

Abstract
Nowadays, multidrug resistance and side effects of drugs limit the effectiveness of chemotherapies in clinics. P-glycoprotein (P-gp) (MDR1), as a member of the ATP-binding cassette family, acts on transporting drugs into cell plasma across the membrane of cancer cells and leads to the occurrence of multidrug resistance, thus resulting in the failure of chemotherapy in cancer. The main aims of this research were to design a nanodelivery system for accomplishing the effective co-delivery of gene and antitumor drug and overcoming multidrug resistance effect. In this study, shMDR1 and gefitinib-encapsulating chitosan nanoparticles with sustained release, small particle size, and high encapsulation efficiency were prepared. The serum stability, protection from nuclease, and transfection efficiency of gene in vitro were investigated. The effects of co-delivery of shMDR1 and gefitinib in nanoparticles on reversing multidrug resistance were also evaluated by investigating the cytotoxicity, cellular uptake mechanism, and cell apoptosis on established gefitinib-resistant cells. The results demonstrated that chitosan nanoparticles entrapping gefitinib and shMDR1 had the potential to overcome the multidrug resistance and improve cancer treatment efficacy, especially toward resistant cells.
Keywords: P-glycoprotein, nanodelivery system, chemotherapy, gene
Introduction
Although chemotherapy is the major route for tumor therapy nowadays, multidrug resistance (MDR) and side effects of drugs limit the effectiveness of chemotherapies in clinic.1–7 Nearly 90% of tumor cells gradually become insensitive and MDR occurs after repeated exposure of drugs to the tumor cells for a certain time.8,9 Tumor cells can survive after exposure to chemotherapy drugs to produce MDR through inhibition of apoptosis and other ways.10–14 Although the mechanism of MDR is complicated, it is well known that transmembrane transporters such as proteins, including permeability glycoprotein (P-glycoprotein [P-gp]), MDR-associated protein (MRP), lung resistance-associated protein (LRP), and breast cancer-resistant protein (BCRP), transport drugs out of cell plasma across the membrane of cancer cells.15–17 P-gp, also known as MDR protein 1 (MDR1), ATP-binding cassette subfamily B member 1 (ABCB1), or cluster of differentiation 243 (CD243), is an important cell membrane protein that pumps many foreign substances out of cells. Some cancer cells also express large amounts of P-gp, rendering these cancer cells multidrug resistant.18,19
The main reason for the failure of chemotherapy is the occurrence of MDR in tumor cells. It is necessary to find effective measures to overcome tumor drug resistance and improve the effect of chemotherapy. Some potential treatments such as the application of MDR reversal agents, immune treatments, cytokines, gene therapy, and the combination of P-gp reversal agents (P-gp inhibitors) and chemotherapeutic agents have important clinical significance in improving chemotherapy efficacy and patient survival.20–23 Unfortunately, P-gp inhibitors such as verapamil, cyclosporine A, and their derivatives not only showed higher cytotoxicity in cells and lack of specificity of tumor cell recognition, but also exhibited poor pharmacokinetic profiles due to extensive metabolic degradation and low water solubility.24
Gene therapy is the therapeutic delivery of living specific genetic material into the cells to change targeted cell phenotype or attack the defected genes at gene level to prevent or cure a particular disease.25,26 The shRNA target in MDR1 as the new method of gene-mediated interference could inhibit the selectively targeted MDR1 gene expression, increase the intracellular accumulation of drugs, and restore the sensitivity of cells to the drug, thereby reversing drug resistance.27–30 Compared to traditional gene-mediated treatment, gene-loaded nanoparticles (NPs) showed promising advantages due to their nano-size and specific body distribution.31,32 NPs can be internalized more specifically and efficiently than free therapeutic agents, and, more importantly, NPs can be easily aggregated and accumulated inside tumor tissues for a long time, known as the enhanced permeability and retention effect.33–37 Genes loaded in NPs were effectively protected from the degradation of DNase I and serum, and this significantly improved the efficiency of transfection of shRNA in vitro in cells and nanovector delivery of gene increased its cytotoxicity and induced more cell apoptosis in cancer therapy.38–41
Gefitinib, as the first selective inhibitor of epidermal growth factor receptor (EGFR) tyrosine kinase domain, is widely used in the chemical therapy of many human cancers. However, chemoresistance occurs in almost all patients and limits the clinical usage of EGFR tyrosine kinase domain. In this work, we prepared chitosan (CS) NPs capable of entrapping the anticancer drug gefitinib and shRNA-expressing plasmid DNA targeting the MDR1 gene (shMDR1) to examine whether they could enhance antitumor effects of anticancer drugs against MDR. In this study, we prepared CS NPs with excellent properties of good drug entrapment, sustained release, small particle size, low polydispersity index, and high encapsulation efficiency. shMDR1 entrapped in NPs was protected effectively from the degradation of DNase I and serum, and the efficiency of transfection of shRNA in vitro in gefitinib-resistant Hela cells (established gefitinib resistant) was significantly improved. More importantly, co-delivery of shMDR1 and gefitinib loaded in CS NPs showed increased cytotoxicity and promoted the apoptosis of resistant gefitinib-resistant Hela cells owing to the fact that shMDR1 prevented P-gp activity by silencing the expression of MDR1. These findings indicate co-encapsulation of the anticancer drug gefitinib and shMDR1 could be more effective in reversing MDR and a nano drug-delivery system could contribute greatly to reversing MDR.
