

Abstract
Background and Aims Sorbus domestica (Rosaceae) is one of the rarest deciduous tree species in Europe and is characterized by a scattered distribution. To date, no large-scale geographic studies on population genetics have been carried out. Therefore, the aims of this study were to infer levels of molecular diversity across the major part of the European distribution of S. domestica and to determine its population differentiation and structure. In addition, spatial genetic structure was examined together with the patterns of historic and recent gene flow between two adjacent populations.
Methods Leaf or cambium samples were collected from 17 populations covering major parts of the European native range from north-west France to south-east Bulgaria. Seven nuclear microsatellites and one chloroplast minisatellite were examined and analysed using a variety of methods.
Key Results Allelic richness was unexpectedly high for both markers within populations (mean per locus: 3·868 for nSSR and 1·647 for chloroplast minisatellite). Moreover, there was no evidence of inbreeding (mean Fis = –0·047). The Italian Peninsula was characterized as a geographic region with comparatively high genetic diversity for both genomes. Overall population differentiation was moderate (FST = 0·138) and it was clear that populations formed three groups in Europe, namely France, Mediterranean/Balkan and Austria. Historic gene flow between two local Austrian populations was high and asymmetric, while recent gene flow seemed to be disrupted.
Conclusions It is concluded that molecular mechanisms such as self-incompatibility and high gene flow distances are responsible for the observed level of allelic richness as well as for population differentiation. However, human influence could have contributed to the present genetic pattern, especially in the Mediterranean region. Comparison of historic and recent gene flow may mirror the progress of habitat fragmentation in eastern Austria.
Keywords: Chloroplast DNA, gene flow, genetic variability, habitat fragmentation, nuclear microsatellites, self-incompatibility, Rosaceae, Sorbus domestica, true service tree
INTRODUCTION
The true service tree (Sorbus domestica), a light-demanding, insect-pollinated, rosaceous species, is distributed in the Mediterranean as well as in parts of France, Switzerland, Germany and Austria (Kutzelnigg, 1995), and also has a very scarce occurrence in the British Isles. It is still unclear to what extent this current distribution is natural in view of the species’ cultivation since Roman times (Rotach, 2003). There is both historic (Kausch-Blecken von Schmeling, 2000) and recent evidence (Termentzi, 2008) for use of the fruits for cider production and for medical purposes, which could have enhanced the species’ dispersal by humans after the last glacial period. However, rareness and scattered distribution characterize its present range, mainly as a consequence of preference for high forests instead of coppices with standards (Frochot et al., 2008) and for dense coniferous forests instead of open broad-leafed stands (Ferrini et al., 2008) during the last century. Theoretically, habitat fragmentation decreases the number of species (Mac Arthur and Wilson, 1967) and erodes genetic variation within and among populations due to increasing genetic drift, elevated inbreeding and reduced gene flow, and eventually increases the probability of local extinction of sub-populations (Young et al., 1996). This has been recently confirmed in Fagus sylvatica (Jump and Penuelas, 2006). Traits conferring adaptation to a fragmented habitat are, among others, long-distance pollen and seed dispersal (Nason et al., 1998; Levey et al., 2005). In S. domestica, rare long-distance dispersal events in a Swiss population were reported (maximum 16 km for pollen and maximum 12 km for seed dispersal, respectively; Kamm et al., 2009). From an evolutionary point of view, it can be assumed that this tree species is well adapted to a patchy habitat structure and that these patches are part of a metapopulation dynamic (Rotach, 2003). These dynamics are characterized through their demographic and genetic stochasticity where the degree of fragmentation and the size of ther remaining populations play a key role for the risk of local extinction (Hanski, 1998). Moreover, the persistence of a few large populations, which could act as refugia, is necessary for protecting genetic variation in the long term and to avoid extinction (Gilpin, 1991). Similar effects have been observed in several studies for the related species Sorbus torminalis, which shares life characteristics in terms of habitat structure and reproduction ecology. A main outcome of these studies was the finding of maintenance of high genetic diversity even in populations of small census size (e.g. Hoebee et al., 2006; Angelone et al., 2007).
Up to now, only a few population genetic and morphometric studies in S. domestica have been carried out, which mainly focused on a small geographical scale (e.g. Kamm et al., 2009, 2010, 2011; Nyari, 2010; Brus et al., 2011). A study which encompasses populations covering the main part of its distribution has been lacking. Hence, this study has two objectives: (1) in a first step we investigate levels of molecular diversity and differentiation in S. domestica by analysing samples from the western to the eastern European distribution at both the chloroplast and the nuclear DNA level; and (2) in the second step we zoom in on two putative natural populations located in Austria, which are geographically close to each other (about 60 km) to find out more about their population history by scrutinizing spatial genetic structure and the extent of historic and recent gene flow between them.
MATERIALS AND METHODS
Population sampling
Leaf or cambium samples were collected from 17 populations of Sorbus domestica L. covering major parts of the European native range from France (including Corsica), Switzerland, Italy (including Sicily and Elba), Slovenia, Croatia, Serbia, Bosnia-Herzegovina, Bulgaria and Austria (Table 1). For some populations it was not possible to obtain an adequate sample size because of the scattered nature of the species, but we considered this in our analysis. One of the Austrian populations – AT (NC) – is not a natural population, but a national collection of individuals originating from different regions including Burgenland (eastern Austria), Lower Austria and Vienna, and was established for long-term ex situ conservation of the species. For samples from the central and southern part of France, we also resorted to clonal archives (French National Collection, clone bank of INRA Avignon). Despite the fact that these individuals were collected across a wider geographical area, we pooled them into two populations (southern France and central France), which seems to be justifiable given the dispersal distances (Kamm et al., 2009) as well as the results of the cluster analysis (see below; Supplementary Data Fig. S1 shows the respective origins of these individuals; the original spatial positions of these trees in the field were reconstructed with data from INRA Avignon). There is a huge number of intermediate species in the genus Sorbus, which are thought to have originated through hybridization. However, until now, no hybrids between S. domestica and any other Sorbus species have been observed (e.g. Nelson-Jones et al., 2002; Ludwig et al., 2013). Therefore, we can exclude a bias through introgression. Localization of individual trees was supported by forest inventory data. Fresh leaf material was sampled from trees and stored in silica gel. For the Austrian populations, we used exclusively cambium samples, because sampling was done outside of the growing season. A piece of approx. 1 cm2 was taken from the stem base and stored in silica gel until DNA extraction. We sampled only trees with a dbh (diameter of breast height) between 30 and 50 cm to obtain an approximately even-aged sample.
Table 1.
