

Abstract
Glioblastoma (GBM) is the most common and most aggressive primary brain tumor in adults. The existence of a small population of stem-like tumor cells that efficiently propagate tumors and resist cytotoxic therapy is one proposed mechanism leading to the resilient behavior of tumor cells and poor prognosis. In this study, we performed an in-depth analysis of the DNA methylation landscape in GBM-derived cancer stem cells (GSCs). Parallel comparisons of primary tumors and GSC lines derived from these tumors with normal controls (a neural stem cell (NSC) line and normal brain tissue) identified groups of hyper- and hypomethylated genes that display a trend of either increasing or decreasing methylation levels in the order of controls, primary GBMs, and their counterpart GSC lines, respectively. Interestingly, concurrent promoter hypermethylation and gene body hypomethylation were observed in a subset of genes including MGMT, AJAP1 and PTPRN2. These unique DNA methylation signatures were also found in primary GBM-derived xenograft tumors indicating that they are not tissue culture-related epigenetic changes. Integration of GSC-specific epigenetic signatures with gene expression analysis further identified candidate tumor suppressor genes that are frequently down regulated in GBMs such as SPINT2, NEFM and PENK. Forced re-expression of SPINT2 reduced glioma cell proliferative capacity, anchorage independent growth, cell motility, and tumor sphere formation in vitro. The results from this study demonstrate that GSCs possess unique epigenetic signatures that may play important roles in the pathogenesis of GBM.
Keywords: Glioblastoma, Cancer stem cells, DNA methylation, SPINT2
Introduction
It is well documented that epigenetic alterations are as common as, if not more frequent than, mutational events in cancer initiation and progression (Baylin, 2005). The malignant cells exhibit overall genomic hypomethylation (primarily in repeat elements and pericentromeric regions), but simultaneously show hypermethylation of normally protected CpG islands (CGIs) when compared to normal cells (Jones and Baylin, 2002, 2007). However, most of the extensive characterization of epigenetic alterations in cancer has been obtained from whole cell populations forming the tumor or cancer cell lines. Given the striking heterogeneity within a tumor, it is of great importance to identify specific epigenetic changes and characterize their effects in the subset of cancer cells that are responsible for tumor growth, namely cancer stem cells (CSCs) (Munoz et al., 2012). Multiple studies have demonstrated the existence of CSCs that are capable of self-renewal and are able to generate the bulk of more differentiated cells that form the tumor. CSCs are more resistant to anti-tumor treatments than the non-stem cancer cells, and may contribute disproportionately to tumor growth, malignant progression and recurrence after conventional treatments (Bao et al., 2006; Eramo et al., 2006). However, the epigenetic programs that govern the generation and maintenance of CSCs remain poorly understood.
Glioblastoma (GBM) is the most common and most aggressive type of malignant primary brain tumor in adults. The median survival of patients with GBM after gross total resection, radiation therapy, and temozolomide chemotherapy is approximately 14 months (Johnson and O'Neill, 2012). This has improved only slightly over the last three decades despite advances in surgery, radiation and chemotherapy, and indeed, for patients unable to tolerate maximal aggressive therapy, survival is shorter. It has been demonstrated that GBM-derived stem cells (GSC) can be cultured from human GBM specimens in defined medium in the presence of mitogens; the methodology was initially designed for the propagation of normal neural stem cells (NSCs), to facilitate the capacity of self-renewal and subsequent differentiation upon mitogen removal (Galli et al., 2004; Singh et al., 2004; Vescovi et al., 2006; Dirks, 2008). Studies have shown that GSCs can efficiently propagate tumor xenografts that recapitulate the complex pathology of the human GBM from which they were derived (Lee et al., 2006a). The GSC lines derived from primary GBMs have also been successfully used as models for drug screening (Wurdak et al., 2010). Another approach to preserve and enrich the GSC population is to utilize xenograft models (Sarkaria et al., 2006; Higgins et al., 2013). For these studies, primary GBM tissue specimens are implanted into the mouse flank to expand the cell population while preserving the primary tumor characteristics, and then subsequently implanted into mouse brains for experiments. Serial transplantation into further generations of mice continues the maintenance of the in vivo xenograft lines (Rath et al., 2013) that are not exposed to tissue culture conditions and essentially maintain the characteristics of the primary GBM tumors from which they were derived. Although studies have demonstrated that the genome-wide gene expression profiles in GSC lines are more similar to those of primary GBM specimens (Lee et al., 2006a), little is known about the epigenetic changes during cell culture or xenograft propagation in immunodeficient mice.
Large-scale epigenomic studies have been conducted to identify abnormalities in genes and pathways (Uhlmann et al., 2003; Cadieux et al., 2006; Martinez et al., 2009; Wu et al., 2010). The Cancer Genome Atlas (TCGA) Research Network conducted the largest genomic and epigenomic study to date in several hundred brain tumors (TCGA, 2008). This systematic, multi-dimensional analysis confirmed previously well-known genetic events, and identified genetic and epigenetic alterations not previously reported in GBMs. Intriguingly, the TCGA study identified a CpG island methylator phenotype (G-CIMP) in glioma that is associated with better survival (Noushmehr et al., 2010). Global studies such as TCGA provide great insight into the molecular mechanisms of GBMs; however, the analysis of bulk tumor populations potentially overlooks the epigenetic profiles of the rare cancer stem cells, and may identify genes involved in the regulation of non-tumorigenic cells (Ward and Dirks, 2007). Epigenetic profiling of tumor specimens is also limited by the inherent cellular heterogeneity of malignant tissue and a lack of reference samples with similar composition of corresponding normal cell types.
In this study, we have isolated three GSC lines and characterized the genomic and epigenomic landscapes of the GSC lines in comparison with primary GBM specimens. The genome-wide single-base resolution methylation analysis revealed the epigenetic signature of GSCs and demonstrated a GSC-specific DNA methylation signature in both primary GBM tumors and GBM xenografts originally established at the Mayo Clinic. We have identified candidate oncogenes and tumor suppressors affected by epigenetic alterations not previously known to be associated with gliomas. The integration of DNA methylation and gene expression analyses also leads to the discovery of gene expression changes associated with aberrant DNA methylation alterations in GBMs. Furthermore, we demonstrate that forced expression of a candidate tumor suppressor gene found to be repressed by promoter hypometylation, SPINT2 (serine protease inhibitor kunitz-type 2), reduced proliferative capacity, anchorage independent growth, cell motility, and tumor sphere formation in vitro.
Results
Characterization of GSCs derived from primary GBMs
We established a normal NSC line from a fetal brain (diencephalic and telencephalic regions) and three GSC lines (1063S, 1133S, and 1142S) from primary GBM specimens (1063T, 1133T and 1142T) as described previously (Inagaki et al., 2007). Normal NSCs aggregated as free-floating cell clusters and proliferated as tight spherical shapes (Fig. 1A, a). Similar to the robust growth of the NSC line, GSC lines 1063S and 1133S proliferated and formed spherical cell clusters (Fig. 1A, b and c). Fluorescence immunostaining revealed expression of the stem cell markers Nestin, SOX2, CD133, and Musashi in the NSC line (Fig. 1A, d, g, j), as well as in the GSC lines 1063S (Fig. 1A, e, h, k) and 1133S (Fig. 1A, f, i, l). The NSC cell line and GSC line 1063S uniformly expressed SOX2, Nestin and CD133, whereas GSC line 1133S showed low-level expression of SOX2 and CD133. For differentiation experiments, the cells from the spheres were dissociated and placed on laminin-coated glass slides in mitogen-free medium. All specimens grew cells, which exhibited neurite outgrowth and had morphologies consistent with differentiated cells (neurons and glia). The majority of the cells in all samples were strongly immunoreactive for the astrocytic marker GFAP (Fig. 1A, m–o) and the mature neuronal cell marker neurofilament (NF) protein (Fig. 1A, p–r), whereas a smaller subset of cells expressed the immature neuronal marker β3-tubulin (Fig.1A, m–o).
Fig. 1.
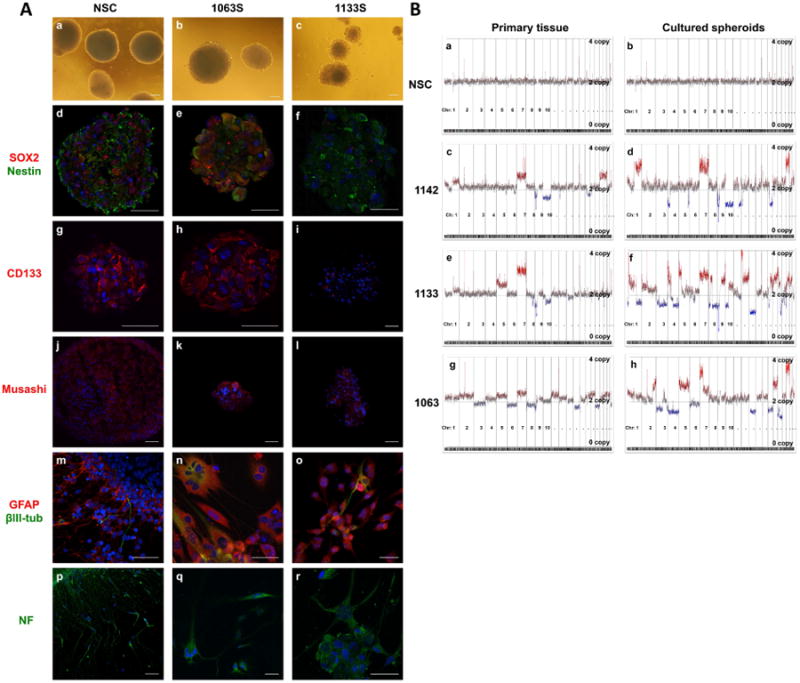
GBM cells cultured in stem-cell selective medium enrich for cells capable of self-renewal and differentiation, similar to neural stem and progenitor cells.