Materials
CS with the deacetylation degree of 80% and molecular weight of approximately 400 kDa was purchased from Haixin Biological Product Co., Ltd (Ningbo, People’s Republic of China); 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and proteinase K were obtained from Sigma-Aldrich Co. (St Louis, MO, USA); and gefitinib was purchased from Eastbang Pharmaceutical Co., Ltd (Guangzhou, People’s Republic of China). pGCsi-U6/Neo/GFP-shRNA-expressing pDNA (pEGFP) and pGCsi-U6/Neo/GFP-MDR1-shRNA-expressing pDNA (shMDR1) which targeted MDR1 mRNA sequence (ACAGAATAGTAACTTGTTT) were purchased from Genechem Co., Ltd. (Shanghai, People’s Republic of China). All other chemicals purchased were of analytical grade and were obtained from a variety of vendors. Gefitinib-resistant Hela cells (established gefitinib resistant) were obtained from Liaoning Medical University (Jinzhou, People’s Republic of China).
Preparation of shMDR1/gefitinib NPs
Ion gelation method was established to prepare shMDR1- and gefitinib-loaded CS NPs (shMDR1/gefitinib NPs). Accurately weighed (20 mg) CS was dissolved in 500 mL acetic acid (2%, v/v), and shMDR1 and gefitinib were also dissolved in sodium tripolyphosphate reserve liquid (0.5 mg/mL). The obtained sodium tripolyphosphate reserve liquid was slowly (30 mL/hr) injected using a micro-syringe into the 500 mL acetic acid (2%, v/v) containing 20 mg CS under magnetic stirring and agitated for 5 hours at room temperature. Finally, shMDR1/gefitinib NPs formed instantaneously and were freeze-dried under vacuum for further analysis. The size, morphology, and zeta potential were characterized by transmission electron microscope (TEM) (JEM-1200EX; JEOL, Tokyo, Japan) and Zetasizer (Nano ZS90; Malvern Instruments, Malvern, UK). The encapsulation ratios of gefitinib and shMDR1 were measured with a UV-Vis spectrometer and the drug release behavior in NPs in vitro was studied using dialysis method.42
Optimization of the mass ratio between shRNA and CS
In order to obtain the best binding ability between shRNA and CS, and to improve the encapsulation efficiency of shRNA in NPs, gel retardation assay determining the loading ability of shRNA in NPs was performed. Using naked shRNA as the control, NPs containing 0.08 nmol shRNA with different mass ratios of CS and shRNA (50:1, 30:1, and 20:1) were mixed with the sample buffer and added into sample loading wells for gel electrophoresis at the voltage of 100 V for 1 hour. Finally, the gel was incubated with buffer containing 0.5 μg/mL of ethidium bromide at room temperature for 30 minutes and analyzed by agarose gel electrophoresis.
DNase I protection test and serum protection test
According to a previous protocol,43 the collected NPs were incubated with the mixture of 10 μL of buffer containing 2 kat of DNase I and 50 μL nuclease-free water at 37°C for 1 hour. shRNA loaded in NPs was extracted with the phenol–chloroform–isoamyl–butyrate method and analyzed by agarose gel electrophoresis. At the same time, the naked shRNA was treated by the same method as a control treatment. In terms of serum protection test, NPs were immersed in cell culture medium containing 10% fetal bovine serum and coincubated at 37°C for 12 hours and shRNA was extracted from NPs according to the procedure described above. At the same time, the naked shRNA was also treated by the same method as a control treatment.
In vitro transfection experiments
Free EGFP as the reporter gene was encapsulated into NPs for evaluating the transfection efficiency of the gene. Gefitinib-resistant Hela cells at a density of 5×104/mL were incubated with free EGFP and EGFP-loaded NPs at different concentrations of EGFP for 48 hours followed by double washing with ice-cold phosphate-buffered saline, and intracellular gene transfection effects were observed using confocal laser scanning microscopy.
Distribution of NPs in cells
Gefitinib-resistant Hela cells (established gefitinib resistant) at logarithmic growth phase were plated into the wells. After adherence to reach a density of 5×104/mL, NPs encapsulating fluorescein isothiocyanate (FITC) were added and incubated with cells. The location and distribution of FITC-labeled NPs in cells was visualized by confocal laser scanning microscopy (FluoView FV10i; Olympus Corporation, Tokyo, Japan) at given time intervals.
Quantitative analysis of cellular uptake of NPs
Cell-internalized FITC-labeled NPs were digested to release FITC, and the intensity of fluorescence from FITC which is excited at 485 nm and emitted at 528 nm in cells was quantified using a microplate reader. Relative fluorescence ratio (RFR) (%) could represent the relative uptake rates of NPs and was calculated using Equation 1:
| (1) |
FItotal is the fluorescence intensity from the initially added FITC-labeled NPs, and FIinternalized is the fluorescence intensity from FITC-labeled NPs internalized in cells.
To study uptake mechanisms involved in the cellular entry of NPs, various endocytic inhibitors were employed. Gefitinib-resistant Hela cells at the logarithmic growth stage were seeded into six-well plates to reach 5×104/mL and were preincubated, respectively, with the following endocytosis inhibitors for 1 hour: chlorpromazine (inhibition of clathrin-mediated uptake) at 10 μg/mL; genistein (caveolae-mediated uptake) at 1 μg/mL; cytochalasin D (30 μM, macropinocytosis); and 20 μg/mL sodium azide (an energy inhibitor). Then, FITC-labeled NPs were added into plates for incubation. After 24 hours, the original medium containing NPs was removed followed by double washing with phosphate-buffered saline, and cells were collected for quantitative analysis of the cellular uptake of NPs. The relative fluorescence ratio was compared between the treatment with added inhibitors and the treatment with added non-inhibitors.
