each temperature for at least 3 h. Amide proton temperature gradients were obtained by linear regression analysis of the amide proton chemical shifts versus temperature. All data were processed using the FELIX 2004 software package (Felix NMR, Inc., San Diego, CA) and/or NMRPipe62 and analyzed with FELIX 2004 or NMRView63 as described before23,56.
RESULTS
MD simulations indicate structural and dynamic perturbations at the Gly substitution sites
MD simulations offer a complement to x-ray and NMR structures by providing models and structures that can be analyzed for conformational flexibility, as well as global and local motions. CMPs have been typically considered to be relatively rigid rod-like molecules and the crystal structure of the G→A peptide (Figure 1a) appears as a straight structure. Our simulations show small but noticeable bending motions, on a time scale of hundreds of picoseconds for the (POG)10 peptide and the G→A peptide (Figure 1b). The distribution of bending angles for both peptides suggests that the triple helix cannot be treated as a rigid rod and notably, the G→A peptide has a slightly larger distribution of bending angles than the (POG)10 peptide. The bending angle calculated from the crystal structure of either (POG)10 or G→A is nearly linear (ca. 176°) and notably different from those observed in the solution MD simulations, where values from 150° to 180° are found. Snapshots corresponding to three different bending angles of G→A peptide are shown in Figure 1c, highlighting the non-negligible conformational fluctuations in solution of the mutation peptide relative to the (POG)10 peptide. Such bending implies a non-equivalence in the three chains.
Figure 1.

(a) Schematic representation of the G→A crystal structure (PDB 1cag18) with Ala residues highlighted as red VDW balls; (b) the distributions of bend angles, θ (shown in (c). The dashed line indicates the angle of 175° and 177° calculated from crystal structure of (POG)10 (PDB 1v7h25) in black and G→A peptide in red, respectively. (c) MD selected snapshots corresponding to three bend angles (~180° in blue, ~160° in red, and ~145° in yellow) for the G→A peptide. (d) Ramachandran plot (ϕ,ψ) from the MD trajectories. Warmer colors represent higher populations.
The local perturbations in conformation induced by the Gly to Ala substitution can be further characterized by comparing the (ϕ,ψ) backbone angles of the three chains at the Ala at the 15th position (referred to as A15) with those of G24 that acts as an internal control for a region that is not disrupted (Figure 1d). The angles in a repeating (G-X-Y) region are centered at (−61°, 148°), characteristic of a PPII conformation. The three A15 and G24 residues have similar ψ angle distributions but adopt different ϕ angles. The Ala residue in the leading chain (LA15) (where “L”, “M” and “T” stand for the leading, middle and trailing strands,) is most similar to the G24 control, whereas the TA15 shows a small population further away from PPII conformation and MA15 adopts dihedral angles closer to the β region of the Ramachandran plot, confirming that the disruption creates an inequivalence between the three chains. The X-ray conformation18,19 also has a more negative ϕ angle for the middle chain, and we show below that these trends are consistent with NMR coupling constants in solution23. The presence of a more extended backbone conformation in the middle chain (relative to the leading and trailing chains) is consistent with a greater propensity for bending, allowing the path-length of this chain to be somewhat longer than those of the other two chains.
A Gly→Ala substitution results in an asymmetric water mediated H-bonding distribution within the three chains
The characteristic H-bonds that exist for Gly residues in the (G-X-Y)n repeating environment is significantly altered at the Gly to Ala substitution site, and modestly altered at the G18 position one triplet C-terminal to it. Typically in the (G-X-Y)n repeating unit there is a direct H-bond between the Gly NH and the O=C of the X residue in the neighboring chain (Figure 2a)18,19,25. In the G→A peptide (Figure 2b), the terminal G6 and G24 residues and the G12 residue that is just N-terminal to A15 have an N-O distance distribution that is identical for all three chains and that is consistent with a typical inter-chain H-bond. By contrast, for A15 the distribution of N-O bond distance is different for the three chains and for each chain the distribution is bi-modal. The peak near 2.8 Å, is consistent with a direct H-bond, whereas the second peak, at 4.5~5 Å, indicates the loss of a direct H-bond. The leading chain, LA15, has an inter-chain H-bond 72% of the time, whereas TA15 and MA15 have inter-chain H-bond occupancies of 30% and 17%. H-bonding disruption is also seen at G18, C-terminal to the mutation site, but not at G12, N-terminal to the mutation site. Therefore in addition to inequivalent H-bonding distributions at the mutation site, the distribution of hydrogen bonds relative to the substitution site is not symmetric.
Figure 2.

Hydrogen bonding topology/patterns in the G→A peptide. (a) One residue staggering of the triple helix leads to leading (L), middle (M), and trailing (T) chains. (b) Distribution of N-O bond distances between N of the indicated residue and the carbonyl oxygen of the Pro in the neighboring chain. Chains L, M, and T are colored as black, red, and green, respectively.