A: Representative bright field images show DN2L cultured NSCs and GBMs growing as spheres (a–c). Neurospheres and tumor spheres were immuonoreactive for neural stem cell markers SOX2, Nestin (d–f), CD133 (g–i), and Musashi (j–l). Upon mitogen withdrawal, the cells extended processes and exhibited morphologies similar to neuronal and astrocytic cells. Differentiated cells expressed mature cell markers GFAP, β3-tub (β3 tubulin) (m–o) and neurofilament (NF) (p–r). Nuclei were counterstained with DAPI. Scale bars for all images are 50 μm. B: Copy number profiles of the primary specimen and corresponding DN2L-derived cultures (spikes at centromeric and telomeric regions are considered artifact). NSCs showed a normal genome with negligible gain/loss, consistent with the DN2L cultured cells (a, b). Primary GBM specimens contained gain/loss hallmarks of GBMs (i.e., gain of chr. 7) (c, e, g). Copy number analysis of DN2L cultured tumorsphere from tumors 1142, 1133, and 1063 showed a cancer genome similar to the primary parental tumor (d, f, h).
Copy number aberrations (CNAs) in primary GBMs and GSCs
The results of the array CGH (comparative genomic hybridization) analysis for the primary GBM specimens and GSC lines are shown in Fig. 1B (spikes at centromeric and telomeric regions of the chromosomes are considered artifact, i.e., chromosome 2, chromosome 14-15). The primary fetal brain tissue sample (Fig. 1B, a) and the cultured NSCs derived from the same tissue (Fig. 1B, b) showed a normal copy number karyotype. Primary GBM specimens 1142T (Fig. 1B, c), 1133T (Fig. 1B, e) and 1063T (Fig. 1B, g) all contained multiple chromosomal gains and losses including common chromosomal alterations in regions of chromosome 1p, 7, 8q, 9p, 10, 12q, 13q, 19q, 20, and 22q, all which were previously identified by a comprehensive analysis of the genetic aberrations in primary GBMs (Bredel et al., 2009). The most frequent GBM alterations are regional amplifications in chromosome 7, observed in each primary tumor, and the regional loss in chromosome 10, observed in GBMs 1142T and 1133T. GSC lines 1142S (Fig. 1B, d), 1133S (Fig. 1B, f), and 1063S (Fig. 1B, h) maintained multiple signature chromosomal aberrations seen in the matched primary tumors but also gained additional chromosomal changes that were not obvious in matched primary tumors.
Genome-wide bisulfite sequencing in the primary GBMs, GSCs and GBM xenografts
The reduced representation bisulfite sequencing (RRBS) approach (Meissner et al., 2008) was used to generate genome-wide single-base resolution CpG methylation profiles of three primary GBMs (1063T, 1133T, and 1142T) and three GSC lines (1063S, 1133S, 1142S) derived from these primary GBM tumors. The NSC line and the normal brain (NB) tissue sample were used as controls for comparison purposes. In addition, we analyzed three GBM xenograft tumor tissue samples (Mayo22, Mayo39, Mayo59) developed by Dr. Jann N. Sarkaria of Mayo Clinic. A summary of the sequencing experiments is provided in Table S1. We generated 21–27 million bisulfite sequencing reads for each sample. The total number of CpG sites measured in each sample was approximately 2.2 million. The average sequencing depth per CpG ranged from 16.4× to 34.2×. More than 39% of CpG sites measured (average 0.88 million) were located in CpG Islands (CGIs). Over 23,000 CGIs, accounting for ∼83% annotated CGIs in the genome, were examined.
Identification of differentially methylated regions (DMRs) in GBMs, GSCs, and GBM xenografts
To identify the DMRs among the four groups: control, primary GBMs, GSCs, and GBM xenografts, we divided the human genome into 200 bp tiled-windows along each chromosome and calculated the average methylation values of all CpGs within each window. We then performed Student's t-test to identify statistically significant methylation differences in a given window between two groups of samples. Using the criteria of P < 0.01 and methylation difference of greater than 0.25, we identified 823, 3231, and 5237 DMRs for primary GBMs, GSC lines and GBM xenografts, respectively, when compared to the control group. A majority of hypomethylated loci (>75%) were located in intergenic and intronic regions (Fig. S1A), while only about half of hypermethylated loci were located in intergenic and intronic regions. The distribution of DMRs in CGIs, and CGI shores (0–2 kb from CGI) were similar between the groups. Hypomethylated DMRs are mainly located outside of CGIs and CGI shores, while a majority of hypermethylated DMRs are located within the CGI (Fig. S1B). The number of hypermethylated promoter DMRs increased sequentially from primary GBMs to GSC cell lines, and to GBM xenografts, and significantly more hypermethylated DMRs were identified in GBM xenografts as compared to the two other groups (Fig. S1). Supervised cluster analysis using the identified DMRs revealed striking differences in DNA methylation patterns among the four groups (Fig. S2).
Aberrant promoter hypermethylation in GSCs
K-mean cluster analysis revealed two groups of DMRs within the 3231 GSC-specific DMRs. One group of hypomethylated loci show a downward trend of decreasing methylation levels from control group to primary GBMs, and then to GSC cell lines, while the second group of hypermethylated loci demonstrate an upward trend of increasing methylation levels (Fig. 2A). For a majority of the GSC-specific DMRs, their methylation levels in primary GBMs fell between the controls and GSCs (Fig. 2A). Interestingly, although the three GBM xenografts were developed independently, they displayed similar methylation profiles as GSCs when the 3231 GSC-specific DMRs were used for cluster analysis (Fig. S2). A total of 675 genes were associated with GSC-specific DNA methylation changes (Table S2). Among them, 202 genes were associated with DMRs in the promoter and 5′-UTR regions. Analysis using DAVID functional annotation tool (http://david.abcc.ncifcrf.gov/) revealed that the 202 differentially methylated genes were enriched in the following molecular function groups: developmental process, regulation of transcription, nervous system development, and regulation of cellular metabolic process (Table 1). This list of genes consists of many hypermethylated genes previously reported in gliomas such as GDNF, NEFM, CHAT, GATA6, TES, PENK, RAX, and DLX2 (Martinez et al., 2009; Wu et al., 2010). Fig. 2B shows representative examples of the promoter hypermethylation in candidate tumor suppressor genes PENK (McTavish et al., 2007), TES (Engstrom et al., 2012), GATA6 (Kamnasaran et al., 2007), and SPINT2 (Kongkham et al., 2008). Gene set enrichment analysis (GSEA) indicated that hypermethylated genes were enriched in the targeted genes of polycomb repressive complexes 2 (PRC2), or one of the PRC2 components (EED, SUZ12), or in genes associated with bivalent histone modifications in human embryonic stem (ES) cells (Lee et al., 2006b). These frequently hypermethylated PRC2 target genes include homeobox genes (DLX2, IRX3, LHX2, PITX2, SHOX2, and SIX2), lineage-specific forkhead box (FOX), SRY-box (SOX), basic Helix-Loop-Helix (bHLH), T-box, and Zinc finger transcription factors such as FOXD3, SOX17, MYOD1, TBX4, GATA3 and GATA6 (Table 1).
Fig. 2.
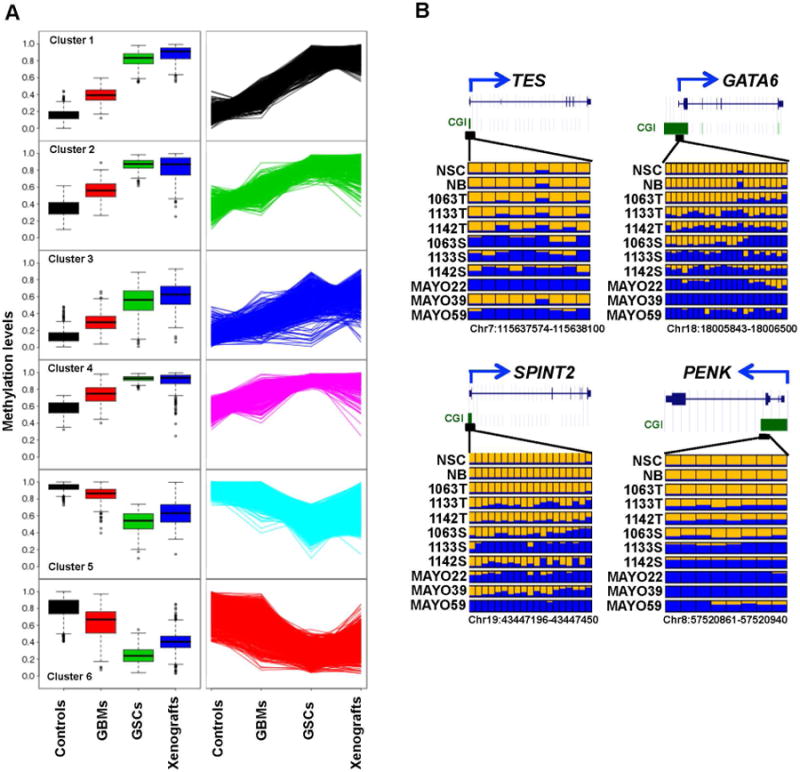
Global DNA methylation changes in primary GBMs, GSC lines and GBM xenografts compared to normal controls (NB and NSC).
A: K-mean cluster analysis of 3231 DMRs identified six distinct clusters that display an upward or downward trend of DNA methylation levels from control group to primary GBMs, and then to GSC cell lines. Box plots in the left panel show the variation and median (horizontal bars) methylation levels among four groups for each cluster. The line graphs in the right panel display the upward or downward trend of DNA methylation values from control group to primary GBMs, and then to GSC cell lines. B: DNA methylation profiles of four candidate tumor suppressor genes. In each panel, each row is the result of an individual tumor sample and each box represents an individual CpG. Yellow, no methylation; blue, methylation. The proportion of yellow and blue in each box represents the methylation level. Only common CpGs shared by all samples are shown. T refers to the primary tumor, S to the spheres of tumor cells in DN2L culture, and Mayo to the xenograft tumors.