Analysed populations of Sorbus domestica
| Population code | Location | Latitude | Longitude | Origin | Sampler | cpDNA | nSSR |
|---|---|---|---|---|---|---|---|
| F (NW) | France (north-west) | 48°01′39·61″ | –01°45′12·90″ | pop | MR | 33 | 27 |
| F (C) | France (central) | col/pop | EC/JT | 38 | 33 | ||
| F (S) | France (south) | col | EC | 24 | 23 | ||
| CH | Switzerland | 47°43′44·63″ | 08°38′23·76″ | pop | UK | 18 | ND |
| EB | Elba | 42°47′50·28″ | 10°18′23·76″ | pop | TK | 14 | 11 |
| CS | Corsica | 42°29′12·21″ | 09°27′24·45″ | pop | JT | 14 | 15 |
| IT (N) | Italy (north) | 43°57′44·18″ | 10°55′22·59″ | pop | LG | 13 | 13 |
| IT (C) | Italy (central) | 42°14″37·58″ | 13°01′24·93″ | pop | FV | 4 | 4 |
| SC | Sicily | 37°34′24·60″ | 14°21′07·20″ | pop | CS | 4 | 3 |
| SLO | Slovenia | 45°47′00·24″ | 14°05′04·20″ | pop | DB | 31 | 18 |
| AT (NC) | Austria (national collection) | 47°47′30·94″ | 16°06′56·04″ | col | HK | 29 | 19 |
| AT (WD) | Austria (Wolkersdorf) | 48°25′25·35″ | 16°36′21·15″ | pop | JPG | 38 | 41 |
| AT (MS) | Austria (Merkenstein) | 47°57′34·42″ | 16°07′16·21″ | pop | JPG | 45 | 45 |
| HR | Croatia | 45°08′23·28″ | 14°46′13·08″ | pop | MI | 34 | 18 |
| BIH | Bosnia Hercegovina | 43°37′50·16″ | 18°56′51·72″ | pop | DB | 49 | 18 |
| SRB | Serbia | 44°05′10·68″ | 22°05′58·56″ | pop | DB | 32 | 7 |
| BG | Bulgaria | 42°05′35·36″ | 27°40′53·39″ | pop | PZ | 21 | 20 |
| total | 441 | 302 |
cpDNA, data from the chloroplast minisatellite; nSSR, data from nuclear microsatellites; ND, no data; pop, forest population; col, national collection with known origin; MR, Michel Rondouin; EC, Eric Collin; JT, Jean Thevenet; UK, Urs Kamm; TK, Thomas Kirisits; LG, Luisa Ghelardini; FV, Federico Vesella; CS, Christian Stauffer; DB, Dalibor Ballian; HK, Heino Konrad; JPG, Jan-Peter George; MI, Marilena Idzojtic; PZ, Petar Zhelev.
DNA extraction, PCR and genotyping
Dried material was shock-frozen in liquid nitrogen for 3 min and crushed for 1 min at 20 Hz using a Qiagen Tissue Lyser device (Qiagen, Germany). DNA was extracted from 40–60 mg of dry leaf or cambium material using a DNeasy Plant Mini Kit (Qiagen, USA). The given protocol provided by the manufacturer was slightly modified by using 600 μL of AP1 buffer and 150 μL of elution buffer (two elution steps instead of one). We determined the concentration and purity of DNA using an ND-1000 spectrophotometer (NanoDrop, USA). DNA was stored at 4 °C. Initially we used eight nuclear microsatellite loci and one chloroplast (cp) minisatellite. The nuclear microsatellite loci used were MSS5, MSS16, BGT23b, MS14h03, CH01h01, CH01h10, CH02c09 and CH02d08, formerly also employed by Kamm et al. (2009). The cp-minisatellite is a 22 bp repeat with a (CATTATATTATTGATTTTAGTT)n motif (see Supplementary Data Table S1 for further information on the used markers). It was found by screening the rps16 region, which had also been described as polymorphic for several other related species (e.g. Oxelmann et al., 1997; Chester et al., 2007). For the nuclear microsatellites each reaction volume contained approx. 10 ng of DNA, 1× PCR buffer, 1 mm MgCl2, 0·15 μm of each primer (forward and reverse), 1·6 mm dNTPs, 0·6 U of polymerase (Peqlab, Germany), and ddH2O to a total volume of 10·75 μL. Primers were combined in two multiplex-PCR amplifications with four primers in each set. The reaction volume for the chloroplast minisatellite contained approx. 10 ng of DNA, 1× reaction buffer, 0·5 mm MgCl2, 1 μm of each primer, 0·8 mm dNTPs, 0·5 U of polymerase, and ddH2O to a total volume of 12·5 μL. PCR conditions for nuclear markers were initialized by a denaturation step at 94 °C for 5 min and followed by 30 cycles of 94 °C for 45 s, 50 or 60 °C (depending on the locus) for 45 s, 72 °C for 1 min and a final extension step at 72 °C for 10 min. For the chloroplast marker, PCR started with denaturation at 94 °C for 5 min, followed by 35 cycles of 94 °C for 1 min, 58 °C for 1 min, 72 °C for 30 s and a final extension step at 72 °C for 10 min. PCRs were carried out on a PTC100 (MJ, USA). Fragment analysis was done through capillary gel electrophoresis using a CEQ8000 sequencer (Beckman-Coulter, USA). For the analysis of the cp-minisatellite, we sequenced a sub-sample of 100 individuals to make sure that haplotypes were identical by descent. No signs of homoplasy were found.
Data analysis
Microsatellite data were checked for the occurrence of null alleles, genotyping errors and large allele dropouts using Micro-Checker (van Oosterhout et al., 2004). Deviations from Hardy–Weinberg proportions were assessed using Genepop (Raymond and Rousset, 1995) with 10 000 dememorizations, 100 batches and 10 000 iterations using Monte Carlo Markov chain (MCMC) simulations. Linkage disequilibrium was tested using Fstat 2.9.3 (Goudet, 1995). Descriptive population genetic statistics such as allele frequencies, effective number of haplotypes/alleles, unbiased haplotype diversity, number of private haplotypes/alleles, observed and expected heterozygosity and Fis values were calculated with Genalex 6·5 (Peakall and Smouse, 2012). Allelic richness with a rarefaction to ten individuals was calculated with Fstat for nuclear microsatellites and using Contrib (Petit et al., 1998) for the cp-minisatellite.
We used two methods to infer the relationship among populations. First we computed an unweighted pair group method arithmetic average dendrogram (UPGMA) from 1000 bootstrapped matrices based on Cavalli-Sforza and Edwards’ chord distances (Cavalli-Sforza and Edwards, 1967) created by Microsatellite Analyser (MSA) (Dieringer and Schlötterer, 2003). To find the ‘lowest common denominator’ we constructed a consensus tree following the majority rule out of the 1000 produced matrices using the programs Neighbour and Consense in the Phylip 3.63 package (Felsenstein, 1989). Additionally, we performed a principal co-ordinate analysis (PCoA) to visualize configurations among interpopulation genetic distances. This was carried out in Genalex using Nei’s standard genetic distance (Nei, 1972).
To infer population structure, we used the software Structure 2.3.4 (Pritchard et al., 2000), which uses a Bayesian clustering algorithm, where individuals are assigned to a pre-defined number of clusters (K). Assignment is done in such a way that deviations from Hardy–Weinberg proportions and gametic phase disequilibrium within clusters are minimized. This analysis was performed with K-values from 2 to 6 and a total run length of 1 000 000 with a burn-in period of 200 000. Each run was repeated four times for reasons of iteration. To find the appropriate number of clusters, we used the ΔK statistic of Evanno et al. (2005) implemented in the software Structure Harvester (Earl and von Holdt, 2012).
