

Abstract
Mitochondrial genome rearrangements that result in control region duplication have been described for a variety of birds, but the mechanisms leading to their appearance and maintenance remain unclear, and their effect on sequence evolution has not been explored. A recent survey of mitochondrial genomes in the Psittaciformes (parrots) found that control region duplications have arisen independently at least six times across the order. We analyzed complete mitochondrial genome sequences from 20 parrot species, including representatives of each lineage with control region duplications, to document the gene order changes and to examine effects of genome rearrangements on patterns of sequence evolution. The gene order previously reported for Amazona parrots was found for four of the six independently derived genome rearrangements, and a previously undescribed gene order was found in Prioniturus luconensis, representing a fifth clade with rearranged genomes; the gene order resulting from the remaining rearrangement event could not be confirmed. In all rearranged genomes, two copies of the control region are present and are very similar at the sequence level, while duplicates of the other genes involved in the rearrangement show signs of degeneration or have been lost altogether. We compared rates of sequence evolution in genomes with and without control region duplications and did not find a consistent acceleration or deceleration associated with the duplications. This could be due to the fact that most of the genome rearrangement events in parrots are ancient, and additionally, to an effect of body size on evolutionary rate that we found for mitochondrial but not nuclear sequences. Base composition analyses found that relative to other birds, parrots have unusually strong compositional asymmetry (AT- and GC-skew) in their coding sequences, especially at four-fold degenerate sites. Furthermore, we found higher AT skew in species with control region duplications. One potential cause for this compositional asymmetry is that parrots have unusually slow mtDNA replication. If this is the case, then any replicative advantage provided by having a second control region could result in selection for maintenance of both control regions once duplicated.
Keywords: mitochondrial genome, control region duplication, evolutionary rate, base composition, nucleotide skew, body size effect
Graphical abstract

1. Introduction
Mitochondrial DNA (mtDNA) sequence evolution in bilaterian animals is rapid compared with that of nuclear DNA (nucDNA) (Lynch et al., 2006), a feature that has made it useful in phylogenetic reconstruction. In spite of this high rate of sequence evolution, the vertebrate mitochondrial genome was long thought to be relatively stable in terms of genome structure, and rearrangements in gene order were thought to be rare events (Boore, 1999; Gissi et al., 2008). However, over the past 15 years, sequence-based studies have revealed that rearrangements are not uncommon, particularly in birds, where several different lineages have undergone repeated rearrangements (Abbott et al., 2005; Bensch and Härlid, 2000; Cho et al., 2009; Eberhard et al., 2001; Gibb et al., 2007; Haring et al., 2001; Mindell et al., 1998a; Morris-Pocock et al., 2010; Roques et al., 2004; Schirtzinger et al., 2012; Singh et al., 2008; Slack et al., 2007; Verkuil et al., 2010; Zhou et al., 2014). Despite the growing list of avian taxa in which changes in mitochondrial gene order have been described, questions persist regarding the mechanisms by which rearrangements occur, the degree to which duplications are retained over evolutionary time, and the effect that these rearrangements have on the function, replication, and evolution of the mitochondrial genome.
The mitochondrial genome of most bilaterian animals includes the same set of 37 genes (two ribosomal RNAs, 13 proteins, and 22 tRNAs) and a non-coding control region (Boore, 1999; Lavrov, 2007). This consistency in gene content across distantly related lineages, as well as the lack of intergenic spacers, suggests that that the mitochondrial genome is under selection for compact size (Rand and Harrison, 1986). The first avian mitochondrial genome to be completely sequenced was that of the chicken (Desjardins and Morais, 1990), and its gene order (hereafter referred to as the “typical” avian gene order) is the one most commonly observed in birds. The typical avian gene order differs from most other vertebrates due to a rearrangement near the control region, resulting in the gene order shown in Figure 1A. In 1998, Mindell et al. reported an alternative avian gene order that contains a non-coding region thought to be a degenerate copy of the control region (Figure 1H). Subsequently, additional gene orders have been reported for a variety of avian taxa (Table 1), indicating that mitochondrial genome rearrangements are taxonomically widespread and more common than previously thought. Gibb et al. (2007) reviewed the gene orders found in avian mitochondrial genomes and concluded that there are at least four distinct gene orders within birds, with the typical (chicken) gene order being the ancestral one. Since then, another gene order variant has been found in ruffs (Verkuil et al., 2010), and a recent study that examined mitochondrial genome sequences of 16 ardeid birds found four distinct gene orders, including two that had not been previously described, all thought to be derived from a single ancestral genome rearrangement (Zhou et al., 2014). In all cases known to date, the novel avian gene orders can be derived from a tandem duplication of the control region and neighboring genes followed by subsequent degeneration and/or loss of some of the duplicate genes (Bensch and Härlid, 2000)(see Figure 1 and Table 1).
Fig. 1.
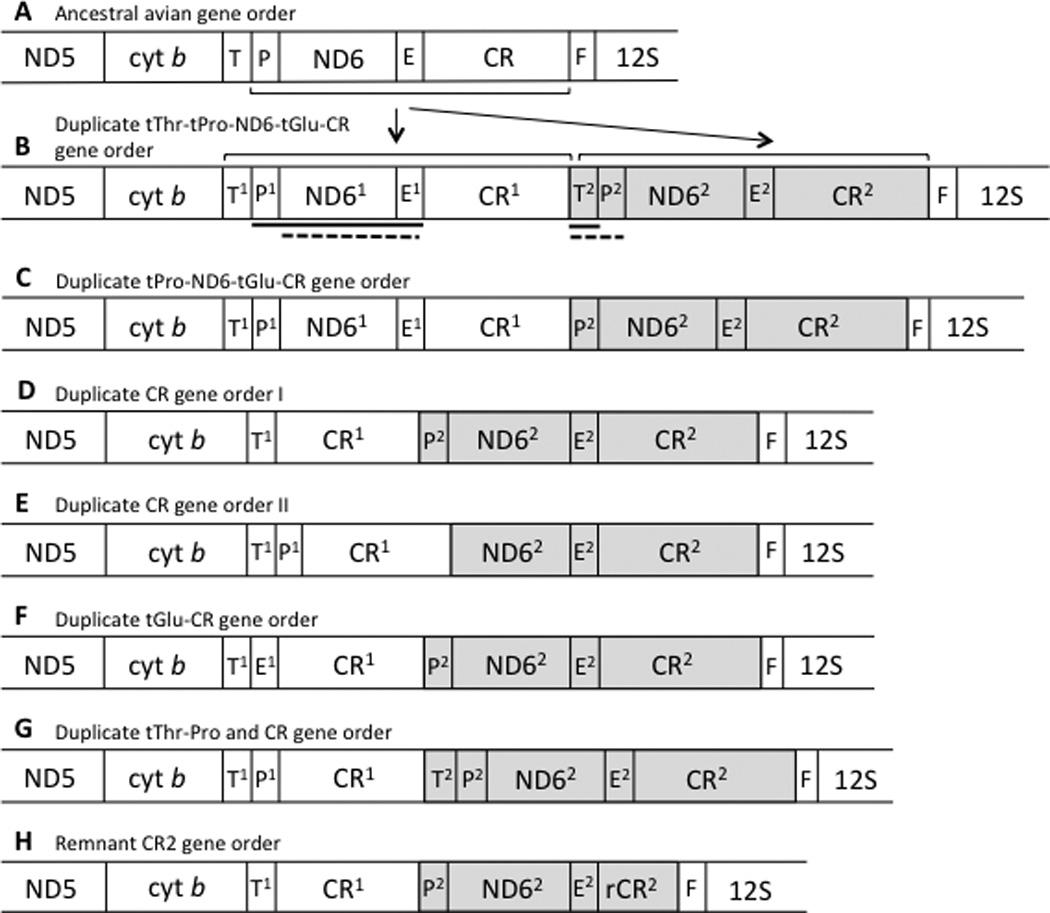
Gene orders surrounding the control region (CR) known for avian mitochondrial genomes. Brackets and arrows indicate the segment involved in a tandem duplication that has been hypothesized to be involved in the conversion between the standard and alternative gene orders. The solid and dashed lines beneath B indicate sections that degenerate (or are lost) in the transitions from B to D and E, respectively, to yield the two gene orders observed in parrots. Gene order H is similar to D, but the second copy of the control region is degenerate. Transfer RNAs are indicated by their single-letter abbreviations, E (Glutamyl), F (Phenylalanine), P (Proline), and T (Threonine). Pseudogenes, which have been described in various taxa for this portion of the mitochondrial genome, are not shown. Shading indicates hypothesized homology between highlighted regions. Taxa for which these different orders have been reported to date are listed in Table 1.
Table 1.
Avian species for which rearranged mitochondrial genomes and gene order have been reported in the literature; genomes sequenced for this study are also included. Gene orders are named following the suggestions of Gibb et al. (2007), and letters indicate the panel in Figure 1 that illustrates the gene arrangement.
| Gene order | Fig. 1 | Species | Reference |
|---|---|---|---|
| Duplicate tRNAThr-CR | B | Thalassarche albatrosses | Abbott et al. 2005 |
| Black-browed albatross (Diomedea melanophris) | Slack et al. 2006 and Gibb et al. 2007 | ||
| Black-faced spoonbill (Platalea minor) | Cho et al. 2009 | ||
| Chinese egret (Egretta eulophotes) | Zhou et al. 2014 | ||
| Eastern reef heron (Egretta sacra) | Zhou et al. 2014 | ||
| Little egret (Egretta garzetta) | Zhou et al. 2014 | ||
| Striated heron (Butorides striatus) | Zhou et al. 2014 | ||
| Chinese pond heron (Ardeola bacchus) | Zhou et al. 2014 | ||
| Black-crowned night heron (Nycticorax nycticorax) | Zhou et al. 2014 | ||
| Eurasian bittern (Botaurus stellaris) | Zhou et al. 2014 | ||
| Yellow bittern (Ixobrychus sinensis) | Zhou et al. 2014 | ||
| Schrenck’s bittern (Ixobrychus eurythmus) | Zhou et al. 2014 | ||
| Black bittern (Ixobrychus flavicollis) | Zhou et al. 2014 | ||
| Brown booby (Sula leucogaster) | Morris-Pocock et al. 2010 | ||
| Red-footed booby (Sula sula) | Morris-Pocock et al. 2010 | ||
| Blue-footed booby (Sula nebouxii) | Morris-Pocock et al. 2010 | ||
| Duplicate tRNAPro-CR | C | Ruffs (Philomachus pugnax) | Verkuil et al. 2010 |
| Duplicate CR I | D | Eastern great egret (Ardea modesta) | Zhou et al. 2014 |
| Intermediate egret (Ardea intermedia) | Zhou et al. 2014 | ||
| Cattle egret (Ardea ibis) | Zhou et al. 2014 | ||
| Osprey (Pandion haliaetus) | Gibb et al. 2007 | ||
| Ivory-billed aracari (Pteroglossus azara) | Gibb et al. 2007 | ||
| Amazona parrots | Eberhard et al. 2001 | ||
| Green-rumped parrotlet (Forpus passerinus) | This study | ||
| African grey parrot (Psittacus erithacus) | This study | ||
| Budgerigar (Melopsittacus undulatus) | Guan et al. unpub. (NC 009134) | ||
| Tachycineta swallows | Cerasale et al. 2012 | ||
| Gray-breasted martin (Progne chalybea) | Cerasale et al. 2012 | ||
| Reed warbler (Acrocephalus scirpaeus) | Singh et al. 2008 | ||
| Blackcap (Sylvia atricapilla) | Singh et al. 2008 | ||
| Duplicate CR II | E | Green racquet-tailed parrot (Prioniturus luconensis) | This study |
| Duplicate tRNAGlu-CR | F | Grey heron (Ardea cinerea) | Zhou et al. 2014 |
| Purple heron (Ardea purpurea) | Zhou et al. 2014 | ||
| Duplicate tRNAThr-tRNAPro and CR | G | Cinnamon bittern (Ixobrychus cinnamomeus) | Zhou et al. 2014 |
| Remnant CR2 | H | Bearded vulture (Gypaetus barbatus) | Roques et al. 2004 |
| Egyptian vulture (Neophron percnopterus) | Roques et al. 2004 | ||
| Bonelli’s eagle (Hieraaetus fasciatus) | Cadahía et al. 2009 | ||
| Common buzzard (Buteo buteo) | Haring et al. 2001 | ||
| Peregrine falcon (Falco peregrinus) | Mindell et al. 1998 | ||
| Forest falcon (Falco gilvicollis) | Gibb et al. 2007 | ||
| American kestrel (Falco sparverius) | Gibb et al. 2007 | ||
| Pileated woodpecker (Dryocopus pileatus) | Gibb et al. 2007 | ||
| Fuscous flycatcher (Cnemotriccus fuscatus) | Slack et al. 2007 | ||
| Superb lyrebird (Menura novaehollandiae) | Slack et al. 2007 | ||
| Petroica robins (P. phoenicea, P. boodang, P. goodenovii) | Cooke et al. 2012 | ||
| Phylloscopus warblers | Bensch and Härlid 2000 | ||
| Eastern orphean warbler (Sylvia crassirostris) | Singh et al. 2008 | ||
In most avian taxa that have been examined to date mitochondrial rearrangements involve retention of both duplicated control regions and degeneration of the neighboring duplicated coding genes (Table 1). Why duplicated control regions are often maintained while neighboring duplicated genes usually degenerate may relate to the function of the control region itself. The control region has long been thought to contain the origin of transcription and replication of the mitochondrial genome (Brown et al., 2005; Shadel and Clayton, 1997), but its precise role in this process is uncertain. In vertebrates, the control region is thought to contain the origin of heavy-strand replication (OH) and transcription promoters (Clayton 1982, 1991). However, a study of chicken mtDNA replication concluded that replication of the mitochondrial genome is bidirectional and can initiate across the entire genome, with the majority of initiation events mapping to the ND6 gene, which in the typical bird gene order is just upstream of the control region (Reyes et al., 2005). The Reyes et al. (2005) study re-casts the avian control region as a genomic feature that may function more as a replication promoter (much as a gene promoter initiates transcription of a nearby gene) rather than the replication initiation site itself, as it has been traditional considered. Given the control region’s involvement in replication, the presence of two control regions could affect rates of evolution in other mitochondrial genes.