An analysis of the H-bond dynamics in the G→A peptide is given in Figure 3. LA15 has direct inter-chain H-bonds between the NH—OC for almost the entire length of the MD simulation (Figure 3b, 3f), except for brief disruptions, e.g. at 2, 12 and 19 ns; G24 in the repeating (G-X-Y)n region is similar, but lacks these brief disruptions (Figure 3a, 3e). By contrast, MA15 exhibits interstitial NH—H2O—OC water bridges with lifetimes of a few nanoseconds (Figure 3c, 3g), and TA15 has NH—H2O dangling water H-bonds in which the waters move in and out on the 100 ps timescale (Figure 3d, 3h). The loss of inter-chain H-bonds appear to be compensated by solvent H-bonds at all times during the simulation. The three chains are inequivalent with regard to the type of H-bonds at the mutation sites and show an asymmetry in terms of the residence times for the water H-bonds. Parallel simulations in the crystalline environment show no evidence for direct hydrogen bonding in the L and M chains, and a partial loss of a dangling water in the T chain (Figure S1). This suggests that the differences we see relative to the crystal configuration are not artifacts of the force field, but are more strongly related to the change in triple-helix environment in going from crystal to solution.
Figure 3.

Left panel: N-O bond distance between amide atom (N) of G24/A15 residues and the nearby oxygen atom (O) either from Pro carbonyl group (inter-chain distance colored as black) or a water molecule (solvent H-bond shown as other colors; water mediated H-bonds are always shown in blue and H-bonds to dangling water molecules are presented with different colors). For G24 in (a) and LA15 in (b), only inter-chain H-bonding is observed. For MA15 in (c) and TA15 in (d), alternate types of H bonds are found. Right panel shows the most representative H bond type corresponding to the residue in the left box and the H bond is indicated with a dashed line: (e) and (f) show inter-chain H bonds corresponding to G24 and LA15, respectively; (g) shows an interstitial water-mediated H bond for MA15 (colored in blue in panel c); (h) shows a dangling solvent H bond for TA15 (colored with many colors indicating different dangling solvent bonds in panel d).
NMR chemical shifts in the G→A peptide are accurately predicted from MD snapshots that contain water mediated H-bonds
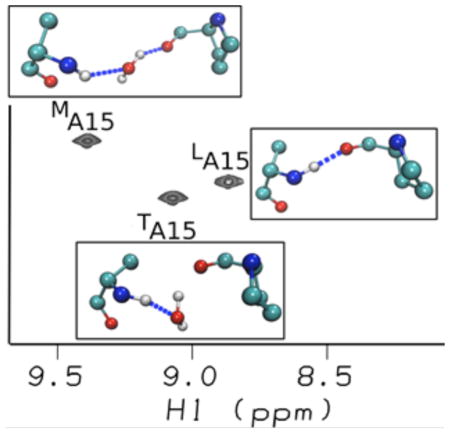
NMR studies on the G→A peptide have shown the non-equivalence of the three Ala residues in solution both in terms of their chemical shifts and their hydrogen bonding patterns22,23,26. The 1H-15N HSQC spectrum of G→A (Figure 4a) indicates that A15 gives rise to three trimer peaks while G24 gives rise to only one trimer peak due to overlapping resonances arising from the similar (POG)4 environment surrounding the Gly NH. The three amide protons from alanine range from 8.8 to 9.4 ppm, and the most downfield-shifted proton is bonded to the least downfield nitrogen (Figure 4a). At the time these measurements were made, it was not possible to make assignments to specific chains, and they were simply labeled “1”, “2” and “3”, as in Figure 4a22,23. A mide proton temperature gradients (NH Δδ/ΔT) can be used to indicate the existence of hydrogen bonding; a value greater than – 4.6 ppb/°C suggests the existence of an internal (non-water) hydrogen bond27–29. The NH Δδ/ΔT measurements (Table 1) show that the H-bonding of the three chains is inequivalent; two Ala residues have a much more negative temperature dependence than the third, suggesting a lack of inter-chain hydrogen bonding in two of the three chains.
Figure 4.

(a) NMR HSQC spectrum23 of G→A peptide measured at 15°C, where three A15 trimers (t1A15, t2A15, and t3A15) are downfield shifted in the amide proton dimension relative to A15 monomer (mA15). (b) The AF-QMMM (black) and SHIFTX2 (red) calculations of chemical shifts on the 400 snapshots along 20-ns-long MD simulations of the G→A peptide in the trimer state.
Table 1.
NMR amide proton temperature gradient (NH Δδ/ΔT), and coupling constants 3JHNNA for residues G24 and A15 in the G→A peptide and the corresponding dihedral angles from experimental data (NMR23 and x-ray18,19) and MD simulations.
| NH temperature gradients (ppb/°C) | 3JHNHA (Hz) | ϕ (degree) | ||||
|---|---|---|---|---|---|---|
|
| ||||||
| Residue | NMR | NMR | MD | NMR | MD | X-ray |
| G24 | −3.5 | 4.7±0.3 | 5.6±1.4 | −65±2 | −71±10 | −72 |
| LA15 | −4.3 | 5.6±0.6 | 5.3±1.6 | −72±5 | −70±14 | −81 |
| MA15 | −12.2 | 8.6±0.5 | 8.5±1.8 | −96±5 | −114±27 | −104 |
| TA15 | −10.7 | 5.6±0.8 | 7.0±1.8 | −72±6 | −85±20 | −61 |
The relation between 3JHNHA and ϕ dihedral angle is based on the parameterized Karplus equation30: 3JHNHA=6.51cos2(ϕ −60)−1.76cos(ϕ −60)+1.6. (±) denotes the standard deviation from the mean value.