To obtain information about the levels of differentiation among populations, we performed an analysis of molecular variance (AMOVA) using Arlequin 3.5 (Excoffier and Lischer, 2010) where global FST and RST values were calculated. Groups for AMOVA were defined according to their geographical proximity and also by the above-mentioned tree construction method, PCoA and individual-based population assignment. Allelic richness, observed and expected heterozygosity, tree construction and PCoA as well as AMOVA were performed only for populations with a sample size ≥10. To extract information also from populations with lower sample size, we included them in another approach and calculated the allelic distance (D0) according to Gregorius (1974) among four groups (Austria, Balkan, France and Mediterranean). D0 is the mean of differences (absolute values) in relative allele frequencies between populations and can vary between 0 and 1 (D0 = 1, populations are genetically completely different; D0 = 0, populations are genetically identical). D0 was not calculated pairwise, but as a distance of one group of populations against the residual gene pool of the remaining groups.
Finally, we inferred spatial genetic structure for the within-population analysis for the two natural Austrian populations using the kinship coefficient of Loiselle (1995) using SPAGeDi 1.4 (Hardy and Vekemans, 2002). Individual sampling locations were permuted 10 000 times. The software Migrate-n (Beerli, 2006) was used to investigate the amount and direction of historic gene flow by calculating the parameters θ and M, where θ is four times the effective population size multiplied by the mutation rate per site and generation, and M is the immigration rate divided by the mutation rate. The latter parameter gives information on how much more important migration is relative to mutation to bring new variants into a population. Multiplying θ by M then gives the number of migrants into a population per generation. In this case ‘historic’ refers back to the date of the most recent common ancestor which is assumed to be 4Ne generations (for diploid organisms). We used the Bayesian inference method implemented in Migrate-n and also the continuous Brownian motion model. Runs were started with FST values to estimate θ and M. We assumed a constant mutation rate and used the following MCMC settings: 500 000 recorded steps, 100 000 discarded trees per chain, and an increment of 20 and 500 bins for θ and M, respectively. In the following runs, we used the newly estimated values for θ and M as calculated by the software until the posterior distributions for both parameters converged. Instead of conventional convergence diagnostics, we used the width of the 95 % confidence interval as the evaluation criterion: if these intervals were overlapping in at least three final runs for both parameters, we assessed the results as accurate. The run with the highest marginal likelihood was used for interpretation.
Recent gene flow was inferred with Bayesass 3.0 (Wilson and Rannala, 2003) where the probability of migration for the last two generations within and between populations is calculated. Bayesass uses a Bayesian method to estimate recent migration rates, population allele frequencies, inbreeding coefficients and individual migrant ancestries. It assumes that levels of differentiation between populations are high and that migration rates are low for an accurate estimation. A further assumption is that all populations which are considered to exchange migrants have been sampled. These assumptions are well fulfilled by our two model populations. We performed 10 000 000 iterations with a burn-in period of 1 00 000 to let the Markov chain reach a steady state, and chose a sampling interval of 1000. To be sure that our results were reliable, we repeated the runs with different mixing parameter values (from 0·1 to 0·5) and also started each time from a different initial random seed. These analyses were performed exclusively for the two Austrian populations, because here we had sufficient sample sizes (>40) and nearly equal census sizes (approx. 150 individuals in both populations), and because we were able to exclude a potential bias from non-sampled populations, that nevertheless might have contributed to gene flow (Beerli, 2004).
RESULTS
Chloroplast and nuclear genetic diversity
The cp-minisatellite revealed a total of five haplotypes with a minimum of two and a maximum of four haplotypes per population. Haplotype 244 was most frequent and occurred in all populations with a sample size >10. Haplotype 222 was absent in northern Italy and Slovenia and most frequent in Bulgaria and central Italy. Haplotype 266 was common in all populations except Switzerland, Serbia and Bulgaria. The shortest and the longest haplotype (200 and 288, respectively) showed peculiarities concerning their occurrences. While haplotype 200 occurred only twice (once each in Croatia and in Austria-Wolkersdorf), haplotype 288 occurred more frequently, but only in northern Italy and in Elba. Private haplotypes were not observed. Haplotype richness was highest in Elba and Croatia as well as in north-western and central France, whereas Bulgaria, Serbia, Slovenia and Switzerland showed low levels of haplotype richness (see Table 2 and Fig. 1).
Table 2.
Genetic diversity indices of the analysed S. domestica populations
| Chloroplast minisatellite |
Nuclear SSR |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Population | Na | Ne | RS | uh | Group | Na | Ne | RS | Ho | He | Fis | Ap |
| F (NW) | 3 | 2·889 | 1·959 | 0·674 | French | 5·000 | 2·740 | 3·774 | 0·603 | 0·602 | −0·008 | 2 |
| F (C) | 3 | 2·971 | 1·971 | 0·681 | French | 5·286 | 3·419 | 3·871 | 0·693 | 0·675 | −0·035 | 0 |
| F (S) | 3 | 2·642 | 1·904 | 0·649 | French | 5·429 | 2·794 | 4·181 | 0·595 | 0·588 | −0·013 | 0 |
| CH | 2 | 1·385 | 0·931 | 0·294 | ND | ND | ND | ND | ND | ND | ND | |
| EB | 4 | 3·161 | 2·702 | 0·736 | Mediteranean | 3·571 | 2·163 | 3·185 | 0·469 | 0·454 | −0·071 | 2 |
| CS | 3 | 2·085 | 1·923 | 0·560 | Mediteranean | 5·143 | 3·229 | 4·108 | 0·575 | 0·610 | 0·035 | 0 |
| IT (N) | 3 | 2·195 | 1·958 | 0·590 | Mediteranean | 5·286 | 3·674 | 4·461 | 0·648 | 0·607 | −0·048 | 2 |
| IT (C) | 2 | 1·600 | 0·500 | Mediteranean | 3·286 | 2·497 | 0 | |||||
| SC | 2 | 1·600 | 0·500 | Mediteranean | 2·857 | 2·506 | 0 | |||||
| SLO | 2 | 1·538 | 0·956 | 0·361 | Balkan | 3·429 | 2·251 | 2·994 | 0·642 | 0·480 | −0·274 | 0 |
| AT (NC) | 3 | 2·046 | 1·343 | 0·530 | Austrian | 4·857 | 2·696 | 3·621 | 0·575 | 0·544 | 0·003 | 0 |
| AT (WD) | 4 | 1·557 | 1·413 | 0·368 | Austrian | 6·000 | 2·867 | 3·895 | 0·604 | 0·612 | 0·016 | 2 |
| AT (MS) | 3 | 2·363 | 1·647 | 0·590 | Austrian | 5·429 | 2·553 | 3·594 | 0·541 | 0·555 | 0·030 | 1 |
| HR | 4 | 3·158 | 2·266 | 0·704 | Balkan | 4·571 | 2·622 | 3·597 | 0·612 | 0·533 | −0·147 | 2 |
| BIH | 3 | 2·498 | 1·764 | 0·612 | Balkan | 5·714 | 3·320 | 4·423 | 0·643 | 0·614 | −0047 | 1 |
| SRB | 2 | 1·679 | 0·982 | 0·417 | Balkan | 4·286 | 3·252 | 2 | ||||
| BG | 2 | 1·800 | 0·997 | 0·467 | Balkan | 6·143 | 3·218 | 4·443 | 0·609 | 0·611 | −0·010 | 3 |
| Mean | 2·828 | 2·186 | 1·647 | 0·543 | 5·055 | 2·874 | 3·868 | 0·601 | 0·574 | −0·047 | ||
Na, number of alleles; Ne, effective number of alleles; RS, allelic richness; uh, unbiased haplotype diversity; Ho, observed heterozygosity; He, expected heterozygosity; Fis, inbreeding coefficient; Ap, number of private alleles; ND, no data.