Table 1. Functional annotation of differentially methylated genes in GSC lines.
| ID | Function | Count | P value | Benjaminia | Gene |
|---|---|---|---|---|---|
| GO:0032502b | Developmental process | 68 | 1.54E-08 | 1.27E-05 | MYOD1, DUOX1, GLI2, GDNF, CXCL12, YBX2, ACVR1C, HOOK1, OSR2, SLC2A4, OSR1, AQP11, GATA3, HLX, PAX8, PIWIL2, PIWIL3, HHIP, SRD5A2, FAM83H, CHAT, PITX2, SOX10, TBX15, GSC, RAX, SCUBE1, SIX2, ZNF141, VAX2, HOXD9, SPAG6, TFAP2A, LAMC2, NSD1, SHROOM1, IRX3, CCK, PCDHB15, FIGLA, OXTR, IGF2BP2, PRDM16, MSX2, TCP11, PCDHB16, KCNE1, GFI1, B3GNT2, WNT8A, NEFM, FOXD4, FOXD3, BGLAP, TBX3, IKZF1, TBX4, DBH, SHOX2, TNFSF11, PKP1, PSMC3, FOXE1, AVPR1A, GHSR, SMPD3, KLF4, NR5A1 |
| GO:0031323 | Regulation of cellular metabolic process | 57 | 0.001584 | 0.051934 | MYOD1, LPAR3, CPEB1, GLI2, GDNF, YBX2, GATA3, HLX, PAX8, ZNF578, PIWIL2, PIWIL3, FOXB1, RTEL1, PITX2, CIITA, SOX10, TBX15, GSC, RAX, SIX2, ZNF141, VAX2, GRHL2, HMGA1, GRM1, FOXR1, HOXD9, TFAP2A, ZNF710, NSD1, IRX3, IRX6, CCK, FIGLA, IGF2BP2, PRDM16, MSX2, HRH1, PRDM14, CDYL, GFI1, FOXD4, FOXD3, BGLAP, TBX3, IKZF1, TBX4, ABCG4, SHOX2, PSMC3, AVPR1A, SP5, FOXE1, GHSR, KLF4, NR5A1 |
| GO:0045449 | Regulation of transcription | 45 | 0.002614 | 0.070496 | MYOD1, IRX3, IRX6, FIGLA, PRDM16, GLI2, GDNF, YBX2, MSX2, PRDM14, CDYL, GATA3, HLX, PAX8, ZNF578, GFI1, FOXB1, FOXD4, FOXD3, PITX2, CIITA, SOX10, BGLAP, RAX, TBX15, GSC, IKZF1, TBX3, TBX4, SIX2, ZNF141, VAX2, ABCG4, GRHL2, HMGA1, FOXR1, HOXD9, SHOX2, FOXE1, SP5, TFAP2A, ZNF710, NSD1, KLF4, NR5A1 |
| GO:0007399 | Nervous system development | 25 | 0.001121 | 0.040257 | IRX3, CCK, PCDHB15, OXTR, GLI2, PRDM16, CXCL12, GDNF, HLX, PCDHB16, B3GNT2, HHIP, GFI1, NEFM, CHAT, SOX10, GSC, RAX, TBX3, IKZF1, VAX2, SHOX2, HOXD9, AVPR1A, TFAP2A |
Benjamini-hochberg adjusted P-values;
Gene Ontology (GO) term ID
Similar to a previous study in astrocytomas (Wu et al., 2010), aberrant hypermethylation changes were observed in all four HOX gene clusters. Figs. 3A and 3B show a snapshot of DNA methylation maps in the HOXA and HOXD clusters, respectively. Most of the CpG sites in these two HOX clusters measured in control group (NSC and NB) were unmethylated or methylated at lower level (less than 20%). Increased methylation levels were observed in 18 out of 39 HOX genes from all four HOX gene families in primary tumors, GSCs, and GBM xenografts compared with the control group (Student's t-test P < 0.05). Fig. 3C shows several examples such as HOXA5, HOXA10, HOXD1 and HOXD12, which reveal increased methylation levels in the order of primary GBMs, GSCs and GBM xenografts. In GSC lines and GBM xenografts, most of HOX genes exhibited hypermethylation that was much denser and more uniform than was observed in primary GBMs.
Fig. 3.
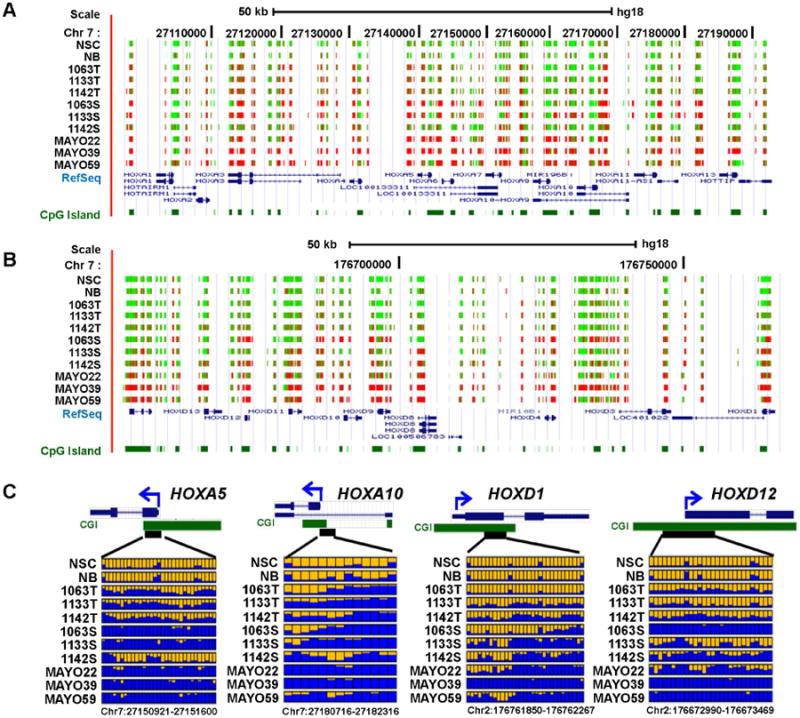
Aberrant DNA hypermethylation in HOX gene families.
A and B: UCSC genome browser screenshot illustrating the RRBS results in the HOXA and HOXD gene clusters, respectively. The tracks shown from the top to bottom in each Figure are the DNA methylation level at each CpG site derived from the bisulfite sequencing reads, RefSeq genes and annotated CpG islands in the UCSC genome browser. Red and green colors indicate methylated and unmethylated CpG sites, respectively. C: Methylation profiles of four representative HOX genes. Color scheme is consistent with in Fig. 2B.
Identification of megabase-size hypomethylated domains in GSCs
Although RRBS enriches for CpG-rich regions of the genome that tend to be hypermethylated in cancer, it also surveys thousands of positions in low CpG-density regions. As shown in Fig. S1B, most of the hypomethylated DMRs are located outside of CGIs. Varley et al. (2013) identified 114 megabase-size hypomethylated domains in a number of human cancer cell lines using RRBS. Using a similar approach, we observed that many of hypomethylated DMRs were co-localized in the genome. We identified 194 independent megabase windows in the genome that have more hypomethylated loci than expected (Chi-square test, P < 0.05) in at least one of the three GSC lines (Fig. 4A and Table S3). Fig. 4B illustrates a UCSC genome browser snapshot of a 40 Mb interval on the chromosome 10 that contains a long-range hypomethylated domain in the GSC line 1133S and in GBM xenograft Mayo59 when compared to the control group. Intriguingly, one of the genes located in this hypomethyated domain is MGMT, whose promoter methylation is a valuable prognostic marker and indicator of clinical response to Temozolomide treatment in GBMs (Hegi et al., 2005). After carefully examining the DNA methylation data, we found that the hypomethylation across the gene body of MGMT is inversely correlated with the promoter hypermethylation. As shown in Fig. 4C, the MGMT promoter was hypermethylated while the multiple hypomethylated loci could be found in the gene body of MGMT in two GSC lines (1133S and 1142S) and two GBM xenografts (Mayo22 and Mayo59). A similar observation was also made in genes AJAP1 and PTPRN2 (Fig. S3).
Fig. 4.
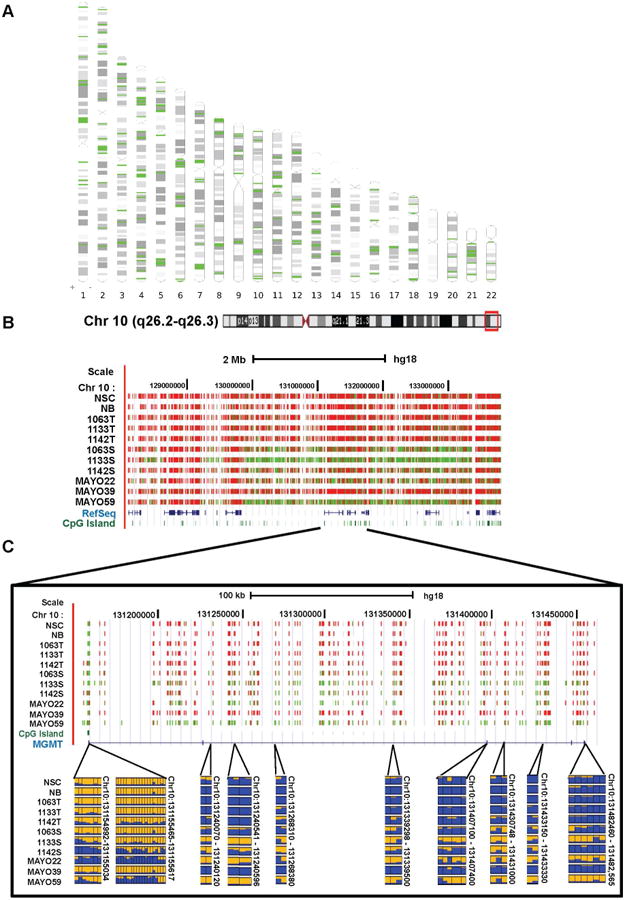
Megabase-size hypomethylated domains identified in GSC lines.
A: The 194 megabase windows in the genome contain significantly more hypomethylated CpG sites than expected. These domains are enriched near the ends and centromeres of chromosomes. B: UCSC genome browser snapshot of a 40-Mb interval on chromosome 10 that contains a long-range hypomethylated domain in the GSC cell line 1133S and GBM xenograft MAYO59 as compared to the control group. The color in the RRBS track indicates the percent of molecules that are methylated at each CpG position. Color scheme is consistent with in Fig. 3A and B. C: The observation of promoter hypermethylation and gene body hypomethylation in the MGMT gene. The upper panel shows a UCSC genome browser snapshot illustrating the RRBS results of MGMT. The lower panels illustrate the DNA methylation profiles of multiple DMRs located in the 5′-end and gene body of MGMT.
Integrated analysis of DNA methylation, CNA and gene expression
In addition to CpG methylation profiling of GSC and NSC lines, Affymetric genechip analysis was carried out to identify the differentially expressed genes. As shown in a volcano plot (Fig. 5A), differential gene expression of the grouped GSC lines (1142S, 1133S, and 1063S) versus two biological replicates of the NSC line identified 119 genes that were up regulated and 190 genes that were down regulated (fold change greater than 2, P-value < 0.05). Initial analysis of the gene expression data indicated the up-regulation of several HOX genes including strong expression of HOXC6, HOXA9, and HOXA10 in GSC lines, all which were confirmed by real time RT-PCR (Fig. 5B). Cluster analysis revealed the up-regulation of many HOX family genes in GSC lines 1063S and 1133S, but fewer in 1142S (Fig. 5C).