Cell apoptosis evaluation by MTT and flow cytometry
In order to determine the role of MDR1 on the cell apoptosis, MTT assay was used to evaluate cell viability. Annexin V–FITC/propidium iodide staining assay was performed and the apoptotic and necrotic cells were quantified by flow cytometry. According to the protocol of our previous study,44 free gefitinib, gefitinib and shMDR1, gefitinib NPs, gefitinib NPs combined with shMDR1 NPs, and shMDR1/gefitinib NPs with the same concentration of gefitinib were used to treat gefitinib-resistant Hela cells for 48 hours at 37°C for further analysis.
Western blot assay
Western blot assay was performed to determine the level of relative proteins when free gefitinib, gefitinib and shMDR1, gefitinib NPs, and shMDR1/gefitinib NPs were incubated with gefitinib-resistant Hela cells for 48 hours. Briefly, the proteins were then transferred to a membrane (typically nitrocellulose or PVDF), where they were stained with antibodies specific to the target protein. The gel electrophoresis step was included in the Western blot analysis to resolve the issue of cross-reactivity of antibodies.
Results
The characterization of shRNA/gefitinib NPs
In order to obtain the best binding ability between shRNA and CS and improve the encapsulation efficiency of shRNA in NPs, gel retardation assay was performed to determine the loading ability of shRNA in NPs. It can be seen in Figure 1 that, after electrophoresis, the band presenting naked DNA was stronger. In contrast, when the proportion of CS and shRNA was increased from 20:1 to 30:1, the band of free shRNA still remained visible, implying that a small amount of free shRNA was not encapsulated in NPs and ran out from the surface of NPs. When the proportion of CS and shRNA was 50:1, the band almost disappeared, suggesting that the binding ability of shRNA with NPs was the strongest among the three groups, more shRNA was entrapped in NPs, and the optimal ratio of CS and shRNA was 50:1.
Figure 1.

Characterization of shMDR1/gefitinib nanoparticles (NPs).
Notes: (A) Gel retardation assay for determining loading ability of shRNA in NPs (lane 1: naked shRNA; lanes 2–4, shRNA-loaded NPs prepared at mass ratios of 50:1, 30:1, and 20:1 chitosan to shRNA, respectively). (B) TEM image of shMDR1/gefitinib NPs (50:1). (C) DLS analysis of the obtained shMDR1/gefitinib NPs. (D) In vitro release profile of shMDR1/gefitinib NPs in phosphate-buffered saline (pH 5.8 and pH 7.4 at 37°C) for 48 hours.
Abbreviations: DLS, dynamic light scattering; TEM, transmission electron microscope.
It can be seen in Figure 1 and Table 1 that the optimal shMDR1/gefitinib NPs were induced by ion gelation preparation with the properties of high encapsulation efficiency (88.3%±7.2% for shMDR1 and 89.8%±7.1% for gefitinib), biphasic sustained release, smaller average particle size (92±11 nm), and low polydispersity index (0.14±0.06). The in vitro release of gefitinib was conducted by dialysis bag method and the results implied that the release of gefitinib from NPs belonged to the biphasic release. Gefitinib release from NPs was faster in the first 2 hours and over approximately 15% of total drugs entered into the release medium during this period; furthermore, the drug could be almost completely released within 48 hours. Interestingly, the drug showed a pH-dependent release pattern and was released rapidly at low pH conditions but slowly at physical condition. This would be a benefit for smart release of gefitinib in acidic condition around tumor cells. NPs also controlled the slow and smooth release of shMDR1 and more than approximately 75% of total shMDR1 slowly leaked out from NPs into medium within 48 hours at different pHs.
Table 1.
Key parameters of shMDR1/gefitinib nanoparticles (NPs) with different mass ratios between shMDR1 and chitosan
| Group | Diameter (nm) | Zeta potential (mV) | Polydispersity index | Encapsulation efficiency (gefitinib) | Encapsulation efficiency (shMDR1) |
|---|---|---|---|---|---|
| shMDR1/gefitinib NPs (20:1) | 50±7 | 15.3±2.2 | 0.16±0.08 | 80.3%±4.3% | 50.6%±4.5% |
| shMDR1/gefitinib NPs (30:1) | 75±8 | 20.8±5.1 | 0.19±0.05 | 85.6%±6.3% | 70.6%±5.6% |
| shMDR1/gefitinib NPs (50:1) | 92±11 | 26.5±3.2 | 0.14±0.06 | 89.8%±7.1% | 88.3%±7.2% |
Note: Data are presented as mean ± SD.
DNase I protection test and serum protection test
The results shown in Figure 2 demonstrate that, compared to naked shRNA, shRNA was protected efficiently from the degradation of DNase I and serum through encapsulating into the core of NPs. After treatment with DNase I for 12 hours, shRNA loaded in NPs still kept its integrated structure and the band representing shRNA was still observed. Conversely, naked shRNA was totally degraded, represented by the disappearance of the band within 4 hours. At the same time, shRNA loaded in NPs treated with serum retained its superhelical structure for 8 hours, while naked shRNA was degraded thoroughly within 4 hours. Taken together, shRNA was protected efficiently from enzyme degradation by encapsulating into NPs.
Figure 2.

Combining ability and protecting effects of shRNA-NPs by agarose gel electrophoresis.
Notes: (A) Serum protection of shRNA nanoparticles (NPs) (lanes 1–4, shRNA-NPs were treated for 1, 4, 8, and 12 hours; lanes 5–8, naked shRNA was treated for 1, 4, 8, and 12 hours). (B) DNase I protection of shRNA NPs (lanes 1–4, naked shRNA was treated for 1, 4, 8, and 12 hours; lanes 5–8, shRNA NPs were treated for 1, 4, 8, and 12 hours).