RS, Ho, He and Fis were only calculated for populationss with n ≥10.
Values in bold indicate significant deviations.
Fig. 1.

cpDNA diversity of Sorbus domestica L. in Europe. Circle size is proportional to sample size. The barplot on the left shows the overall frequency of each allele. The green layer indicates the actual distribution of S. domestica according to the EUFORGEN-network.
In the nuclear microsatellite analysis, locus CH02d08 showed evidence for the occurrence of null alleles in some populations and was excluded from further analysis. The remaining seven loci showed signs neither of null alleles or large allele dropouts nor of genotyping errors due to the occurrence of stutter bands. Furthermore, all populations were in linkage equilibrium. All samples within populations originated from sexual reproduction as no identical multilocus genotypes were found.
A total of 78 alleles with a minimum of five and a maximum of 19 alleles per locus were observed. Only populations in Elba and northern Italy were fixed for one allele at locus MSS16. Allelic richness was slightly higher in northern Italy, in Corsica, in southern France as well as in Bosnia-Herzegovina and Bulgaria. In contrast to high cpDNA diversity, lower levels of allelic richness were observed in Elba. In Slovenia, low allelic richness was congruent with low levels of cpDNA diversity. Additionally, only the population in Slovenia showed a significant excess of heterozygotes (see Fis values in Table 2). Private alleles were found in nine populations with a maximum of three private alleles in Bulgaria. Frequencies of private alleles ranged from 0·013 (north-western France) to considerably high values of 0·214 (in Elba). From the 17 observed private alleles, 11 had a frequency <0·05 (data not shown). Allelic richness within populations was not correlated between both genomes (R2 = 0·006).
Genetic differentiation among populations
The UPGMA dendrogram based on Cavalli-Sforza and Edwards (1967) chord distance revealed distances between populations corresponding to geographic adjacency in the case of the French and Austrian group. Populations from the Mediterranean and Balkan group (except Bosnia-Herzegovina) were pooled in an unresolved clade when the majority rule was used as the decision criterion (Fig. 2). A slightly different result was provided by PCoA using Nei’s standard genetic distance (Nei, 1972), where the population from southern France clustered more clearly with populations from northern Italy, Corsica, Slovenia, Croatia and Bosnia-Herzegovina compared with the UPGMA dendrogram. Populations from Bulgaria and Elba were not assigned explicitly to this group (Fig. 3). The first two axes of the PCoA explained 74·1 % of variation (58·26 and 15·84 %, respectively). Allelic distance according to Gregorius (1974) revealed an average of 0·297. Populations of the Mediterranean group showed the least differentiation from the remaining gene pool (0·199), followed by populations from the Balkan region (0·253). The Austrian and French populations were most dissimilar from the residual gene pool (0·328 and 0·355, respectively; Fig. 4). Structure analysis showed consistent high assignment values among different runs and the results were partly in contrast to geographical proximity, but supporting the results from the UPGMA dendrogram as well as from PCoA. Populations from Austria and France built more distinct clusters than populations from Balkan and Mediterranean regions (Fig. 5). Based on the rate of change of the likelihood distribution, a value of K = 3 was chosen as the most likely number of clusters (Supplementary Data Fig. S2).
Fig. 2.

UPGMA consensus tree created out of 1000 bootstrapped matrices using Cavalli-Sforza and Edwards chord distance. Numbers beside the branches indicate bootstrap values.
Fig. 3.

Results of the principal co-ordinate analysis (PCoA) using Nei’s standard genetic distance. The first two axes are shown and explain 74·1 % of the observed variation.
Fig. 4.
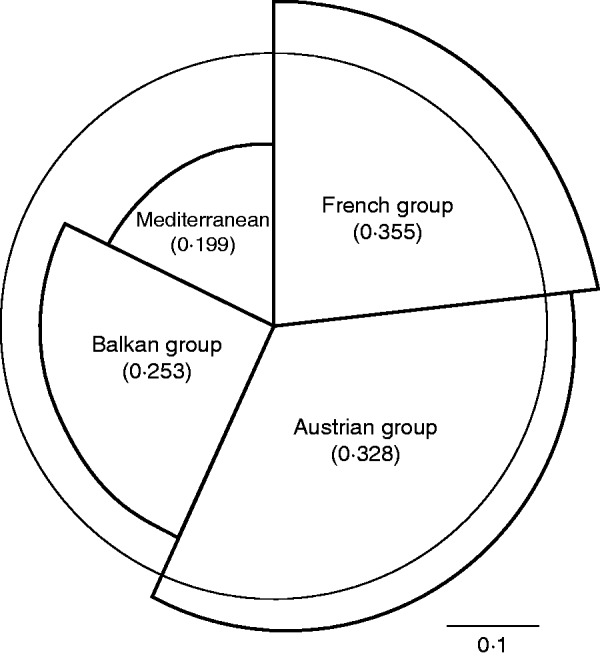
Allelic distance according to Gregorius (1974). The distance of each group to the residual gene pool is represented by the radius and the indicated numbers in parentheses. The angle of each segment shows the relative sample size. The thin line shows the average allelic distance.
Fig. 5.

Genetic clustering barplot using the software Structure. The results for K-values from 2 to 4 are shown.
The AMOVA revealed an FST value of 0·138 and an RST value of 0·102 when groups were defined according to their geographical proximity. For this arrangement, the percentage variation among groups decreased roughly when taking into account squared allele size differences and even reached a negative value, suggesting that stepwise mutations did not play a key role for differentiation even on a large geographical scale (Table 3). When groups were rearranged according to the results of the chord distance of Cavalli-Sforza and Edwards, Nei’s standard genetic distance as well as individual population assignment, variation patterns among and within groups changed slightly, but RST never exceeded FST, indicating the absence of a phylogeographic structure that is based on stepwise mutations (Supplementary Data Tables S2–S4).
Table 3.
AMOVA (analysis of molecular variance) results of seven nuclear microsatellites for populations with a sample size ≥10 based on FST and RST
| Source of variation |
FST |
RST |
||||
|---|---|---|---|---|---|---|
| Sum of squares | Variance components | Percentage variation | Sum of squares | Variance components | Percentage variation | |
| Among groups | 108·17 | 0·18 | 7·22 | 125561·39 | –36·14 | –0·46 |
| Among populations within groups | 85·87 | 0·16 | 6·62 | 404973·10 | 843·10 | 10·66 |
| Within populations | 1261·64 | 2·14 | 86·16 | 4185151·61 | 7105·52 | 89·80 |
| Total | 1455·69 | 2·49 | 4715686·10 | 7912·48 | ||
Groups correspond to those groups defined in Table 2.
Spatial genetic structure and historic vs. recent gene flow
Mean values of distance intervals for spatial genetic analysis reached from 207 to 3690 m in Merkenstein and from 397 to 4598 m in Wolkersdorf, and seven distance classes were defined for both populations. Kinship coefficients fluctuated in the same order of magnitude in both populations (–0·015 to 0·015), but did not deviate significantly from the permuted mean value in any of the seven distance classes (Supplementary Data Figs S3 and S4).