Automated fragment quantum mechanics/molecular mechanics (AF-QMMM) calculations31,32 were used to calculate NMR chemical shifts for the G24 and A15 residues using 400 MD snapshots from the last 20 nanoseconds of the simulation (Figure 4b). For comparison, the same snapshots were analyzed using the SHIFTX2 empirical model33, which has been calibrated against observed shifts in globular proteins (Figure 4b). (Details are given in the Methods section.) The calculated chemical shifts from the AF-QMMM method show a similar pattern to that observed in the experimental NMR spectrum22,23. MA15 is significantly downfield shifted relative to TA15 and LA15, and G24 in the (POG)4 region is shifted relative to LA15. In contrast, the SHIFTX2 calculations show that the three Ala residues have a very narrow range of chemical shifts relative to each other and G24 is centered at the same 1H chemical shift as the three Ala residues. The amide proton shifts in A15 in the three chains differ by only 0.2 ppm in the SHIFTX2 results, compared to 0.8 ppm in the AF-QMMM model, and 0.6 ppm in the observed spectrum. These data can be understood by looking at the distribution of amide chemical shifts for the individual snapshots using both the AF-QMMM and SHIFTX2 calculations. Figure 5a shows that individual snapshots using AF-QMMM have a wide range (up to 5 ppm) of proton shifts, and underscore the well-known difficulties of interpreting amide-proton shifts32,34–36. One must have both accurate estimates of shifts of individual conformers and of relative populations in order to obtain a useful estimate of the average resonance position. The QM/MM calculated 1H chemical shifts are systematically underestimated by ~1.4 ppm, a pattern that has been seen in studies of folded proteins as well32,37. The empirical SHIFTX2 models are trained to give average shifts from a single conformation, and thus have a bias towards understating conformational variability (Figure 4b). The variation among snapshots (Figure 5b) is much smaller, about 1 ppm for the SHIFTX2. In spite of these differences, the pattern of proton shifts (L<T<M chain) is the same in both calculations, as shown in Figure 4b. Calculations using the SHIFTS model35 also understate the differences between the A15 resonances in the three chains, although the distribution of shifts is wider than that seen in Fig. 5b for SHIFTX2; (see Fig. S4 in Supporting Information). This wider distribution is in line with expectations, since SHIFTS was calibrated against quantum calculations whereas SHIFTX2 was fit to experimental data.
Figure 5.

Distribution of amide proton 1H chemical shifts using (a) AF-QMMM and (b) SHIFTX2 calculations on the 400 snapshots along a 20-ns-MD simulation. Note that the range of chemical shifts is significantly wider with the AF-QMMM calculations than with SHIFTX2 prediction. See Fig. S4 in Supporting Information for results using the SHIFTS program.
To explore the basis for the relationship between the values of the calculated chemical shifts and the asymmetric hydrogen bonding distributions, correlations between the 1H chemical shift and the distance to either a direct H-bond or a water mediated H-bond were examined. The AF-QMMM simulations indicate that the amide proton chemical shift is sensitive to the type of H-bond acceptor particularly for short H-bond distances (Figure 6a) and that hydrogen bonding to water results in downfield shifted 1H resonances relative to direct H-bonds of the same length. This trend is not observed for the 15N chemical shift (Figure 6b). For the direct H-bonds between the Ala NH and the Pro O=C, the 1H shift is very sensitive to the distance between donor and acceptor and decreases linearly with a steep slope for values between 2.0 and 2.5 Å. Representative examples of the amide proton shift resulting from a direct hydrogen bond for LA15 and a water-mediated hydrogen bond for MA15 are shown in Figure 6c and Figure 6d. These results suggest that, at a given distance, hydrogen bonding to water is more electron de-shielding than direct hydrogen bonding to a peptide carbonyl, and that the enhanced downfield 1H chemical shift is related to the distance of the closest hydrogen bond acceptor. These data are consistent with the fact that the distributions of chemical shifts for the different residues seen in Figure 5a resembles the population distribution for the hydrogen bonding topology seen in Figure 2b. Both the distribution of distances (Figure 2b) of the amide proton shifts (Figure 5a) are roughly bimodal for the three Ala residues: the relatively infrequent breaking of H-bonds in the leading chain (noted above, cf. Figure 3b) is evident in the shift distribution, and the dynamic swapping of direct and water-mediated H-bonds seen in Figure 3c and 3d is also present in the shift distributions.
Figure 6.

AF-QMMM chemical shift for the amide hydrogen in (a) and nitrogen in (b) of A15, demonstrating the dependence of chemical shift on the distance to the closest oxygen atom either from the carbonyl group (black dot) of a neighboring chain or from a water molecule (red dot) (c)&(d): representative snapshots with high downfield 1H chemical shifts attributed to H-bonding interactions shown by solid lines for (c) a direct H-bond with a 1H shift = 9.46 ppm for LA15; (d) for a water mediated H-bond with a 1H shift = 10.02 ppm for MA15.
The theoretical HSQC spectra can be used in order to assign the chain identity and stagger to the three Ala in the HSQC spectrum. Residue 1A15 is assigned to MA15, residue 2A15 to TA15 and residue 3A15 to LA15 (Figure 4). The calculations predict that the most-downfield NMR chemical shift observed experimentally corresponds to the MA15 amide proton hydrogen bond. Having the chains assignments in the NMR spectrum allows us to examine the relationship between the NMR temperature dependence and the nature of the hydrogen bonding observed in the MD simulations. The hydrogen bond analyses of the Ala residues from the MD simulation are in excellent agreement with the NMR amide proton temperature dependent chemical shifts. The data suggest that the LA15 is consistent with formation of direct H-bonds, while other two Ala residues (MA15 and TA15) are consistent with the lack of direct H bonds.