In vitro transfection evaluation
EGFP transfected into cells was observed using confocal laser scanning microscopy, as shown in Figure 3. It was demonstrated that the transfection efficiency of gene in cells was a concentration-dependent process and, with the increase of loaded gene, the green fluorescence intensity from EGFP was significantly enhanced, indicating that more EGFP was transfected into cells. When incubated with cells, naked EGFP was easily degraded before entering the cells, therefore weakening the transfecting effects represented by the slight green color throughout the whole cytoplasm. On the contrary, after incubation with EGFP-loaded NPs, the transfection efficiency of gene was greatly improved through the protection of gene loaded in NPs from the degradation and easy intracellular transportation by the endocytosis of NPs.
Figure 3.

In vitro transfection efficiency of EGFP-loaded NPs.
Notes: Free EGFP and EGFP-loaded nanoparticles (NPs) were incubated with cells for 24 and 48 hours. In vitro transfection efficiency of EGFP-loaded NPs was evaluated by the change on the intracellular green fluorescence intensity. The scale bar is 50 μm and applies to all figure parts.
Distribution of NPs in cells
The cellular uptake of FITC-labeled NPs by cells was observed by confocal laser scanning microscopy, as shown in Figure 4. With the continuous incubation of cells with NPs, the green fluorescence intensity inside the cells gradually increased. Only a small amount of green spots was concentrated and distributed around the cell membrane in the first 3 hours of incubation, and, with the extension of time, NPs began to diffuse into the cytoplasm at 6 hours and displayed enhanced green fluorescence. Finally, a majority of NPs accomplished their cellular uptake within 6 hours, and this suggests that the uptake of NPs was a time-dependent internalization process and the initial rapid uptake maintained a higher drug concentration in a short time, facilitating shMDR1 and gefitinib to enhance their cytotoxic activities significantly and promoting the apoptosis of drug in resistant gefitinib-resistant Hela cells, and thus reversing MDR in cancer therapy.
Figure 4.

Confocal images of (A) FITC labeled blank NPs and (B) FITC labeled shMDR1 NPs incubated with cells for 12 hours.
Notes: The nucleus was stained with Hoechst (blue) for 15 minutes at 37°C and all NPs were labeled with FITC (green). The lysosome was stained with LysoTracker Red DND-99. The scale bar is 50 μm and applies to all figure parts.
Abbreviation: FITC, fluorescein isothiocyanate.
Quantitative analysis of cellular uptake of NPs
As MDR1 was overexpressed in gefitinib-resistant Hela cells and promoted drug to outflow from the interior of cells, the uptake of drugs could be reduced and produced the drug resistance. As can be seen in Figure 5A, NPs were internalized into cells and the RFR observed in cells treated with NPs was increased gradually from approximately 30.5% in the initial 3 hours to over 60.1% at 6 hours and finally significantly decreased to 43.2% at 12 hours. This could be credited to the fact that, with the extension of time, MDR1 could play an important role in pumping drugs out from the cells and resulted in the reduction in uptake of drug. In contrast, shMDR1 loaded in NPs as a potential inhibitor by silencing the expression of MDR1 attenuated the efflux of free drug induced by MDR1, therefore leading to the retention of drug in cells. The RFR observed in cells treated with shMDR1 NPs was increased gradually from approximately 37.7% in the initial 3 hours to over 73.4% at 6 hours and finally slightly decreased to 66.5% at 12 hours.
Figure 5.

Analysis of the uptake mechanism of nanoparticles (NPs) in gefitinib-resistant Hela cells.
Notes: (A) Fluorescence spectrum analysis of the uptake of NPs in gefitinib-resistant Hela cells. Results are expressed as means ± standard deviation (n=3). ***P<0.001 vs fluorescein isothiocyanate (FITC)-labeled NPs treated with cells at 3 hours. ###P<0.001 vs the FITC-labeled shMDR1 NPs treated with cells at 3 hours. (B) Effects of endocytic inhibitors on the uptake ability of the two NPs in gefitinib-resistant Hela cells. Results are expressed as means ± standard deviation (n=3). ***P<0.001 vs FITC-labeled NPs treated with chlorpromazine. ###P<0.001 vs the FITC-labeled shMDR1 NPs treated with chlorpromazine.
In order to study the mechanism underlying the uptake of NPs, different inhibitors were used to analyze the intracellular uptake pattern of NPs. The results (Figure 5B) showed that when the energy inhibitor sodium azide was added, the relative uptake rates were decreased to 43.5% for NPs and 59.7% for shMDR1 NPs, respectively. This indicated that the uptake of both NPs was energy dependent and inhibition of energy could significantly limit the internalization of the two kinds of NPs. On the contrary, the uptake of both NPs had no significant change before and after treatment with the addition of chlorpromazine, an inhibitor of clathrin-mediated endocytosis. Cytochalasin D as the inhibitor of macropinocytosis can inhibit actin polymerization and cell membrane flow. The relative uptake rates after treatment with cytochalasin D were 55.6% for NPs and 65.4% for shMDR1 NPs. Interestingly, through the incubation of genistein as a tyrosine kinase inhibitor, the actin network was destructed and protein aggregation was inhibited, thus preventing caveolae-mediated endocytosis, and the uptake of NPs and shMDR1 NPs in gefitinib-resistant Hela cells pretreated with genistein was 60.4% and 70.6%, respectively. Taken together, both NPs showed a similar intracellular uptake mechanism in cells and transportation of NPs in the cells was energy dependent and mainly relied on caveolae-mediated endocytosis and macropinocytosis uptake.