Analysis of recent gene flow showed that the majority of individuals originated from self-recruitment in both populations (96·8 and 96·5 %, respectively) and only one individual in Wolkersdorf was found to be a potential first-generation migrant from Merkenstein (posterior probability of migrant ancestry 72 %, data not shown). We did not find potential second-generation migrants from Merkenstein to Wolkersdorf or the reverse (Table 4). In contrast, by analysing historic gene flow, there was strong evidence that both populations previously exchanged a considerable number of migrants during many generations and that the direction of migration had been highly asymmetric (approx. 12 individuals per generation from Merkenstein to Wolkersdorf vs. only three in the opposite direction) (Table 5).
Table 4.
Pattern of recent gene flow between Wolkersdorf and Merkenstein
| Proportion of migrants (non-migrants) |
First-generation migrants (P > 0·5) |
Second-generation migrants (P > 0·5) |
||||
|---|---|---|---|---|---|---|
| Population | AT (WD)→ | AT (MS)→ | AT (WD)→ | AT (MS)→ | AT (WD)→ | AT (MS)→ |
| AT (WD) | 0·968 (0·0227) | 0·032 (0·0251) | 1 | 0 | ||
| AT (MS) | 0·035 (0·0251) | 0·965 (0·0227) | 0 | 0 | ||
P, posterior probability of migrant ancestry; the standard error is given in parentheses; receiving populations in rows.
Table 5.
Historic gene flow between Wolkersdorf and Merkenstein
| θ |
M (m/μ) |
n | ||
|---|---|---|---|---|
| AT (WD)→ | AT (MS)→ | |||
| AT (WD) | 3·3 (1·9–5·4) | 15·1 (11·8–18·8) | 12 | |
| AT (MS) | 1·9 (0·0–3·6) | 8·3 (5·4–11·2) | 3 | |
M, mutation-scaled migration rate; μ, mutation rate; m, migration rate; θ, 4Neμ; n, 1/4 × θ × M (approximate number of migrants received per generation); the credibility interval is given in parentheses; receiving populations in rows; values in bold for θ and M represent the modus of the posterior distribution.
DISCUSSION
Range-wide genetic pattern and diversity
The analysed populations had nuclear diversity levels similar to those reported for other tree species with similar life characteristics, in particular with a comparable scattered population structure such as Pyrus pyraster (Volk et al., 2006), Malus sylvestris (Cornille et al., 2012) or Sorbus torminalis (Demesure et al., 2000; Hoebee et al., 2006). Furthermore, almost all populations showed evidence of outbreeding (Fis values, Table 2). Considering that the degree of scattered occurrence is even higher for S. domestica compared with the mentioned species, we may explain this pattern by the existence of molecular mechanisms of self-incompatibility (SI) systems, observed in many rosaceous species (De Nettancourt, 2001; Cheng et al., 2006; Schüler et al., 2006), and in combination with its long-distance dispersal strategy (Kamm et al., 2009). These traits could have been responsible for maintaining genetic diversity at a high level. Until now, however, pre-zygotic SI systems have not been investigated in S. domestica. Nevertheless, we can assume its existence, considering data obtained from seed orchards, where numerous potentially mating trees had significantly more viable seeds per fruit compared with isolated trees (Bariteau et al., 2006). Even when pre-zygotic SI breaks down and selfing occurs, embryonic lethal factors such as albinism may effectively prevent inbreeding (Kamm et al., 2012).
Based on the data from the nuclear microsatellites, we found regions of comparatively high genetic diversity in southern and south-eastern Europe (Italy, southern France, Serbia and Bulgaria). However, this is not surprising, because these areas were potential refugia during the last glacial maximum for a wide range of taxa (e.g. Hewitt, 1996; Taberlet et al., 1998; Leroy and Arpe, 2004). An interesting result is that the decline in allelic richness from these potential refugia to the northern distribution limit (e.g. from southern France to north-western France) or to at least more northerly located populations (e.g. from Serbia to Austria) was only weak. This is in contrast to other studies that describe a clear pattern of ‘southern richness and northern purity’ [see Hewitt (1999) for a general overview and Comps et al. (2001) for an example dealing with tree species]. Unfortunately, we cannot confirm whether this pattern also holds true for other populations from the northern distribution limit, because some important populations (e.g. from northern Germany or Great Britain) were missing in our data set. However, cpDNA data showed a slightly opposite pattern. Only the northern Italian population showed high molecular diversity on both the nuclear and chloroplast level, while cpDNA diversity in the other two potential refugia (southern France and south-eastern Europe) was lower than in more distantly located populations. How can this pattern and the fact that even a small island like Elba encompassed the highest number of haplotypes be explained? An appropriate scenario is that historic trade of S. domestica fruits has shaped the current distribution and has been partially responsible for the diversity pattern. The Apennine Peninsula and also Elba, located in the central Mediterranean area and also as the centre of the Roman Empire, may have acted as a receiver rather than as a donor of fruits from western and south-eastern Europe, especially from countries with direct or indirect access to the Mediterranean Sea. There is strong evidence that the Romans were heavily dependent on imports of food to feed the growing population and that these imports came partly from distant provinces (Temin, 2006; Kessler and Temin, 2007). Support for this hypothesis is also provided by the results from PCoA and individual population assignment which revealed that one cluster contains nearly all populations near to the Mediterranean Sea (except Slovenia). This cluster is also evident in the UPGMA dendrogram, but represents an unresolved clade and we presume that historic admixture through humans (20–50 generations ago) could be the reason. Considering the mutation rates for microsatellites (10–6–10–2 per generation) (Schlötterer, 2000) and assuming an average generation time of 80–100 years for S. domestica, we may conclude that this time frame is insufficient for splitting into further clades. The Mediterranean group also had the lowest allelic distance to the residual gene pool (Fig. 4), even though it had the smallest sample size. Similar relationships among populations from distant regions within the Mediterranean were also observed for other tree species with long-standing economic importance such as Olea europaea (Belaj et al., 2002) or Castanea sativa (Villani et al., 1991).
Haplotype 288 was private to the Italian region, it occurred neither in central Europe nor in the south-east. Three possible scenarios can be discussed: (1) this haplotype persisted during the last glacial maximum in the southern part of the Apennine Peninsula and spread northward during more favourable conditions, but was not able to pass the Alps; (2) given the repeat number of this haplotype, we can assume that it is the youngest of all observed haplotypes (Schlötterer, 2000) and may have developed rapidly, when the former land bridge of the Adriatic Sea had already vanished; and (3) it is of unknown origin from a region, which was not sampled. Considering the low mutation rate for the chloroplast genome (Provan et al. 1999) we may exclude hypothesis (2), but it remains unclear if the presence of this haplotype is of natural or anthropogenic origin.
While the French populations showed a slight decrease in nuclear allelic richness from south to north, the cpDNA diversity pattern was different, with higher allelic richness in the central and in the north-western part of the country. A similar picture can be drawn for the Austrian populations compared with those of southern Europe. A ‘melting pot hypothesis’ as suggested by Petit et al. (2003) must be at least in part rejected as the explanation. Despite their higher cpDNA diversity, these populations harboured no haplotypes which were not also found in the southern regions.
It was obvious that the population in Slovenia is peculiar. Unexpectedly low levels of both nuclear and chloroplast diversity and a significant heterozygote excess compared with Hardy–Weinberg expectations were found. We cannot deduce its population history, but the data strongly support an anthropogenic origin.