A comparison of the experimental NMR23 and theoretical 3JHNHA coupling constants (Table 1) provides further support for the assignments described above. G24 and LA15 have similar coupling constants that are consistent with a PPII conformation, whereas MA15 (both experimentally and computationally from the MD simulation) show a larger coupling constant that is more consistent with conformational sampling of the β region. As noted above, this feature of the middle chain is also present in the G→A peptide crystal structure18.
DISCUSSION
Gly substitutions in the repeating (G-X-Y)n sequence of collagen result in connective tissue disease that can range from lethal to non-lethal phenotype; characterization of the triple helix structure is complex and here we are interested in understanding the effect on structure and dynamics of a G to A disruption in the canonical (Gly-X-Y)n sequence. X-ray crystallography18,19 has provided insight into such sequences via the detailed description of the simplest model peptide of OI (G→A peptide). X-ray data has shown that the central zone that surrounds the mutation site has a small bulge and that the hydrogen bonding network is altered. All three alanine residues have a water mediated hydrogen bond, rather than a direct hydrogen bond, from the alanine NH to the carbonyl oxygen of the proline on the X residue. As opposed to the x-ray structure, solution NMR studies22,23 have shown that the disruption at the alanine substitution site leads to an asymmetry of the three chains; the alanine amide proton in one of the chains is significantly downfield shifted relative to the other two and the chemical shift temperature dependence suggests that only a single chain contains a direct H-bond. Due to the anisotropic shape of the peptide, the one residue stagger and the screw symmetry of the three supercoiled PPII chains, it has been impossible to assign the resonances to the individual chains and interpret these unusual chemical shifts and temperature dependent data using NMR alone22–24.
MD simulations have been performed on CMPs to provide a structural and dynamic view of the collagen triple helix in order to better understand recognition regions and the nature of Gly interruptions38–43. The G→A peptide was studied in early MD simulations by Klein and Huang40. These used an earlier version of the Amber force field, included a small shell of 267 water molecules, and were carried out for 0.5 to 1ns of simulation time. Hydrogen-bonding patterns changed very little during the course of the simulation, and almost no bending was seen. Improvements in programs and computers now permit calculations like those presented here, which employ periodic boundary conditions, about 27,000 water molecules, and simulation times that are two orders of magnitude longer than those available in 1999. In particular, we note that much of the transition to three inequivalent chains took place only after 40 ns of simulation, as illustrated in Figure S2 in the Supporting Information.
Other recent MD simulations of triple helices have focused on hydration effects in a variety of sequence contexts44–49. A striking difference between the crystal structure and the MD solution structures is the water density and its mobility around the mutation sites (Figure 7). In the crystal, the amide groups of all three Ala residues (and one Gly, which is one triplet C-terminal to Ala) do not have direct H-bonds, but instead have interstitial waters bridges (Figure 7a). In contrast to the equivalent environment in the crystal structure of the three Ala residues, the solution MD simulations indicate an inequivalent distribution of solvent around the three Ala residues such that LA15 has no water occupancy and MA15 has the highest-density (>75%) contour of water occupancy (Figure 7b). A schematic of the H-bonding in the crystal and MD simulations indicates the differences in water-mediated hydrogen bonding patterns (Figure 7c, 7d).
Figure 7.

Water distribution around the Gly substitution sites: (a) crystal structure18 and (b) MD solution from top view of the N terminal triple helix. In (a), W1 bridges the amide group of LA15 in the L chain and carbonyl group of Pro in the M chain. W2 and W3 are the water bridges of MA15 and TA15, respectively. W4 represents a water bridge for LG18. In (b), the MD simulated water density is shown with contour lines, which are coded by the total occupancy level of the volume. Volumes occupied > 75% are shown in red, > 50% in orange, and > 25% in blue. Crystal interstitial waters (W1–W3) are marked as red spheres; the three Ala are represented as licorice; Pro and Hyp are colored in orange and gray, respectively. Schematic of the hydrogen bonding topology around the Gly substitution sites (gray area) of a G→A peptide in the (c) crystal form and (d) solution form. Direct inter-chain hydrogen bonds are indicated as solid lines, hydrogen bonding to water is represented with the W label in the circle.
A direct interpretation of the unusual NMR chemical shifts and temperature dependent data is obtained from the MD results for the G→A peptide reported here. The H-bonding patterns in the NMR temperature dependent chemical shift experiments are mirrored in the MD simulations and together support the view that different chains display different H-bonding occupancies. H-bonds at the Ala mutation site and one-triplet C-terminal to it have complex dynamics in which water is moving in and out to break or form inter-chain H-bonds. Specifically, one Ala NH is involved in water-mediated hydrogen bonding while another has a direct hydrogen bond showing that the H-bonding configuration is different at the three axial levels and that one Ala can maintain a direct H-bond in the context of a G→A mutation while the other two cannot.