Cell apoptosis and necrosis
The MTT results (Figure 6A) showed that, compared with the other four groups, free gefitinib induced the lowest cell inhibition effects and the IC50 (half maximal inhibitory concentration) values in gefitinib-treated gefitinib-resistant Hela cells within 48 hours was 22.3 μg/mL, demonstrating that overexpression of MDR1 in gefitinib-resistant Hela cells could prohibit cell apoptosis by transporting more drugs out of cells and suggesting a big role of MDR1-mediated MDR. Although the naked shMDR1 for silencing MDR1 was combined with free gefitinib for treating cells, due to its low transfection effects and rapid degradation by enzyme, there was no significant difference in the improvement of cell cytotoxicity between free gefitinib and the co-delivery of shMDR1 and gefitinib, and the IC50 values in gefitinib and shMDR1-treated gefitinib-resistant Hela cells within 48 hours was 21.0 μg/mL. By contrast, when cells were treated with either gefitinib alone loaded in NPs or the combination of gefitinib and shMDR1 encapsulated in NPs, the cell viability was significantly decreased and the IC50 values in gefitinib NP- and shMDR1/gefitinib NP-treated cells within 48 hours was 16.7 μg/mL and 12.2 μg/mL, respectively. In addition, the IC50 value of gefitinib NPs combined with shMDR1 NPs was 14.4 μg/mL.
Figure 6.

Cell apoptosis determined by MTT and flow cytometer analysis.
Notes: (A) Viability of gefitinib-resistant Hela cells after incubation with free gefitinib, gefitinib and shMDR1, gefitinib nanoparticles (NPs), gefitinib NPs combined with shMDR1 NPs, and shMDR1/gefitinib NPs for 48 hours (n=3). (B) Cell apoptosis determined by Annexin V–fluorescein isothiocyanate/propidium iodide staining.
Annexin V–FITC/propidium iodide staining assay was performed and the results shown in Figure 6B also further confirm the findings obtained by MTT assay. Free gefitinib and the combination of shMDR1 and gefitinib demonstrated lower nontoxicity and lower sensitivity in Hela/gefitinib cell and resulted in less cell apoptosis. With the co-delivery of gefitinib and shMDR1 encapsulated in NPs, MDR1 was downregulated by silencing MDR1 and the highest cell apoptosis was induced. Similarly, gefitinib NPs and gefitinib NPs combined with shMDR1 NPs also increased the sensitivity of cells to the apoptosis signal, suggesting that, compared with free drugs, more drug could be transferred into cells with the mediation of internalization of NPs, leading to the retention of drug in cells and increasing the cytotoxic effects.
Western blot assay
To detect the expression of relative proteins, Western blot assay was performed. Figure 7 shows that after gefitinib-resistant Hela cells were treated with free drug or drug-loaded NPs, the cell apoptosis effects were improved, represented by the upregulation of expression of cleaved caspase-3, the main marker for playing a central role in the execution phase of cell apoptosis. In the meantime, the ratio of LC3-II to LC3-I was slightly increased, thus indicating that the autophagy effects were strengthened to a slight degree to degrade unnecessary or dysfunctional cellular components through the actions of lysosomes. With the mediation of shMDR1, the expression of MDR1 protein was reduced and inhibited the MDR. Therefore, drugs could not conjugate to an opening within the inner leaflet of the membrane and were effectively protected from being excreted from the cell. Furthermore, the sensitivity of cells to gefitinib was enhanced and the apoptosis effects of cells were promoted through the increasing expression of caspase-3 proteins. Taken together, co-delivery of shMDR1 and gefitinib encapsulated in NPs induced the highest cell apoptosis effects and overcame the MDR by silencing MDR1 with shMDR1.
Figure 7.
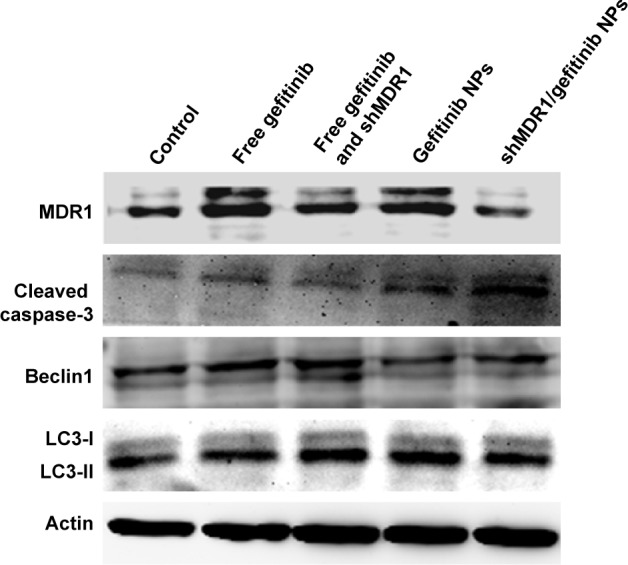
Western blot analyses for investigating apoptotic effects of various gefitinib formulations on gefitinib-resistant Hela cells.
Abbreviation: NPs, nanoparticles.
Discussion
Chemotherapy is one of the most effective tools in many cancer treatments. However, MDR severely blocks the successful management of cancer therapy. The MDR1/P-gp plays an important role in chemotherapy and acts as an efflux pump in cell surface. The function of MDR1 is to eliminate cytotoxic drugs and substances by binding of the hydrophobic drugs with neutral charge as the substrate with the transmembrane region of MDR1. In tumor cells, commonly used chemotherapeutic agents such as doxorubicin, etoposide, and trichostatin A are all P-gp substrates, and their clinical efficacies are thus greatly reduced. Since MDR1 plays a vital role in resistance to chemotherapy, regulating MDR1 is now employed as a therapeutic strategy to conquer the drug resistance in tumors.