Population differentiation and structure
Overall population differentiation based on allele identity was moderate (FST = 0·138) and is in accordance with values reported for tree species with similar life characteristics concerning long-distance gene flow such as S. torminalis (Oddou-Muratorio et al., 2001a; Angelone et al., 2007) or M. sylvestris (Cornille et al., 2012) on a comparable geographical scale. When taking into account the allele size under the assumption of a stepwise mutation model, RST could not explain more of the variation than FST either on a large (among groups) or on a relatively small geographical scale (among populations within groups). Although there has been considerable criticism about the comparison of FST and RST concerning gene flow estimation, mutation model assumptions and sampling variance (e.g. Whitlock and McCauley, 1999; Balloux and Lugon-Moulin, 2002; Hardy et al., 2003), this pattern might be explained through the extensive gene flow ability of the species (Kamm et al., 2009) in combination with human-mediated seed transfer at an even larger scale. As a consequence, migration (either natural or anthropogenic) always counteracts population differentiation based on stepwise mutations. This is also mirrored by the relatively low number of private alleles, which was expected to be higher, especially in the potential refugia. The fact that our whole sample set was differentiated into only three clusters with detectable transitions provides further support for extensive gene flow. Only the Austrian populations and north-western and central France formed more or less distinct clusters (Fig. 5). This is not surprising, as these populations probably have been genetically well connected during their population history.
Spatial genetic structure and historic vs. recent gene flow in the Austrian populations
Spatial genetic structure did not deviate significantly from a random permuted pattern, indicating that nearby individuals are not more related to each other than more distant individuals. While in barochorous broad-leafed species, clusters of related individuals are common (e.g. Geburek and Tripp-Knowles, 1994), a weak spatial genetic structure of a zoochorous species is very likely. In consequence, we may presume effective pollen and seed dispersal in these two populations.
The pattern of historic gene flow showed frequent exchange of migrants with conspicuous asymmetry (approximately three individuals per generation from Wolkersdorf to Merkenstein, but 12 individuals in the opposite direction). It can be expected that both populations were of greater size in the past and also better connected by single individuals that might have acted as stepping stones. Thus, it would have been much easier for migrants to bridge the current distribution gap of approx. 60 km. Since there are no significant natural landscape barriers between these populations except the river Danube (the landscape is characterized by low elevations and is of undulating shape), which were more likely to have been passed in one direction, we presume that this asymmetric pattern is to some extent the result of human-mediated seed transfer. This is also mirrored in the cpDNA haplotype distribution, which shows significant differences between these two populations, while it seems to be well balanced among populations across France for instance. Fruits of S. domestica had some former importance for the production of wine and cider, which has a long tradition in eastern Austria, in particular around the area of Wolkersdorf.
Compared with historic gene flow, the pattern of recent gene flow was different. Only one individual had a moderate probability to be a first-generation migrant. Its migration direction confirmed the asymmetry towards the northern located population Wolkersdorf. The decline in recent gene flow can have several explanations: habitat fragmentation took place intensively in this area during the last 200 years and a considerable part of the land use changed towards agriculture and urban development, resulting in a severe reduction of mature S. domestica trees and reduced movement ranges for both pollinators and seed dispersers (e.g. wild boar, roe deer, martens and foxes). Usually, dispersal distances achieved through these species are far below the distance that actually lies between Merkenstein and Wolkersdorf [e.g. Ellenberg (1978) reported 1–50 ha home ranges for roe deer, and Briedermann (1990) stated that wild boar regularly moves 5 km d–1]. Moreover, these species would have to cross extensive distances across an open area, because the size and number of forest patches left in between have been small. The only frugivores that are theoretically able to cover such distances are birds, mainly migratory species, for which mean distances of 60 km per day were reported (Ellegren, 1993). Another factor probably is the change in forest management within remaining patches. The early successional S. domestica in both populations is indeed under strong competition from late-successional and economically more important tree species such as beech, oak and pine. Hence, dominant individuals with well developed crowns are rare in the forest. This has led to reduced fruiting success and thus to a lower probability of being visited by frugivores. Oddou-Muratorio et al. (2005) have shown for S. torminalis that mating success of single trees is heavily dependent on tree size and competition status (i.e. the larger the individuals the larger is their fecundity).
Conclusions
Our study showed high levels of molecular diversity for S. domestica despite its scattered nature, with a likely human influence on its present diversity pattern. The key results of this study are: (1) S. domestica populations form three distinct groups in Europe (French, Mediterranean/Balkan and Austrian); (2) the Apennine Peninsula encompasses comparatively high genetic diversity in both genomes; and (3) the Austrian populations build a distinct cluster in Europe and showed significant decline in their exchange of migrants during the last two generations in comparison with several previous generations. Additional population samples from the Iberian Peninsula, the Carpathians and from south-eastern Europe, but also from further north, could help to provide a more definitive picture concerning diversity and the post-glacial colonization history of S. domestica in Europe.
SUPPLEMENTARY DATA
Supplementary data are available online at www.aob.oxfordjournals.org and consist of the following. Figure. S1: origin of individuals from the French National Collection and scattered single trees from central and southern France. Figure S2: change of the log probability between k-values 2–4. Figure S3: microspatial genetic structure for the population Merkenstein. Figure S4: microspatial genetic structure for the population Wolkersdorf. Table S1: overview of all markers used in the study. Table S2: AMOVA results according to groups from PCoA based on Nei’s standard genetic distance. Table S3: AMOVA results according to groups from UPGMA based on Cavalli-Sforza and Edwards chord distance. Table S4: AMOVA results based on genetic clustering calculated with Structure.
ACKNOWLEDGEMENTS
We thank Manfred and Herfried Steiner and Andreas Himmelbauer for providing information about the spatial positions of trees, and Thomas Kirisits, Christian Stauffer, Michel Rondouin, Luisa Ghelardini and Federico Vesella for providing additional samples. We also would like to thank two anonymous reviewers for their remarks and suggestions on how to improve the manuscript.