Fig. 5.
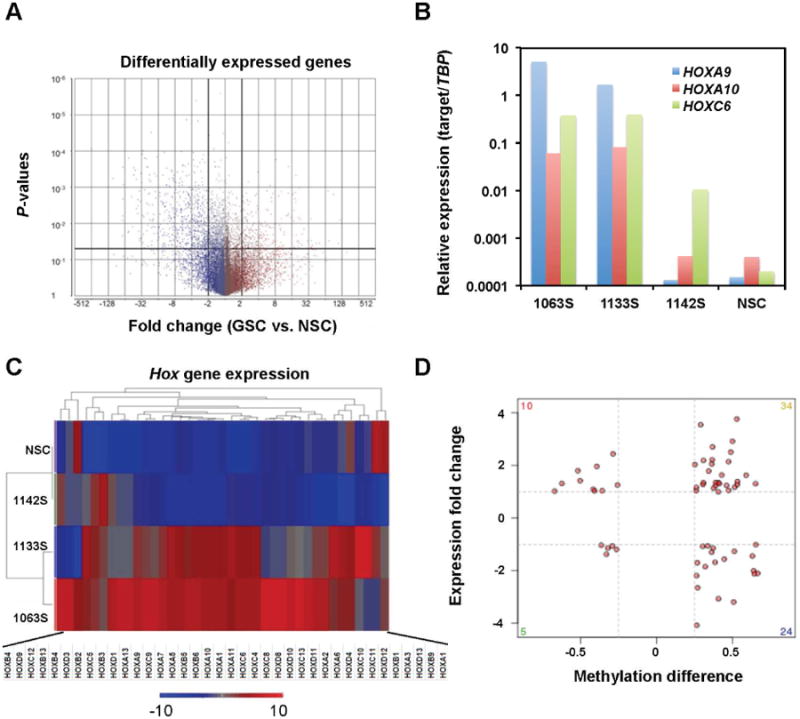
Differential DNA methylation correlates with differential expression in a small subset of genes.
A: Volcano plot highlights the differentially expressed genes. Each gene is represented by a single dot. Genes that have a 2-fold expression change and meet the P-value threshold (<0.05) are considered differentially expressed. B: qRT-PCR confirmation of the expression of four HOX genes in NSC and GSC lines. C: Heat map analysis identifies many HOX genes in GSC 1133 and 1063 as being up-regulated. D: Integrated analysis of gene expression and promoter methylation changes between NSC and GSC lines. Gene expression differences between NSC and GSC lines are plotted against the DNA methylation differences for each gene. Red points highlight those genes whose methylation values are positively or negatively correlated with gene expression.
The gene expression data were integrated with RRBS methylation data to identify the aberrant gene expression patterns in GSC lines 1142S, 1133S, and 1063S that may be a result of the alterations in DNA methylation. Of the 190 down-regulated genes in GSCs, 24 genes were hypermethylated in the promoter regions including several candidate tumor suppressor genes such as SPINT2, NEFM, PENK, and CCNA1 (Fig. 5D and Table S4). Of the 119 up-regulated genes in GSCs, 10 genes were hypomethylated in the promoter regions including several genes involved in tumor invasion and metastasis (CARD10, CTSB, CLIC3 and HMGA1) and glucose metabolism (G6PD and INSIG1).
The integration of DNA methylation and copy number data yielded complicated results. The Venn diagrams in Fig. S4 illustrate the number of genes affected by both genetic and epigenetic alterations. Although very few genes were consistently affected by the same types of genetic and epigenetic alterations across samples (Table S5), the integrative analysis did identify several candidate oncogenes (i. e., EGFR, NTSR1, LIMK1) that were affected by hypomethylation and copy number gain, as well as tumor suppressor genes (i.e., BCOR, SOX17, CASZ1) that were affected by hypermethylation and copy number loss. However, comparison with gene expression data revealed that only NTSR1 was significantly up-regulated in all three GSC lines and correlated with promoter hypomethylation and copy number gain.
We surveyed the Oncomine microarray database for a subset of genes, which were differentially expressed as a result of methylation status in the 5′ regulatory region (i.e., over-expressed and hypomethylated, or under-expressed and hypermethylated). The Oncomine microarray database revealed that these genes were also up- or down-regulated between GBMs and normal controls across multiple datasets, respectively. For instance, SPINT2, NEFM, PENK, CCNA1 were down-regulated in GBMs compared to normal controls in all six microarray datasets (data not shown). Using R2 microarray analysis and visualization platform (http://hgserver1.amc.nl/cgi-bin/r2/main.cgi), we examined whether the epigenetically regulated genes identified in GSCs were associated with patient survival in a previously published dataset (GSE16011) (Gravendeel et al., 2009) consisting of 276 gliomas (of all grades) and 8 controls. The Kaplan-Meier survival plots demonstrate that the expression of CTSB and G6PD are associated with patients' overall survival in GBM patients (Fig. S5).
Bisulfite pyrosequencing analysis of the hypermethylated region in SPINT2 promoter confirmed the RRBS results in the GSC, GBM and controls samples (Fig. 6A). The hypermethylation of SPINT2 was inversely correlated with its mRNA expression as RT-PCR analysis demonstrated that SPINT2 was significantly down-regulated in GSC samples as compared with the NSC cell line as well as several normal brain tissue samples (Fig. 6B). Analysis of TCGA data showed that SPINT2 was ranked number 52 (P = 1.6×10-25) among the top 1% of down-regulated genes in GBMs (Fig. 6C). This down-regulation was also inversely correlated with promoter methylation determined by Illumina 450K methylation array (Fig. 6D).
Fig. 6.
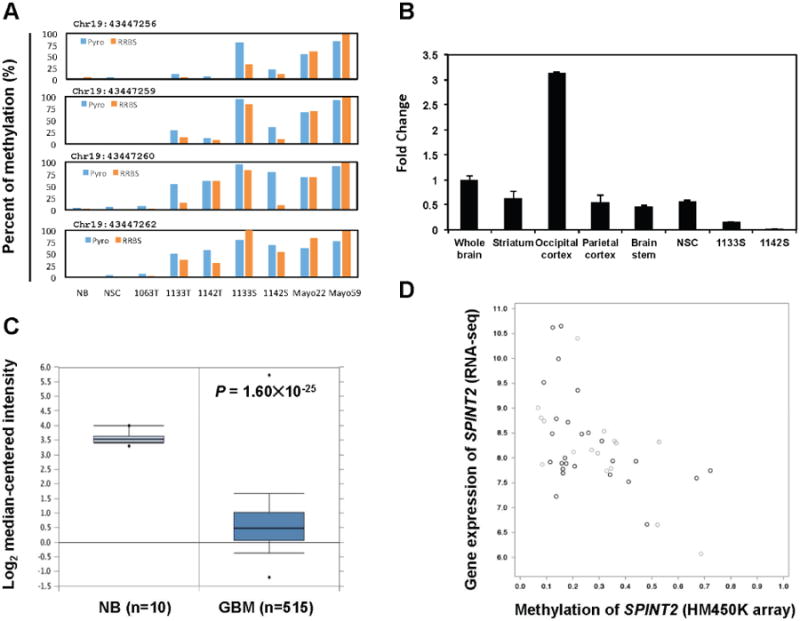
SPINT2 is silenced by DNA methylation in GBM.
A: Bisulfite pyrosequencing confirmed the RRBS methylation results at four CpG sites within the promoter of SPINT2. Each panel shows the comparison between bisulfite sequencing and RRBS data for the corresponding CpG site. B: qRT-PCR analysis of SPINT2 mRNA level revealed that SPINT2 was expressed at various parts of normal brain tissue and the NSC line, but significantly down-regulated in GSC lines 1133S and 1142S. C: SPINT2 was significantly down-regulated in GBMs compared to normal brain tissue samples in TCGA data set. The graph was generated using Oncomine (https://www.oncomine.org/). D: The SPINT2 mRNA expression level was inversely correlated with its promoter methylation status in primary GBM samples based on analyses using TCGA RNA-seq and Illumina 450K methylation array data sets. The correlation plot was generated using cBioPortal (http://www.cbioportal.org/).
SPINT2 is a candidate tumor suppressor gene in GBM
To determine if SPINT2 has tumor suppressor function in GBM, we overexpressed SPINT2 in GBM cell line U87G (Figs. 7A and S6). U87G cells stably expressing SPINT2 had a markedly reduced cell proliferation compared to the empty vector control as assessed by MTT assay (Fig. 7B). To determine the effect of SPINT2 re-expression on the ability of the GBM cells to grow in anchorage-independent conditions, we performed soft agar colony formation assay using stably transduced U87G cells. U87G cells transduced with empty vector produced a mean number of 900.3 ± 50.1 colonies, while SPINT2 overexpressing cells produced mean colony counts of 274.0 ± 30.8 (Fig. 7C and D). Similar to a previous report (Hamasuna et al., 2001), forced re-expression of SPINT2 also reduced cell mobility in U87G cells as accessed by artificial wound healing analysis (data not shown).
Fig. 7.