We note the strong agreement between the calculated chemical shifts from the MD simulations and the experimental NMR chemical shifts. Using AF-QMMM calculations31,32, the calculated chemical shifts are able to reproduce both the unusually downfield shifted Ala 1H chemical shift and the wide range of chemical shifts that arise for the three distinct Ala residues. Attempts at predicting the chemical shifts for the triple helix using empirical approaches such as SHIFTX233 results in a significantly weaker correlation with experimental data; notably the range of chemical shifts in the empirical calculations is extremely narrow. The AF-QMMM calculations provide insight into the reasons for the unusually downfield shifted A15 resonance and the range of chemical shifts observed at the A15 position. They suggest that there is a strong dependence of the chemical shift on the population of interstitial water relative to the population of backbone NH amide protons that have direct H-bonds. The better agreement of the AF-QMMM calculations to the experimental data relative to SHIFTX2 suggests that inclusion of the solvent is critical for obtaining more accurate chemical shift calculations32 and highlights the sensitivity of the 1H chemical shift to the complex dynamics of the H-bonding occupancies.
NMR conformation and dynamics of the G→A peptide can be compared with a G→S peptide [T1-898(G901S)] that models a natural glycine replaced with serine at site 901 associated with a mild case of OI23. Peptide T1-898(G901S) (sequence : Ac-(GPO)4-GPV-SPA-GAR-(GPO)4GY-CONH2) contains the collagen sequence GPV-SPA-GAR from position 898-906 in the α1 chain, thereby making this a more realistic model of OI. As in the G→A peptide, T1-898(G901S) shows an asymmetry in the solution behavior of the three chains and increased flexibility C-terminal to the mutation site relative to the N-terminal site. Strikingly, the chemical shift and temperature dependent chemical shift profile for T1-898(G901S) is similar to that of the G→A peptide. One serine resonance (trailing chain) is highly downfield shifted relative to the other two and another serine (middle chain) shows temperature dependent chemical shift data suggestive of a direct hydrogen bond. We are investigating whether the downfield shifted resonance in T1-898(G901S) is due to the presence of interstitial water but the MD simulations of the G→A peptide and the similarity of the NMR parameters is highly suggestive. In both NMR studies and MD simulations, we observe an asymmetry in the dynamics of the C-terminus relative to the N-terminus. This is consistent with previous suggestions that clinical severity is related to dynamics immediately adjacent to the mutation site24,50 and to the existence of destabilizing sequences C-terminal to the mutation site22,50–52.
OI is a disease that arises from the substitution of a single glycine to another residue in fibrillar collagens, which have an absolute requirement for Gly as every third residue. However non-fibrillar collagens, such as type IV collagen in basement membranes, also contain breaks in the (Gly-X-Y)n repeating pattern53,54. In non-fibrillar collagens, these breaks are not associated with disease but rather may be involved in molecular recognition or flexibility54. Interruptions in non-fibrillar collagens may be of variable length and are classified by the number of residues between the repeating Gly residues; for example, Gly residues that are separated by four residues are referred to as G4G interruptions53. Single Gly substitutions can be considered G5G interruptions (for example: GPOAPOG) suggesting that the properties that are observed for the OI substitution peptides may be relevant to peptides that model interruptions26,53,55. We have previously shown that peptides53,56,57 that model natural interruptions in the (Gly-X-Y)n sequence show similar NMR patterns to peptides that model G to X mutations. NMR studies56,57 on the GAAVMG peptide (G4G) show a highly localized structural perturbation with close packing of the Val residue near the central axis, similarly to the G→A and T1-898(G901S) peptides. The intriguing trends in chemical shifts that we have observed in peptides that contain Gly substitutions are also observed in the GAAVM peptide; asymmetry in the behavior of the three chains at the (GXY)n break is observed along with 1H chemical shifts that are significantly downfield shifted for one chain relative to the other two. Based on the MD simulations for the G→A peptide, we suggest that the downfield shifted resonance arises from water-mediated hydrogen bonding at a one of the Val residues in the interruption region. This is consistent with hypotheses by Bella26, based on the existing crystal structure for a G1G interruption, Hyp− (…POGPGPOG…), that the hydrogen bonding topology for a G4G peptide (which is commensurate with a G1G peptide,) would contains water mediated hydrogen bonds. The complementary NMR and MD data suggest that disruption of the repeating (Gly-X-Y)n sequence either through Gly to X mutations or through natural interruptions may result in the introduction of interstitial water hydrogen bonds at the non-canonical (G-X-Y)n sites. We can explore in this unique system the differences between direct (interchain) hydrogen bonding and water-mediated interactions in ways that are generally not possible with globular proteins. These results, combined with NMR data for other peptides, suggest a general model for the structural and dynamic consequences of glycine interruptions in collagen.
Supplementary Material
Acknowledgments
Funding Sources
This work was supported by NIH grants GM45811 (DAC) and GM45302 (JB).
We thank Helen Berman for many helpful discussions.