LITERATURE CITED
- Angelone S, Hilfiker K, Holderegger R, Bergamini A, Hoebee S. 2007. Regional population dynamics define the local genetic structure in Sorbus torminalis. Molecular Ecology 16: 1291–1301. [DOI] [PubMed] [Google Scholar]
- Balloux F, Lugon-Moulin N. 2002. The estimation of population differentiation with microsatellite markers. Molecular Ecology 11: 155–165. [DOI] [PubMed] [Google Scholar]
- Bariteau M, Brahic P, Thevenet J. 2006. Comment domestiquer le Cormier (Sorbus domestica)? Bilan des recherchessur la multiplication sexuee et vegetative . Foret mediterraneenne 27: 17–30. [Google Scholar]
- Beerli P. 2006. Comparison of Bayesian and maximum-likelihood inference of population genetic parameters. Bioinformatics 22: 341–345. [DOI] [PubMed] [Google Scholar]
- Beerli P. 2004. Effects of unsampled populations on the estimation of population sizes and migration rates between sampled populations. Molecular Ecology 13: 827–836. [DOI] [PubMed] [Google Scholar]
- Belaj A, Satovic Z, Rallo L, Trujillo I. 2002. Genetic diversity and relationships in olive (Olea europaea L.) germplasm collections as determined by randomly amplified polymorphic DNA. Theoretical and Applied Genetics 105: 638–644. [DOI] [PubMed] [Google Scholar]
- Briedermann L. 1990. Schwarzwild, 2nd edn Berlin: VEB Landwirtschaftsverlag. [Google Scholar]
- Brus R, Ballian D, Bogunic F, Bobinac M, Idzojtic M. 2011. Leaflet morphometric variation of service tree (Sorbus domestica L.) in the Balkan Peninsula. Plant Biosystems 145: 278–285. [Google Scholar]
- Cavalli-Sforza LL, Edwards AWF. 1967. Phylogenetic analysis: models and estimation procedures. American Journal of Human Genetics 19: 233–257. [PMC free article] [PubMed] [Google Scholar]
- Cheng J, Han Z, Xu X, Li T. 2006. Isolation and identification of the pollen-expressed polymorphic F-Box genes linked to the S-locus in apple (Malus×domestica). Sexual Plant Reproduction 19: 175–183. [Google Scholar]
- Chester M, Cowan RS, Fay MF, Rich TC. 2007. Parentage of endemic Sorbus L. (Rosaceae) species in the British Isles: evidence from plastid DNA. Botanical Journal of the Linnean Society 154: 291–304. [Google Scholar]
- Comps B, Gömöry D, Letouzey J, Thiebaut B, Petit RJ. 2001. Diverging trends between heterozygosity and allelic richness during postglacial recolonization in the European Beech. Genetics 157: 389–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornille A, Giraud T, Bellard C, et al. 2012. Postglacial recolonization history of the European crabapple (Malus sylvestris Mill.), a wild contributor to the domesticated apple. Molecular Ecology 22: 2249–2263. [DOI] [PubMed] [Google Scholar]
- Demesure B, Le Guerroue B, Lucchi G, Prat D, Petit RJ. 2000. Genetic variability of a scattered temperate forest tree: Sorbus torminalis L. (Crantz). Annals of Forest Science 57: 63–71. [Google Scholar]
- De Nettancourt D. 2001. Incompatibility and incongruity in wild and cultivated plants, 2nd edn Berlin: Springer. [Google Scholar]
- Dieringer D, Schlötterer C. 2003. Microsatellite Analyzer (MSA): a platform independent analysis tool for large microsatellite data sets. Molecular Ecology Notes 3: 167–169. [Google Scholar]
- Earl DA, vonHoldt BM. 2012. Structure Harvester: a website and programme for visualizing structure output and implementing the Evanno method. Conservation Genetics Resources 4: 359–361. [Google Scholar]
- Ellegren H. 1993. Speed of migration and migratory flight lengths of passerine birds ringed during autumn migration in Sweden. Ornis Scandinavica 24: 220–228. [Google Scholar]
- Ellenberg H. 1978. Zur Populationsökologie des Rehes (Capreolus capreolus L., Cervidae) in Mitteleuropa. Spixiana Supplement 2: 1–211. [Google Scholar]
- Evanno G, Regnaut S, Goudet J. 2005. Detecting the numbers of clusters of individuals using the software STRUCTURE: a simulation study. Molecular Ecology 14: 2611–2620. [DOI] [PubMed] [Google Scholar]
- Excoffier L, Lischer HEL. 2010. Arlequin suite ver. 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Molecular Ecology Resources 10: 564–567. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. 1989. Phylip – phylogeny inference package vers. 3.2. Cladistics 5: 164–166. [Google Scholar]
- Ferrini F, Sartorato I, Sanesi G. 2008. Italy. In: Willoughby I, Ballandier P, Bentsen NS, McCarthy N, Claridge J, eds. Forest vegetation management in Europe – current practices and future needs. Brussels: COST office, 75–84. [Google Scholar]
- Frochot H, Ballandier P, Michalet R, van Lerberghe P. 2008. France. In: Willoughby I, Ballandier P, Bentsen NS, McCarthy N, Claridge J, eds. Forest vegetation management in Europe – current practices and future needs. Brussels: COST office, 33–42. [Google Scholar]
- Geburek T, Tripp-Knowles P. 1994. Genetic architecture in bur oak, Quercus macrocarpa (Fagaceae), inferred by means of spatial autocorrelation analysis. Plant Systematics and Evolution 189: 63–74. [Google Scholar]
- Gianfranceschi L, Seglias N, Tarchini R, Komjanc M, Gessler C. 1998. Simple sequence repeats for the genetic analysis of apple. Theoretical and Applied Genetics 96: 1069–1076. [Google Scholar]
- Gilpin M. 1991. The genetic effective size of a metapopulation . Biological Journal of the Linnean Society 42: 165–175. [Google Scholar]
- Goudet J. 1995. Fstat vers. 1.2: a computer programme to calculate F-statistics. Journal of Heredity 86: 485–486. [Google Scholar]
- Gregorius HR. 1974. Genetischer Abstand zwischen Populationen – I. Zur Konzeption der genetischen Abstandsmessung. Silvae Genetica 23: 22–27. [Google Scholar]
- Hanski I. 1998. Metapopulation dynamics. Nature 396: 41–49. [Google Scholar]
- Hardy OJ, Vekemans X. 2002. SPAGeDI: a versatile computer programme to analyse spatial genetic structure at the individual or population level. Molecular Ecology Notes 2: 618–620. [Google Scholar]
- Hardy OJ, Charbonnel N, Freville H, Heuertz M. 2003. Microsatellite allele sizes: a simple test to assess their significance on genetic differentiation. Genetics 163: 1467–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewitt GM. 1996. Some genetic consequences of ice ages and their role in divergence and speciation. Biological Journal of the Linnean Society 58: 247–276. [Google Scholar]
- Hewitt GM. 1999. Post-glacial re-colonization of European Biota. Biological Journal of the Linnean Society 68: 87–112. [Google Scholar]
- Hoebee SE, Menn C, Rotach P, Finkeldey R, Holderegger R. 2006. Spatial genetic structure of Sorbus torminalis: the extent of clonal reproduction in natural stands of a rare tree species with a scattered distribution. Forest Ecology and Management 226: 1–8. [Google Scholar]
- Jump AS, Penuelas J. 2006. Genetic effects of chronic habitat fragmentation in a wind-pollinated tree . Proceedings of the National Academy of Sciences, USA 103: 8096–8100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamm U, Rotach P, Gugerli F, Siroky M, Edwards P, Holderegger R. 2009. Frequent long-distance gene flow in a rare temperate forest tree (Sorbus domestica) at the landscape scale. Heredity 103: 476–482. [DOI] [PubMed] [Google Scholar]
- Kamm U, Gugerli F, Rotach P, Edwards P, Holderegger R. 2010. Open areas in a landscape enhance pollen-mediated gene flow of a tree species: evidence from northern Switzerland. Landscape Ecology 25: 903–911. [Google Scholar]