One aspect of sequence evolution that could give further insight into the potential role of duplicated control regions is the degree of base composition asymmetry in the DNA strands that compose the mitochondrial genome. In a study of mammalian mtDNA, Reyes et al. (1998) proposed that the asymmetrical base composition (measured as AT and GC skew) is a result of deamination of C and A on the H strand that occurs when the H strand is in a single-stranded state during replication. Species with slower replication would potentially have greater asymmetry because their H strand is in a single-stranded form for longer periods of time. This asymmetry is expected to be strongest at four-fold degenerate sites, which are under weak (if any) selection, so an observed difference between skew for all sites versus skew for four-fold degenerate sites implicates asymmetrical directional mutation pressure, rather than selection, as a mechanism causing the base composition asymmetry (Reyes et al., 1998). In snakes, duplicate control regions have been shown to be associated with (though perhaps not the direct cause of) changes in the rates of sequence evolution and base composition of the mitochondrial genome (Jiang et al., 2007), but their effect on rates of evolution in birds have yet to be examined.
Parrots (the avian order Psittaciformes) provide a particularly rich group in which to examine mitochondrial genome rearrangements and their effects on sequence evolution. Duplicate control regions were first found in the genus Amazona by Eberhard et al. (2001). Schirtzinger et al. (2012) followed by conducting the only systematic survey to date of genome rearrangements within a large avian group, using diagnostic PCR fragments to screen 117 parrot species from 79 of the 82 extant genera for the presence of control region duplications. Of these, about half of the species (belonging to 26 different genera) were found to have duplicate control regions. When control region status was mapped onto a phylogeny, the typical avian gene arrangement with a single control region was found to be ancestral and control region duplications arose at least six times in the parrots, with no apparent reversions from duplicate to single control regions (Schirtzinger et al., 2012).
The Schirtzinger et al. (2012) analysis was designed to detect the presence of duplicate control regions, but did not determine the gene order across the mitochondrial genomes in the taxa where duplicate control regions were found. Therefore, it is not known whether rearrangements leading to control region duplications always result in the same gene order, whether entire control region copies are maintained in a convergent state (as found in snakes), or whether the gene orders in the rearranged genomes are consistent with the tandem duplication model illustrated in Figure 1. Also, there has been no examination of whether the presence of duplicate control regions affects rates of sequence evolution and base composition in the rest of the mitochondrial genome. To investigate these questions, we sequenced the complete mitochondrial genomes of selected parrot taxa that were chosen to provide phylogenetically paired lineages with single vs. duplicate control regions. Our goal was to obtain at least one pair of taxa (one with and one without the control region duplication) spanning each of the duplication origins identified by Schirtzinger et al. (2012). The genome sequences that we obtained were analyzed in conjunction with other parrot mitochondrial genomes that are available in GenBank, in order to characterize the pattern of genome evolution associated with control region duplications in this large avian group.
2. Methods
2.1 Genome sequencing
Taxa were selected for genome sequencing based on preliminary results obtained during Schirtzinger et al’s (2012) study, and to complement genome sequences available in GenBank (Table 2). Our goal was to obtain pairs of taxa that span each origin of duplicate control regions reconstructed on a parrot phylogeny.
Table 2.
Mitochondrial genomes examined for this study. Genomes with duplicate control regions are indicated with an asterisk; partial genome sequences are indicated with a superscript and sections missing from them are indicated below the table. Diamonds (◆) indicate taxa that were included in the base composition analyses. Nomenclature for species formerly included in the genus Aratinga follows the revision by Remsen et al. (2008).
| Species | Accession number | Reference | |
|---|---|---|---|
| Nestor notabilis | ◆ | KM611472 | This study |
| Strigops habroptilusa | ◆ | NC_005931 | Harrison et al. 2004 |
| Nymphicus hollandicus | ◆ | NC_015192 | Pacheco et la. 2011 |
| Calyptorhynchus baudinii | JF414242 | White et al. 2011 | |
| Calyptorhynchus lathami | JF414241 | White et al. 2011 | |
| Calyptorhynchus latirostris | JF414243 | White et al. 2011 | |
| Cacatua moluccensis | ◆ | JF414239 | White et al. 2011 |
| Cacatua pastinator | JF414240 | White et al. 2011 | |
| Psittacus erithacus* | ◆ | KM611474 | This study |
| Ara militaris | ◆ | KM611466 | This study |
| Rhyncopsitta terrisi | KF010318 | Urantowka et al. 2013d | |
| Thectocercus [Aratinga] acuticaudata | JQ782214 | Urantowka et al. 2013a | |
| Psittacara [Aratinga] brevipes | KC936100 | Urantowka et al. 2013c | |
| Eupsittula [Aratinga] pertinax | ◆ | NC_015197 | Pacheco et al. 2011 |
| Amazona barbadensis* | JX524615 | Urantowka et al. 2013b | |
| Amazona o. ochrocephala* | ◆ | KM611467 | This study |
| Forpus passerinus* | ◆ | KM611470 | This study |
| Forpus modestus (sclateri)*b | ◆ | HM755882 | Pacheco et al. 2011 |
| Brotogeris cyanoptera | ◆ | NC_015530 | Pacheco et al. 2011 |
| Myiopsitta monachus | ◆ | KM611471 | This study |
| Pionites leucogasterc | KM611478, KM611479 | This study | |
| Deroptyus accipitrinus*d | ◆ | KM611476 | This study |
| Psittrichas fulgidus | ◆ | KM611475 | This study |
| Coracopsis vasa | ◆ | KM611468 | This study |
| Neophema chrysogaster | JX133087 | Miller et al. 2013 | |
| Melopsittacus undulatus* | ◆ | NC_009134 | Guan et al. unpublished |
| Melopsittacus undulatus*e | KM611477 | This study | |
| Agapornis roseicollis | ◆ | NC_011708 | Pratt et al. 2009 |
| Prioniturus luconensis* | ◆ | KM611473 | This study |
| Tanygnathus lucionensisf | ◆ | KM611480 | This study |
| Eclectus roratus | ◆ | KM611469 | This study |
S. habroptilus CR
F. modestus tPro/CR1/ND6/tGlu/CR2
P. leucogaster partial 16S/tLeu/ND1/tIle/tGln/tMet/partial ND2; tPro/partial ND6
D. accipitrinus CR1/ND6/tGlu/CR2
M. undulatus ND4L/ND4/tHis
T. luconensis partial ND6/tGlu/CR/tPhe/12S
Template DNA was extracted from tissue samples using DNEasy kits (Qiagen Inc., Valencia, CA) following the manufacturer’s protocol. Mitochondrial DNA was amplified in large (3–4.5kb) segments using long-range PCR (LA-Taq, TaKaRa) and degenerate primers designed for avian mtDNA (Sorenson et al., 1999). These long segments were then used as templates for subsequent PCR and sequencing of shorter, overlapping segments. The long templates were used to minimize the chances of sequencing nuclear mitochondrial DNA inserts (NUMTs), since the average size of NUMTs has been found to be well below 1kb (Richly and Leister, 2004). For PCR and sequencing, we used primers designed for general avian use (Sorenson et al., 1999, 2003), primers used in previous studies of parrot mtDNA (Cheng et al., 1994; Eberhard et al., 2001; Eberhard and Bermingham, 2004; Miyaki et al., 1998; Palumbi, 1996; Tavares et al., 2004), and primers designed for this study (Supplemental Table 1). PCR fragments were run in agarose gels to estimate their size and check for the presence of secondary bands. For several taxa, we were unable to eliminate secondary bands in long-range PCRs spanning the control region; in those cases, the segment from cytb to 12S was amplified and sequenced as a series of overlapping fragments. PCR products were cleaned using polyethylene glycol (PEG) precipitation, and sequenced both strands using BigDye v.3.1 (Applied Biosystems Inc., Foster City, CA) cycle sequencing chemistry on an ABI 3100 automated sequencer.
For each genome, sequence fragments were aligned manually using Sequencher 4.1.2 (Gene Codes Corp., Ann Arbor, MI). Once assembled, the genome sequences were aligned to the Agapornis roseicollis mitochondrial genome from GenBank (Pratt et al., 2009) in order to examine gene order and check for indels. We searched for pseudogenes in rearranged genomes by attempting to align functional coding sequences (of genes expected to be involved in the rearrangements) from that same genome to the control region and surrounding spacer sequence. If at least ~50 bp of a coding sequence could be aligned with >50% similarity, it was considered to be a pseudogene. In taxa with rearranged genomes, the tRNAs and pseudo-tRNAs were folded using tRNASCAN-LE (Lowe and Eddy, 1997) to assess whether tRNAs fold appropriately and code for the correct amino acids.