Footnotes
Figure S1, illustrating the distribution of N-O bond distance for A15 in the crystal simulations of a G→A peptide; Figure S2, tracking the H-O bond distance between Ala NH and the carbonyl oxygen of Pro in the neighbor for each A15 residue over a-170-ns-MD solution simulations; Figure S3, showing backbone fluctuations for a 1 μs solution simulation. This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.Myllyharju J, Kivirikko KI. Collagens, Modifying enzymes and their mutations in humans, flies and worms. Trends Genet. 2004;20:33–43. doi: 10.1016/j.tig.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 2.Shoulders MD, Raines RT. Collagen structure and stability. Annu Rev Biochem. 2009;78:929–958. doi: 10.1146/annurev.biochem.77.032207.120833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heino J. The collagen family members as cell adhesion proteins. Bioessays. 2007;29:1001–1010. doi: 10.1002/bies.20636. [DOI] [PubMed] [Google Scholar]
- 4.Ramachandran GN, Kartha G. Structure of collagen. Nature. 1955;176:593–595. doi: 10.1038/176593a0. [DOI] [PubMed] [Google Scholar]
- 5.Rich A, Crick FH. The structure of collagen. Nature. 1955;176:915–916. doi: 10.1038/176915a0. [DOI] [PubMed] [Google Scholar]
- 6.Marini JC, Forlino A, Cabral WA, Barnes AM, San Antonio JD, Milgrom S, Hyland JC, Korkko J, Prockop DJ, De Paepe A. Consortium for Osteogenesis Imperfecta mutations in the helical domain of type I collagen: regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Hum Mutat. 2007;28:209–221. doi: 10.1002/humu.20429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kuivaniemi H, Tromp G, Prockop DJ. Mutations in fibrillar collagens (types I, II, III, and XI), fibril-associated collagen (type IX), and network-forming collagen (type X) cause a spectrum of diseases of bone, cartilage, and blood vessels. Hum Mutat. 1997;9:300–315. doi: 10.1002/(SICI)1098-1004(1997)9:4<300::AID-HUMU2>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 8.Byers PH, Cole WG. In: Osteogenesis Imperfecta, in Connective Tissue and Its Heritable Disorders: Molecular, Genetic, and Medical Aspects. 2. Royce PM, Steinmann B, editors. John Wiley & Sons, Inc; Hoboken, NJ, USA: 2002. [DOI] [Google Scholar]
- 9.Forlino A, Cabral WA, Barnes AM, Marini JC. New perspectives on Osteogenesis Imperfecta. Nat Rev Endocrinol. 2011;7:540–557. doi: 10.1038/nrendo.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Makareeva E, Cabral WA, Marini JC, Leikin S. Molecular mechanism of alpha1(I)-Osteogenesis imperfecta/Ehlers-Danlos syndrome: unfolding of an N-anchor domain at the N-terminal end of the type I collagen triple helix. J Biol Chem. 2006;281:6463–6470. doi: 10.1074/jbc.M511830200. [DOI] [PubMed] [Google Scholar]
- 11.Brodsky B, Persikov AV. Molecular structure of the collagen triple helix. Adv Protein Chem. 2005;70:301–339. doi: 10.1016/S0065-3233(05)70009-7. [DOI] [PubMed] [Google Scholar]
- 12.Baum J, Brodsky B. Folding of peptide models of collagen and misfolding in disease. Curr Opin Struct Biol. 1999;9:122–128. doi: 10.1016/s0959-440x(99)80016-5. [DOI] [PubMed] [Google Scholar]
- 13.Fields GB. Synthesis and biological applications of collagen-model triple-helical peptides. Org Biomol Chem. 2010;8:1237–1258. doi: 10.1039/b920670a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fallas JA, O’Leary LER, Hartgerink JD. Synthetic collagen mimics: self-assembly of homotrimers, heterotrimers and higher order structures. Chem Soc Rev. 2010;39:3510–3527. doi: 10.1039/b919455j. [DOI] [PubMed] [Google Scholar]
- 15.Xu Y. Thermal stability of collagen triple helix. Methods Enzymol. 2009;466:211–232. doi: 10.1016/S0076-6879(09)66009-2. [DOI] [PubMed] [Google Scholar]
- 16.Long CG, Li MH, Baum J, Brodsky B. Nuclear magnetic resonance and circular dichroism studies of a triple-helical peptide with a glycine substitution. J Mol Biol. 1992;225:1–4. doi: 10.1016/0022-2836(92)91020-p. [DOI] [PubMed] [Google Scholar]
- 17.Gauba V, Hartgerink JD. Synthetic collagen heterotrimers: structural mimics of wild-type and mutant collagen type I. J Am Chem Soc. 2008;130:7509–7515. doi: 10.1021/ja801670v. [DOI] [PubMed] [Google Scholar]
- 18.Bella J, Eaton M, Brodsky B, Berman HM. Crystal-structure and molecular-structure of a collagen-like peptide at 1.9-angstrom resolution (Gly to Ala peptide) Science. 1994;266:75–81. doi: 10.1126/science.7695699. [DOI] [PubMed] [Google Scholar]
- 19.Bella J, Brodsky B, Berman HM. Hydration structure of a collagen peptide structure. Structure. 1995;3:893–906. doi: 10.1016/S0969-2126(01)00224-6. [DOI] [PubMed] [Google Scholar]
- 20.Xiao J, Baum J. Structural insights from 15N relaxation data for an anisotropic collagen peptide. J Am Chem Soc. 2009;131:18194–18195. doi: 10.1021/ja9056823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bhate M, Wang X, Baum J, Brodsky B. Folding and conformational consequences of glycine to alanine replacements at different positions in a collagen model peptide. Biochemistry. 2002;41:6539–6547. doi: 10.1021/bi020070d. [DOI] [PubMed] [Google Scholar]