- Kamm U, Gugerli F, Rotach P, Edwards P, Holderegger R. 2011. Genetic consequences of local tree densities – implications for the management of naturally rare, insect pollinated species in temperate forests . Forest Ecology and Management 262: 1047–1053. [Google Scholar]
- Kamm U, Gugerli F, Rotach P, Edwards P, Holderegger R. 2012. Rare and scattered: consequences for the mating success of the service tree . Schweizer Zeitschrift für Forstwesen 163: 130–136. [Google Scholar]
- Kausch-Blecken von Schmeling W. 2000. Der Speierling, 2nd edn Bovenden: private publishing. [Google Scholar]
- Kessler D, Temin P. 2007. The organization of the grain trade in the early Roman Empire. Economic History Review 60: 313–332. [Google Scholar]
- Kutzelnigg H. 1995. Sorbus domestica. In: Connert HH, Jäger EJ, Kadereit JW, Schultze-Motel W, Wagenitz G, Weber HE, eds. Gustav-Hegi – Illustrierte Flora von Mitteleurop, Berlin: Blackwell, 383–343. [Google Scholar]
- Leroy SA, Arpe K. 2004. Glacial refugia for summer-green trees in Europe and south-west Asia as proposed by ECHAM3 time-slice atmospheric model simulations. Journal of Biogeography 34: 2115–2128. [Google Scholar]
- Levey DJ, Bolker BM, Tewksbury JJ, Sargent S, Haddad NM. 2005. Effects of landscape corridors on seed dispersal by birds. Science 309: 146–148. [DOI] [PubMed] [Google Scholar]
- Liebhard R, Gianfranceschi L, Koller B, et al. 2002. Development and characterization of 140 new microsatellites in apple (Malus×domestica Borkh.). Molecular Breeding 10: 217–241. [Google Scholar]
- Loiselle BA, Sork VL, Nason J, Graham C. 1995. Spatial genetic structure of a tropical understory shrub, Psychotria officinalis (Rubiaceae). American Journal of Botany 82: 1420–1425. [Google Scholar]
- Ludwig S, Robertson A, Rich TCG, et al. 2013. Breeding systems, hybridization and continuing evolution in Avon Gorge Sorbus. Annals of Botany 111: 563–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mac Arthur R, Wilson EO. 1967. The theory of island biogeography. Princeton, NJ: Princeton University Press. [Google Scholar]
- Nason JD, Herre EA, Hamrick JL. 1998. The breeding structure of a tropical keystone plant resource. Nature 391: 685–687. [Google Scholar]
- Nei M. 1972. Genetic distance between populations. American Naturalist 106: 283–392. [Google Scholar]
- Nelson-Jones EB, Briggs D, Smith AG. 2002. The origin of intermediate species of the genus Sorbus. Theoretical and Applied Genetics 105: 953–963. [DOI] [PubMed] [Google Scholar]
- Nyari L. 2010. Genetic variability of service tree (Sorbus domestica L.) in the Hungarian middle mountains – based on cp-DNA analysis in two regions. Acta Silvatica & Lignaria Hungarica 6: 17–32. [Google Scholar]
- Oddou-Muratorio S, Petit RJ, Le Guerroue B, Guesnet D, Demesure B. 2001a. Pollen- versus seed-mediated gene flow in a scattered forest tree species. Evolution 55: 1123–1135. [DOI] [PubMed] [Google Scholar]
- Oddou-Muratorio S, Aligon C, Decroocq S, Plomion C, Lamant T, Mush DB. 2001b. Microsatellite primers for Sorbus torminalis and related species. Molecular Ecology Notes 1: 297–299. [Google Scholar]
- Oddou-Muratorio S, Klein EK, Austerlitz F. 2005. Pollen flow in the wild service tree, Sorbus torminalis (L.) Crantz. II. Pollen dispersal and heterogeneity in mating success inferred from parent–offspring analysis. Molecular Ecology 14: 4441–4452. [DOI] [PubMed] [Google Scholar]
- Oxelmann B, Magnus L, Berglund D. 1997. Chloroplast rps16 intron phylogeny of the tribe Sileneae (Caryophylaceae). Plant Systematics and Evolution 206: 393–410. [Google Scholar]
- Peakall R, Smouse PE. 2012. GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research – an update . Bioinformatics 28: 2537–2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit RJ, El Mousadik A, Pons O. 1998. Identifying populations for conservation on the basis of genetic markers. Conservation Biology 12: 844–855. [Google Scholar]
- Petit RJ, Aguinadalde I, de Beaulieu JL, et al. 2003. Glacial refugia: hotspots but not melting pots of genetic diversity. Science 300: 1563–1565. [DOI] [PubMed] [Google Scholar]
- Pritchard JK, Stephens P, Donnelly P. 2000. Inference of population structure using multilocus genotype data. Genetics 155: 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provan J, Soranzo N, Wilson NJ, Goldstein DB, Powell W. 1999. A low mutation rate for chloroplast microsatellites . Genetics 153: 943–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond M, Rousset F. 1995. Genepop: population genetics software for exact tests and ecumenicism. Journal of Heredity 86: 248–249. [Google Scholar]
- Rotach P. 2003. EUFORGEN Technical Guidelines for genetic conservation and use for service tree (Sorbus domestica). Rome, Italy: International Plant Genetic Ressources Institute. [Google Scholar]
- Schlötterer C. 2000. Evolutionary dynamics of microsatellite DNA. Chromosoma 109: 365–371. [DOI] [PubMed] [Google Scholar]
- Schüler S, Tusch A, Scholz F. 2006. Comparative analysis of the within-population genetic structure of wild cherry (Prunus avium L.) at the self-incompatibility locus and nuclear microsatellites. Molecular Ecology 15: 3231–3243. [DOI] [PubMed] [Google Scholar]
- Taberlet P, Fumagalli L, Wust-Saucy AG, Cosson JF. 1998. Comparative phylogeography and postglacial colonization routes in Europe. Molecular Ecology 7: 453–464. [DOI] [PubMed] [Google Scholar]
- Temin P. 2006. The economy of the early Roman Empire. Journal of Economic Perspectives 20: 133–151. [Google Scholar]
- Termentzi A, Alexiou P, Demopoulos VJ, Kokkalou E. 2008. The aldose reductase inhibitory capacity of Sorbus domestica fruit extracts depends on their phenolic content and may be useful for the control of diabetic complications . Pharmazie 63: 693–696. [PubMed] [Google Scholar]
- Van Oosterhout C, Hutchinson WF, Wills DP, Shipley P. 2004. Micro-Checker: software for identifying and correcting genotyping errors in microsatellite data. Molecular Ecology Notes 4: 535–538. [Google Scholar]
- Villani F, Pigliucci M, Benedettelli S, Cherubini M. 1991. Genetic differentiation among Turkish chestnut (Castanea sativa Mill.) populations. Heredity 66: 131–136. [Google Scholar]
- Volk GM, Richards CM, Henk AD, Reilley AA. 2006. Diversity of wild Pyrus communis based on microsatellite analysis. Journal of the American Society for Horticultural Science 131: 408–417. [Google Scholar]
- Wilson GA, Rannala B. 2003. Bayesian inference of recent migration rates using multilocus genotypes. Genetics 163: 1177–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitlock MC, McCauley D. 1999. Indirect measures of gene flow and migration: FST≠1/(4Nm+1). Heredity 82: 117–125. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Kimura T, Shoda M, Ban Y, Hayashi T, Matsuta N. 2002. Development of microsatellite markers in the Japanese pear (Pyrus pyrifolia Nakai). Molecular Ecology Notes 2: 14–16. [Google Scholar]
- Young A, Boyle T, Brown T. 1996. The population genetic consequences of habitat fragmentation for plants . Trends in Ecology and Evolution 11: 413–418. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.