Due to the presence of pseudogenes in some taxa, and our limited understanding of control region function and sequence evolution, identifying the boundaries of the control region sensu stricto is not straightforward. For the purpose of comparing control region lengths across taxa, we used boundaries that could reliably be identified across taxa. As a more inclusive length measure, the boundaries of control regions were defined by the flanking functional genes, but this region could potentially contain pseudogenes in rearranged genomes (see Figure 2). One of the few readily identifiable control region features of all known parrot control regions is a poly-C sequence located near the 5’ end of the control region, and sometimes called the “goose hairpin” due to its similarity to a sequence at the start of the goose control region (Quinn and Wilson, 1993). We used this landmark as a proxy for the 5’ end of the control region sensu stricto in order to estimate the length of the 5’ spacer that sometimes contains pseudogenes.
Fig. 2.
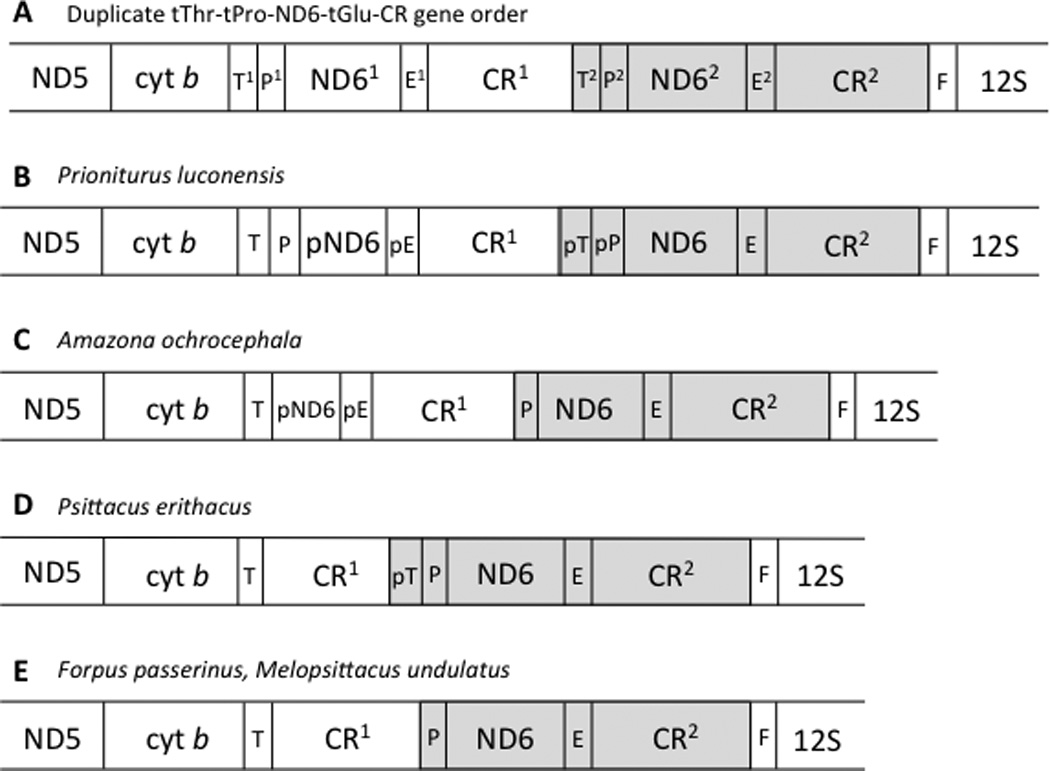
A: Gene order resulting from a tandem duplication of the tThr to CR segment of the mitochondrial genome, and thought to be ancestral to the gene orders shown below. B–E: Gene orders surrounding the control region (CR) for five parrot species with duplicate control regions, each representing an independent origin of the duplicate control region state. Transfer RNAs are indicated by their single-letter abbreviations, E (Glutamyl), T (Threonine), P (Proline) and F (Phenylalanine). Pseudogenes are indicated with a “p” preceding the gene’s abbreviation. The gene order shown in A is that resulting from the tandem duplication illustrated in Figure 1, and which is the hypothesized precursor to the gene orders shown in B–E. Shading indicates hypothesized homology between highlighted regions.
Sequence variation within the control region—within an individual as well as across taxa—was assessed with a sliding window analysis using a custom R script (K. Harms, pers. comm.; available from authors upon request). We counted the number of variable nucleotide positions observed within non-overlapping 25-nt windows along the aligned control region sequences. One set of analyses examined interspecific variation in control region sequences by comparing the control regions of three sets of closely related species: four species of Amazona (A. o. ochrocephala, A. o. auropalliata, A. o. oratrix, A. barbadensis, and A. farinosa), three species of parakeet formerly classified as members of the genus Aratinga (Thectocercus acuticaudata, Psittacara holochlora brevipes, and Eupsittula pertinax), and five species of cockatoos (Cacatua moluccensis, Cacatua pastinator, Calyptorhynchus baudinii, Calyptorhynchus lathami, and Calyptorhynchus latirostris). Broader taxonomic comparisons were not possible because control regions could not be aligned across genera. Because Amazona parrots have duplicate control regions, only the CR2 sequences were used for this control region alignment, to provide results comparable to the other two inter-specific alignments from taxa with single control regions. The second set of sliding window analyses compared the two control regions (CR1 and CR2) for each of the species with duplicate control regions. In all of these analyses, the 5’ end of a given alignment began at the poly-C sequence, and the 3’ end of the longer control region was trimmed so that it matched the end of the shortest control region in that alignment. This analysis was performed for Amazona ochrocephala, Forpus passerinus, Melopsittacus undulatus, Prioniturus luconensis, and Psittacus erithacus; in addition we analyzed the control regions of three Amazona taxa for which complete CR1 and CR2 sequences are available in GenBank: A. ochrocephala auropalliata (AF338819), A. ochrocephala oratrix (AF338820) and A. farinosa (AF338821).
2.2 Rates of evolution
To examine rates of evolution across the genome within a phylogenetic context, we used PhyML (Guindon and Gascuel, 2003) to estimate branch lengths on a user-defined tree. We used the Bayesian tree obtained by Schirtzinger et al. (2012), pruned to include only the taxa for which whole-genome sequence data were available at the time that these analyses were performed (taxa indicated with diamonds in Table 2; tree shown in Figure 3). The tree was only used to define the topology used in the analysis; PhyML used the sequence data from a given gene region(s) to optimize branch lengths on the specified tree. Branch lengths were calculated based on several different sequence matrices (as described below), and for each dataset the appropriate model of evolution was selected using jModelTest 2.1.1 (Darriba et al., 2012). Because our analyses of base composition revealed differences in nucleotide skew among lineages (see Section 3.4), which could potentially impact our evolutionary rate estimates (Phillips et al., 2004), the sequence data were RY-coded using the program FASconCAT-G (Kück and Meusemann, 2010). We also conducted these evolutionary rate analyses using a dataset that was restricted to four-fold degenerate sites, which are expected to be the least constrained by selection, and therefore the most likely to accumulate changes (Reyes et al., 1998).
Fig. 3.

Cladogram defining phylogenetic relationships among taxa for which complete mitochondrial genomes were analyzed using PhyML. The relationships represent a pruned version of the phylogeny obtained by Schirtzinger et al. (2012) in a Bayesian analysis of 117 parrot taxa. Black circles indicate taxa with duplicate control regions. Numbered brackets indicate pairs of taxa used in comparisons of taxa with vs. without duplicate control regions. One taxon, Eupsittula pertinax, appears in the Schirtzinger et al. (2012) phylogeny under its previous name, Aratinga pertinax.
Two sets of branch length analyses were conducted using data from the complete genomes. First, we used a concatenated mitochondrial gene dataset, which included 11 protein coding genes (ND1 and ND6 were omitted due to missing data for several taxa) and six pairs of taxa. Second, we estimated branch lengths using sequence data from one protein coding gene at a time for each of the light-strand coding regions (i.e. all coding genes except ND6). For these two sets of analyses, we compared the rate of sequence evolution (branch length) between taxa in phylogenetically matched pairs in which one taxon has a single control region and the other has two control regions. The pairs of taxa used in these comparisons are shown in Figure 3. For each pair of taxa, we recorded their branch lengths from the node representing their most recent common ancestor. To determine whether the presence of a duplicate control region affects the rate of sequence evolution in the mitochondrial genome, we conducted paired t-tests to determine whether branch lengths differed between taxa with duplicate control regions and those without. A paired t-test was done for the concatenated mitochondrial gene dataset, and also for each of the protein coding gene datasets (in this latter case, a Bonferroni correction for multiple tests was applied). In addition, for each taxon pair, we conducted a paired t-test to compare the branch lengths calculated using each of the protein coding genes. All t-tests were two-tailed.
We conducted a third set of branch length analyses using a data set from Schirtzinger et al. (2012) with much broader taxon sampling (100 species) but fewer mitochondrial genes (ND2 and CO1). As a baseline comparison, we also used Schirtzinger et al’s data to examine rates of evolution in two nuclear genes, transforming growth factor beta 2 intron one (TGFB2) and tropomyosin intron five (TROP), which would presumably be unaffected by a mitochondrial genome rearrangement. Branch lengths were optimized using PhyML as described above, and we then compared branch lengths of taxa belonging to clades A–F in Figure 5 of Schirtzinger et al. (2012), which represent independent origins of control region duplication, with those of taxa in the nearest sister clade with single control regions (see Supplemental Figure 1). For each pair of clades, we compared node-to-tip branch lengths from the node representing their most recent common ancestor. A two-tailed Mann-Whitney U-test was done for each pair of clades to determine whether the presence of a duplicate control region is associated with either longer or shorter branch lengths. For reasons indicated above, this set of analyses was also conducted using RY-coding of the COI and ND2 datasets. This third analysis of evolutionary rates does not control for phylogeny as precisely as the first two above (Rothfels and Schuettpelz, 2014), but has the advantage of including a larger (and perhaps more representative) sample of taxa bracketing each origin of duplicate control regions.
Fig. 5.
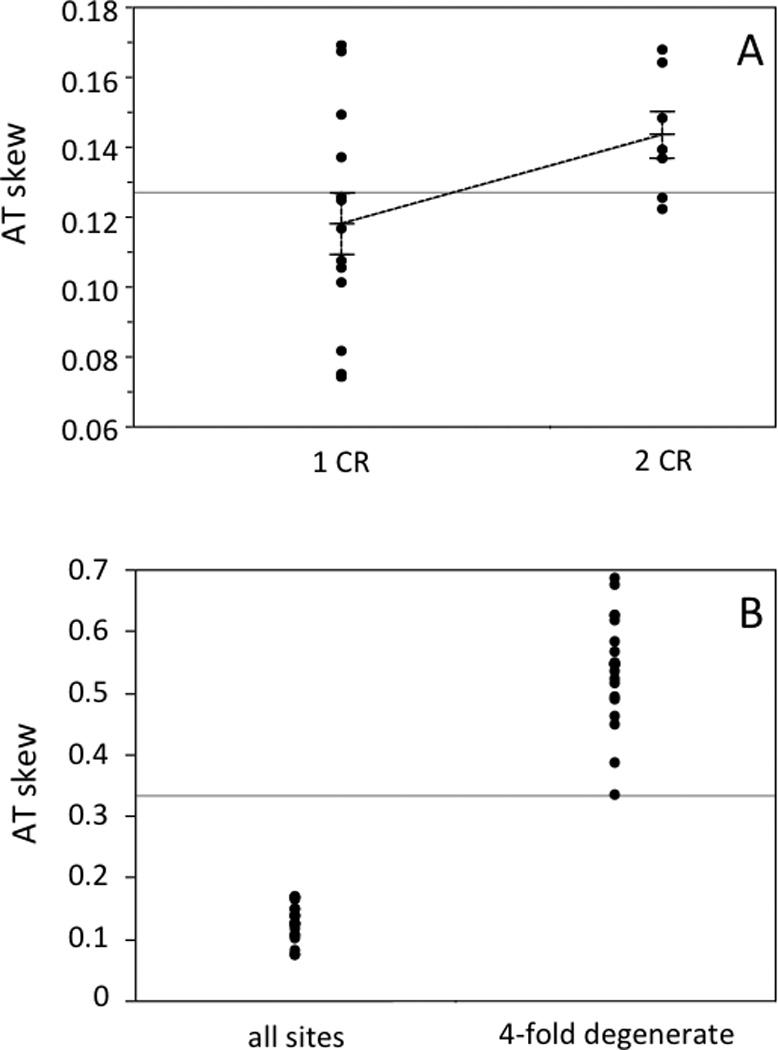
AT skew for the H-strand protein coding genes of 20 parrot mitochondrial genomes. A) Nucleotide skew was calculated using the formulae of Perna and Kocher (1995) for all sites. Data are grouped according to the number of control regions in a given genome, and each group’s mean is indicated, along with standard error bars based on the standard error of the mean. B) AT skew values are shown for all genomes, calculated using all sites and using only 4-fold degenerate sites.