- 22.Buevich A, Baum J. Nuclear magnetic resonance characterization of peptide models of collagen-folding diseases. Philos Trans R Soc Lond B Biol Sci. 2001;356:159–168. doi: 10.1098/rstb.2000.0761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Y, Brodsky B, Baum J. NMR conformational and dynamic consequences of a Gly to Ser substitution in an Osteogenesis Imperfecta collagen model peptide. J Biol Chem. 2009;284:20660–20667. doi: 10.1074/jbc.M109.018077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xiao J, Cheng H, Silva T, Baum J, Brodsky B. Osteogenesis Imperfecta missense mutations in collagen: structural consequences of a glycine to alanine replacement at a highly charged site. Biochemistry. 2011;50:10771–10780. doi: 10.1021/bi201476a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Okuyama K, Hongo C, Fukushima R, Wu G, Narita H, Noguchi K, Tanaka Y, Nishino N. Crystal structures of collagen model peptides with Pro-Hyp-Gly repeating sequence at 1.26 A resolution: implications for proline ring puckering. Biopolymers. 2004;76:367–377. doi: 10.1002/bip.20107. [DOI] [PubMed] [Google Scholar]
- 26.Bella J. A first census of collagen interruptions: collagen’s own stutters and stammers. J Struct Biol. 2014;186:438–450. doi: 10.1016/j.jsb.2014.03.022. [DOI] [PubMed] [Google Scholar]
- 27.Cierpicki T, Otlewski J. Amide proton temperature coefficients as hydrogen bond indicators in proteins. J Biomol NMR. 2001;21:249–261. doi: 10.1023/a:1012911329730. [DOI] [PubMed] [Google Scholar]
- 28.Cierpicki T, Zhukov I, Byrd RA, Otlewski J. Hydrogen bonds in human ubiquitin reflected in temperature coefficients of amide protons. J Magn Reson. 2002;157:178–180. doi: 10.1006/jmre.2002.2597. [DOI] [PubMed] [Google Scholar]
- 29.Tomlinson JH, Williamson MP. Amide temperature coefficients in the protein G B1 domain. J Biomol NMR. 2012;52:57–64. doi: 10.1007/s10858-011-9583-4. [DOI] [PubMed] [Google Scholar]
- 30.Vuister GW, Bax A. Quantitative J correlation: a new approach for measuring homonuclear three-bond J(HNHA) coupling constants in 15N-enriched proteins. J Am Chem Soc. 1993;115:7772–7777. [Google Scholar]
- 31.He X, Wang B, Merz KM. Protein NMR chemical shift calculations based on the automated fragmentation QM/MM approach. J Phys Chem B. 2009;113:10380–10388. doi: 10.1021/jp901992p. [DOI] [PubMed] [Google Scholar]
- 32.Zhu T, Zhang JZH, He X. Automated fragmentation QM/MM calculation of amide proton chemical shifts in proteins with explicit solvent model. J Chem Theory Comput. 2013;9:2104–2114. doi: 10.1021/ct300999w. [DOI] [PubMed] [Google Scholar]
- 33.Han B, Liu Y, Ginzinger SW, Wishart DS. SHIFTX2: Significantly improved protein chemical shift prediction. J Biomol NMR. 2011;50:43–57. doi: 10.1007/s10858-011-9478-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.He X, Wang B, Merz KM. Protein NMR chemical shift calculations based on the automated fragmentation QM/MM approach. J Phys Chem B. 2009;113:10380–10388. doi: 10.1021/jp901992p. [DOI] [PubMed] [Google Scholar]
- 35.Case DA. Chemical shifts in biomolecules. Curr Opin Struct Biol. 2013;23:172–179. doi: 10.1016/j.sbi.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sitkoff D, Case DA. Density functional calculations of proton chemical shifts in model peptides. J Am Chem Soc. 1997;119:12262–12273. [Google Scholar]
- 37.Jiang XN, Wang CS. Rapid prediction of the hydrogen bond cooperativity in N-methylacetamide chains. Chem Phys Chem. 2009;10:3330–3336. doi: 10.1002/cphc.200900591. [DOI] [PubMed] [Google Scholar]
- 38.Stultz CM. Localized unfolding of collagen explains collagenase cleavage near imino-poor sites. J Mol Biol. 2002;319:997–1003. doi: 10.1016/S0022-2836(02)00421-7. [DOI] [PubMed] [Google Scholar]
- 39.Stultz CM. The folding mechanism of collagen-like model peptides explored through detailed molecular simulations. Protein Sci. 2006;15:2166–2177. doi: 10.1110/ps.062124606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aliev AE, Courtier-Mouias D, Bhandal S, Zhou S. A combined NMR/MD/QM approach for structure and dynamics elucidation in the solution state: pilot studies using tetrapeptides. Chem Commun. 2010;46:695–697. doi: 10.1039/b910499b. [DOI] [PubMed] [Google Scholar]; Aliev AE, Kulke M, Khaneja HS, Chudasama V, Sheppard TD, Lanigan RM. Motional timescale predictions by molecular dynamics simulations: Case study using proline and hydroxyproline sidechain dynamics. Proteins. 2014;82:195–215. doi: 10.1002/prot.24350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mooney SD, Huang CC, Kollman PA, Klein TE. Computed free energy differences between point mutations in a collagen-like peptide. Biopolymers. 2001;58:347–353. doi: 10.1002/1097-0282(200103)58:3<347::AID-BIP1011>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 42.Mooney SD, Klein TE. Structural models of Osteogenesis Imperfecta-associated variants in the COL1A1 Gene. Mol Cell Proteomics. 2002;1:868–875. doi: 10.1074/mcp.m200064-mcp200. [DOI] [PubMed] [Google Scholar]