Nowadays, modulators or inhibitors of MDR1/P-gp and gene therapy have been used to reverse MDR. As for free drugs, the efflux of drugs mediated by MDR1 could involve the following process: drug diffuses into the cell passively, followed by binding with MDR1 in the cytoplasm, and is then pumped out of the cell. Especially, many drugs with high lipid solubility can combine with MDR1 situated at the surface of the lipid bilayer and diffuse out before entering the cells. Taken together, it is necessary to inhibit the activity and the expression of MDR1 for overcoming MDR. In this study, NPs made of CS were successfully formulated as delivery systems for the anticancer drug gefitinib. Moreover, we showed that, compared with traditional chemical MDR1 inhibitor, shMDR1 in NPs provided specific and long-lasting gene silencing by delivery of plasmids for overcoming MDR effects. The results showed that shMDR1 was integrated into the core of CS NPs, avoiding its degradation by enzyme and serum, thus leading to greater gene silencing. Not only that, but shMDR1 transfection with gefitinib obviously decreased the resistance to gefitinib in gefitinib-resistant Hela cells. This indicates that the inhibition of MDR1 did attenuate MDR effects in gefitinib-resistant Hela cells and the cytotoxicity and apoptosis of cells treated with drugs were significantly enhanced. Figure 3 shows that the NPs could introduce EGFP into the cytosol and do not influence the cell motility. Moreover, the NPs containing shMDR1 and gefitinib could increase the cytotoxicity of gefitinib in cells (Figure 6). As resistance to gefitinib is related to tumor cell autophagy, we also detected the expression of LC3 and Beclin1 in Western blot analysis (Figure 7). This showed that either free gefitinib or NPs containing both shMDR1 and gefitinib could trigger the autophagy process to a slight extent in gefitinib-resistant Hela cells.
We conclude that CS NPs with a small particle size and positive charge could effectively promote co-delivery of gefitinib and shMDR1 into cells and control the sustained release of drugs to prolong the acting time. Importantly, different from simple intracellular diffusion, NPs could transfer more gefitinib and shMDR1 into cells depending on caveolae-mediated endocytosis and the macropinocytosis uptake and prevent drugs from pumping out of cells by effective suppression of MDR1 proteins, leading to the further enhancement on cytotoxicity and apoptosis effects and overcoming MDR effects. Therefore, these nanocarriers represent a promising strategy for the administration of the antineoplastic gefitinib with shMDR1 to tumor cells, which merits further investigation.
Conclusion
Compared to free gefitinib which caused lower sensitivity and low cytotoxicity to resistant cell strains, shMDR1 as a new technology for gene-meditated interference was loaded in NPs to improve shRNA anti-DNA enzyme degradation capacity and inhibit the MDR1 gene expression efficiently. NPs with co-delivery of gefitinib and shMDR1 increased intracellular accumulation of drugs and restored the sensitivity of cells to the drug, thereby reversing the MDR.
Acknowledgments
This work was supported by Liaoning Educational Committee (number L2014339), Natural Science Foundation of Liaoning Province (number 2013022035), and Grant of Liaoning Medical University (number XZJJ20130104-05).
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Joyce H, McCann A, Clynes M, Larkin A. Influence of multidrug resistance and drug transport proteins on chemotherapy drug metabolism. Expert Opin Drug Metab Toxicol. 2015;11(5):795–809. doi: 10.1517/17425255.2015.1028356. [DOI] [PubMed] [Google Scholar]
- 2.Kathawala RJ, Gupta P, Ashby CR, Jr, Chen ZS. The modulation of ABC transporter-mediated multidrug resistance in cancer: a review of the past decade. Drug Resist Updat. 2015;18:1–17. doi: 10.1016/j.drup.2014.11.002. [DOI] [PubMed] [Google Scholar]
- 3.Merk J, Rolff J, Dorn C, Leschber G, Fichtner I. Chemoresistance in non-small-cell lung cancer: can multidrug resistance markers predict the response of xenograft lung cancer models to chemotherapy? Eur J Cardiothorac Surg. 2011;40(1):e29–e33. doi: 10.1016/j.ejcts.2011.02.010. [DOI] [PubMed] [Google Scholar]
- 4.Selle F, Wittnebel S, Biron P, et al. A phase II trial of high-dose chemotherapy (HDCT) supported by hematopoietic stem-cell transplantation (HSCT) in germ-cell tumors (GCTs) patients failing cisplatin-based chemotherapy: the Multicentric TAXIF II study. Ann Oncol. 2014;25(9):1775–1782. doi: 10.1093/annonc/mdu198. [DOI] [PubMed] [Google Scholar]
- 5.Tirmazy SH, Barthakur U, El-Modir A, Anwar S, Fernando I. Chemotherapy for advanced endometrial cancer with carboplatin and epirubicin. Anticancer Res. 2014;34(7):3793–3798. [PubMed] [Google Scholar]