None of the control region duplication events in parrots are recent (see Schirtzinger et al., 2010; Figure 3 and Supplemental Figure 1), and as a consequence, some of our comparisons involved taxa with very different body sizes. Since body size has been found to be negatively correlated with rates of sequence evolution in birds (e.g., Nunn and Stanley, 1998; Eo et al., 2011; Thomson et al., 2014), we examined our data for evidence that body size affects rates of evolution in parrots. We used the phylogenetic approach described by Nunn and Stanley (1998) to analyze mitochondrial sequences (ND2) and nuclear sequences (concatenated TGFB2+TROP) from Schirtzinger et al. (2012), which allowed us to include a large enough number of taxa for the test. Briefly, we used the Schirtzinger et al. phylogeny (Figure 3 in Schirtzinger et al., 2012) to select terminal paired sister taxa with nonzero branch lengths, and obtained body mass data for those taxa from the literature. Most body mass data were from Dunning (2008); in a few cases we used data from Forshaw (1989), and the weight for Strigops was read from a graph in Moorhouse and Powesland (1991). If both male and female weights were available, we used female weights as mtDNA is maternally inherited. If members of a terminal sister pair did not differ in mass, the next closest taxon was used in the comparison. This yielded a total of 27 phylogenetically independent comparisons for the ND2 dataset, and 23 comparisons for the nuclear dataset. For each set of comparisons, we counted the number of times the larger taxon from each pair was associated with a longer branch, and the number of times the larger taxon in a given pair was associated with a shorter branch. We used a one-tailed binomial test to determine whether there was a greater number of pairs where the larger-bodied species had a shorter branch length (Nunn and Stanley, 1998; Eo et al., 2011; Thomson et al., 2014).
2.3 Base Composition
Using the set of 20 parrot mitochondrial genomes indicated in Table 2, we calculated base composition across the complete genome and also for the H-strand protein coding genes (ND6, which is coded on the L-strand, was omitted) using MEGA 5.10 (Tamura et al., 2011). Nucleotide skew was calculated for these same regions using the formulae of Perna and Kocher (1995), which provides an index of compositional asymmetry between strands, with values between −1 and 1, where 0 indicates an absence of strand bias (Reyes et al., 1998); the sign of the skew value is only meaningful with reference to a particular strand, since the skew calculated for the opposite strand would take the same value with the opposite sign (Perna and Kocher, 1995). Skew was calculated for all sites, and also for four-fold degenerate sites, which are expected to be less constrained (Reyes et al., 1998). We then compared these measures of nucleotide skew in taxa with single control regions versus those with duplicate control regions in two ways: using the data from i) all of the parrot genomes (two-tailed t-test) and ii) the phylogenetically paired genomes (Figure 3, pairs 1–6) analyzed in the PhyML analysis described above (two-tailed paired t-test).
3. Results and Discussion
We used 10 mitochondrial genomes newly sequenced for this study in combination with those available in GenBank to investigate structural and evolutionary consequences of repeated control region duplications within the Psittaciformes. In addition to characterizing the genome structure, we examined patterns of sequence evolution across lineages with and without duplicate control regions, and relate our results to the larger question of how control region duplications evolve and why they persist in parrots.
3.1 Sequencing and structure of complete mitochondrial genomes
We sequenced the complete mitochondrial genomes for 10 parrot species; in addition, nearly complete genomes were sequenced for another four species (see Table 2 for GenBank accession numbers). The latter four species were not sequenced completely due to various technical problems, including unsuccessful PCRs across certain sections and sequencing failures which we were not able to resolve using modified PCR profiles, primer redesign, or use of alternative primer pairs. In two species, Pionites leucogaster and Deroptyus accipitrinus, the problematic regions span one of the origins of control region duplication identified by Schirtzinger et al. (2012); because of uncertainty regarding the control region status of P. leucogaster (Schirtzinger et al., 2012), it was omitted from the evolutionary rate and base composition analyses. As of November, 2012, there were 12 additional complete or nearly complete parrot genome sequences available in GenBank and these were included in our analyses of control region structure, rates of sequence evolution and base composition (Table 2); five more sequences were available by mid-2013, and are included in Table 2 and Supplemental Table 2, but not included in our analyses.
No unexpected frame-shift mutations were found within the protein-coding regions, with the exception of a single 1 bp mutation in ND3, which was found in all parrot taxa, and which has been reported in many birds (Mindell et al., 1998b). In a few cases, 3 bp indels were found, indicating the gain/loss of an amino acid, as expected for a coding sequence. Such indels were observed in ATP8 (Cacatua moluccensis), CO1 (Coracopsis vasa), cytb (Brotogeris cyanoptera), ND4 (Pionites leucogaster), and ND5 (Deroptyus accipitrinus, Forpus modestus, Pionites leucogaster, Psittacus erithacus); ND6 was highly variable in length, and though all taxa had an ATG start codon, three different stop codons (TAA, TAG, and AGG) were found, all of which have been noted in other avian taxa (Slack et al., 2003). The sizes and start/stop codons for the 13 mitochondrial protein-coding regions are summarized in Supplemental Table 2.
Five of the species that we sequenced (Amazona ochrocephala, Deroptyus accipitrinus, Forpus passerinus, Prioniturus luconensis, and Psittacus erithacus) and one from GenBank (Melopsittacus undulatus) have rearranged genomes with duplicate control regions (Schirtzinger et al., 2012). In most of these, the gene order around the control regions was the same as that reported by Eberhard et al. (2001) for Amazona, with the duplicate control region copy inserted between tRNA-Thr and tRNA-Pro, but with some minor variation due to the presence of pseudogenes in some taxa (Figure 2). The only exception was Prioniturus luconensis, whose gene order retains functional copies of tRNA-Thr and tRNA-Pro between cytb and CR1. In P. luconensis, the pGlu sequence forms a cruciform structure in tRNASCAN-LE (Lowe and Eddy, 1997), but a TTT anticodon is predicted (corresponding to tRNA-Lys), whereas the putative tRNA-Glu sequence also folds appropriately and predicts the TTC anticodon specific to tRNA-Glu. Due to PCR and sequencing difficulties, we were unable to confirm the gene order around the control region for Deroptyus accipitrinus, but limited sequence data reported in Schirtzinger et al. (2012) suggest that it has a potentially functional tRNA-Pro between tRNA-Thr and CR1. Based on PCR fragment length analyses (Schirtzinger et al., 2012), D. accipitrinus was considered to have two control regions for the analyses reported here.
Both of the rearrangements that we found can be explained by a tandem duplication of the section of the genome between cyt b and 12S, followed by loss of the duplicate coding genes and tRNAs (Bensch and Härlid, 2000)(see Figure 1). Gibb et al. (2007) proposed a framework for naming gene orders, but the scheme does not provide a way to differentiate between the two gene orders that occur in parrots, so we have called them “duplicate control region gene order I” (found in most parrots to date) and “duplicate control region gene order II” (found in P. luconensis). The fact that the independently derived genome rearrangements in parrots, as well as those reported for other birds, are all consistent with the tandem duplication model illustrated in Figure 1 suggests that a single mechanism is likely responsible the origins of avian genome rearrangements. Following such a rearrangement event, stochasticity in the loss of duplicated paralogs could result in the different gene orders observed in different lineages.
In parrots, mitochondrial genome rearrangements consistently result in the maintenance of duplicate control regions, while other duplicated segments of DNA (e.g., ND6 and tRNAs) degenerate and either disappear or persist as nonfunctional pseudogenes (present results and Schirtzinger et al., 2012). This pattern suggests that there is some selective advantage to having a duplicate control region, but not to having duplicate coding regions. Some potential advantages to having duplicate control regions have been suggested; these include a replicative advantage for the mtDNA genome (Kumazawa et al., 1996), control region involvement in transcriptional control of adjacent tRNAs (Arndt and Smith, 1998), and protection against age-related deterioration of mitochondrial function (Wright et al., 2011). Interestingly, a general pattern in other birds is that following genome reorganization, if one of the duplicate control regions shows signs of degeneration or loss, it is always CR2 (for examples, see Remnant CR2 taxa in Table 1). This is surprising, since CR2 is the copy that is in the ancestral control region position between tRNA-Glu and tRNA-Phe.
3.2 Structure of the Control Regions
The lengths of parrot control regions vary across taxa, as do the lengths of the spacers that lie 5’ of the poly-C section (Supplemental Table 3). Lengths of control regions located between tRNA-Glu and tRNA-Phe range from 1117 bp (Psittacus erithacus CR2) to 1867 bp (Brotogeris cyanoptera), with an average length of 1383 bp ± 177 bp (mean ± standard deviation). The average 5’ spacer length is 21 bp ± 13 bp. (Note: these statistics do not include Neophema chrysogaster, which has a deviant control region, as discussed below.) The fact that all of the Calyptorhynchus control regions (from three different species; White et al., 2011) are exactly the same size seems surprising, given that we found within-species variation in control region length for Melopsittacus (one individual sequenced by Guan et al., and a second for this study; see Table 2 and Supplemental Table 3). Duplicated control region lengths (CR1 in genomes with two control regions) are slightly longer, ranging from 1307 bp (Melopsittacus undulatus) to 1756 bp (Prioniturus luconensis), with an average length of 1533 bp ± 213 bp and an average 5’ spacer length of 139 bp ± 107 bp. For genomes with two control regions, if only the control region sensu stricto lengths (without the 5’ spacer) are considered, the mean lengths of CR1 and CR2 are not significantly different (paired t-test: t=0.590, P = 0.5766, d.f.=6). In some cases, pseudogenes could be detected in the 5’ spacer regions, or in the 3’ end of CR1, but in other cases no pseudogenes could be detected (see Figure 2). Descriptive data for the control region sequences are summarized in Supplemental Table 3.
A poly-C sequence was found near the 5’ end of all parrot control regions. In most taxa, the sequence was CCCCCCCTACCCCCCC; a few taxa had TT in the middle, and a few differed slightly in the number of Cs. Only one species, Neophema chrysogaster (Miller et al., 2013) has two poly-C sequences—one near the 5’ end of the control region, and a second near the middle, in each case at the 5' end of a 242 bp section that was duplicated exactly. In this species another 341 bp section was also duplicated within the control region, and between these repeated sections there is a 60 bp segment tandemly repeated six times. This combination of features deviates strongly from all other available parrot control region sequences, and we suspect they may be artifacts of the assembly process (it was sequenced using next generation sequencing and de novo assembly techniques) (Miller et al., 2013); for this reason N. chrysogaster was not included in the summary statistics above.