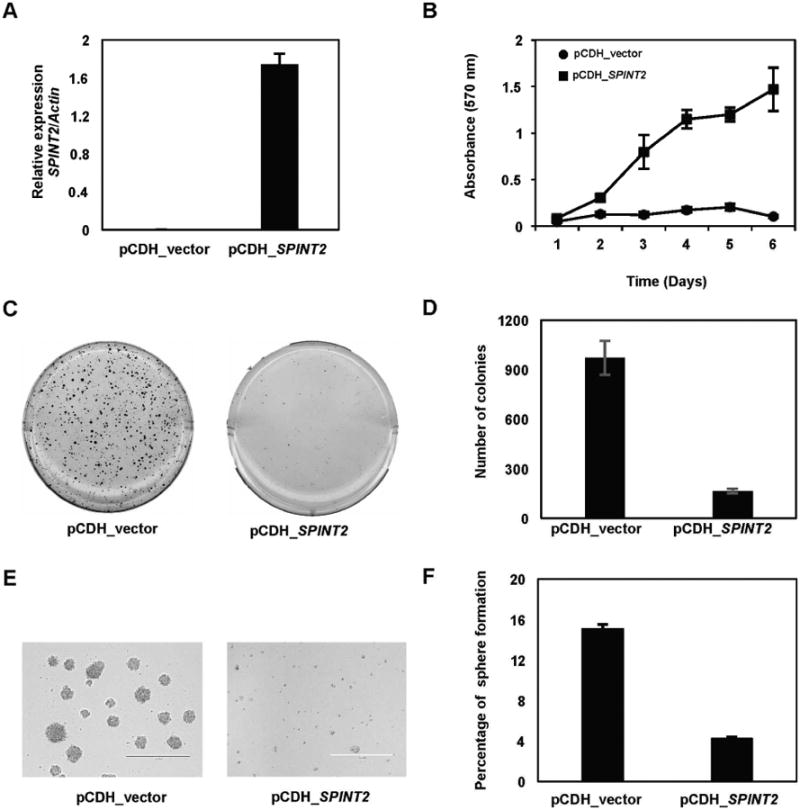
SPINT2 is a tumor suppressor of GBM.
A: qRT-PCR results demonstrated that SPINT2 was not expressed in U87G cells, but significantly up-regulated in cells transduced with a lentiviral vector expressing SPINT2 B: Forced expression of SPINT2 in U87G cells significantly inhibit cell proliferation as determined by MTT assays. C and D: Soft agar assay results demonstrated that forced expression of SPINT2 inhibited anchorage independent growth of U87G cells. E and F: Force expression of SPINT2 also inhibited formation of primary (E) and secondary tumor spheres (F) from U87G cells.
When placed in the serum free media (DN2L), U87G cells with empty vector control formed tumor spheres nicely (Fig. 7E). However, U87G cells stably transduced with SPINT2 showed significantly reduced ability to form tumor spheres (Fig. 7E). We collected the primary tumor spheres and performed sphere reformation assay. The primary tumor spheres were dissociated and the single cell suspensions were then seeded in 96-well plates at one cell per well. The percentage of sphere reformation was significantly reduced in SPINT2 re-expressing U87G cells as compared to the empty vector control (Fig. 7F), suggesting that forced expression of SPINT2 reduces the self-renewing capacity of U87G tumor stem cells.
Discussion
Numerous studies have demonstrated the presence of GSCs in primary GBM specimens. These GSCs can be cultured from human GBM specimens in defined medium in the presence of mitogens, express stem cell markers (CD133, SOX2, and Nestin), and have the capacity of self-renewal and subsequent differentiation upon mitogen removal (Galli et al., 2004; Singh et al., 2004; Vescovi et al., 2006; Dirks, 2008). In this study, we conducted an in-depth analysis of DNA methylation in GSCs derived from primary GBMs in relation to an untransformed karyotypically normal NSC line and a non-neoplastic brain tissue sample. The single-base resolution analysis revealed unique DNA methylation signatures in GSCs compared to normal controls. Interestingly, we identified two groups of GSC-specific DMRs whose methylation levels display an increasing or decreasing trend from normal controls to primary GBMs, then to GSC cell lines, respectively (Fig. 2A). For most of the GSC-specific DMRs, both hyper- and hypomethylated, primary GBMs displayed intermediate methylation levels between normal controls and GSCs. DNA methylation profiles of primary GBM tissues contain mixed signals due to nonneoplastic cell components of tumors (including endothelial and inflammatory cells), and the heterogeneous population of cancer cells, only some of which contribute to tumor progression and maintenance. Therefore, our results suggest that the three GSC lines are indeed derived from a subset of cells within the primary tumor tissues. The neurosphere culture condition might have enabled the clonal expansion of a subpopulation of cells with the GSC-specific DNA methylation signatures within primary GBMs. The copy-number analysis by array CGH also supports the notion that these three GSC lines represent sub-clones of original primary tumors. The three GSC lines possess the signature genomic abnormalities of primary tumors such as regional amplifications in chromosome 7 and the loss of regions in chromosome 10; however, they also gained additional chromosomal changes that are not detected in the original tumors. To address the concern that tissue culture may cause epigenetic artifacts (Baysan et al., 2014), we also studied the xenograft model of primary GBMs (Higgins et al., 2013). Since the tumor cells in the xenograft tumors were not exposed to long-term tissue culture conditions, the aberrant methylation changes identified in GBM xenografts were unlikely to be tissue culture-related changes. As expected, the GSC-specific methylation signatures can be found in GBM xenograft tumors derived from different patient samples. Therefore, the GSC-specific signatures identified in this study are unlikely to be caused by cell culture conditions. The increased number of DMRs identified in GSCs and GBM xenografts is likely due to two reasons. Firstly, the primary GBM tumors are not pure tumor cells, and the presence of non-tumor cells like immune cells and endothelial cells may have diluting effects on DNA methylation levels, therefore rendering some DMRs to fall below the cut off value used for detecting DMRs in primary GBM tissues. Secondly, the tumor stem cell population can acquire the dominance during continued proliferation, therefore leading to the increased detection of methylated genes unique to the GSCs or to a more aggressive clonal population of tumor cells. Most prior genome-wide DNA methylation studies in GBM have profiled tumor sections in relation to non-neoplastic brain tissues (Uhlmann et al., 2003; Cadieux et al., 2006; Martinez et al., 2009; TCGA, 2008; Wu et al., 2010). Our results strongly argue that methylation profiling in GSCs and GBM xenografts may better capture the true picture of epigenetic changes in GBMs.
Most GSC-specific differentially methylated genes are associated with biological functions in developmental processes, transcription regulation, differentiation, and brain-related neurogenesis. Among them, the hypermethylated genes were enriched in the PRC2 targeted genes, which is consistent with previous findings that de novo promoter hypermethylation in cancer cells was frequently identified among a subset of genes associated with PRC2 complex in ES cells (Ohm et al., 2007; Widschwendter et al., 2007). Recently, Stricker and colleagues (2013) have shown that the differentially methylated genes in GSC cells were significantly enriched in PRC2 target genes using Illumina 450K methylation arrays. Several studies have also demonstrated the importance of polycomb proteins such as BMI1 and EZH2 in maintaining the stemness of the GSC cells (Abdouh et al., 2009; Natsume et al., 2013). We identified candidate tumor suppressor genes silenced by promoter hypermethylation in GSCs. For instance, SPINT2 has been established as a tumor suppressor gene silenced by promoter methylation in medulloblastoma (Kongkham et al., 2008). SPINT2 is a negative regulator of the hepatocyte growth factor (HGF)/MET signaling pathway and its downregulation has already been implicated in other human cancers (Parr et al., 2004; Morris et al., 2005; Dong et al., 2010). Increasing evidence indicates that c-Met expression marks tumor-initiating stem-like cells and that c-Met signaling drives GSC cell stemness in vitro (Li et al., 2011; De Bacco et al., 2012; Joo et al., 2012;) Recently, we have shown that all three GBM-xenograft tumors (Mayo 22, Mayo39 and Mayo59) had active HGF and c-Met activities (Rath et al., 2013), and therefore SPINT2 promoter hypermethylation may have contributed to the activation of c-MET signaling in the three GBM xenograft tumor samples. A survey of the Oncomine microarray databases revealed that SPINT2 was frequently down-regulated in GBMs compared to normal controls in multiple microarray datasets, suggesting that epigenetic silencing of SPINT2 is a common event in GBMs. Forced re-expression of SPINT2 in GBM cell line U87G reduced proliferative capacity, anchorage-independent growth, cell motility, and tumor sphere formation in vitro, suggesting that SPINT2 may exert its tumor suppressor function through inhibiting the self-renewal capacity of GSCs.
Global hypomethylation is a hallmark in cancer. Recent whole genome bisulfite sequencing analyses in primary tumor and tumor cell lines have identified hypomethylation in megabase-scale domains in cancer cells (Varley et al., 2013). We have identified 194 of these megabase domains in GSC lines and GBM xenograft tumors. Interestingly, MGMT is located in one of the hypomethylated domains located on chromosome 10. We observed promoter hypermethylation and gene-body hypomethylation of MGMT as well as several other genes like AJAP1 and PTPRN2 (Fig. S4), both of which are frequently affected by promoter hypermethylation in cancer (Selamat et al., 2011; Lin et al., 2012). However, this mega-base domain hypomethylation is largely absent in the primary GBMs. The mechanism of global hypomethylation remains unclear in cancer epigenetics. It was speculated that a passive mechanism, whereby methylation is gradually lost over successive cell divisions, is responsible for global hypomethylation (Wild and Flanagan, 2010). That cancer cells grow faster than methylation can be copied from the replicating parental DNA, resulting in progressive loss of DNA methylation. Our observation of mega-base hypomethylation in GSC lines and GBM xenografts could be due to the significantly increased growth and cell division in cultures and immunodeficient mice. On the other hand, the mixed signals from the stromal cell components and the heterogeneous population of cancer cells may hide the hypomethylation signals in the primary GBM tumors. Similarly, whole genome bisulfite sequencing identified so-called partially methylated domains in primary colon cancer tissues (Hansen et al., 2011).
Integrative analysis of gene expression and DNA methylation identified a small number of genes whose expression is inversely correlated with DNA methylation. Several genes down-regulated by promoter hypermethylation in GSCs, specifically SPINT2, NEFM, PENK, and CCNA1, are candidate tumor suppressors, while hypomethylated and highly up-regulated genes are involved in tumor invasion and metastasis including CARD10, CTSB, CLIC3 and HMGA1. Examination of several published gene expression array studies demonstrated that the expression of SPINT2, NEFM, PENK, CCNA1, and CTSB is frequently altered in GBMs compared to normal controls, suggesting that these genes may play important roles in gliomagenesis. HOX genes encode for a group of dynamically regulated transcription factors that modulate cellular processes such as growth and communication during development. However, in neoplasms, some HOX genes are aberrantly expressed and have been shown to contribute to cancer progression (Abate-Shen, 2002; Abdel-Fattah et al., 2006). Particularly in GBMs, HOXA6, HOXA7, HOXA9, HOXA13, HOXB13, HOXD4, HOXD9, and HOXD13 have been reported to be substantially up regulated and may contribute to malignancy (Abdel-Fattah et al., 2006; Cillo et al., 1996). In another study, an expression signature dominated by HOX genes emerged as a predictor for poor survival in patients treated with concomitant chemo- or radiotherapy (Murat et al., 2008). Our results indicate that HOX family genes are frequently hypermethylated in primary GBM tissues, which is consistent with a previous report (Martinez et al., 2009). Their methylation levels further increased in GBM-derived GSC lines and xenografts. At the same time, the array CGH analysis indicates that HOX genes, particularly HOXA genes, are amplified in all three GBMs and GSC lines. Gene expression data also revealed significant up-regulation of HOX genes in the GSC cell lines, which is in good agreement with their copy number status in these samples. Although promoter and/or first exon hypermethylation is generally correlated with a reduction in gene expression, these results show the complexity of the tumor genome and it is possible that the copy number amplification has a greater significance on the regulation of the expression than DNA methylation. Because the sample size of each group in our study is relatively small, future study using a large patient population is needed to further the underlying molecular mechanisms.