- 6.Ratti F, Cipriani F, Catena M, Paganelli M, Aldrighetti L. Liver failure in patients treated with chemotherapy for colorectal liver metastases: role of chronic disease scores in patients undergoing major liver surgery. A case-matched analysis. Eur J Surg Oncol. 2014;40(11):1550–1556. doi: 10.1016/j.ejso.2014.06.011. [DOI] [PubMed] [Google Scholar]
- 7.Beusterien K, Grinspan J, Kuchuk I, et al. Use of conjoint analysis to assess breast cancer patient preferences for chemotherapy side effects. Oncologist. 2014;19(2):127–134. doi: 10.1634/theoncologist.2013-0359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kunická T, Souček P. Importance of ABCC1 for cancer therapy and prognosis. Drug Metab Rev. 2014;46(3):325–342. doi: 10.3109/03602532.2014.901348. [DOI] [PubMed] [Google Scholar]
- 9.Wu CP, Hsieh CH, Wu YS. The emergence of drug transporter-mediated multidrug resistance to cancer chemotherapy. Mol Pharm. 2011;8:1996–2011. doi: 10.1021/mp200261n. [DOI] [PubMed] [Google Scholar]
- 10.Azzariti A, Porcelli L, Quatrale AE, Silvestris N, Paradiso A. The coordinated role of CYP450 enzymes and P-gp in determining cancer resistance to chemotherapy. Curr Drug Metab. 2011;12:713–721. doi: 10.2174/138920011798357042. [DOI] [PubMed] [Google Scholar]
- 11.Chen Y, Tang Y, Guo C, Wang J, Boral D, Nie D. Nuclear receptors in the multidrug resistance through the regulation of drug-metabolizing enzymes and drug transporters. Biochem Pharmacol. 2012;83:1112–1126. doi: 10.1016/j.bcp.2012.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Singh N, Sarkar J, Sashidhara KV, Ali S, Sinha S. Anti-tumour activity of a novel coumarin-chalcone hybrid is mediated through intrinsic apoptotic pathway by inducing PUMA and altering Bax/Bcl-2 ratio. Apoptosis. 2014;19(6):1017–1028. doi: 10.1007/s10495-014-0975-2. [DOI] [PubMed] [Google Scholar]
- 13.Kontos CK, Christodoulou MI, Scorilas A. Apoptosis-related BCL2-family members: key players in chemotherapy. Anticancer Agents Med Chem. 2014;14(3):353–374. doi: 10.2174/18715206113139990091. [DOI] [PubMed] [Google Scholar]
- 14.Klukovits A, Krajcsi P. Mechanisms and therapeutic potential of inhibiting drug efflux transporters. Expert Opin Drug Metab Toxicol. 2015;11(6):907–920. doi: 10.1517/17425255.2015.1028917. [DOI] [PubMed] [Google Scholar]
- 15.Natarajan K, Xie Y, Baer MR, Ross DD. Role of breast cancer resistance protein (BCRP/ABCG2) in cancer drug resistance. Biochem Pharmacol. 2012;83:1084–1103. doi: 10.1016/j.bcp.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kovalev AA, Tsvetaeva DA, Grudinskaja TV. Role of ABC-cassette transporters (MDR1, MRP1, BCRP) in the development of primary and acquired multiple drug resistance in patients with early and metastatic breast cancer. Exp Oncol. 2013;35(4):287–290. [PubMed] [Google Scholar]
- 17.Yang JJ, Milton MN, Yu S, et al. P-glycoprotein and breast cancer resistance protein affect disposition of tandutinib, a tyrosine kinase inhibitor. Drug Metab Lett. 2010;4(4):201–212. doi: 10.2174/187231210792928279. [DOI] [PubMed] [Google Scholar]
- 18.Ferreira RJ, dos Santos DJ, Ferreira MJ. P-glycoprotein and membrane roles in multidrug resistance. Future Med Chem. 2015;7(7):929–946. doi: 10.4155/fmc.15.36. [DOI] [PubMed] [Google Scholar]
- 19.Seebacher NA, Richardson DR, Jansson PJ. Glucose modulation induces reactive oxygen species and increases P-glycoprotein-mediated multidrug resistance to chemotherapeutics. Br J Pharmacol. 2015;172(10):2557–2572. doi: 10.1111/bph.13079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Binkhathlan Z, Lavasanifar A. P-glycoprotein inhibition as a therapeutic approach for overcoming multidrug resistance in cancer: current status and future perspectives. Curr Cancer Drug Targets. 2013;13(3):326–346. doi: 10.2174/15680096113139990076. [DOI] [PubMed] [Google Scholar]
- 21.Xue P, Yang X, Liu Y, Xiong C, Ruan J. A novel compound RY10-4 downregulates P-glycoprotein expression and reverses multidrug-resistant phenotype in human breast cancer MCF-7/ADR cells. Biomed Pharmacother. 2014;68(8):1049–1056. doi: 10.1016/j.biopha.2014.10.004. [DOI] [PubMed] [Google Scholar]
- 22.Santos SA, Paulo A. Small molecule inhibitors of multidrug resistance gene (MDR1) expression: preclinical evaluation and mechanisms of action. Curr Cancer Drug Targets. 2013;13(8):814–828. doi: 10.2174/15680096113139990082. [DOI] [PubMed] [Google Scholar]
- 23.Kuete V, Efferth T. African flora has the potential to fight multidrug resistance of cancer. Biomed Res Int. 2015;2015:914813. doi: 10.1155/2015/914813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nobili S, Landini I, Giglioni B, Mini E. Pharmacological strategies for overcoming multidrug resistance. Curr Drug Targets. 2006;7(7):861–879. doi: 10.2174/138945006777709593. [DOI] [PubMed] [Google Scholar]