The sliding window analysis of three sets of closely related species reveals a consistent pattern of sequence variation along the control region, making it possible to roughly divide the control region into three domains that reflect different levels of variability, as observed in other birds (Baker and Marshall, 1997). Domain I is moderately variable, the central Domain II is the most conserved, and Domain III is highly variable (see Figure 4a). Within Domain III, most parrot control regions have a repeat region that contains tandem repeats of motifs that vary in length. In most cases the motifs are relatively short, ranging in size from 4 bp to 23 bp, and repeated up to 37 times. Some taxa have tandem repeats of more than one, and as many as three, different motifs. In Coracopsis vasa, an exceptionally long (60 bp) motif was found, repeated seven times. The control regions of closely related species (i.e., congeners) are alignable over Domains I and II, but the alignment breaks down in Domain III, often in the repeat region (see Figure 4a).
Fig. 4.
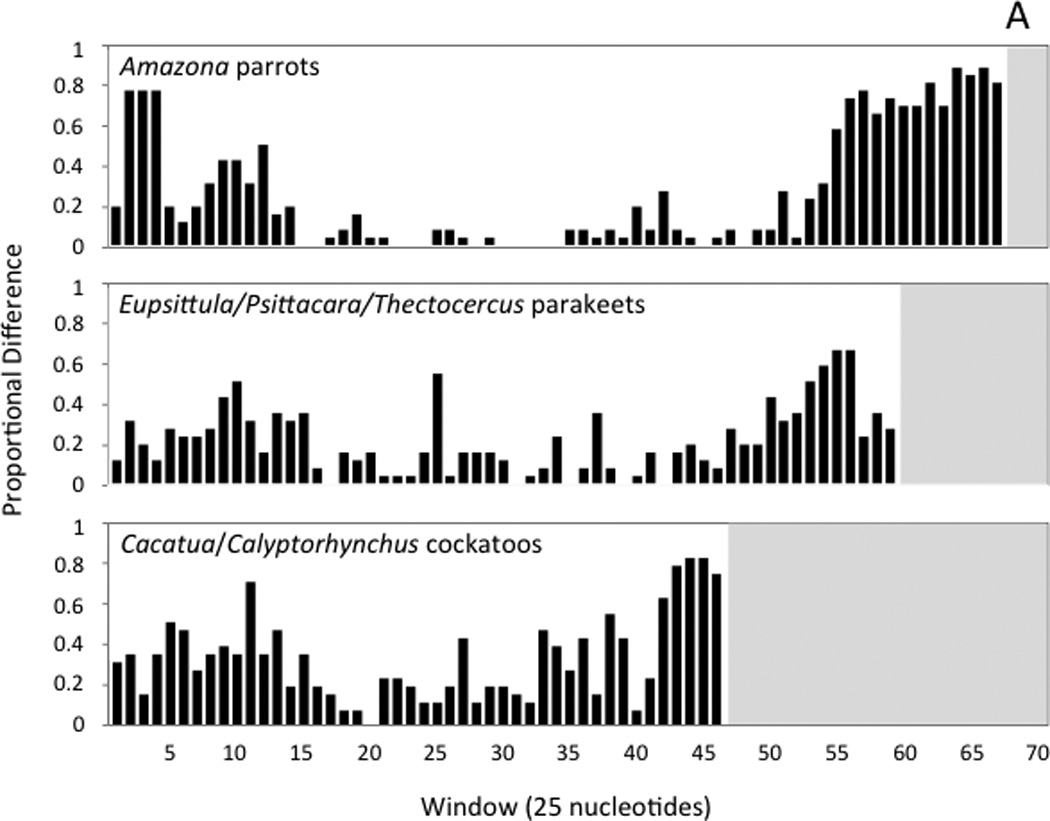
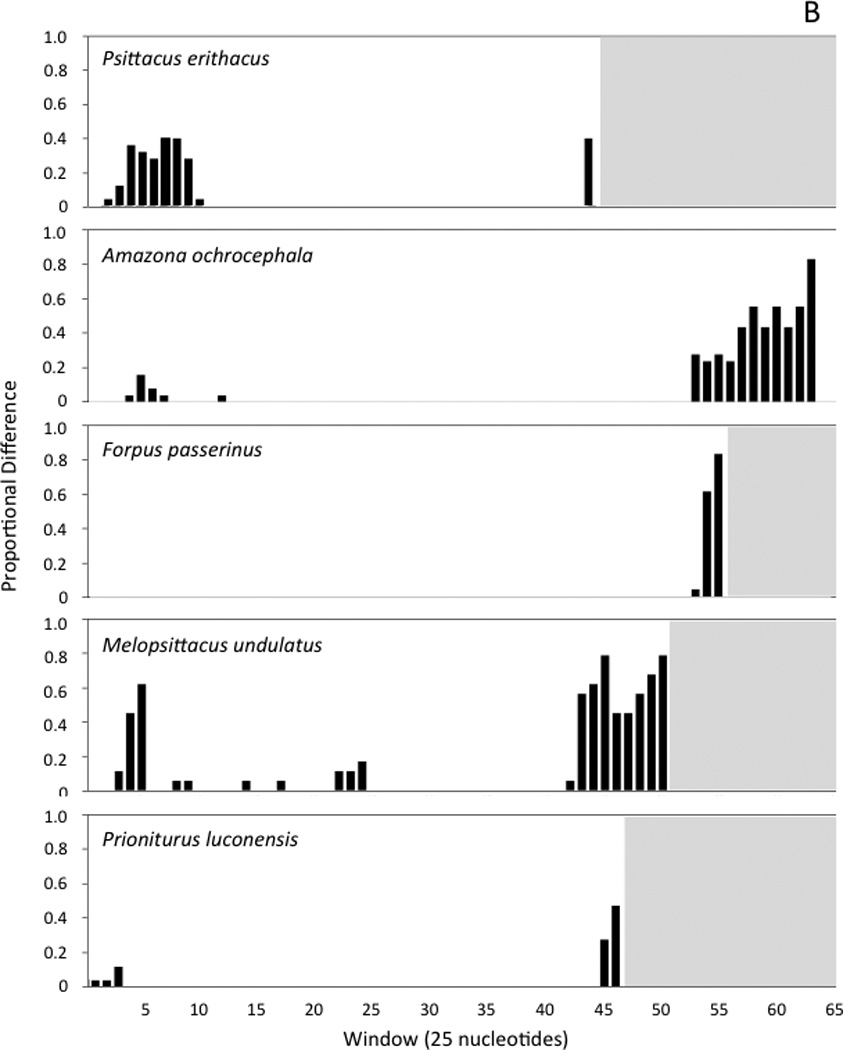
Histograms showing the number variable nucleotide positions as a proportion of the changes possible in each 25-nucleotide window of the sliding-window analysis. Windows are numbered, with the first window starting at the poly-C sequence near the 5’ end of the control region. Panel A shows the results of sliding window analyses performed with the control regions of three sets of closely related taxa (see text). Panel B shows the comparisons of the two control region copies found within individual genomes for five taxa that have duplicate control regions. In each histogram, the white background indicates the interval spanned by a given control region; since control regions differ in length, the number of 25-nucleotide windows is different in each case.
In all nine parrot taxa with duplicate control regions, both control regions appear to be complete, or at least the lengths of both control regions are similar to each other and to the lengths of control regions in single-control region taxa (see previous section). We used a sliding window analysis to examine patterns of sequence divergence between control region copies within taxa with duplications (Figure 4b). Duplicate control regions can be easily aligned to each other, though the alignment breaks down to varying degrees in Domain III (Figure 4b; histograms for Amazona ochroephala oratrix, A. o. auropalliata, A. barbadensis, and A. farinosa are not shown, but are qualitatively similar to A. o. ochrocephala). Similarity scores between control region copies (i.e., within-genome comparison of control regions) for the section stretching from the poly-C sequence near the beginning of Domain I through the point at which the alignment breaks down in Domain III are very high, ranging from 94.8% (Psittacus erithacus) to 100% (Forpus passerinus). In most taxa, the unalignable section begins within the tandem repeat region, though the 3’ ends of control regions are also unalignable in species without detectable repeat regions (i.e., Psittacus erithacus and Prioniturus luconensis). Psittacus erithacus diverges from this general pattern, with mismatches concentrated in Domain I rather than Domain III (Figure 4b).
The fact that sequence variation between control regions in different taxa (paralogs) mirrors the variation observed between duplicate copies within an individual (orthologs), and the high similarity between orthologs (at least in Domains I and II), indicate that the duplicate copies do not evolve independently following a duplication event. One simple explanation for high sequence similarity between control region orthologs could be that the duplications in each species of parrot are recently derived, but two observations suggest that this is not the case. First, if the duplications were recent, equally high sequence similarity would be expected across the duplicated section (not only in the duplicate control regions); and second, Schirtzinger et al.’s analyses (2012) indicate that in at least two of the clades with rearranged genomes, the rearrangements are clearly not recent; the clade of taxa with duplicate control regions that includes M. undulatus diverged from its nearest relatives at least 28 Mya, and the clade containing P. erithacus split off from its nearest relatives at least 33 Mya (Wright et al., 2008).
Alternatively, the similarity between paralogs could reflect concerted evolution of the duplicate control regions, possibly via a mechanism such as gene conversion. Although this mechanism was proposed to explain concerted evolution of snake mitochondrial genomes (Kumazawa et al., 1996, see also Tatarenkov and Avise, 2007), there is to date no direct evidence of its action. Concerted evolution could be driven by selection on control region functionality, as first suggested by Kumazawa et al. (1996). In parrots, this hypothesis is consistent with the fact that in all genomes examined, the least sequence divergence between control region copies is observed in Domain II, which is responsible for the formation of the 3-strand displacement loop during mtDNA replication according to the prevailing strand-displacement model (Clayton, 1991; Brown et al., 2005; but see Reyes et al., 2005) and contains several conserved sequence blocks that have been noted in both birds and mammals (Eberhard et al., 2001; Marshall and Baker, 1997; Randi and Lucchini, 1998; Ruokonen and Kvist, 2002). The hypothesis of selection on control region functionality is also consistent with the previously noted observation that, across all instances of mitochondrial genome rearrangement in parrots (and in many other avian taxa), the duplicate control region copies are maintained even as duplicate copies of tRNAs and coding genes show signs of degeneration.
3.3 Rates of evolution
We did not find evidence that the presence of a duplicate control region is associated with a consistent change in the rate of sequence evolution in the mitochondrial genome. Using the concatenated mitochondrial gene dataset of RY-coded sequences, we compared branch lengths between phylogenetically matched pairs of taxa that differ in control region number, and found no difference across the six pairs (paired t-test: t = 0.12, P = 0.910, d.f. = 5); the same was true for branch lengths estimated using four-fold degenerate sites (paired t-test: t = 0.20, P = 0.850, d.f. = 5). This indicates that genomes with a duplicate control region do not show significantly increased or decreased rates of sequence evolution across the entire mitochondrial genome. The same pattern was observed when we examined rates, from one protein coding gene at a time using the both RY-coded sequences and four-fold degenerate sites; for all genes, comparison of branch lengths across the six taxon pairs showed no consistent acceleration or deceleration of evolutionary rate associated with control region duplication (none of the statistical tests indicated significant differences, even before applying a Bonferroni correction; see Supplemental Table 4). When we took the taxon pairs one at a time, and compared branch lengths calculated across the different coding genes, we found that the rate of sequence evolution differed significantly between single- and dual-control region lineages for four of the six taxon pairs (Table 3), but there is not a consistent association between control region number and increased evolutionary rate: in two pairs the species with duplicated control regions had significantly longer gene branch lengths, but in the other two pairs, the species with single control regions had longer branch lengths. Concordant results were obtained in analyses of both RY-coded sequences and four-fold degenerate sites (Table 3). Interestingly, in three of the four taxon pairs that differed significantly in evolutionary rate (pairs 3, 4, and 6), the faster-evolving lineage was the one represented by a smaller-bodied species. This pattern suggests that the differences in evolutionary rate that we found might be due to, or at least confounded by, body size effects. We further investigated this hypothesis below.