The comparison among primary GBM tumors, GSC cell lines, GBM-xenografts and normal controls allowed us to identify the cancer-specific DNA methylation alterations in GBM and avoided the influence of the impurity and heterogeneous nature of primary GBM tumor tissues. Several genes aberrantly methylated in GSC lines have not previously been implicated in glioma, but are known to play a role in other neoplasms or in cellular processes related to malignancy. The complexity of tumor genomes cannot be neglected when comparing the DNA methylation changes with gene expression. Despite the widespread DNA methylation alterations in the HOX gene clusters, we found significant up-regulation of HOX genes in GSC. Our results generally support the use of GSC cells as a relevant model for investigating the epigenetic basis of glioblastoma, and comparison with NSC cell lines provides an important baseline in this setting. Finally, integrative molecular profiling was very useful to gain insight into the cancer genome of GBM stem-like cells and to understand the interplay of multiple mechanisms and aberrations that may contribute to malignant progression.
Materials and Methods
Patient tissue
Tissue samples from GBM specimens and fetal brain-derived neural stem cells from diencephalic and telencephalic regions were provided via appropriate Institutional Review Board-approved protocols of the Department of Surgery, Division of Neurological Surgery, and the Department of Pathology and Anatomical Sciences in the School of Medicine of the University of Missouri. The primary GBM samples were labeled as 1063T, 1133T, and 1142T (Fig. 1 and Table S6). The three corresponding GSC cell lines grown as spheres from the primary tumors were labeled as 1063S, 1133S, and 1142S. One normal brain tissue sample purchased from BioChain (Newark, CA, USA) was used as an additional control. In addition, we analyzed three GBM xenograft lines (Mayo22, Mayo39, Mayo59 in Table S7) developed by Dr. Jann N. Sarkaria, from the Mayo Clinic, Minnesota (Carlson et al., 2011).
Cell culture
GBM specimens 1063, 1133, and 1142 were snap-frozen in liquid nitrogen upon removal from the patients, and were stored in liquid nitrogen until use. To establish the neurosphere or tumor sphere cultures, primary tissue samples were mechanically dissociated and washed with PBS; red blood cells were removed with Histopaque (Sigma, USA). The single-cell suspensions were then grown as non-adherent cultures (spheres) in un-coated petri dishes with serum-free defined medium containing DMEM-F12 supplemented with 20 ng/mL epidermal growth (EGF; Invitrogen, USA), 20 ng/mL basic fibroblast growth factor (bFGF; Invitrogen), 1:50 B27 supplement (Invitrogen), 1:100 N2 supplement (Invitrogen), 10 ng/mL leukemia inhibitory factor (LIF; Millipore, USA), as a stem-cell permissive medium to facilitate the growth of neurospheres and tumor spheres, hereafter known as “DN2L medium” (Inagaki et al., 2007).
Immunofluorescence
Neurospheres derived from non-neoplastic controls and tumor spheres derived from GBMs were frozen embedded in OCT, and 10 μm cryostat sections were l abeled with antibody as previously described (Bleau et al., 2008). For differentiated cells, the samples were labeled and processed as described in the Human NS Cell Characterization Kit (Millipore, USA). Primary antibodies used were anti-Nestin, anti-SOX2, anti-Musashi, anti-β3 Tubulin, anti-GFAP, anti-Neurofilament 150 kD (all from Millipore), and anti-CD133 (Miltenyi Biotec, USA). Secondary antibodies used were anti-mouse Alexa Fluor 488, anti-rabbit Alexa Fluor 546, and anti-rabbit Alexa Fluor 488 (all from Invitrogen). Sections were counterstained with Vectashield (Vector Labs, USA) mounting medium that contains the DNA counter stain DAPI, before visualization. Negative controls were processed as described above with no primary antibody.
Imaging
Brightfield images of neurospheres and tumor spheres were captured with a Nikon D100 camera through an Olympus CKx41 upright microscope. Confocal images were captured with a Zeiss 510 META NLO and a Zeiss 5 live, and processed using LSM 5 Image Examiner software.
Base resolution DNA methylation analysis
RRBS analysis was performed as described previously (Pei et al., 2012). Briefly, 1 mg of genomic DNA was digested with Msp I (NEB, USA), end-repaired and ligated with methylated sequencing adaptors (Illumina). The ligated DNA was size-selected on a 3% NuSieve 3:1 agarose gel and treated with sodium bisulfite using Zymo Research EZ Methylation kit. The bisulfite-converted sequencing libraries were amplified by PCR using PfuTurbo Cx polymerase (Agilent Technologies, USA). PCR products were purified using AMpure beads (Agencourt, USA) and sequenced using Illumina GAIIx and HiSeq2000. The raw sequencing reads were trimmed to remove sequencing adapters, low quality bases (Q < 67 in Illumina 1.5) in the 3′ end and ambiguous bases in both ends. The cleaned bisulfite sequencing reads were mapped to the human genome using Bowtie (Langmead et al., 2009) as described previously (Pei et al., 2012). The methylation level of each CpG site was defined as the fraction of methylated reads to that of methylated and unmethylated reads combined. We used windows of length 200 bp with an overlap of 100 bp between to identify DMRs by summing the numbers of methylated and unmethylated CpGs in reads, respectively. The DMR was identified pairwisely between each tumor group versus the control, respectively (i.e., GBM vs. Control; GSC vs. Control). Cluster analysis was performed in Partek Genomics Suite (Partek, USA) using the DMRs identified by the Student's t-test. The DMRs were compared with RefSeq genes, CGIs, and repeats in the UCSC genome browser and classified into functional categories such as promoter [−1500 bp to transcription start sites (TSS)], 5′-untranslated region (UTR), coding sequences (CDS), 3′-UTR, intron, and intergenic regions.
Microarray analysis of gene expression and copy number variation
Total DNA and RNA were isolated (DNeasy, RNeasy kits; Qiagen, USA) from primary tissue and DN2L cultured cells upon disruption with the TissueLyser apparatus (Qiagen). For gene expression experiments, specimens were profiled using the Human W-6 v3.0 Expression BeadChips (Illumina, USA) which assays >48,000 transcripts and covers >25,400 genes in the NCBI RefSeq database. For array CGH experiments, specimens were profiled using Affymetrix Human SNP 5.0 array (Affymetrix, USA), which contains over 500,000 SNP probes and 420,000 non-polymorphic copy number probes. Both platforms are whole genome arrays and assays were performed in accordance with the respective manufacturer's protocol. Raw microarray data have been uploaded to Gene Omnibus database and can be accessed via accession number (pending upon acceptance) (http://www.ncbi.nlm.nih.gov/geo/). Gene expression and array CGH analyses were performed using Partek Genomics Suite. Briefly, genetic CN events from Affymetrix chips were reported based on CN values using Partek's Genomic Segmentation algorithm. SNP genotype data were generated through Affymetrix's Genotyping Console, then imported into Partek and compared against a 270 HapMap genotype sample dataset. Gene expression values from Illumina chips were generated by ANOVA (analysis of variance) to create a list of differentially expressed genes.
Lentivirus-mediated SPINT2 expression in U87G cells
A plasmid containing full-length human SPINT2 cDNA was purchase from Open Biosystems (USA). The cDNA was subcloned into the pCDH-CMV-MCS-EF1-Puro lentiviral vector (System Biosciences, USA). Empty vector was used as a negative control. Once sequence-verified, the lentiviral expression vectors were cotransfected with the lenti-packaging plasmids (Addgene, USA) into HEK293FT cells, and the pseudovirus-containing supernatant was collected 48 h post-transfection. Virus supernatant was used to infect U87G cells cultured in 6-well plate. Transduced cells were then selected with puromycin (1 μg/mL) to establish a stable cell line.
Soft agar colony formation assay
3000 cells were resuspended in DMEM media with 0.35% agar and overlaid on the top of the pre-solidified base layer containing DMEM media with 0.5% agar in a 6-well plate, and subsequently kept in a humidified chamber at 37°C for 2 weeks. Plates were stained with MTT, and colony counts was performed using NIH ImageJ 1.48 software package.
Tumor sphere reformation assay
Puromycin selected stable cell lines were placed into DN2L media in non-adherent tissue culture plate. After 5 days, the tumor spheres are dissociated with Accutase (Millipore). For sphere reformation assay, single-cell suspensions prepared form the primary tumor spheres were plated in the DN2L medium in non-adherent 96-well plate at one cell per well. After 11 days of culture, the number of spheres in each well was counted. Spheres were defined as morphologically characteristic three-dimensional structures of approximately > 400 μm.
Immunoblot analysis
The protein lysates were prepared from pCDH vector and pCDH-SPINT2 stable cell lines in RIPA buffer. Then, 30 μg of the protein lysates were separated on a 10% SDS-PAGE gel. The separated proteins were transferred to nitrocellulose membranes and probed with primary antibody and secondary antibody, respectively. Membranes were scanned, and bands were quantified using the Odyssey infrared imaging system (LI-COR, USA). The antibodies used were: anti-Flag M2 (F1804, Sigma), anti-β-Actin (C4) (sc-47778, Santa Cruz, USA), and goat anti-mouse IgG IRDye800CW (827-0836, LI-COR).
Supplementary Material
Fig. S1. Genome-wide distribution of hyper- and hypomethylated DMRs between GSC cell lines and the control group.
Fig. S2. Supervised cluster analysis of DNA methylation using DMRs between control and GSCs.
Fig. S3. The observation of promoter hypermethylation and gene body hypomethylation in PTPRN2 and AJAP1.