- 25.Wirth T, Parker N, Ylä-Herttuala S. History of gene therapy. Gene. 2013;525(2):162–169. doi: 10.1016/j.gene.2013.03.137. [DOI] [PubMed] [Google Scholar]
- 26.Kaufmann KB, Büning H, Galy A, Schambach A, Grez M. Gene therapy on the move. EMBO Mol Med. 2013;5(11):1642–1661. doi: 10.1002/emmm.201202287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang F, Li T, Zhang B, et al. MicroRNA-19a/b regulates multidrug resistance in human gastric cancer cells by targeting PTEN. Biochem Biophys Res Commun. 2013;434(3):688–694. doi: 10.1016/j.bbrc.2013.04.010. [DOI] [PubMed] [Google Scholar]
- 28.Li JM, Zhang W, Su H, et al. Reversal of multidrug resistance in MCF-7/Adr cells by codelivery of doxorubicin and BCL2 siRNA using a folic acid-conjugated polyethylenimine hydroxypropyl-β-cyclodextrin nanocarrier. Int J Nanomedicine. 2015;10:3147–3162. doi: 10.2147/IJN.S67146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martinez-Quintanilla J, Cascallo M, Gros A, Fillat C, Alemany R. Positive selection of gene-modified cells increases the efficacy of pancreatic cancer suicide gene therapy. Mol Cancer Ther. 2009;8(11):3098–3107. doi: 10.1158/1535-7163.MCT-09-0350. [DOI] [PubMed] [Google Scholar]
- 30.Wu Z, Li X, Zeng Y, et al. In vitro and in vivo inhibition of MRP gene expression and reversal of multidrug resistance by siRNA. Basic Clin Pharmacol Toxicol. 2011;108(3):177–184. doi: 10.1111/j.1742-7843.2010.00642.x. [DOI] [PubMed] [Google Scholar]
- 31.Li M, Deng H, Peng H, Wang Q. Functional nanoparticles in targeting glioma diagnosis and therapies. J Nanosci Nanotechnol. 2014;14(1):415–432. doi: 10.1166/jnn.2014.8757. [DOI] [PubMed] [Google Scholar]
- 32.Ikoba U, Peng H, Li H, Miller C, Yu C, Wang Q. Nanocarriers in therapy of infectious and inflammatory diseases. Nanoscale. 2015;7(10):4291–4305. doi: 10.1039/c4nr07682f. [DOI] [PubMed] [Google Scholar]
- 33.Xu H, Li Z, Si J. Nanocarriers in gene therapy: a review. J Biomed Nanotechnol. 2014;10(12):3483–3507. doi: 10.1166/jbn.2014.2044. [DOI] [PubMed] [Google Scholar]
- 34.Yang Y, Wang S, Wang Y, Wang X, Wang Q, Chen M. Advances in self-assembled chitosan nanomaterials for drug delivery. Biotechnol Adv. 2014;32(7):1301–1316. doi: 10.1016/j.biotechadv.2014.07.007. [DOI] [PubMed] [Google Scholar]
- 35.Ding F, Deng H, Du Y, Shi X, Wang Q. Emerging chitin and chitosan nanofibrous materials for biomedical applications. Nanoscale. 2014;6(16):9477–9493. doi: 10.1039/c4nr02814g. [DOI] [PubMed] [Google Scholar]
- 36.Song F, Li X, Wang Q, Liao L, Zhang C. Nanocomposite hydrogels and their applications in drug delivery and tissue engineering. J Biomed Nanotechnol. 2015;11(1):40–52. doi: 10.1166/jbn.2015.1962. [DOI] [PubMed] [Google Scholar]
- 37.Li MH, Yu H, Wang TF, et al. Tamoxifen embedded in lipid bilayer improves the oncotarget of liposomal daunorubicin in vivo. J Mater Chem B Mater Biol Med. 2014;2(12):1619–1625. doi: 10.1039/c3tb21423k. [DOI] [PubMed] [Google Scholar]
- 38.Shen H, Sun T, Ferrari M. Nanovector delivery of siRNA for cancer therapy. Cancer Gene Ther. 2012;19(6):367–373. doi: 10.1038/cgt.2012.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bates K, Kostarelos K. Carbon nanotubes as vectors for gene therapy: past achievements, present challenges and future goals. Adv Drug Deliv Rev. 2013;65(15):2023–2033. doi: 10.1016/j.addr.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 40.Hu FQ, Chen WW, Zhao MD, Yuan H, Du YZ. Effective antitumor gene therapy delivered by polyethylenimine-conjugated stearic acid-g-chitosan oligosaccharide micelles. Gene Ther. 2013;20(6):597–606. doi: 10.1038/gt.2012.72. [DOI] [PubMed] [Google Scholar]
- 41.Raftery R, O’Brien FJ, Cryan SA. Chitosan for gene delivery and orthopedic tissue engineering applications. Molecules. 2013;18(5):5611–5647. doi: 10.3390/molecules18055611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Su C, Li H, Shi Y, et al. Carboxymethyl-β-cyclodextrin conjugated nanoparticles facilitate therapy for folate receptor-positive tumor with the mediation of folic acid. Int J Pharm. 2014;474(1–2):202–211. doi: 10.1016/j.ijpharm.2014.08.026. [DOI] [PubMed] [Google Scholar]
- 43.Liu M, Feng B, Shi Y, Su C, Song H, Cheng W, Zhao L. Protamine nanoparticles for improving shRNA-mediated anti-cancer effects. Nanoscale Res Lett. 2015;10:134. doi: 10.1186/s11671-015-0845-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhao L, Su R, Cui W, Shi Y, Liu L, Su C. Preparation of biocompatible heat-labile enterotoxin subunit B-bovine serum albumin nanoparticles for improving tumor-targeted drug delivery via heat-labile enterotoxin subunit B mediation. Int J Nanomedicine. 2014;9:2149–2156. doi: 10.2147/IJN.S60764. [DOI] [PMC free article] [PubMed] [Google Scholar]