Table 3.
Evolutionary rate (branch length) comparisons between phylogenetically paired taxa with single versus duplicate control regions. For each taxon pair shown in Figure 3, a paired t-test (two-tailed) was used to compare branch lengths across 12 mitochondrial protein coding genes; ND6 sequence data for one taxon (D. accipitrinus) was not available, so only the twelve H-strand protein coding genes were included in the comparisons. Branch lengths were estimated using RY-coded sequence data and four-fold degenerate (4fd) sites for each of the genes. Comparisons that showed a significant difference between paired taxa across the 12 coding genes are highlighted in bold type, and the directionality of the difference between the rate of sequence evolution in the taxon with a single control region (1CR) and the taxon with the duplicate control region (2CR) is indicated.
| Taxon pair | t-statistic | P value | d. f. | Rate comparison | |
|---|---|---|---|---|---|
| RY-coding | 1 | −3.323 | 0.007 | 11 | 1CR > 2CR |
| 2 | 0.975 | 0.350 | 11 | n. s. | |
| 3 | 2.840 | 0.016 | 11 | 2CR > 1 CR | |
| 4 | 4.447 | 0.001 | 11 | 2CR > 1 CR | |
| 5 | −1.153 | 0.154 | 11 | n. s. | |
| 6 | −4.932 | 0.0004 | 11 | 1CR > 2CR | |
| 4fd sites | 1 | −2.281 | 0.043 | 11 | 1CR > 2CR |
| 2 | 1.687 | 0.120 | 11 | n. s. | |
| 3 | 2.835 | 0.016 | 11 | 2CR > 1 CR | |
| 4 | 2.930 | 0.014 | 11 | 2CR > 1 CR | |
| 5 | −0.405 | 0.694 | 11 | n. s. | |
| 6 | −2.521 | 0.028 | 11 | 1CR > 2CR | |
When we expanded our branch length analyses to the large taxon sample from Schirtzinger et al. (2012), only one pair of differed in rates of mitochondrial sequence evolution (see Supplemental Figure 1), but only for the ND2 dataset (Supplemental Table 5). We also detected a significant difference in rate for the two nuclear loci, TROP and TGFB2 for the same pair of clades. However, the rate differences were not consistent—TGFB2 evolves more quickly in the Agapornis et al. clade than in Clade F, but the reverse is true for TROP. In all other pairs of clades, there was no difference in the rate of sequence evolution for either locus (Supplemental Table 5).
The analysis of evolutionary rate with respect to body size showed that in phylogenetically matched terminal taxon pairs, larger-bodied parrots are associated with shorter branch lengths (Sign test: P = 0.015, n = 27), indicating that they have significantly lower rates of mitochondrial (ND2) sequence evolution than smaller-bodied parrots. We found no evidence of a similar effect of body size on the rate of nuclear (TGFB2+TROP) sequence evolution (Sign test: P = 0.10 n = 23). This is consistent with other studies that have found slower rates of mitochondrial sequence evolution in large-bodied taxa for birds (Eo et al., 2011; Nunn and Stanley, 1998; Thomson et al., 2014). A similar body size effect has been observed in mammals, for which multiple regression analyses found that the best predictor the rate of mitochondrial sequence evolution is lifespan, which is correlated with body size (Welch at al., 2008). This may also be the case of parrots, since larger-bodied parrots have longer lifespans (Young et al., 2011). While this pattern could at least partially explain our observation of accelerated mitochondrial sequence evolution in Myiopsitta monachus (mean body weight 120 g) relative to Amazona ochrocephala (440 g), Forpus passerinus (23 g) relative to Ara militaris (1134 g), and Psittacus erithacus (mean body weight 333 g) relative to Coracopsis vasa (525 g), it does not account for the high rate of evolution in Agapornis roseicollis (55 g) relative to Melopsittacus undulatus (29 g). Further analysis would be required to examine the relative effects of body size and life span on rates of sequence evolution in parrots.
3.4 Base Composition
The GC content of 20 complete parrot mitochondrial genomes ranges from 43.2% (Strigops habroptilus) to 48.9% (Agapornis roseicollis). If only the 12 H-strand coding genes are considered, GC content ranges from 42.9% (S. habroptilus) to 49.5% (A. roseicollis). For the complete genomes, AT skew ranges from 0.108 (Cacatua moluccensis) to 0.195 (Tanygnathus lucionensis) with a mean of 0.145, and GC skew ranges from −0.367 (C. moluccensis) to −0.438 (Prioniturus luconensis) with a mean of −0.405. These skew values fall within the range of whole-genome skews reported for a set of 30 avian genomes (AT skew range: 0.068 to 0.180; GC skew range: −0346 to −0.438; Kan et al., 2010). However, parrots have unusually strong compositional asymmetry in their coding genes relative to other birds: for the 13 coding genes, mean AT skew is 0.145 and the mean GC skew is −0.487, which are outliers relative to the range of skews reported for 30 avian mitochondrial genomes (AT skew range: 0.004 to 0.126; GC skew range: −0.369 to −0.465)(Kan et al., 2010). The asymmetric replication of mtDNA, in which the parental H strand is exposed to mutation while it is in a single-stranded state, is likely responsible for the strong compositional asymmetry in mitochondrial genomes (Reyes et al., 1998). Following this logic, a possible explanation for the marked nucleotide skew in parrots could be that mtDNA replication in parrots is slower than in other birds, exposing the H strand to deamination for longer periods.
The presence of a duplicate control region affects the compositional asymmetry of the H-strand coding genes, but not whole genome sequences. In the set of 20 genomes that we examined, AT skew for the entire genome is the same in parrot genomes with two control regions compared with those having a single control region (t-test: t = 0.037, P = 0.971, d. f. = 12.96); the same is true for GC skew (t-test: t = −0.94, P = 0.366, d. f. = 10.76). However, if only the H-strand coding genes are considered and all of the analyzed genomes are compared, AT skew is significantly greater in taxa with two control regions (Figure 5a; t-test: t = 2.29, P = 0.034, d.f. = 17.92), while GC skews do not differ (t-test: t = −0.94 P = 0.366, d.f. = 10.76). This pattern is not as strong when analysis is restricted to four-fold degenerate sites in the H-strand coding genes, which are presumed to be under weaker selection, with only a trend toward greater AT skew in taxa with two control regions (t-test: t = 1.76. P = 0.096, d.f. = 17.98), and no difference in GC skew (t-test: t = −0.43, P = 0.676, d.f. = 9.30). For the concatenated dataset of the H-strand coding genes, AT and GC skew are much stronger at four-fold degenerate sites compared to skews calculated for all sites (paired t-tests: t = 33.27, P < 0.0001, d.f. = 19 for AT; t = −45.50, P < 0.0001, d.f. = 19 for GC skew; Figure 5b).
The finding that skews are much greater for four-fold degenerate sites (Figure 5b) indicates that strand asymmetry in base composition is stronger at weakly constrained sites, which is thought to result from deamination of the H strand when it is in a single-stranded state during replication (Reyes et al. 1998). We also compared the nucleotide skew in taxa with single versus duplicate control regions using only the six phylogenetically matched genome pairs used in the evolutionary rates analysis (above). In this smaller dataset we did not detect significant differences in either AT skew or GC skew; however, for five of the six comparisons, AT skew was greater in the taxon with two control regions, consistent with our analysis of all genomes. Similar results were obtained for four-fold degenerate sites.
Interestingly, the AT skews for the control regions (analyzing only the control regions in the ancestral position, immediately upstream of tRNA-Phe) were all negative, in contrast to the uniformly positive skews observed in coding sequences, and they tended to be more negative for taxa with duplicate control regions (t-test: t = −2.02, P = 0.068, d.f. = 11.19). These more negative AT skews in the control regions of taxa with two control regions offset the greater skews observed in the H-strand coding sequences of those taxa, resulting in the observed lack of difference between the AT skews for complete genomes of single- and duplicate control region taxa.
Our data on the compositional asymmetry of single- and duplicate-control region taxa suggest that the presence of a duplicate control region may affect replication mechanics. The marked compositional asymmetry in parrot genomes may be indicative of slow mitochondrial genome replication in the group, and the greater AT skew in taxa with duplicate control regions could be due to slower mtDNA replication (Reyes et al., 1998). Alternatively, it could be a consequence of the H strand spending a larger amount of time in a single-stranded state (and exposed to deamination (Reyes et al. 1998)) because duplicate control regions increase the number/extent of replication initiation zones and, in turn, the number of nascent H strands per genome. Duplicate control regions could provide a replicative advantage (Holt and Reyes, 2014; Kumazawa et al., 1996), not by increasing the speed of replication, but by providing additional replication initiation zones, which could increase the number of genome replicates per template genome (Jiang et al., 2007). This would be especially advantageous if parrot mtDNA replication is particularly slow, as suggested by the unusually large nucleotide skew that we found in parrot mitochondrial genomes. One way to test this idea would be with studies to map replication initiation sites to see whether parrot mitochondrial genomes with duplicate control regions have replication initiation zones flanking both control region copies.
5. Conclusions
Our analysis of mitochondrial genome sequences from 20 parrot species found that two different gene orders with duplicate control regions have arisen in the group. Rates of sequence evolution are not consistently faster or slower in genomes with duplicate control regions, and since most of the genome rearrangements in parrots are ancient, their effect on the rate of sequence evolution is likely confounded with a significant negative correlation that we found between body size and the rate of mitochondrial sequence evolution. In contrast, we found that base compositional asymmetry is strong in parrots relative to other birds, and species with duplicated control region had increased AT skew in H-strand coding genes compared to species with single control regions. These results suggest that mtDNA replication is relatively slow in parrots, and the H strand spends more time in the single-stranded state required for genome replication. If so, then any genomic rearrangements that increased the efficiency of genome replication might be favored. Together, our results suggest the hypothesis that duplicated control regions are maintained in a functional state in many parrots because they serve to increase the number of initiation zones for mitochondrial replication and thus counteract an otherwise unusually slow rate of genome replication in this avian group.
Supplementary Material
Redrawn and annotated version of Figure 5 from Schirtzinger et al. (2012), showing the ancestral state reconstruction of mitochondrial control region duplications in the Psittaciformes. White circles indicate taxa/nodes with a single control region; black circles indicate taxa/nodes with a rearranged mitochondrial genome containing duplicate control regions; gray circles indicate unscorable or ambiguous states. Clades A–F identify clades with independently derived mitochondrial genome rearrangements, and comparisons 1a–6a identify the clades involved in analyses of evolutionary rates (see text).
Highlights.
Repeated mitochondrial genome rearrangements in parrots resulted in two different gene orders and duplicate control regions.
Control region duplications don’t affect rates of sequence evolution.
Rate of mtDNA sequence evolution is negatively correlated with body size in parrots.
Nucleotide skew in parrots is unusually strong, possibly indicating slow replication.
Duplicated control regions may be retained due to a replicative advantage.