Fig. S4. Venn diagrams displaying the overlaps between genes consistently affected by genetic and epigenetic alterations in GSCs.
Fig. S5. Kaplan-Meier analysis of overall survival for CTSB and G6PD using the French dataset with the log-rank test P value indicated.
Fig. S6. Immunoblot analysis of Flag-tagged SPINT2 fusion protein expression in pCDH-SPINT2 stable cell line.
Table S1. Statistic summary of RRBS analyses performed in this study.
Table S2. List of genes differentially methylated at the promoter or gene body in GSC cell lines compared with the controls (NS and NB).
Table S3. List of hypomethylated megabase domains in GSC cell lines.
Table S4. Genes showing positive or negative correlation between gene expression and DNA methylation levels in the regions up or downstream of TSS (±1000 bp).
Table S5. Genes affected by both genetic and epigenetic changes.
Table S6. Demographical and diagnostic information of GBM specimens.
Table S7. Molecular characteristic of GBM xenograft tumors.
Acknowledgments
This work was supported in part by the National Institute of Health (Grant Nos. CA134304 and DA025779 to H.S., NS073611 and NS076759 to J.L.). H.S. is a Georgia Cancer Coalition Distinguished Cancer Scientist. We thank Dr. Jann N. Sarkaria for providing the GBM-xenograft models.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abate-Shen C. Deregulated homeobox gene expression in cancer: cause or consequence? Nat Rev Cancer. 2002;2:777–785. doi: 10.1038/nrc907. [DOI] [PubMed] [Google Scholar]
- Abdel-Fattah R, Xiao A, Bomgardner D, Pease CS, Lopes MB, Hussaini IM. Differential expression of HOX genes in neoplastic and non-neoplastic human astrocytes. J Pathol. 2006;209:15–24. doi: 10.1002/path.1939. [DOI] [PubMed] [Google Scholar]
- Abdouh M, Facchino S, Chatoo W, Balasingam V, Ferreira J, Bernier G. BMI1 Sustains Human Glioblastoma Multiforme Stem Cell Renewal. J Neurosci. 2009;29:8884–8896. doi: 10.1523/JNEUROSCI.0968-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- Baylin SB. DNA methylation and gene silencing in cancer. Nat Clin Pract Oncol. 2005;2(Suppl 1):S4–11. doi: 10.1038/ncponc0354. [DOI] [PubMed] [Google Scholar]
- Baysan M, Woolard K, Bozdag S, Riddick G, Kotliarova S, Cam MC, Belova GI, Ahn S, Zhang W, Song H, Walling J, Stevenson H, Meltzer P, Fine HA. Micro-environment causes reversible changes in DNA methylation and mRNA expression profiles in patient-derived glioma stem cells. PLoS ONE. 2014;9:e94045. doi: 10.1371/journal.pone.0094045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleau AM, Howard BM, Taylor LA, Gursel D, Greenfield JP, Lim Tung HY, Holland EC, Boockvar JA. New strategy for the analysis of phenotypic marker antigens in brain tumor-derived neurospheres in mice and humans. Neurosurg Focus. 2008;24:E28. doi: 10.3171/FOC/2008/24/3-4/E27. [DOI] [PubMed] [Google Scholar]
- Bredel M, Scholtens DM, Harsh GR, Bredel C, Chandler JP, Renfrow JJ, Yadav AK, Vogel H, Scheck AC, Tibshirani R, Sikic BI. A network model of a cooperative genetic landscape in brain tumors. JAMA. 2009;302:261–275. doi: 10.1001/jama.2009.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadieux BÆ, Ching TT, VandenBerg SR, Costello JF. Genome-wide hypomethylation in human glioblastomas associated with specific copy number alteration, methylenetetrahydrofolate reductase allele status, and increased proliferation. Cancer Res. 2006;66:8469–8476. doi: 10.1158/0008-5472.CAN-06-1547. [DOI] [PubMed] [Google Scholar]
- Carlson BL, Pokorny JL, Schroeder MA, Sarkaria JN. Establishment, maintenance and in vitro and in vivo applications of primary human glioblastoma multiforme (GBM) xenograft models for translational biology studies and drug discovery. Curr Protoc Pharmacol. 2011;52:1–14. doi: 10.1002/0471141755.ph1416s52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cillo C, Cantile M, Mortarini R, Barba P, Parmiani G, Anichini A. Differential patterns of HOX gene expression are associated with specific integrin and ICAM profiles in clonal populations isolated from a single human melanoma metastasis. Int J Cancer. 1996;66:692–697. doi: 10.1002/(SICI)1097-0215(19960529)66:5<692::AID-IJC18>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- De Bacco F, Casanova E, Medico E, Pellegatta S, Orzan F, Albano R, Luraghi P, Reato G, D'Ambrosio A, Porrati P, Patane M, Maderna E, Pollo B, Comoglio PM, Finocchiaro G, Boccaccio C. The MET oncogene is a functional marker of a glioblastoma stem cell subtype. Cancer Res. 2012;72:4537–4550. doi: 10.1158/0008-5472.CAN-11-3490. [DOI] [PubMed] [Google Scholar]
- Dirks PB. Brain tumor stem cells: bringing order to the chaos of brain cancer. J Clin Oncol. 2008;26:2916–2924. doi: 10.1200/JCO.2008.17.6792. [DOI] [PubMed] [Google Scholar]
- Dong WJ, Chen XB, Xie J, Sun PH, Wu YL. Epigenetic inactivation and tumor suppressor activity of HAI-2/SPINT2 in gastric cancer. Int J Cancer. 2010;127:1526–1534. doi: 10.1002/ijc.25161. [DOI] [PubMed] [Google Scholar]
- Engstrom PG, Tommei D, Stricker SH, Ender C, Pollard SM, Bertone P. Digital transcriptome profiling of normal and glioblastoma-derived neural stem cells identifies genes associated with patient survival. Genome Med. 2012;4:76. doi: 10.1186/gm377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eramo A, Ricci-Vitiani L, Zeuner A, Pallini R, Lotti F, Sette G, Pilozzi E, Larocca LM, Peschle C, De Maria R. Chemotherapy resistance of glioblastoma stem cells. Cell Death Differ. 2006;13:1238–1241. doi: 10.1038/sj.cdd.4401872. [DOI] [PubMed] [Google Scholar]
- Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, De Vitis S, Fiocco R, Foroni C, Dimeco F, Vescovi A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004;64:7011–7021. doi: 10.1158/0008-5472.CAN-04-1364. [DOI] [PubMed] [Google Scholar]
- Gravendeel LAM, Kouwenhoven MCM, Gevaert O, de Rooi JJ, Stubbs AP, Duijm JE, Daemen A, Bleeker FE, Bralten LBC, Kloosterhof NK, De Moor B, Eilers PHC, van der Spek PJ, Kros JM, Sillevis Smitt PAE, van den Bent MJ, French PJ. Intrinsic Gene Expression Profiles of Gliomas Are a Better Predictor of Survival than Histology. Cancer Res. 2009;69:9065–9072. doi: 10.1158/0008-5472.CAN-09-2307. [DOI] [PubMed] [Google Scholar]
- Hamasuna R, Kataoka H, Meng JY, Itoh H, Moriyama T, Wakisaka S, Koono M. Reduced expression of hepatocyte growth factor activator inhibitor type-2/placental bikunin (HAI-2/PB) in human glioblastomas: implication for anti-invasive role of HAI-2/PB in glioblastoma cells. Int J Cancer. 2001;93:339–345. doi: 10.1002/ijc.1349. [DOI] [PubMed] [Google Scholar]
- Hansen KD, Timp W, Bravo HC, Sabunciyan S, Langmead B, McDonald OG, Wen B, Wu H, Liu Y, Diep D, Briem E, Zhang K, Irizarry RA, Feinberg AP. Increased methylation variation in epigenetic domains across cancer types. Nat Genet. 2011;43:768–775. doi: 10.1038/ng.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani L, Bromberg JE, Hau P, Mirimanoff RO, Cairncross JG, Janzer RC, Stupp R. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- Higgins DM, Wang R, Milligan B, Schroeder M, Carlson B, Pokorny J, Cheshier SH, Meyer FB, Weissman IL, Sarkaria JN, Henley JR. Brain tumor stem cell multipotency correlates with nanog expression and extent of passaging in human glioblastoma xenografts. Oncotarget. 2013;4:792–801. doi: 10.18632/oncotarget.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki A, Soeda A, Oka N, Kitajima H, Nakagawa J, Motohashi T, Kunisada T, Iwama T. Long-term maintenance of brain tumor stem cell properties under at non-adherent and adherent culture conditions. Biochem Biophys Res Commun. 2007;361:586–592. doi: 10.1016/j.bbrc.2007.07.037. [DOI] [PubMed] [Google Scholar]
- Johnson D, O'Neill B. Glioblastoma survival in the United States before and during the temozolomide era. J Neurooncol. 2012;107:359–364. doi: 10.1007/s11060-011-0749-4. [DOI] [PubMed] [Google Scholar]
- Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3:415–428. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo KM, Jin J, Kim E, Ho Kim K, Kim Y, Gu Kang B, Kang YJ, Lathia JD, Cheong KH, Song PH, Kim H, Seol HJ, Kong DS, Lee JI, Rich JN, Lee J, Nam DH. MET signaling regulates glioblastoma stem cells. Cancer Res. 2012;72:3828–3838. doi: 10.1158/0008-5472.CAN-11-3760. [DOI] [PubMed] [Google Scholar]
- Kamnasaran D, Qian B, Hawkins C, Stanford WL, Guha A. GATA6 is an astrocytoma tumor suppressor gene identified by gene trapping of mouse glioma model. Proc Natl Acad Sci USA. 2007;104:8053–8058. doi: 10.1073/pnas.0611669104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kongkham PN, Northcott PA, Ra YS, Nakahara Y, Mainprize TG, Croul SE, Smith CA, Taylor MD, Rutka JT. An Epigenetic Genome-Wide Screen Identifies SPINT2 as a Novel Tumor Suppressor Gene in Pediatric Medulloblastoma. Cancer Res. 2008;68:9945–9953. doi: 10.1158/0008-5472.CAN-08-2169. [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Kotliarova S, Kotliarov Y, Li A, Su Q, Donin NM, Pastorino S, Purow BW, Christopher N, Zhang W, Park JK, Fine HA. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell. 2006a;9:391–403. doi: 10.1016/j.ccr.2006.03.030. [DOI] [PubMed] [Google Scholar]