- 43.Radmer RJ, Klein TE. Severity of Osteogenesis Imperfecta and structure of a collagen-like peptide modeling a lethal mutation site. Biochemistry. 2004;43:5314–5323. doi: 10.1021/bi035676w. [DOI] [PubMed] [Google Scholar]
- 44.Vitagliano L, Berisio R, De Simone A. Role of hydration in collagen recognition by bacterial adhesins. Biophys J. 2011;100:2253–2261. doi: 10.1016/j.bpj.2011.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ravikumar KM, Hwang W. Region-specific role of water in collagen unwinding and assembly. Proteins. 2008;72:1320–1332. doi: 10.1002/prot.22026. [DOI] [PubMed] [Google Scholar]
- 46.Gurry T, Nerenberg PS, Stultz CM. The contribution of interchain salt bridges to triple-helical stability in collagen. Biophys J. 2010;98:2634–2643. doi: 10.1016/j.bpj.2010.01.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Raman SS, Gopalakrishnan R, Wade RC, Subramanian V. Structural basis for the varying propensities of different amino acids to adopt the collagen conformation. J Phys Chem B. 2011;115:2593–2607. doi: 10.1021/jp109133v. [DOI] [PubMed] [Google Scholar]
- 48.Singam ERA, Balamurugan K, Gopalakrishnan R, Subramanian SR, Subramanian V, Ramasami T. Molecular dynamic simulation studies on the effect of one residue chain staggering on the structure and stability of heterotrimeric collagen-like peptides with interruption. Biopolymers. 2012;97:847–863. doi: 10.1002/bip.22085. [DOI] [PubMed] [Google Scholar]
- 49.Teng X, Hwang W. Chain registry and load-dependent conformational dynamics of collagen. Biomacromolecules. 2014;15:3019–3029. doi: 10.1021/bm500641f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bodian DL, Madhan B, Brodsky B, Klein TE. Predicting the clinical lethality of Osteogenesis Imperfecta from collagen glycine mutations. Biochemistry. 2008;47:5424–5432. doi: 10.1021/bi800026k. [DOI] [PubMed] [Google Scholar]
- 51.Hyde TJ, Bryan MA, Brodsky B, Baum J. Sequence dependence of renucleation after a Gly mutation in model collagen peptides. J Biol Chem. 2006;281:36937–36943. doi: 10.1074/jbc.M605135200. [DOI] [PubMed] [Google Scholar]
- 52.Xiao J, Madhan B, Li Y, Brodsky B, Baum J. Osteogenesis Imperfecta model peptides: incorporation of residues replacing Gly within a triple helix achieved by renucleation and local flexibility. Biophys J. 2011;101:449–458. doi: 10.1016/j.bpj.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Thiagarajan G, Li Y, Mohs A, Strafaci C, Popiel M, Baum J, Brodsky B. Common interruptions in the repeating tripeptide sequence of non-fibrillar collagens: sequence analysis and structural studies on triple-helix peptide models. J Mol Biol. 2008;376:736–748. doi: 10.1016/j.jmb.2007.11.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Miles AJ, Knutson JR, Skubitz AP, Furcht LT, McCarthy JB, Fields GB. A peptide model of basement membrane collagen alpha 1 (IV) 531–543 binds the alpha 3 beta 1 integrin. J Biol Chem. 1995;270:29047–29050. doi: 10.1074/jbc.270.49.29047. [DOI] [PubMed] [Google Scholar]
- 55.Hwang ES, Thiagarajan G, Parmar AS, Brodsky B. Interruptions in the collagen repeating tripeptide pattern can promote supramolecular association. Protein Sci. 2010;19:1053–1064. doi: 10.1002/pro.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li Y, Brodsky B, Baum J. NMR shows hydrophobic interactions replace glycine packing in the triple helix at a natural break in the (Gly-X-Y)n repeat. J Biol Chem. 2007;282:22699–22706. doi: 10.1074/jbc.M702910200. [DOI] [PubMed] [Google Scholar]
- 57.Mohs A, Popiel M, Li Y, Baum J, Brodsky B. Conformational features of a natural break in the type IV collagen Gly-X-Y repeat. J Biol Chem. 2006;281:17197–17202. doi: 10.1074/jbc.M601763200. [DOI] [PubMed] [Google Scholar]
- 58.Hornak V. Comparison of multiple amber force fields and development of improved protein backbone parameters. Proteins. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Case DA, Darden TA, Cheatham TE, III, Simmerling CL, Wang J, Duke RE, Luo R, Crowley M, Walker RC, Zhang W, Merz KM, Wang B, Hayik S, Roitberg A, Seabra G, Kolossváry I, Wong KF, Paesani F, Vanicek J, Wu X, Brozell SR, Steinbrecher T, Gohlke H, Yang L, Tan C, Mongan J, Hornak V, Cui G, Mathews DH, Seetin MG, Sagui C, Babin V, Kollman PA. AMBER 10. University of California; San Francisco: 2008. [Google Scholar]
- 60.Roe DR, Cheatham TE. PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J Chem Theory Comput. 2013;9:3084–3095. doi: 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- 61.Tang S, Case DA. Calculation of chemical shift anisotropy in proteins. J Biomol NMR. 2011;51:303–312. doi: 10.1007/s10858-011-9556-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 63.Johnson BA, Blevins RA. NMR View: a computer program for the visualization and analysis of NMR Data. J Biomol NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.