Acknowledgments
We thank the Louisiana State University Museum of Natural Science, the National Museum of Natural History, and the Field Museum of Natural History for providing tissues that were used in this study. This work was supported by the National Institutes of Health grant number S06 GM008136 to TFW. We also thank K. Harms for writing the R code used in the sliding window analysis and D. Bailey for discussion of the analysis of rate heterogeneity. JRE would like to thank F. Sheldon and R. Brumfield for access to laboratory facilities.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbott CL, Double MC, Trueman JWH, Robinson A, Cockburn A. An unusual source of apparent mitochondrial heteroplasmy: duplicate mitochondrial control regions in Thalassarche albatrosses. Mol. Ecol. 2005;14:3605–3613. doi: 10.1111/j.1365-294X.2005.02672.x. [DOI] [PubMed] [Google Scholar]
- Albring M, Griffith J, Attardi G. Association of a protein structure of probable membrane derivation with HeLa cell mitochondrial DNA near its origin of replication. Proc. Nat. Acad. Sci. USA. 1977;74:1348–1352. doi: 10.1073/pnas.74.4.1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arndt A, Smith MJ. Mitochondrial gene rearrangement in the sea cucumber genus Cucumaria. Mol. Biol. Evol. 1998;15:1009–1016. doi: 10.1093/oxfordjournals.molbev.a025999. [DOI] [PubMed] [Google Scholar]
- Baker AJ, Marshall HD. Mitochondrial control region sequences as tools for understanding evolution. In: Mindell DP, editor. Avian molecular evolution and systematics. San Diego: Academic Press; 1997. pp. 51–83. [Google Scholar]
- Bensch S, Härlid A. Mitochondrial genomic rearrangements in songbirds. Mol. Biol. Evol. 2000;17:107–113. doi: 10.1093/oxfordjournals.molbev.a026223. [DOI] [PubMed] [Google Scholar]
- Boore JL. Animal mitochondrial genomes. Nucl. Acids Res. 1999;27:1767–1780. doi: 10.1093/nar/27.8.1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown TA, Cecconi C, Tkachuk AN, Bustamante C, Clayton DA. Replication of mitochondrial DNA occurs by strand displacement with alternative light-strand origins, not via a strand-coupled mechanism. Genes Dev. 2005;19:2466–2476. doi: 10.1101/gad.1352105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadahía L, Pinsker W, Negro JJ, Pavlicev M, Urios V, Haring E. Repeated sequence homogenization between the control region and pseudo-control regions in the mitochondrial genomes of the subfamily Aquilinae. J. Exp. Zool. B-Mol. Develop. Evol. 2009;312B:171–185. doi: 10.1002/jez.b.21282. [DOI] [PubMed] [Google Scholar]
- Castoe TA, Gu W, de Koning APJ, Daza JM, Jian ZJ, Parkinson CL, Pollock DD. Dynamic nucleotide mutation gradients and control region usage in squamate reptile mitochondrial genomes. Cyt. Genome Res. 2009;127:112–127l. doi: 10.1159/000295342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerasale DJ, Dor R, Winkler DW, Lovette IJ. Phylogeny of the Tachycineta genus of New World swallows: insights from complete mitochondrial genomes. Mol. Phylog. Evol. 2012;63:64–71. doi: 10.1016/j.ympev.2011.12.014. [DOI] [PubMed] [Google Scholar]
- Cheng S, Higuchi R, Stoneking M. Complete mitochondrial genome amplification. Nature Genetics. 1994;7:350–351. doi: 10.1038/ng0794-350. [DOI] [PubMed] [Google Scholar]
- Cho HJ, Eda M, Nishida S, Yasukochi Y, Chong JR, Koike H. Tandem duplication of mitochondrial DNA in the black-faced spoonbill Platalea minor. Genes, Genet. Syst. 2009;84:297–305. doi: 10.1266/ggs.84.297. [DOI] [PubMed] [Google Scholar]
- Clayton DA. Replication and transcription of vertebrate mitochondrial DNA. Annu. Rev. Cell Biol. 1991;7:453–478. doi: 10.1146/annurev.cb.07.110191.002321. [DOI] [PubMed] [Google Scholar]
- Clayton DA. Replication of animal mitochondrial DNA. Cell. 1982;28:693–705. doi: 10.1016/0092-8674(82)90049-6. [DOI] [PubMed] [Google Scholar]
- Cooke GM, King AG, Johnson RN, Boles WE, Major RE. Rapid characterization of mitochondrial genome rearrangements in Australian songbirds using next-generation sequencing technology. J. Hered. 2012;103:882–886. doi: 10.1093/jhered/ess091. [DOI] [PubMed] [Google Scholar]
- Darriba D, Taboada GL, Doallo R, Posada D. jModelTest2: more models, new heuristics and parallel computing. Nature Methods. 2012;9:772. doi: 10.1038/nmeth.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desjardins P, Morais R. Sequence and gene organization of the chicken mitochondrial genome: a novel gene order in higher vertebrates. J. Mol. Biol. 1990;212:599–634. doi: 10.1016/0022-2836(90)90225-B. [DOI] [PubMed] [Google Scholar]
- Dunning JG. CRC Handbook of Avian Body Masses. Boca Raton: CRC Press; 2008. [Google Scholar]
- Eberhard JR, Bermingham E. Phylogeny and biogeography of the Amazona ochrocephala (Aves Psittacidae) complex. Auk. 2004;121:318–332. [Google Scholar]
- Eberhard JR, Wright TF, Bermingham E. Duplication and concerted evolution of the mitochondrial control region in the parrot genus Amazona. Mol. Biol. Evol. 2001;18:1330–1342. doi: 10.1093/oxfordjournals.molbev.a003917. [DOI] [PubMed] [Google Scholar]
- Eo SH, Doyle JM, DeWoody JA. Genetic diversity in birds is associated with body mass and habitat type. J. Zool. 2011;283:220–226. [Google Scholar]
- Forshaw JM. Parrots of the world. 3rd ed. Melbourne: Landsdowne Editions; 1989. [Google Scholar]
- Gibb GC, Kardailsky O, Kimball RT, Braun EL, Penny D. Mitochondrial genomes and avian phylogeny: complex characters and resolvability without explosive radiations. Mol. Biol. Evol. 2007;24:269–280. doi: 10.1093/molbev/msl158. [DOI] [PubMed] [Google Scholar]
- Gissi C, Iannelli F, Pesole G. Evolution of the mitochondrial genome of Metazoa as exemplified by comparison of congeneric species. Heredity. 2008;101:301–320. doi: 10.1038/hdy.2008.62. [DOI] [PubMed] [Google Scholar]
- Guindon S, Gascuel O. A siple, fast and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- Haring E, Kurckenhauser L, Gamauf A, Riesing MJ, Pinsker W. The complete sequence of the mitochondrial genome of Buteo buteo (Aves, Accipitridae) indicates an early split in the phylogeny of raptors. Mol. Biol. Evol. 2001;18:1892–1904. doi: 10.1093/oxfordjournals.molbev.a003730. [DOI] [PubMed] [Google Scholar]
- Harrison GL, McLenachan PA, Phillips MJ, Slack KE, Cooper A, Penny D. Four new avian mitochondrial genomes help get to basic evolutionary questions in the late cretaceous. Mol. Biol. Evol. 2004;21:974–983. doi: 10.1093/molbev/msh065. [DOI] [PubMed] [Google Scholar]
- Holt IJ, Reyes A. Human mitochondrial DNA replication. Cold Spring Harb. Perspect. Biol. 2014;4:a012971. doi: 10.1101/cshperspect.a012971. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang ZJ, Castoe TA, Austin CC, Burbrink FT, Herron MD, McGuire JA, Parkinson CL, Pollock DD. Comparative mitochondrial genomics of snakes: extraordinary substitution rate dynamics and functionality of the duplicate control region. BMC Evol. Biol. 2007;7:123. doi: 10.1186/1471-2148-7-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kan XZ, Li XF, Zhang LQ, Chen L, Qian CJ, Zhang XW, Wang L. Characterization of the complete mitochondrial genome of the Rock pigeon, Columba livia (Columbiformes: Columbidae) Genet. Mol. Res. 2010;9:1234–1249. doi: 10.4238/vol9-2gmr853. [DOI] [PubMed] [Google Scholar]
- Kumazawa Y, Ota H, Nishida M, Ozawa T. Gene rearrangements in snake mitochondrial genomes: highly concerted evolution of control-region-like sequences duplicated and inserted into a tRNA gene cluster. Mol. Biol. Evol. 1996;13:1242–1254. doi: 10.1093/oxfordjournals.molbev.a025690. [DOI] [PubMed] [Google Scholar]
- Kumazawa Y, Ota H, Nishida M, Ozawa T. The complete nucleotide sequence of a snake (Dinodon semicarinatus) mitochondrial genome with two identical control regions. Genetics. 1998;150:313–329. doi: 10.1093/genetics/150.1.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kück P, Meusemann K. FASconCAT: Convenient handling of data matrices. Mol. Phylogenet. Evol. 2010;56:1115–1118. doi: 10.1016/j.ympev.2010.04.024. [DOI] [PubMed] [Google Scholar]
- Lavrov DV. Key transitions in animal evolution: a mitochondrial DNA perspective. Integ. Comp. Biol. 2007;47:734–743. doi: 10.1093/icb/icm045. [DOI] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucl. Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, Koskella B, Schaack S. Mutation pressure and the evolution of organelle genomic architecture. Science. 2006;311:1727–1730. doi: 10.1126/science.1118884. [DOI] [PubMed] [Google Scholar]
- Marshall HD, Baker AJ. Structural conservation and variation in the mitochondrial control region of fringilline finches (Fringilla spp.) and the Greenfinch (Carduelis chloris) Mol. Biol. Evol. 1998;14:173–184. doi: 10.1093/oxfordjournals.molbev.a025750. [DOI] [PubMed] [Google Scholar]
- Miller AD, Good RT, Coleman RA, Lancaster ML, Weeks AR. Microsatellite loci and the complete mitochondrial DNA sequence characterized through next generation sequencing and de novo genome assembly for the critically endangered orange-bellied parrot, Neophema chrysogaster. Mol. Biol. Rep. 2013;40:35–42. doi: 10.1007/s11033-012-1950-z. [DOI] [PubMed] [Google Scholar]
- Mindell DP, Sorenson MD, Dimcheff DE. Multiple independent origins of mitochondrial gene order in birds. Proc. Natl. Acad. Sci. USA. 1998a;95:10693–10697. doi: 10.1073/pnas.95.18.10693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mindell DP, Sorenson MD, Dimcheff DE. An extra nucleotide is not translated in mitochondrial ND3 of some birds and turtles. Mol. Biol. Evol. 1998b;15:1568–1571. doi: 10.1093/oxfordjournals.molbev.a025884. [DOI] [PubMed] [Google Scholar]
- Miyaki CY, Matioli SR, Burke T, Wajntal A. Parrot evolution and paleogeographical events: mitochondrial DNA evidence. Mol. Biol. Evol. 1998;15:544–551. [Google Scholar]
- Moorhouse RJ, Powesland RG. Aspects of the ecology of kakapo Strigops habroptilus liberated on Little Barrier Island (Hauturu) N. Z. Biol. Cons. 1991;56:349–365. [Google Scholar]
- Morris-Pocock JA, Taylor SA, Birt TP, Friesen VL. Concerted evolution of duplicated mitochondrial control regions in three related seabird species. BMC Evol. Biol. 2010;10:14. doi: 10.1186/1471-2148-10-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunn GB, Stanley SE. Body size effects and rates of cytochrome b evolution in tube-nosed seabirds. Mol. Biol. Evol. 1998;15:1360–1371. doi: 10.1093/oxfordjournals.molbev.a025864. [DOI] [PubMed] [Google Scholar]