- Lee TI, Jenner RG, Boyer LA, Guenther MG, Levine SS, Kumar RM, Chevalier B, Johnstone SE, Cole MF, Isono K, Koseki H, Fuchikami T, Abe K, Murray HL, Zucker JP, Yuan B, Bell GW, Herbolsheimer E, Hannett NM, Sun K, Odom DT, Otte AP, Volkert TL, Bartel DP, Melton DA, Gifford DK, Jaenisch R, Young RA. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell. 2006b;125:301–313. doi: 10.1016/j.cell.2006.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Li A, Glas M, Lal B, Ying M, Sang Y, Xia S, Trageser D, Guerrero-Cazares H, Eberhart CG, Quinones-Hinojosa A, Scheffler B, Laterra J. c-Met signaling induces a reprogramming network and supports the glioblastoma stem-like phenotype. Proc Natl Acad Sci USA. 2011;108:9951–9956. doi: 10.1073/pnas.1016912108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin N, Di C, Bortoff K, Fu J, Truszkowski P, Killela P, Duncan C, McLendon R, Bigner D, Gregory S, Adamson DC. Deletion or epigenetic silencing of AJAP1 on 1p36 in glioblastoma. Mol Cancer Res. 2012;10:208–217. doi: 10.1158/1541-7786.MCR-10-0109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez R, Martin-Subero JI, Rohde V, Kirsch M, Alaminos M, Fernandez AF, Ropero S, Schackert G, Esteller M. A microarray-based DNA methylation study of glioblastoma multiforme. Epigenetics. 2009;4:255–264. doi: 10.4161/epi.9130. [DOI] [PubMed] [Google Scholar]
- McTavish N, Copeland LA, Saville MK, Perkins ND, Spruce BA. Proenkephalin assists stress-activated apoptosis through transcriptional repression of NF-κ- and p53-regulated gene targets. Cell Death Differ. 2007;14:1700–1710. doi: 10.1038/sj.cdd.4402172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, Zhang X, Bernstein BE, Nusbaum C, Jaffe DB, Gnirke A, Jaenisch R, Lander ES. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008;454:766–770. doi: 10.1038/nature07107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris MR, Gentle D, Abdulrahman M, Maina EN, Gupta K, Banks RE, Wiesener MS, Kishida T, Yao M, Teh B, Latif F, Maher ER. Tumor suppressor activity and epigenetic inactivation of hepatocyte growth factor activator inhibitor type 2/SPINT2 in papillary and clear cell renal cell carcinoma. Cancer Res. 2005;65:4598–4606. doi: 10.1158/0008-5472.CAN-04-3371. [DOI] [PubMed] [Google Scholar]
- Munoz P, Iliou MS, Esteller M. Epigenetic alterations involved in cancer stem cell reprogramming. Mol Oncol. 2012;6:620–636. doi: 10.1016/j.molonc.2012.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murat A, Migliavacca E, Gorlia T, Lambiv WL, Shay T, Hamou MF, de Tribolet N, Regli L, Wick W, Kouwenhoven MC, Hainfellner JA, Heppner FL, Dietrich PY, Zimmer Y, Cairncross JG, Janzer RC, Domany E, Delorenzi M, Stupp R, Hegi ME. Stem cell-related “self-renewal” signature and high epidermal growth factor receptor expression associated with resistance to concomitant chemoradiotherapy in glioblastoma. J Clin Oncol. 2008;26:3015–3024. doi: 10.1200/JCO.2007.15.7164. [DOI] [PubMed] [Google Scholar]
- Natsume A, Ito M, Katsushima K, Ohka F, Hatanaka A, Shinjo K, Sato S, Takahashi S, Ishikawa Y, Takeuchi I, Shimogawa H, Uesugi M, Okano H, Kim SU, Wakabayashi T, Issa JPJ, Sekido Y, Kondo Y. Chromatin regulator PRC2 is a key regulator of epigenetic plasticity in glioblastoma. Cancer Res. 2013;73:4559–4570. doi: 10.1158/0008-5472.CAN-13-0109. [DOI] [PubMed] [Google Scholar]
- Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP, Verhaak RGW, Hoadley KA, Hayes DN, Perou CM, Schmidt HK, Ding L, Wilson RK, Van Den Berg D, Shen H, Bengtsson H, Neuvial P, Cope LM, Buckley J, Herman JG, Baylin SB, Laird PW, Aldape K. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17:510–522. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohm JE, McGarvey KM, Yu X, Cheng L, Schuebel KE, Cope L, Mohammad HP, Chen W, Daniel VC, Yu W, Berman DM, Jenuwein T, Pruitt K, Sharkis SJ, Watkins DN, Herman JG, Baylin SB. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat Genet. 2007;39:237–242. doi: 10.1038/ng1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parr C, Watkins G, Mansel RE, Jiang WG. The hepatocyte growth factor regulatory factors in human breast cancer. Clin Cancer Res. 2004;10:202–211. doi: 10.1158/1078-0432.ccr-0553-3. [DOI] [PubMed] [Google Scholar]
- Pei L, Choi JH, Liu J, Lee EJ, McCarthy B, Wilson JM, Speir E, Awan F, Tae H, Arthur G, Schnabel JL, Taylor KH, Wang X, Xu D, Ding HF, Munn DH, Caldwell C, Shi H. Genome-wide DNA methylation analysis reveals novel epigenetic changes in chronic lymphocytic leukemia. Epigenetics. 2012;7:567–578. doi: 10.4161/epi.20237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rath P, Lal B, Ajala O, Li Y, Xia S, Kim J, Laterra J. In vivo c-met pathway inhibition depletes human glioma xenografts of tumor-propagating stem-like cells. Transl Oncol. 2013;6:104–111. doi: 10.1593/tlo.13127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkaria JN, Carlson BL, Schroeder MA, Grogan P, Brown PD, Giannini C, Ballman KV, Kitange GJ, Guha A, Pandita A, James CD. Use of an orthotopic xenograft model for assessing the effect of epidermal growth factor receptor amplification on glioblastoma radiation response. Clin Cancer Res. 2006;12:2264–2271. doi: 10.1158/1078-0432.CCR-05-2510. [DOI] [PubMed] [Google Scholar]
- Selamat SA, Galler JS, Joshi AD, Fyfe MN, Campan M, Siegmund KD, Kerr KM, Laird-Offringa IA. DNA methylation changes in atypical adenomatous hyperplasia, adenocarcinoma in situ, and lung adenocarcinoma. PLoS ONE. 2011;6:e21443. doi: 10.1371/journal.pone.0021443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- TCGA. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlmann K, Rohde K, Zeller C, Szymas J, Vogel S, Marczinek K, Thiel G, Nurnberg P, Laird PW. Distinct methylation profiles of glioma subtypes. Int J Cancer. 2003;106:52–59. doi: 10.1002/ijc.11175. [DOI] [PubMed] [Google Scholar]
- Varley KE, Gertz J, Bowling KM, Parker SL, Reddy TE, Pauli-Behn F, Cross MK, Williams BA, Stamatoyannopoulos JA, Crawford GE, Absher DM, Wold BJ, Myers RM. Dynamic DNA methylation across diverse human cell lines and tissues. Genome Research. 2013;23:555–567. doi: 10.1101/gr.147942.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vescovi AL, Galli R, Reynolds BA. Brain tumour stem cells. Nat Rev Cancer. 2006;6:425–436. doi: 10.1038/nrc1889. [DOI] [PubMed] [Google Scholar]
- Ward RJ, Dirks PB. Cancer stem cells: at the headwaters of tumor development. Annu Rev Pathol. 2007;2:175–189. doi: 10.1146/annurev.pathol.2.010506.091847. [DOI] [PubMed] [Google Scholar]
- Widschwendter M, Fiegl H, Egle D, Mueller-Holzner E, Spizzo G, Marth C, Weisenberger DJ, Campan M, Young J, Jacobs I, Laird PW. Epigenetic stem cell signature in cancer. Nat Genet. 2007;39:157–158. doi: 10.1038/ng1941. [DOI] [PubMed] [Google Scholar]
- Wild L, Flanagan JM. Genome-wide hypomethylation in cancer may be a passive consequence of transformation. Biochim Biophys Acta. 2010;1806:50–57. doi: 10.1016/j.bbcan.2010.03.003. [DOI] [PubMed] [Google Scholar]
- Wu X, Rauch TA, Zhong X, Bennett WP, Latif F, Krex D, Pfeifer GP. CpG island hypermethylation in human astrocytomas. Cancer Res. 2010;70:2718–2727. doi: 10.1158/0008-5472.CAN-09-3631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurdak H, Zhu S, Romero A, Lorger M, Watson J, Chiang CY, Zhang J, Natu VS, Lairson LL, Walker JR, Trussell CM, Harsh GR, Vogel H, Felding-Habermann B, Orth AP, Miraglia LJ, Rines DR, Skirboll SL, Schultz PG. An RNAi screen identifies TRRAP as a regulator of brain tumor-initiating cell differentiation. Cell Stem Cell. 2010;6:37–47. doi: 10.1016/j.stem.2009.11.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Genome-wide distribution of hyper- and hypomethylated DMRs between GSC cell lines and the control group.
Fig. S2. Supervised cluster analysis of DNA methylation using DMRs between control and GSCs.
Fig. S3. The observation of promoter hypermethylation and gene body hypomethylation in PTPRN2 and AJAP1.
Fig. S4. Venn diagrams displaying the overlaps between genes consistently affected by genetic and epigenetic alterations in GSCs.
Fig. S5. Kaplan-Meier analysis of overall survival for CTSB and G6PD using the French dataset with the log-rank test P value indicated.
Fig. S6. Immunoblot analysis of Flag-tagged SPINT2 fusion protein expression in pCDH-SPINT2 stable cell line.
Table S1. Statistic summary of RRBS analyses performed in this study.
Table S2. List of genes differentially methylated at the promoter or gene body in GSC cell lines compared with the controls (NS and NB).
Table S3. List of hypomethylated megabase domains in GSC cell lines.
Table S4. Genes showing positive or negative correlation between gene expression and DNA methylation levels in the regions up or downstream of TSS (±1000 bp).
Table S5. Genes affected by both genetic and epigenetic changes.
Table S6. Demographical and diagnostic information of GBM specimens.
Table S7. Molecular characteristic of GBM xenograft tumors.