- Pacheco MA, Battistuzzi FU, Lentino M, Aguilar R, Kumar S, Escalante AA. Evolution of modern birds revealed by mitogenomics: timing the radiation and origin of major orders. Mol. Biol. Evol. 2011;28:1927–1942. doi: 10.1093/molbev/msr014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palumbi SR. Nucleic acids II: The polymerase chain reaction. In: Hillis DM, Moritz C, Mable BK, editors. Molecular Systematics. 2nd ed. Sunderland: Sinauer Associates; 1996. pp. 205–247. [Google Scholar]
- Perna NT, Kocher TD. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995;41:353–358. doi: 10.1007/BF00186547. [DOI] [PubMed] [Google Scholar]
- Phillips MJ, Delsuc F, Penny D. Genome-scale phylogeny and the detection of systematic biases. Mol. Biol. Evol. 2004;21:1455–1458. doi: 10.1093/molbev/msh137. [DOI] [PubMed] [Google Scholar]
- Pratt RC, Gibb GC, Morgan-Richards J, Phillips MJ, Hendy MD, Penny D. Toward resolving deep neoaves phylogeny: data, signal enhancement, and priors. Mol. Biol. Evol. 2009;26:313–326. doi: 10.1093/molbev/msn248. [DOI] [PubMed] [Google Scholar]
- Quinn TW, Wilson AC. Sequence evolution in and around the mitochondrial control region in birds. J. Mol. Evol. 1993;37:417–425. doi: 10.1007/BF00178871. [DOI] [PubMed] [Google Scholar]
- Rand DM, Harrison RG. Mitochondrial DNA transmission in crickets. Genetics. 1986;114:955–970. doi: 10.1093/genetics/114.3.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randi E, Lucchini V. Organization and evolution of the mitochondrial DNA control region in the avian genus Alectoris. J. Mol. Evol. 1998;47:449–462. doi: 10.1007/pl00006402. [DOI] [PubMed] [Google Scholar]
- Remsen JV, Schirtzinger EE, Ferraroni A, Silveira LF, Wright TF. DNA-sequence data require revision of the parrot genus Aratinga (Aves: Psittacidae) Zootaxa. 2008;3641:296–300. doi: 10.11646/zootaxa.3641.3.9. [DOI] [PubMed] [Google Scholar]
- Reyes A, Gissi C, Pesole G, Saccone C. Asymmetrical directional mutation pressure in the mitochondrial genome of mammals. Mol. Biol. Evol. 1998;15:957–966. doi: 10.1093/oxfordjournals.molbev.a026011. [DOI] [PubMed] [Google Scholar]
- Reyes A, Yang MY, Bowmaker M, Holt IJ. Bidirectional replication initiates at sites throughout the mitochondrial genome of birds. J. Biol. Chem. 2005;280:3242–3250. doi: 10.1074/jbc.M411916200. [DOI] [PubMed] [Google Scholar]
- Richly E, Leister D. NUMTs in sequenced eukaryotic genomes. Mol. Biol. Evol. 2004;21:1081–1084. doi: 10.1093/molbev/msh110. [DOI] [PubMed] [Google Scholar]
- Roques S, Godoy A, Negro JJ, Hiraldo F. Organization and variation of the mitochondrial control region in two vulture species, Gypaetus barbatus and Neophron percnopterus. J. Hered. 2004;95:332–337. doi: 10.1093/jhered/esh047. [DOI] [PubMed] [Google Scholar]
- Rothfels CJ, Schuettpelz E. Accelerated rate of molecular evolution for Vittarioid ferns is strong and not driven by selection. Syst. Biol. 2014;63:31–54. doi: 10.1093/sysbio/syt058. [DOI] [PubMed] [Google Scholar]
- Ruokonen M, Kvist L. Structure and evolution of the avian mitochondrial control region. Mol. Phylogenet. Evol. 2002;23:422–432. doi: 10.1016/s1055-7903(02)00021-0. [DOI] [PubMed] [Google Scholar]
- Sbisà E, Tanzariello F, Reyes A, Pesole G, Saccone C. Mammalian mitochondrial d-loop region structural analysis: identification of new conserved sequences and their functional and evolutionary implications. Gene. 1997;205:125–140. doi: 10.1016/s0378-1119(97)00404-6. [DOI] [PubMed] [Google Scholar]
- Schirtzinger EE, Tavares ES, Gonzales LA, Eberhard JR, Miyaki CY, Sanchez JJ, Hernandez A, Müeller H, Graves GR, Fleischer RC, Wright TF. Multiple independent origins of mitochondrial control region duplications in the order Psittaciformes. Mol. Phylogenet. Evol. 2012;64:342–356. doi: 10.1016/j.ympev.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweizer M, Seehausen O, Güntert M, Hertwig ST. The evolutionary diversification of parrots supports a taxon pulse model with multiple trans-oceanic dispersal events and local radiations. Mol. Phylogenet. Evol. 2010;54:984–994. doi: 10.1016/j.ympev.2009.08.021. [DOI] [PubMed] [Google Scholar]
- Shadel GS, Clayton DA. Mitochondrial DNA maintenance in vertebrates. Annu. Rev. Biochem. 1997;66:409–435. doi: 10.1146/annurev.biochem.66.1.409. [DOI] [PubMed] [Google Scholar]
- Singh TR, Shneor O, Huchon D. Bird mitochondrial gene order: insight from 3 warbler mitochondrial genomes. Mol. Biol. Evol. 2008;25:475–477. doi: 10.1093/molbev/msn003. [DOI] [PubMed] [Google Scholar]
- Slack KE, Janke A, Penny D, Arnason U. Two new avian mitochondrial genomes (penguin and goose) and a summary of bird and reptile mitogenomic features. Gene. 2003;302:43–52. doi: 10.1016/s0378111902010533. [DOI] [PubMed] [Google Scholar]
- Slack KE, Jones CM, Ando T, Harrison GL, Fordyce RE, Arnason U, Penny D. Early penguin fossils, plus mitochondrial genomes, calibrate avian evolution. Mol. Biol. Evol. 2006;23:1144–1155. doi: 10.1093/molbev/msj124. [DOI] [PubMed] [Google Scholar]
- Slack KE, Delsuc F, Mclenachan PA, Arnason U, Penny D. Resolving the root of the avian mitogenomic tree by breaking up long branches. Mol. Phylogenet. Evol. 2007;42:1–13. doi: 10.1016/j.ympev.2006.06.002. [DOI] [PubMed] [Google Scholar]
- Sorenson MD, Ast JC, Dimcheff DE, Yuri T, Mindell DP. Primers for a PCR-based approach to mitochondrial genome sequencing in birds and other vertebrates. Mol. Phylogenet. Evol. 1999;12:105–114. doi: 10.1006/mpev.1998.0602. [DOI] [PubMed] [Google Scholar]
- Sorenson MD. Avian mtDNA primers. 2003 http://people.bu.edu/msoren/primers.html. [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA 5: Molecular Evolutionary Genetics Analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatarenkov A, Avise JC. Rapid concerted evolution in animal mitochondrial DNA. Proc. R. Soc. B. 2007;274:1795–1798. doi: 10.1098/rspb.2007.0169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavares ES, Yamashita C, Miyaki CY. Phylogenetic relationships among some Neotropical parrot genera (Psittacidae) based on mitochondrial sequences. Auk. 2004;121:230–242. [Google Scholar]
- Thomson CE, Gilbert JDJ, Brooke MD. Cytochrome b divergence between avian sister species is linked to generation length and body mass. PLoS ONE. 2014;9(2):e85006. doi: 10.1371/journal.pone.0085006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urantowka AD, Grabowski KA, Strzala T. Complete mitochondrial genome of Blue-crowned Parakeet (Aratinga acuticaudata)—phylogenetic position of the species among parrots group called Conures. Mitochondrial DNA. 2013a;24:336–338. doi: 10.3109/19401736.2012.760080. [DOI] [PubMed] [Google Scholar]
- Urantowka AD, Hajduk K, Kosowska B. Complete mitochondrial genome of endangered Yellow-shouldered Amazon (Abazona barbadensis): Two control region copies in parrot species of the Amazona genus. Mitochondrial DNA. 2013a;24:411–413. doi: 10.3109/19401736.2013.766177. [DOI] [PubMed] [Google Scholar]
- Urantowka AD, Kroczak AM, Strzala T. Complete mitochondrial genome of endangered Socorro Conure (Aratinga brevipes)—taxonomic position of the species and its relationship with Green Conure. Mitochondrial DNA. 2013c doi: 10.3109/19401736.2013.803095. [DOI] [PubMed] [Google Scholar]
- Urantowka AD, Strzala T, Grabowski KA. Complete mitochondrial genome of endangered Maroon-fronted Parrot (Rhynchopsitta terrisi)—conspecific relation of the species with Thick-billed parrot (Rhynchopsitta pachyrhyncha) Mitochondrial DNA. 2013d doi: 10.3109/19401736.2013.809440. [DOI] [PubMed] [Google Scholar]
- Verkuil YI, Piersma T, Baker AJ. A novel mitochondrial gene order in shorebirds (Scolopacidae, Charadriiformes) Mol. Phylogenet. Evol. 2010;57:411–416. doi: 10.1016/j.ympev.2010.06.010. [DOI] [PubMed] [Google Scholar]
- Welch JJ, Bininda-Emonds ORP, Bromham L. Correlates of substitution rate variation in mammalian protein-coding genes. BMC Evol. Biol. 2008;8:53. doi: 10.1186/1471-2148-8-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White NE, Phillips MJ, Gilbert MTP, Alfaro-Nuñez A, Willerslev E, Mawson PR, Spencer PBS, Bunce M. The evolutionary history of cockatoos (Aves: Psittaciformes: Cacatuidae) Mol. Phylogenet. Evol. 2011;59:615–622. doi: 10.1016/j.ympev.2011.03.011. [DOI] [PubMed] [Google Scholar]
- Wright TF, Schirtzinger EE, Matsumotos T, Eberhard JR, Graves GR, Sanchez JJ, Capelli S, Müller H, Scharpegge J, Chambers GK, Fleischer RC. A multilocus molecular phylogeny of the parrots (Psittaciformes): support for a Gondwanan origin during the Cretaceous. Mol. Biol. Evol. 2008;25:2141–2156. doi: 10.1093/molbev/msn160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright TF, Schirtzinger EE, Young AM, Hobson EA, Eberhard JR. Mitochondria and aging: do duplicated mtDNA control regions confer longer lifespans in parrots? Society for Integrative and Comparative Biology, Salt Lake City, UT. Integrative and Comparative Biology. 2011;51(S1):E154. [Google Scholar]
- Young AM, Hobson EA, Bingaman Lackey L, Wright TF. Survival on the ark: life-history trends in captive parrots. Animal Conservation. 2011 doi: 10.1111/j.1469-1795.2011.00477.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng C, Nie L, Wang J, Zhou H, Hou H, Wang H, Liu J. Recombination and evolution of duplicate control regions in the mitochondrial genome of the Asian bigheaded turtle, Playsternon megacephalum. PLOS ONE. 2013;8(12):e82854. doi: 10.1371/journal.pone.0082854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Lin Q, Fang W, Chen X. The complete mitochondrial genomes of sixteen ardeid birds revealing the evolutionary process of the gene rearrangements. BMC Genomics. 2014;15:573. doi: 10.1186/1471-2164-15-573. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Redrawn and annotated version of Figure 5 from Schirtzinger et al. (2012), showing the ancestral state reconstruction of mitochondrial control region duplications in the Psittaciformes. White circles indicate taxa/nodes with a single control region; black circles indicate taxa/nodes with a rearranged mitochondrial genome containing duplicate control regions; gray circles indicate unscorable or ambiguous states. Clades A–F identify clades with independently derived mitochondrial genome rearrangements, and comparisons 1a–6a identify the clades involved in analyses of evolutionary rates (see text).